

Abstract
Thalassemia free survival after allogeneic stem cell transplantation (SCT) is about 80–90% with either matched related or unrelated donors. However, the probability of finding a HLA-compatible donor is less than 50%. We explored the use of a mismatched related (“Haplo-”) donor. All patients received two courses of pre-transplant immunosuppression therapy (PTIS) with fludarabine (Flu) and dexamethasone (Dxm) to facilitate engraftment. After two courses of PTIS, a reduced-toxicity conditioning regimen of rabbit anti-thymocyte globulin (ATG), Flu, and IV Busulfan (Bu) was given followed by T-cell replete peripheral blood progenitor cells (PBPC). GVHD prophylaxis consisted of cyclophosphamide (Cy) on days SCT +3 and +4 (Post-Cy), and on day SCT +5 tacrolimus or sirolimus was started together with a short course of mycophenolate mofetil. Thirty-one patients underwent haplo-SCT. Their median age was ten years (range, 2 to 20 years). Twenty-nine patients engrafted with 100% donor chimerism. Two of three patients with high titers of donor-specific anti-HLA antibodies suffered primary graft failure. Median time to neutrophil engraftment was 14 days (range, 11 to 18 days). Five patients developed mild to moderate, reversible veno-occlusive disease, while nine patients developed acute GVHD grade II, that quickly responded to steroid therapy. Only five patients developed limited chronic GVHD. Projected overall and event-free survival rates at two years are 95% and 94%, respectively. The median follow up time is 12 months (range; 7 to 33 months). This haplo-SCT protocol may yield excellent outcomes for thalassemia patients, and provide a treatment option for patients lacking a HLA-matched donor.
Introduction
Thalassemia is a hemoglobinopathy which in its more severe forms has a quite poor prognosis. Patients with severe thalassaemia commonly suffer disease-related morbidities and their survival is on average about 20 years without state of the art supportive care (1). The only curative treatment is allogeneic hematopoietic stem cell transplantation (allo-SCT) (2, 3). Allo-SCT is cost-effective compared with the conventional transfusion support and chelation therapy for severe thalassemia patients (4, 5). However, the probability of finding a histocompatible related or unrelated donor is less than 50%. These patients also have an active, or even hyperactive, immune system, and the use of chronic blood transfusions as part of standard management contribute to allo-immunization against donor-specific HLA-antigens. This translates into a high risk for both regimen-related mortality and for graft rejection, typically in the range of 5–30% even if a highly immunosuppressive, myeloablative conditioning program is used (2, 6–9).
We recently reported an alternative strategy; we hypothesized, that a pharmacological pre-transplant immunosuppressive (PTIS) program, based on fludarabine (Flu), given in combination with dexamethasone (Dxm), would immunosuppress the patients to facilitate engraftment when it was followed by a reduced-toxicity conditioning (RTC) regimen consisting of “early” rabbit anti-thymocyte globulin (ATG) and Flu with IV busulfan (Bu) to prepare high risk thalassemia patients for allo-SCT. Further, we employed a high-dose of peripheral blood progenitor cells (PBPC) rather than bone marrow to be able to consistently target a large number of CD-34+ progenitor cells in the graft. This strategy has been working well; so far all patients (n=26) who had at most a one HLA-antigen mismatched donor engrafted (10, 11), and ultimately it resulted in an event-free survival (EFS) of over 90%. In contrast to previous reports, we found no increased risk for (serious) treatment-related complications associated with unrelated donors (10, 11). Our data indicated, that this new approach would be an improvement over the existing allo-SCT standard of care when applied with HLA-compatible donors.
In addition, there is a rapidly increasing interest in using alternative-donor stem-cell sources, primarily cord blood cells or grafts from haplo-identical related donors (Haplo-SCT). This strategy has mostly been investigated for advanced leukemia/lymphoma patients lacking matched donors. In a later development, some investigators reported excellent outcomes in patients with hematologic malignancies using various conditioning programs followed by T-cell replete/unmanipulated marrow or peripheral blood progenitor cells (15–19) and post-transplant GVHD prophylaxis based on cyclophosphamide (“Post-Cy”) (15–18).
Until now there are only two studies that reported on haplo-SCT in patients with hemoglobinopathies; in one, the investigators used reduced-intensity conditioning with Haplo-SCT and GVHD prophylaxis with post-Cy in patients with sickle cell anemia (SCA) (17). This trial had a high incidence of graft failures and unstable mixed chimerism, necessitating long-term immunosuppressive therapy. It was still deemed successful, since no patient died acutely after the conditioning or in the early post-transplant phase (17). In the second investigation, a myeloablative regimen was followed by T-cell depleted PBPC for thalassemia patients (18, 19). Both investigations reported an event-free survival (EFS) of around 40–60%, and described more than 30% graft failures. The overall conclusion was, that haplo-SCT is feasible in SCA and thalassemia.
We hypothesized, that our PTIS-based strategy could be extended to Haplo-SCT, using T-cell replete grafts, and suitably modified to include Post-Cy-based GVHD prophylaxis. This would allow a parent or mismatched sibling to serve as stem cell donor, securing access to transplant therapy for virtually all patients with severe thalassemia. We further postulated, that our modified strategy would allow consistent engraftment, yet carry a low risk for GVHD, and that it should provide long-term disease control. Initially we explored this approach to Haplo-SCT in severe thalassemia; PTIS was followed by a modified Flu-IV Bu RTC regimen (adapted from 20,21), incorporating post-Cy GVHD prophylaxis, the details are outlined in Fig. 1. This manuscript summarizes our experience with this novel approach for the first 31 consecutive, severe thalassemia patients who did not have an available HLA-matched donor. We did not exclude any patient based on age alone, nor did we exclude any class 3 patients with hepatomegaly or iron overload (Table 1).
