

Abstract
A series of ABC triblock copolypeptoids [i.e., poly(N-allyl glycine)-b-poly(N-methyl glycine)-b-poly(N-decyl glycine) (AMD)] with well-defined structure and varying composition have been synthesized by sequential primary amine-initiated ring-opening polymerization of the corresponding N-substituted N-carboxyanhydride monomers (Al-NCA, Me-NCA, and De-NCA). The ABC block copolypeptoids undergo sol-to-gel transitions with increasing temperature in water and biological media at low concentrations (2.5–10 wt %). The sol–gel transition is rapid and fully reversible with a narrow transition window, evidenced by the rheological measurements. The gelation temperature (Tgel) and mechanical stiffness of the hydrogels are highly tunable: Tgel in the 26.2–60.0 °C range, the storage modulus (G′) and Young’s modulus (E) in the 0.2–780 Pa and 0.5–2346 Pa range, respectively, at the physiological temperature (37 °C) can be readily accessed by controlling the block copolypeptoid composition and the polymer solution concentration. The hydrogel is injectable through a 24 gauge syringe needle and maintains their shape upon in contact with surfaces or water baths that are kept above the sol–gel transition temperature. The hydrogels exhibit minimal cytotoxicity toward human adipose derived stem cells (hASCs), evidenced from both alamarBlue and PicoGreen assays. Furthermore, quantitative PCR analysis revealed significant up-regulation of the Col2a1 gene and down-regulation of ANGPT1 gene, suggesting that the hydrogel exhibit biological activity in inducing chondrogenesis of hASCs. It was also demonstrated that the hydrogel can be used to quantitatively encapsulate water-soluble enzymes (e.g., horseradish peroxidase) by manipulating the sol–gel transition. The enzymatic activity of HRP remain unperturbed after encapsulation at 37 °C for up to 7 d, suggesting that the hydrogel does not adversely affect the enzyme structure and thereby the enzymatic activity. These results suggest that the polypeptoid hydrogel a promising synthetic platform for tissue engineering or protein storage applications.
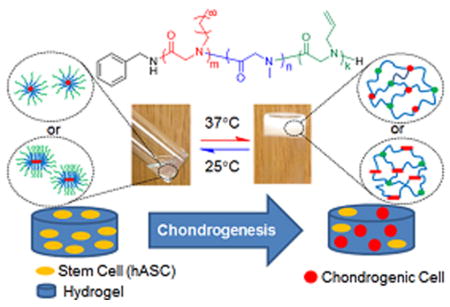
Graphical abstract
INTRODUCTION
Stimuli-sensitive polymer hydrogels, which can respond to various environmental changes such as temperature,1 pH,2,3 light,4 biochemical cues,5 and electronic or magnetic field,6,7 have received considerable attention due to their promising application in drug delivery and tissue engineering.8–10 Temperature is a commonly exploited stimulus, and thermoreversible polymer hydrogels that form gels at human physiological temperature are of particular appeal due to their relevance for in vivo uses. In contrast to hydrogels based on naturallyl occurring biopolymers (e.g., collagen,11,12 alginate,13 and hyaluronic acid14), which have potential immunogenic responses and poor mechanical properties,15,16 synthetic polymer-based hydrogels offer the advantages of adjustable mechanical strength, degradation rate, chemical composition, and gel morphologies, making it possible to tailor the materials for specific biomedical and biotechnological applications.8–10
Thermoreversible hydrogels based on AB and ABA block copolymers, where A and B signify the hydrophobic/thermoresponsive and hydrophilic blocks, respectively, have been widely investigated in the past.1,2,9,17,18 These hydrogels often exhibit broad or slow sol–gel transitions or high critical gelation concentration (cgc, where cgc ≥ 10 wt %).19–30 In the ABA-type hydrogel, the inefficient sol–gel transition characteristic has been attributed to the formation of loops, flower micelles, and dangling ends in the formation of micellar network.1,2,29 By contrast, the sol–gel transition in ABC-type hydrogel, where A, B, and C refer to thermoresponsive block with lower critical solution temperature (LCST), hydrophilic and hydrophobic block, respectively, is much sharper and occurs at lower cgc. With the formation of separate A and C domains, the intramolecular association of the end blocks to form loop configurations is significantly suppressed, thereby giving rise to a rapid and sharp thermoreversible sol–gel transition at low concentration (≤ 5 wt %) upon a temperature increase.29,31