Figure 1.
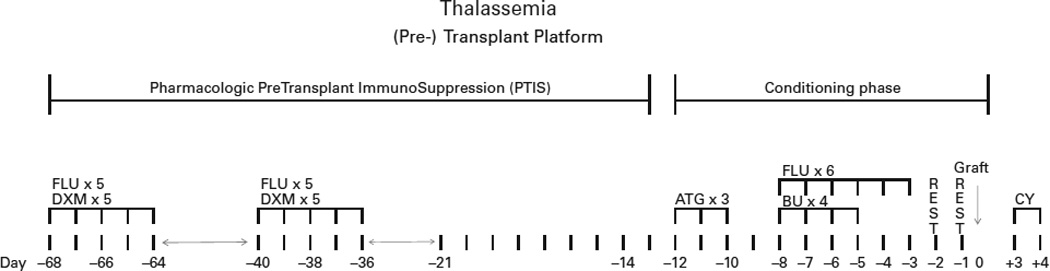
Depiction of the transplant program with first pharmacological Preconditoning Therapy with ImmunoSuppressive drugs (PTIS), followed by reduced-toxicity conditioning with ATG, Fludarabine and IV Busulfan. For the haplo-identical donors we use post-Cy based GvHD prophylaxis and delayed calcineurin inhibitor-/ sirolimus therapy and short-course MMF, the latter two starting on day SCT +5. Please see Patients and Methods section for details.
Table 1.
Patient characteristics at transplantation for 31 thalassemia patients
| UPN | Age (years) |
Sex | Donor, HLA- match |
Anti-HLA antibody |
Ferritin (ng/mL) |
Risk | CD34+ cell dose (×106/kg) |
Outcome | Chimerism (%) | F/U time (months) |
Off IS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 19.9 | F | Father, 5/10 | Negative | 1495 | Class 3 HR | 8.9 | Aa | 100% donor | 33 | Yes |
| 2 | 15.8 | F | Mother, 7/10 | Negative | 3613 | Class 3 HR | 11.6 | A | 100% donor | 28 | Yes |
| 3 | 1.7 | M | Mother, 7/10 | Negative | 1466 | Class 1 | 10.3 | A | 100% donor | 24 | Yes |
| 4 | 19.5 | M | Mother, 6/10 | Negative | 2552 | Class 3 HR | 11.6 | A | 100% donor | 23 | Yes |
| 5 | 14.3 | F | Mother, 5/10 | Negative | 2276 | Class 3 HR | 14.8 | A | 100% donor | 22 | Yes |
| 6 | 4.3 | M | Father, 7/10 | Negative | 1915 | Class 1 | 13.0 | A | 100% donor | 22 | Yes |
| 7 | 17.4 | F | Father, 5/10 | Positive | 2645 | Class 3 HR | 4.0 | Da | 100% donorb | 8 | N/A |
| 8 | 7.4 | M | Mother, 5/10 | Negative | 994 | Class 2 | 11.5 | A | 100% donor | 19 | Yes |
| 9 | 9.6 | M | Mother, 6/10 | Negative | 1599 | Class 2 | 13.0 | A | 100% donor | 19 | Yes |
| 10 | 18.6 | F | Mother, 6/10 | Positive | 3200 | Class 3 HR | 10.0 | A | 100% donor | 18 | Yes |
| 11 | 2.4 | F | Father, 6/10 | Negative | 956 | Class 1 | 15.0 | A | 100% donor | 15 | Yes |
| 12 | 7.5 | M | Mother, 5/10 | Positive | 1799 | Class 2 | 19.0 | A | Graft failure- autologous reconstitution |
15 | Rejected |
| 13 | 13.3 | M | Father, 5/10 | Negative | 3116 | Class 3 HR | 10.0 | A | 100% donor | 14 | Yes |
| 14c | 12.9 | F | Father, 5,10 | Negative | 2948 | Class 3 HR | 11.5 | A | 100% donor | 14 | Yes |
| 15 | 7.2 | M | Mother, 6/10 | Negative | 887 | Class 2 | 10.4 | A | 100% donor | 13 | Yes |
| 16 | 11.7 | M | Mother, 5/10 | Negative | 1540 | Class 3 HR | 8.5 | A | 100% donor | 12 | Yes |
| 17 | 10.8 | F | Mother, 5/10 | Negative | 2915 | Class 3 HR | 13.8 | A | 100% donor | 12 | Yes |
| 18 | 6.9 | F | Mother, 5/10 | Negative | 2487 | Class 1 | 15.0 | A | 100% donor | 11 | Yes |
| 19 | 14.2 | M | Mother, 7/10 | Negative | 1729 | Class 3 HR | 12.6 | A | 100% donor | 10 | Yes |
| 20 | 9.7 | M | Mother, 5/10 | Negative | 1756 | Class 2 | 13.0 | A | 100% donor | 10 | Yes |
| 21 | 7.3 | F | Mother, 5/10 | Negative | 1669 | Class 2 | 9.1 | A | 100% donor | 10 | Yes |
| 22 | 6.8 | F | Father, 6/10 | Negative | 1695 | Class 2 | 11.7 | A | 100% donor | 10 | Yes |
| 23 | 7.7 | F | Mother, 7/10 | Negative | 4341 | Class 2 | 12.4 | A | 100% donor | 10 | Yes |
| 24 | 9.8 | M | Father, 5/10 | Negative | 1131 | Class 3 HR | 10.6 | A | 100% donor | 9 | Yes |
| 25 | 6.6 | F | Father, 6/10 | Negative | 2894 | Class 2 | 13.9 | A | 100% donor | 9 | Yes |
| 26 | 11.0 | M | Mother, 6/10 | Negative | 1796 | Class 3 HR | 10.6 | A | 100% donor | 7 | Yes |
| 27 | 3.8 | M | Mother, 5/10 | Negative | 1460 | Class 1 | 11.6 | A | 100% donor | 7 | No |
| 28 | 2.1 | M | Father, 8/10 | Negative | 905 | Class 1 | 13.3 | A | 100% donor | 7 | No |
| 29 | 3.1 | M | Mother, 7/10 | Negative | 729 | Class 1 | 12.4 | A | 100% donor | 7 | No |
| 30 | 16.0 | F | Mother, 7/10 | Negative | 5100 | Class 3 HR | 9.2 | A | 100% donor | 7 | No |
| 31 | 15.0 | M | Father, 5/10 | Negative | 1518 | Class 3 HR | 11.5 | A | 100% donor | 7 | No |
Abbreviations: Class 3 HR = Class 3 high risk (age ≥ 7 years and hepatomegaly (liver ≥ 5 cms below right costal margin);9,22 F = female; IS = immunosuppression; M = male; N/A = not applicable.
A = Alive; D = dead.
100% donor chimerism; initial graft failure, engrafted after additional CD34 + cells were administered, please see text for details.
Haploidentical transplantation as a second transplantation.
Patients and Methods
Inclusion criteria
Severe thalassemia patients, defined as thalassemia with onset of transfusion dependence during the first 3 years of age, a pre-transfusion hemoglobin level ≤7 gm/dL, hepatosplenomegaly and thalassemia facies, and who did not have an HLA-compatible related or unrelated donor available, were eligible for treatment in this study. All severe thalassemia patients were confirmed by genotype study and had a history of blood transfusion more than 225 mL/kg/year. Therefore, both homozygous β thalassemia and β thalassemia/hemoglobin E patients who met those criteria were included in this study. The class 3 patients were classified according to the criteria proposed by Lucarelli et al (2). Patients who were ≥ 7 years old and had a liver size ≥ 5 cm below the costal margin constitute what Mathews et al. and the Center for International Blood and Marrow Transplant Research (CIMBTR) working group defined as a very high risk subset of a conventional high risk class 3 group (9, 22). This subset of class 3 high risk (HR) patients has an excessive risk of graft rejection and regimen-related toxicity, especially veno-occlusive disease (VOD), leading to multi-organ failure and death (9, 22). However, based on our favorable results using HLA-matched donors (10, 11), we decided to include classe 3 HR patients in this trial. All patients’ care givers/parents gave written informed consent to participate in this trial according to institutional guidelines.
Either parent, who did not have a clinical hemoglobinopathy, and who was generally in good health was volunteered as stem cell donor. The parent with fewest HLA-allele mismatches was favored (HLA high resolution typing; A,B,C, DRB1 and DQB1). If the degree of allele mismatch between the patient and both parents was the same, the mother was favored as donor (23, 24). Samples for donor-specific anti-HLA antibodies were obtained and analyzed from all of the patients; blood component transfusion therapy is part of conventional management for thalassemia, and frequently contributes to development of donor-specific anti-HLA antibodies that contribute to graft failure (25, 26). We did not make any modification to the program based on HLA-antibody titers, since we did not have any data supporting such alteration beyond the modifications described, including the PTIS to accommodate the patients’ generally high immunoreactivity, and that necessitated by using a T-cell replete haplo-SCT with Post-Cy. Detection of antibodies was performed using a single antigen bead assay (SAB)(Lambda, Canoga Park, CA, USA) according to the manufacturer’s instructions. The beads were analyzed on the Luminex (Which is what? This is a trade name, so please give some detail). The cut-off for a positive reaction was set at the normalized mean fluorescence intensity (MFI) value ≥ 1000.
Peripheral blood progenitor cells (PBPC) were collected after filgrastim administration as per institutional routine (27 Suradej: Is this really a good reference for PBPC-collection?). The PBPC collection target was set at 10×106 CD34+ cells/kg recipient body weight.
Pharmacological Pre-Transplant Immunosuppression (PTIS) and Reduced-Toxicity Conditioning Therapy
All high risk class 3 patients received hydroxyurea 20 mg/kg/day daily for at least three months to decrease erythroid marrow expansion, and chelation therapy for iron overload prior to starting the PTIS.
The PTIS, the RTC regimen, and all supportive care procedures have been described (10, 11). The program was modified to facilitate the use of highly HLA-disparate donors (Fig. 1) as follows:
-
Phase I: Sequential Pharmacological PTIS:
The PTIS consisted of Flu 40 mg/m2/day IV together with Dxm 25 mg/m2/day IV, each for a total of 5 days, and administered in two courses on days SCT -68 to -64 and on days SCT -40 to -36, to suppress recipient T-cell function and facilitate engraftment. Figure 1 summarizes the timing and sequence of events during the program.