Although ABC triblock copolymers are attractive structural motif for thermoreversible gelation, studies on the design, characterization, and investigation of potential biomedical uses of ABC hydrogelators have been limited. Poly(N-isopropylacrylamide) (PNIPAAM), showing a LCST in water close to body temperature(~32 °C), is the most commonly investigated thermoresponsive polymer in the design of ABC hydrogelators. 29,32,33 For example, the earlier reported PNIPAM–PMPC–PPO triblock copolymer exhibit sol–gel transition around 37 °C at high concentration (≥ 20 wt %) with low mechanical strength (G′ ~ 25 Pa).34 Recently, a 5 wt % aqueous solution of PNIPAM–PEO–PEP triblock copolymers was shown to undergo thermoreversible sol–gel transition at 42 °C,29 which is higher than the physiological temperature. Another recently reported cell protective ABC copolymer (PNIPAM–PDMA–PPS) was shown to undergo rapid thermoreversible gelation in PBS buffer at 2.5 wt % with a gelation temperature well below 37 °C.33 The reported hydrogel is mechanically soft with storage modulus (G′) lower than 1 kPa at 7.5 wt % polymer concentration. The reported ABC synthetic hydrogels are based on nondegradable polymers.29,33
There is an increasing need to develop thermoreversible hydrogels that are cytocompatible and biodegradable for biomedical applications such as tissue engineering. In addition, synthetic hydrogels often do not exhibit any substantial biological activity, in contrast to naturally derived hydrogels. 8,15,16 Yet, for applications such as tissue engineering, hydrogels that can influence the cell differentiation and tissue development in a controlled manner are highly desirable. Naturally derived hydrogels, which often provide appropriate differentiation cues, suffer from limited mechanical stability in the in vivo environment as a result of enzymatic degradation. 14,35,36 Importing biological cues such as components of specific tissues (e.g., chondroitin sulfate, hyaluronic acid), growth factors (e.g., TGF-β1), extracellular matrix protein-derived cell-adhesive peptide (e.g., RGD) to the synthetic hydrogels through covalent linkage is a common strategy to confer biological activities to synthetic hydrogels.37–43 This strategy, though effective, has the drawbacks of enhanced synthetic complexity, potential emergence of cytotoxicity, and altered physicochemical properties of the hydrogels. By contrast, synthetic hydrogels that are inherently biologically active and can modulate cell function and differentiation de novo without additional biological cues or factors will not only facilitate the preparation of tissue engineering scaffold but also provide an improved platform to investigate the factors that give rise to the biological activities.42 For tissue engineering applications, it is desirable that the hydrogel scaffold not only have tunable mechanical stiffness and morphologies that can be made to match those of the native tissues but also exhibit biological activities that can modulate the cell migration, proliferation, and differentiation.15,16,22,37
Poly(N-substituted glycine) (a.k.a., polypeptoids), with an N-substituted glycine backbone, lacks extensive hydrogen bonding and backbone chirality as compared to polypeptides. This gives rise to excellent thermal processability, good solubility in many common solvents, as well as enhanced protease stability.44,45 In addition, polypeptoids exhibit good cytocompatibility47,48 and can be degraded under oxidative conditions that mimics tissue inflammation.50 These combined attributes make polypeptoids an attractive material of biomedical and biotechnological relevance.45,49–53 Recent development in the organo-mediated controlled polymerization has enabled access to a suite of well-defined polypeptoid homo and block copolymers.44,46,48,54,55 Amphiphilic block copolymers comprised of hydrophilic polypeptoid segments have also been investigated for biomedical applications such as drug carriers.56–58
Taking advantage of the synthetic development, for the first time, we reported here the design and characterization of a series of ABC triblock copolypeptoids that undergo a rapid thermoreversible sol-to-gel transition with increasing temperature well below the body temperature at concentrations as low as 2.5 wt % in water. The gelation temperature and mechanical stiffness of the hydrogels can be readily tuned by controlling the chemical composition of the triblock copolypeptoids. The hydrogel shows minimal cytotoxicity toward human adipose-derived stem cells (hASCs), evidenced by both AlamarBlue and PicoGreen assays. The hydrogels also exhibit biological activities in regulating the chondrogenesis biomarker gene expression of hASCs. In addition, we have also demonstrated that water-soluble enzymes (e.g., horseradish peroxidase) can be readily encapsulated in the hydrogel for extended period of time without adversely affecting the enzymatic activity. Furthermore, the allyl group in the polymer can be further functionalized by photoinitiated thiol-ene addition chemistry to introduce biologically active ligands and enhance hydrogel stiffness through chemical cross-linking.59,60 These results suggest the potential use of the polypeptoid hydrogel as tissue engineering scaffold.