-
Phase II: Reduced toxicity conditioning regimen and graft versus host disease (GVHD) prophylaxis based on post-transplant Cy:
We administered “early” antithymocyte globulin (ATG) (Thymoglobulin™; Sanofi-Genzyme Canada, Ontario, CA), 1.5 mg/kg/day on days SCT -12 to -10. The conditioning chemotherapy consisted of Flu 35 mg/m2/day IV over 60 min once daily on days SCT -8 to -3, each dose of Flu was followed by IV Bu 130 mg/m2 IV over three hours once daily on days -8 to -5. The GVHD prophylaxis consisted of Cy 50 mg/kg/day on days SCT +3 and SCT +4, and on day SCT +5 tacrolimus or sirolimus was started (total administration time: 6–12 months), together with mycophenolate mofetil (MMF), 15 mg/kg orally twice daily for 60 days.
Supportive care
All patients received penicillin V and ciprofloxacin as antibacterial prophylaxis, together with an antifungal agent, starting on day SCT +5, and continued through the end of immunosuppressive medications. Standard Pneumocystis jiroveci and anti-HSV/VZV prophylaxis was given for one year. Patients were monitored for cytomegalovirus (CMV), adenovirus and BK virus reactivation through PCR measurement of viral copy numbers in plasma and urine (Suradej: One of the reviewers asked for detail regarding the PCR-assay for CMV). Preemptive therapy with ganciclovir was initiated when CMV reactivation was detected and cidofovir was used when adenovirus or BK virus was detected in plasma, or in urine together with clinical symptoms of hemorrhagic cystitis. Patients with a viral load ≥ 1,000 copies/mL were preemptively treated as described above. Chimerism studies were performed on peripheral blood on the day of engraftment, then every two weeks till day SCT +100, monthly until one year post-transplantation and then every three months. Chimerism was measured by PCR-based analysis of variable number of nucleotide tandem repeats. Alternatively, when there was a sex-mismatch between patient and donor we employed FISH analysis of XX/XY ratios in a few patients who had a sex-mismatched donor (27).
Survival probability after transplantation was estimated as described (28). The significance of difference between survival curves was estimated by log-rank test.
Results
Thirty-one patients received haplo-SCT. The clinical characteristics of patients and donors are reported in Table 1. The patients’ median age was ten years (range, 2–20 years), and only nine were younger than seven years. Four patients had homozygous β-thalassemia and 27 had β-thalassemia/hemoglobin E. Since the incidence of β-thalassemia/hemoglobin E in Thailand and Southeast Asia is higher than homozygous β-thalassemia, most of our patients were β-thalassemia/hemoglobin E. Fifteen subjects had clinical high risk class 3 disease, their median age was 15 years (range, 10–20 years) (2, 3, 9–11, 22). One of the 31 patients had haplo-SCT as a second SCT. This patient had endured a matched related donor transplant complicated by primary graft failure with autologous marrow reconstitution three years before the current haplo-SCT (Table 1, patient UPN 14). We decided to perform haplo-SCT for this patient since her previously used matched related donor was unavailable and, likewise, there was no matched unrelated donor available. Eleven patients received PBPC from the father and twenty patients from the mother. Fifteen patients received a 5/10 HLA-allele matched graft, 8 patients received a 6/10 HLA-allele matched graft, 7 patients received a 7/10 HLA-allele matched graft and 1 patient received a 8/10 HLA-allele matched graft.
No patient developed any complications/side effects during or following the PTIS treatment phase. During this time, they were evaluated weekly in the out-patient clinic. All patients were admitted to the in-patient unit on day -13 prior to starting ATG as part of the conditioning program.
After completion of the conditioning program the T cell-replete PBPC were given. The median CD34+ cell dose was 11.6×106 cells/kg body weight (range; 4.0 to 19.0×106). The median time to neutrophil engraftment in the 29 evaluable patients was 14 days (range, 11 to 18 days). Median time to platelet engraftment of 20×109/L was 30 days (range, 20 to 45 days). All patients had donor-specific anti-HLA antibody testing performed and three of them had high antibody titers (>1:3,000), two of whom suffered primary graft failure. Both patients received additional immunosuppression with Flu and Cy followed by a boost of PBPC to facilitate engraftment. One of them engrafted successfully, but unfortunately developed grade IV GVHD and later died. The second patient had autologous marrow recovery and is alive, stable, fourteen months post allo-SCT.
Eleven patients got grade I-II mucositis and three grade III mucositis. Five patients developed mild to moderate veno-occlusive disease (VOD) (29, 30), all of which resolved with supportive care measures. Six patients had cytomegalovirus (CMV) reactivation. Four patients got BK-virus cystitis and two patients developed adenovirus cystitis. Finally, one patient reactivated herpes zoster. All viral infectious episodes resolved with therapy. Two patients had gram negative septicemia, which responded to appropriate antibiotic therapy.
Nine patients developed grade II acute GVHD that resolved with steroids, one patient had severe, grade IV, acute GVHD, following a second PBPC-infusion as described above. Five patients developed limited chronic GVHD. All surviving patients who developed GVHD were off systemic immunosuppression within 6 to 12 months after SCT.
Survival
The 2-year overall (OS) and event-free survival (EFS) were 95% (95% CI; 69.5%–99.3%) and 94% (95% CI; 76.6%–98.4%), respectively (Fig. 2). One patient died, of GVHD complications, as described above. The median follow up of surviving patients is 12 months (range; 7 to 33 months). No other patient died of complications, and we have so far not had any secondary graft failure(s).
Figure 2.
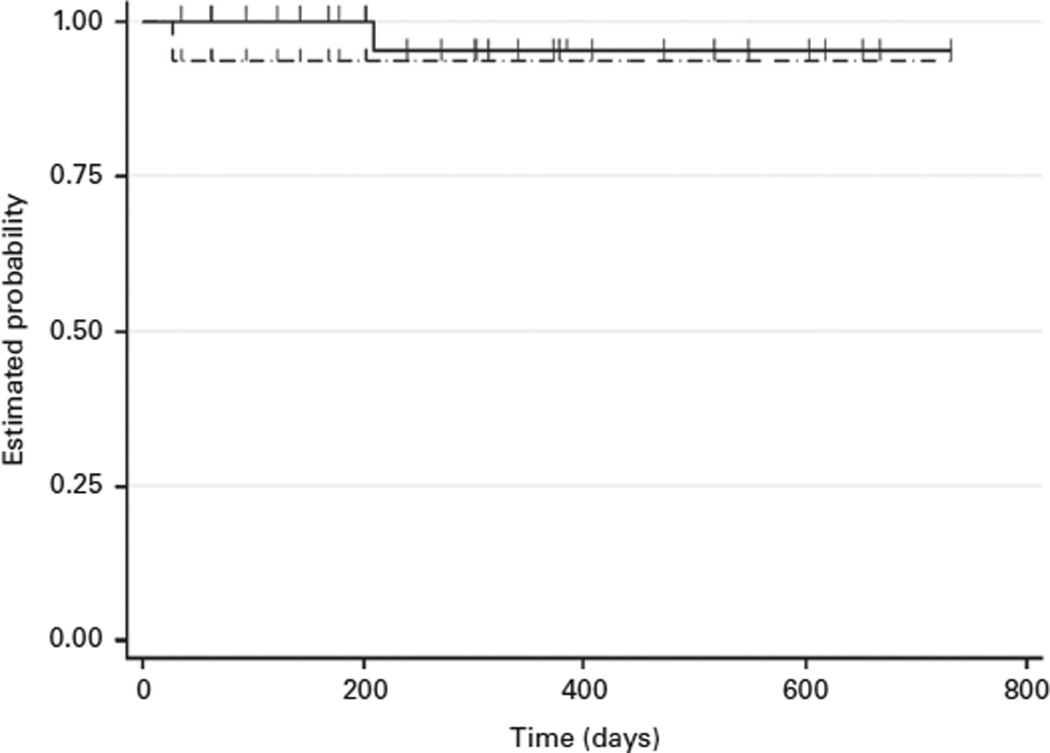
Event free survival (EFS) and Overall survival (OS) of 31 thalassemia patients undergoing haploidentical hematopoietic stem cell transplantation (Haplo-SCT)
Discussion
Allo-SCT provides the only definitive solution to the serious, long-term complications of severe Thalassemia. It is also cost-effective in view of recent developments that lower the risk for serious post-transplant complications (4, 5, 10, 11). Most of the literature reports a thalassemia-free survival after allo-SCT of Class 3 patients with complicating features, such as higher age and hepatomegaly, that ranges from about 50% to 80–90%, depending on whether matched-related or -unrelated donors were used (2, 6, 8, 9, 31–33). Early TRM typically ranges from 5 to 20% and the risk of graft failure is of a similar magnitude. Because of the risk for TRM and for graft failure in patients who have a serious, albeit non-malignant disease, we introduced a conceptually new approach; we use an initial pharmacological PTIS phase consisting of Flu and Dxm, followed by a reduced-toxicity Flu-IV Bu conditioning program (10, 11). All patients in our initial cohort who had matched related or unrelated donors engrafted, assessed by full donor chimerism within the first 60 days. The only deaths among those first 26 patients were from a fungal infection (one patient) and a motor cycle accident (one patient). Both events occurred several months after stable engraftment and we have not encountered any secondary graft failures. The conditioning regimen itself was adapted from our standard nucleoside analog-alkylating agent platform (20, 21). The excellent tolerance to the program and consistent, stable engraftment encouraged us to revise it and incorporate Post-Cy based GVHD prophylaxis to allow the use of related haplo-identical donors (12, 14). We use T-cell replete grafts, which facilitate rapid engraftment and convenient processing of the progenitor cell product. The preamble of our new strategy included that we should have less than 10% graft failures, and low TRM, preferably less than 5% in the first year post transplantation. These assumptions were adapted from the EBMT expert panel’s recommendations regarding management and treatment outcome after allo-SCT for severe thalassemia (3). Our anticipation was, that the use of an alternative donor should not decrease the program’s expected success rate. Thus, we must demonstrate “non-inferiority” when using haplo-identical- compared with HLA-matched donors, similar to what is being advocated for using alternative donors for leukemia patients (16, 34, 35). This will be necessary for the proposed strategy to be considered a valid alternative to more commonly used treatments. Further, our program demonstrates two features that contrast with what commonly accepted views regarding allo-SCT for severe thalassemia; First, older patients (more than seven years), with hepatomegaly and other comorbid conditions, who were hitherto considered as being at too high risk for TRM (9, 22), can now be safely treated even if there is only a haplo-identical related donor available. Second, the risks of graft failure and other serious complications appears no higher than what is considered acceptable for standard risk patients having a matched related donor (3, 10, 11, 32, 33). When we compared our outcomes of haplo-SCT with that of our patients transplanted with matched related or unrelated donors (10, 11), the results are very similar. Our results obtained with PTIS and Flu-IV Bu overall support the notion of a significantly decreased host-immunologic function that facilitates engraftment regardless of donor type.