RESULTS AND DISCUSSION
Synthesis and Characterization of ABC Triblock Copolypeptoids
A series of ABC triblock copolypeptoids have been synthesized by benzyl amine-initiated ring-opening polymerization of the corresponding N-substituted N-carboxyanhydrides (R-NCAs) in a sequential manner (Scheme 1). Representative 1H and 13C{1H} NMR spectra of the monomers were shown in Figure S1–S6. The hydrophobic C segment consists of poly(N-alkyl glycine) where the alkyl groups is varied from butyl (B), octyl (O), to decyl (D) with increasing hydrophobicity. The hydrophilic B segment consists of poly(N-methyl glycine) (M), or poly(N-methoxyethyl glycine) (m), or poly(N-methoxyethoxyethyl glycine)(d) with increasing hydrophilicity. The M, m, and d homopolymers are highly water-soluble with solubility in the 20–200 mg/mL range. The thermoresponsive A segment is based on poly(N-allyl glycine) (A) for all samples. Poly(N-allyl glycine) has previously been demonstrated to be thermoresponsive with cloud point in the 27–54 °C range, which is dependent on the chain length and concentration.59
Scheme 1.

All polymerization reactions were conducted in 50 °C anhydrous acetonitrile and were allowed to reach complete conversion prior to the addition of another monomer. The resulting triblock copolymers were purified by precipitation in hexane and dried under vacuum prior to further analysis. The ABC triblock copolypeptoid compositions were determined by 1H NMR spectroscopy (Figure S7–S11). For example, the number-averaged degree of polymerization (DPn) of the AMD polymers (Entry 1–6, Table 1) was determined by the integrations at 0.91, 3.0, and 5.8 ppm due to the methyl protons in D and M blocks as well as the terminal alkenyl protons in the A block relative to the integration of signals at 7.3 ppm due to the benzyl end-group. The triblock copolypeptoids composition can be systematically adjusted by controlling the initial monomer to initiator feed ratio (Table 1). The weight fraction of individual block is varied in the 0.31–0.50, 0.34–0.54, and 0.13–0.21 range for the A, B, and C segments, respectively (Table 1). Size exclusion chromatographic (SEC) analysis of the polymer products obtained after the growth of each block revealed monomodal peaks that are consistently shifted toward lower elution time, in agreement with the block copolymer formation (Figure S12). The polymer molecular weight distribution remains narrow with low polydispersity indices (PDI) in the 1.03–1.15 range (Figure S13), consistent with the formation of well-defined block copolypeptoid polymers. The molecular parameters of the triblock copolypeptoid samples are summarized in Table 1.
Table 1.
Molecular Parameters of ABC Triblock Copolypeptoids and the Corresponding Hydrogel Properties
| entry # | sample compositiona | [M1]0:[M2]0:[M3]0:[I]0b | Mn(theor.) (kDa)c | Mn(NMR) (kDa)d | Mn(SEC) (kDa)e | PDIe | f1f | f2f | f3f | Tgel (°C)g | G″ (Pa)h | G′ (Pa)h | E (Pa)i |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A98M98D18 | 100:100:20 | 20.8 | 20.0 | 36.7 | 1.04 | 0.47 | 0.35 | 0.18 | 26.2 ± 1.2 | 36 ± 4 | 251 ± 23 | 762 ± 68 |
| 2 | A92M94D12 | 100:100:10 | 18.9 | 18.1 | 36.4 | 1.09 | 0.50 | 0.36 | 0.14 | 26.6 ± 0.6 | 58 ± 8 | 780 ± 46 | 2346 ± 139 |
| 3 | A94M158D16 | 100:150:20 | 26.0 | 23.6 | 41.4 | 1.09 | 0.39 | 0.48 | 0.13 | 30.7 ± 1.0 | 32 ± 1 | 189 ± 8 | 573 ± 22 |
| 4 | A43M92D9 | 50:100:10 | 14.0 | 12.6 | 29.8 | 1.08 | 0.33 | 0.52 | 0.14 | 40.9 ± 1.3 | j | j | j |
| 5 | A45M45D10 | 50:50:10 | 10.5 | 9.64 | 24.1 | 1.03 | 0.46 | 0.34 | 0.21 | 31.0 ± 0.3 | 11 ± 1 | 125 ± 8 | 378 ± 23 |
| 6 | A23M25D5 | 25:25:5 | 5.29 | 5.10 | 10.3 | 1.15 | 0.45 | 0.36 | 0.20 | > 60.0 | j | j | j |
| 7 | A45M47O10 | 50:50:10 | 10.2 | 9.50 | 26.5 | 1.06 | 0.46 | 0.35 | 0.18 | 31.4 ± 0.4 | 4 ± 1 | 25 ± 9 | 76 ± 27 |
| 8 | A41M47B11 | 50:50:10 | 9.64 | 8.66 | 28.2 | 1.06 | 0.47 | 0.39 | 0.14 | 32.7 ± 0.7 | 0.07 ± 0.01 | 0.2 ± 0.04 | 0.5 ± 0.1 |
| 9 | A50m55D11 | 50:50:10 | 12.7 | 13.5 | 26.5 | 1.07 | 0.31 | 0.54 | 0.15 | 30.6 ± 0.6 | 8 ± 1 | 60 ± 8 | 182 ± 25 |
| 10 | A46d42D9 | 50:50:10 | 14.9 | 13.0 | 26.8 | 1.07 | 0.36 | 0.51 | 0.14 | 42.6 ± 1.1 | j | j | j |
The numbers in subscripts correspond to the DPn of individual block determined by end-group analysis using 1H NMR spectroscopy in CD2Cl2.