We hypothesize, that the two patients who suffered primary graft failure had this event caused primarily by their high titers of donor-specific anti-HLA antibodies (25, 26). In the first patient this may have been compounded by excessive busulfan clearance, indirectly testified to by his autologous marrow recovery, which is otherwise something not typically seen at myeloablative IV Bu dosing. However, it has been described in thalassemia patients who have significantly altered Glutathione-S-Transferase(s) activity, leading to increased Bu clearance (36). The second patient got appropriately myeloablated, but received the by far lowest CD34+ cell dose in her first infusion (Table 1). She engrafted rapidly after a PBPC boost, but unfortunately developed severe, lethal, acute GVHD. We suggest that the combined effects of high titers of donor-specific HLA-antibodies and these additional factors may be responsible for primary graft failure. Prospectively we intend to implement pharmacokinetic dose-guidance of Bu, such that a consistent and reproducible systemic target exposure can be ascertained regardless of individual differences in metabolic Bu handling (7, 8, 14,37–39). Such a strategy should allow optimization of the delivered dose to assure long-term stable engraftment while minimizing the risk for excessive toxicity (38–40). We will also implement strategies to more effectively eliminate host plasma-cells and memory B-cells and to eliminate preformed anti-HLA antibodies during the PTIS phase.
In conclusion, the present study demonstrated that outcomes after Haplo-SCT in patients with severe thalassemia are acceptable. The key success factors in our new program are pharmacological pretransplant immunosuppression (PTIS), followed by a Flu-IV Bu reduced-toxicity conditioning program, supplemented with “early” ATG and Post-Cy based GVHD prophylaxis. Engraftment is further facilitated by a large number of T-cell replete PBPC. This resulted in a low incidence of serious complications, with rapid and durable engraftment, yet a low risk for serious aGVHD in this group of mostly high risk Class 3 thalassemia patients. Once the early findings are confirmed in a larger series of patients, our program design will expand the patient population that can be offered SCT by several-fold. Additionally, the program can be adapted to accommodate patients with SCA (41), and other non-malignant diseases (Hongeng et al, in progress, 2015).
Acknowledgments
This work was supported in part by grants from the Ramathibodi Foundation, the National Research University Grant, the Mahidol University Research Grant, the Office of the Higher Education Commission and Mahidol University under the National Research University Initiative, Thailand Research Fund, Research Chair Grant from the National Science and Technology Development Agency (NSTDA) and the National Institutes of Health of United States (CCSG CA16672).
Footnotes
The authors declare no financial conflict(s) of interest.
Authorship
Contribution: SH and BSA designed the research. SH, SP and UA performed the research.
SH, UA, RS and BSA analyzed the data, and performed the statistical analyses.
SH,SP,UA,RS and BSA wrote the manuscript.
NS,DS,AC,PC,AJ,KS,PR,AM,YL,PI,PS,WS,SS,PA,AU and SI enrolled the patients.
References
- 1.Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR, et al. Survival and complications in thalassemia. Ann N Y Acad Sci. 2005;1054:40–47. doi: 10.1196/annals.1345.006. [DOI] [PubMed] [Google Scholar]
- 2.Lucarelli G, Clift RA, Galimberti M, Polchi P, Angelucci E, Baronciani D, et al. Marrow transplantation for patients with thalassemia: results in class 3 patients. Blood. 1996;87:2082–2088. [PubMed] [Google Scholar]
- 3.Angelucci E, Matthes-Martin S, Baroninciani D, Bernaudin F, Bonanoni S, Cappelleni MD, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99:811–820. doi: 10.3324/haematol.2013.099747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leelahavarong P, Chaikledkaew U, Hongeng S, Kasemsup V, Lubell Y, Teerawattananon Y. A cost-utility and budget impact analysis of allogeneic hematopoietic stem cell transplantation for severe thalassemic patients in Thailand. BMC Health Serv Res. 2010;10:209–220. doi: 10.1186/1472-6963-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sruamsiri R, Chaiyakunapruk N, Pakakasama S, Sirireung S, Sripaiboonkij N, Bunworasate U, et al. Cost utility analysis of reduced intensity hematopoietic stem cell transplantation in adolescence and young adult with severe thalassemia compared to hypertransfusion and iron chelation program. BMC Health Serv Res. 2013;13:45–57. doi: 10.1186/1472-6963-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sodani P, Gaziev D, Polchi P, Erer B, Giardini C, Angelucci E, et al. New approach for bone marrow transplantation in patients with class 3 Thalassemia aged younger than 17 years. Blood. 2004;104:1201–1203. doi: 10.1182/blood-2003-08-2800. [DOI] [PubMed] [Google Scholar]
- 7.Gaziev J, Nguyen L, Puozzo C, Mozzi AF, Casella M, Perrone Donnorso M, et al. Novel pharmacokinetic behavior of intravenous busulfan in children with thalassemia undergoing hematopoietic stem cell transplantation: a prospective evaluation of pharmacokinetic and pharmacodynamics profile with therapeutic drug monitoring. Blood. 2010;115:4597–4604. doi: 10.1182/blood-2010-01-265405. [DOI] [PubMed] [Google Scholar]
- 8.Chiesa R, Capelli B, Crocchiolo R, Frugnoli L, Biral E, Noe A, et al. Unpredictability of intravenous busulfan pharmacokinetics in children undergoing hematopoietic stem cell transplantation for advanced beta thalassemia: limited toxicity with a dose-adjustment policy. Biol Blood and Marrow Transplant. 2010;16:622–628. doi: 10.1016/j.bbmt.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 9.Mathews V, George B, Deotare U, Lakshmi KM, Viswabandya A, Daniel D, et al. A new stratification strategy that identifies a subset of class III patients with an adverse prognosis among children with beta thalassemia major undergoing a matched related allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2007;13:889–894. doi: 10.1016/j.bbmt.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Anurathapan U, Pakakasama S, Rujkijyanont P, Sirachainan N, Songdej D, Chuansumrit A, et al. Pharmacologic Immunoablation followed by reduced-toxicity conditioning and stem cell transplantation in high-risk thalassemi: a safe approach to disease control. Biol Blood Marrow Transplant. 2013;19:1259–1262. doi: 10.1016/j.bbmt.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anurathapan U, Pakakasama S, Mekjaruskul P, Sirachainan N, Songdej D, Chuansumrit A, et al. Outcomes of thalassemia patients undergoing hematopoietic stem cell transplant by using a standard myeloablative versus a novel reduced toxicity conditioning regimen according to new risk stratification. Biol Blood Marrow Transplant. 2014;20:2066–2071. doi: 10.1016/j.bbmt.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luznik L, O’Donnell PV, Fuchs EJ. Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin Oncol. 2012;39:683–693. doi: 10.1053/j.seminoncol.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanakry JA, Kasamon YL, Gocke CD, Tsai HL, Davis-Sproul J, Ghosh N, et al. Outcomes of related donor HLA-identical or HLA-haploidentical allogeneic blood or marrow transplantation for peripheral T cell lymphoma. Biol Blood Marrow Transplant. 2013;19:602–606. doi: 10.1016/j.bbmt.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanakry CG, O’Donnell PV, Furlong T, de Lima MJ, Wei W, Medeot M, et al. Multi-institutional study of post-transplantation cyclophosphamide as single-agent graft-versus-host disease prophylaxis after allogeneic bone marrow transplantation using myeloablative busulfan and fludarabine conditioning. J Clin Oncol. 2014;32:3497–3505. doi: 10.1200/JCO.2013.54.0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanakry CG, Tsai HL, Bolanos-Meade J, Smith BD, Gojo I, Kanakry JA, et al. Single-agent GVHD prophylaxis with posttransplantation cyclophosphamide after myeloablative, HLA-compatible BMT for AML ALL, and MDS. Blood. 2014;124:3817–3827. doi: 10.1182/blood-2014-07-587477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raiola AM, Dominietto A, di Grazia C, Gualandi F, Ibatici A, Bregante S, et al. Unmanipulated haploidentical transplants compared with other alternative donors and matched sibling grafts. Biol Blood Marrow Transplant. 2014;20:1573–1579. doi: 10.1016/j.bbmt.2014.05.029. [DOI] [PubMed] [Google Scholar]
- 17.Bolanos-Meade J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120:4285–4291. doi: 10.1182/blood-2012-07-438408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isgro A, Marziali M, Sodani P, Gaziev J, Erer B, Polchi P, et al. Immunohematologic reconstitution in pediatric patients after T cell-depleted HLA-haploidentical stem cell transplantation for thalassemia. Biol Blood Marrow Transplant. 2010;16:1557–1566. doi: 10.1016/j.bbmt.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Sodani P, Isgro A, Gaziev J, Polchi P, Paciaroni K, Marziali M, et al. Purified T-depleted, CD34+ peripheral blood and bone marrow cell transplantation from haploidentical mother to child with thalassemia. Blood. 2010;115:1296–1302. doi: 10.1182/blood-2009-05-218982. [DOI] [PubMed] [Google Scholar]
- 20.Russell JA, Tran HT, Quinlan D, Chaudhry A, Duggan P, Brown C, et al. Once-daily intravenous busulfan given with fludarabine as conditioning for allogeneic stem cell transplantation: study of pharmacokinetics and early clinical outcomes. Biol Blood Marrow Transplant. 2002;8:468–476. doi: 10.1053/bbmt.2002.v8.pm12374451. [DOI] [PubMed] [Google Scholar]
- 21.De Lima M, Couriel D, Thall PF, Wang X, Madden T, Jones R, et al. Once-daily intravenous busulfan and fludarabine: clinical and pharmacokinetic results of a myeloablative, reduced-toxicity conditioning regimen for allogeneic stem cell transplantation in AML and MDS. Blood. 2004;104:857–864. doi: 10.1182/blood-2004-02-0414. [DOI] [PubMed] [Google Scholar]