Initial monomer to initiator ratio.
Theoretical molecular weights were calculated from the initial monomer to initiator ratio.
Determined by 1H NMR analysis.
determinde by the SEC-DRI method using polystyrene standards (0.1 M LiBr/DMF, room temperature).
f1, f2, and f3 refer to the weight fraction of the thermoresponsive A end block, the hydrophilic middle block, and the hydrophobic end block, respectively.
Tgel is the crossover point of G′ and G″ in the plot of G′ and G″ versus temperature: average of two measurements.
G′ and G″ in the gel state at physiological temperature (37 °C): average of two measurements.
Young’s modulus is calculated using E = 2G(1 + ν), where G = (G′2+G″2)1/2, ν = 0.5, and ν is the Poisson’s ratio.61
The polymer solution did not form a gel at 37 °C.
Preparation of the ABC hydrogels
All aqueous solutions of ABC triblock copolymers were prepared by the “thin film hydration” method, as reported by Zhou et al.29 All samples underwent thermoreversible gelation in DI water, evidenced by rheological measurements (vide infra). The 5 wt % solutions of selected polymers (Entry 1, 2, 3, 5, 7, and 9, Table 1) form free-standing opaque gels at close-to-body temperature, and return to a free-flowing liquid when cooled down to room temperature (Figure S14–15). The ABC triblock copolymers also underwent sol-to-gel transition in biological media (stromal media) at the same concentrations. The sol-to-gel transition is rapid with the formation of free-standing gels in less than 30 s. Repeated heating and cooling experiments indicate that the sol-to-gel transition is fully reversible. The hydrogels appear opaque, suggesting the occurrence of phase separation to some extent during gelation. The hydrogels are injectable through a 24 gauge syringe needle and maintain the shape upon contact with 37 °C surfaces or DI water bath (Videos 1 and 2 in Supporting Information).
Rheological Characterization of the Sol–Gel Transition
Rheological measurements of the ABC polypeptoid hydrogels were conducted to quantify the gelation temperature and assess the relative mechanical stiffness of the hydrogels. Two measurements were conducted for each sample (see representative data in Figures S16 and S17). Temperature-dependent dynamic shear moduli (storage moduli G′ and loss moduli G″) of the polypeptoid aqueous solution were recorded at a frequency of 10 rad/s and heating rate of 1 °C/min over a 15–60 °C temperature range. A representative evolution of G′ and G″ with increasing temperature is shown in Figure 1 for a 5 wt % aqueous solution of A98M98D18 (Entry 1, Table 1). The storage (G′) and loss modulus (G″) are both low with G″ larger than G′ at low temperature range, which indicates the viscous liquid-like behavior of the solution. As the temperature increases to the transition point, the G′ and G″ of the sample increase sharply to reach a crossover point at which the G′ starts to exceed the G″, indicating an elastic solid-like behavior of the solution beyond the critical temperature point. Here, we define the crossover point of the G′ and G″ as the gelation temperature (Tgel). With further increase of the temperature, the G′ and G″ values continue to increase and eventually plateau, suggesting the formation of stable hydrogels with a certain mechanical stiffness. To investigate the reversibility of the gelation, a second temperature sweep of the G′ and G″ was conducted after the sample was cooled down to the starting temperature (Figure 2). The two measurements are nearly overlapped, indicating that the sol–gel transition is reversible.
Figure 1.

Plots of storage (G′, filled symbols) and loss moduli (G″, open symbols) versus temperature for the A98M98D18 (Entry 1, Table 1) polymer solutions at 1 wt % (G′, ▲; G″ △), 2.5 wt % (G′, ■; G″, □), and 5 wt % (G′, ●; G″, ○). Inset shows the plot of Tgel versus polymer concentration.