- 22.Sabloff M, Chandy M, Wang Z, Logan BR, Ghavamzadeh A, Li CK, et al. HLA-matched sibling bone marrow transplantation for β-thalassemia major. Blood. 2011;117:1745–1750. doi: 10.1182/blood-2010-09-306829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ichinohe T, Teshima T, Matsuoka K, Maruya E, Saji H. Fetal-maternal microchimerism: impact on hematopoietic stem cell transplantation. Curr Opin Immunol. 2005;17:546–552. doi: 10.1016/j.coi.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Patriarca F, Luznik L, Medeot M, Zecca M, Bacigalupo A, Di Bartolomeo P, et al. Experts’ considerations on HLA-haploidentical stem cell transplantation. Eur J Haematology. 2014;93:187–197. doi: 10.1111/ejh.12322. [DOI] [PubMed] [Google Scholar]
- 25.Cutler C, Kim HT, Sun L, Sese D, Glotzbecker B, Armand P, et al. Donor-specific anti-HLA antibodies predict outcome in double umbilical cord blood transplantation. Blood. 2011;118:6691–6697. doi: 10.1182/blood-2011-05-355263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshihara S, Maruya E, Taniguchi K, Kaida K, Kato R, Inoue T, et al. Risk and prevention of graft failure in patients with preexisting donor-specific HLA antibodies undergoing unmanipulated haploidentical SCT. Bone Marrow Transplant. 2012;47:508–515. doi: 10.1038/bmt.2011.131. [DOI] [PubMed] [Google Scholar]
- 27.Socie G, Lawler M, Gluckman E, McCann SR, Brison O. Studies on hemopoietic chimerism following allogeneic bone marrow transplantation in the molecular biology era. Leuk Res. 1995;19:497–504. doi: 10.1016/0145-2126(95)00026-k. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan EL, Meier P. Nonparametric estimator from incomplete observations. J American Statistical Association. 1958;53:457–481. [Google Scholar]
- 29.McDonald GB, Hinds MS, Fisher LD, Schoch HG, Wolford JL, Banaji M, et al. Venoocclusive disease of the liver and multiorgan failure after bone marrow transplantation: a cohort study of 355 patients. Ann Intern Med. 1993;118:255–267. doi: 10.7326/0003-4819-118-4-199302150-00003. [DOI] [PubMed] [Google Scholar]
- 30.Bearman SI. The syndrome of hepatic veno-occlusive disease after marrow transplantation. Blood. 1995;85:3005–3020. [PubMed] [Google Scholar]
- 31.Chandy M, Balasubramanian SV, Ramachandran SV, Mathews V, George B, Dennison D, et al. Randomized trial of two different conditioning regimens for bone marrow transplantation in thalassemia--the role of busulfan pharmacokinetics in determining outcome. Bone Marrow Transplant. 2005;36:839–845. doi: 10.1038/sj.bmt.1705151. [DOI] [PubMed] [Google Scholar]
- 32.Bernado ME, Piras E, Vacca A, Giorgiani G, Zecca M, Bertaina A, et al. Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood. 2012;120:473–476. doi: 10.1182/blood-2012-04-423822. [DOI] [PubMed] [Google Scholar]
- 33.Mathews V, George B, Viswabandya A, Abraham A, Ahmed R, Ganapule A, et al. Improved clinical outcomes of high risk beta thalassemia major patients undergoing a HLA matched related allogeneic stem cell transplant with a treosulfan based conditioning regimen and peripheral blood stem cell grafts. PloS One. 2013;8:e61637. doi: 10.1371/journal.pone.0061637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggeri A, Labopin M, Sanz G, Piemontese S, Arcese W, Bacigalupo A, et al. Comparison of outcomes after unrelated cord blood and unmanipulated Haploidentical stem cell transplantation in adults with acute leukemia. Leukemia. 2015;29:1891–1900. doi: 10.1038/leu.2015.98. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Liu QF, Xu LP, Liu KY, Zhang XH, Ma X, et al. Haploidentical- versus identical-sibling transplant for AML in remission: a multi-centre, prospective study. Blood. 2015;125:3956–3962. doi: 10.1182/blood-2015-02-627786. [DOI] [PubMed] [Google Scholar]
- 36.Poonkuzhali B, Chandy M, Srivastava A, Dennison D, Krishnamoorthy R. Glutathione S-transferase activity influences busulfan pharmacokinetics in patients with beta thalassemia major undergoing bone marrow transplantation. Drug Metab Dispos. 2001;29:264–267. [PubMed] [Google Scholar]
- 37.Malar R, Sjoo F, Rentsch K, Hassan M, Gungor T. Therapeutic drug monitoring is essential for intravenous busulfan therapy in pediatric hematopoietic stem cell recipients. Pediatr. Transplant. 2011;15:580–588. doi: 10.1111/j.1399-3046.2011.01529.x. [DOI] [PubMed] [Google Scholar]
- 38.Andersson BS, Thall PF, Madden T, Couriel D, Wang X, Tran HT, et al. Busulfan systemic exposure relative to regimen-related toxicity and acute graft-versus-host disease: defining a therapeutic window for i.v. BuCy2 in chronic myelogenous leukemia. Biol Blood Marrow Transplant. 2002;8:477–485. doi: 10.1053/bbmt.2002.v8.pm12374452. [DOI] [PubMed] [Google Scholar]
- 39.Andersson BS, de Lima MJ, Saliba RM, Shpall EJ, Popat U, Jones R, et al. Pharmacokinetic dose guidance of IV busulfan with fludarabine with allogeneic stem cell transplantation improves progression free survival in patients with AML and MDS; results of a randomized phase III study. Blood. 2011;118:892. [Google Scholar]
- 40.Popat U, Bassett R, Chen J, Alousi AM, Anderlini P, Ciurea SO, et al. Allogeneic transplantation for myelofibrosis: final analysis of a prospective study after a median follow up of 5 years. J Clin Oncol. 2015 (suppl;abstr 7008) [Google Scholar]
- 41.Furst S, Bernit E, El Cheikh J, Granata A, Harbi S, Mohty B, et al. Successful engraftment and clearance of donor specific antibodies after haploidentical related stem cell transplantation for an adult patient with sickle cell disease. Blood. 2014;124:5792. [Google Scholar]