Figure 2.

Plots of storage (G′, filled symbols) and loss moduli (G″, open symbols) versus temperature for the A98M98D18 (5 wt %): first heating (G′, ■; G″, □) and second heating (G′, ■; G″, □).
To further characterize the rheological properties of the sol and the gel based on the A98M98D18 triblock copolypeptoid (Entry 1, Table 1), measurement of dynamic shear moduli as a function of angular shearing frequency was conducted (Figure 3). At 22 °C, the loss modulus (G″) is larger than the storage modulus (G′) and both moduli are close to zero at low frequency and adapt a terminal rheological behavior indicative of a viscous liquid. At 25 °C, the G′ started to superimpose on the G″ and approaches the critical gelation temperature when G′ and G″ follow a power law with an exponential of approximately 1/2 (G′ ≈ G″ ~ ω0.5) (Figure 3).29 At 37 °C, G′ is large than G″ through the whole frequency range and is nearly frequency independent, indicating the formation of a stable elastic solid-like hydrogel.
Figure 3.

Plots of storage modulus (G′, filled symbols) and loss modulus (G″, open symbols) versus angular frequency (ω) for the 5 wt % aqueous solution of A98M98D18 (Entry 1, Table 1) at different temperatures: 37 °C (G′, ■; G″, □), 25 °C (G′, ●; G″, ○), and 22 °C (G′, ▲; G″ △).
Microscopic Characterization of the Hydrogel
To investigate the microscopic structure of the hydrogel, cryo-SEM was conducted on the A92M94D12 hydrogel (Entry 2, Table 1, 5 wt %) freshly formed at 37 °C. The cryo-SEM microgram revealed a highly porous structure with large mesh size in the micron regime (>3 μM) (Figure 4), consistent with a gel structure formed by phase separation.
Figure 4.

Cryo-SEM images of the 5 wt % A92M94D12 hydrogel (Entry 2, Table 1). The scale bar in (A) and (B) is 50.0 and 10.0 μm, respectively.
Characterization of Micellation of ABC Triblock Copolypeptoids in Dilute Solution
We hypothesize that the ABC triblock copolypeptoids undergo a thermoreversible sol–gel transition through the formation of micellar networks (Figure 5) similar to early studies on other ABC block copolymer systems.29,31,33 As the three blocks are mutually immiscible, the C block is hydrophobic and the B and A blocks are water-soluble below the sol–gel transition temperature, the triblock copolymers are expected to form core–shell–corona micelles below Tgel. Upon temperature increases, the corona blocks (A) undergo cloud point transition and become dehydrated to form hydrophobic domains. This results in the formation of a three-dimensional micellar network.
Figure 5.

Schematic showing the proposed gelation mechanism of aqueous solutions of the ABC triblock copolypeptoids.
The cloud point of the AMD triblock copolypeptoids, as determined from the turbidity measurement, was shown to be higher than of the poly(N-allyl glycine) homopolymer (A) itself by 8 °C (Figure S21). It is attributed to the attachment of hydrophilic M block at the A terminal, resulting in an increased cloud point of the A segment.
To verify the micelle formation, TEM analysis of the diluted triblock copolypeptoids solutions were conducted. Spherical micelles with a uniform diameter (13.4 ± 1.1 nm) were observed for the A92M94D12 sample (Entry 2, Table 1) (Figure 6A); for the A98M98D18 sample with slightly increased D segmental length (Entry 1, Table 1), rod-shape micelles with moderately uniform diameter (16.6 ± 1.7 nm) become notably present (Figure 6C). Consistent with the TEM analysis on the dried and uranyl acetate stained micellar samples, Cryo-TEM analysis of a dilute aqueous solution (1 wt %) of the same A92M94D12 and A98M98D18 sample also confirm the formation of spherical and rod-shape micelles, respectively (Figure 6B and D). In addition, DLS analysis of the dilute solution of A98M98D18 (0.5 wt %) at 25 °C revealed the presence of particles of a much larger hydrodynamic size than that of A92M94D12 (0.5 wt %, Figure S18), in accordance with their different micellar morphologies observed by TEM. These results together with the cryo-SEM analysis of the polypeptoid hydrogel (Figure 4) suggest that the hydrogel structure is highly hierarchical with spatial features ranging from micron down to nanometer in dimension.
Figure 6.

(A, C) TEM images of the micelles based on A92M94D12 and A98M98D18 polymers, respectively (stained with uranyl acetate), and (B, D) cryo-TEM image of 1 wt % aqueous solution of the same A92M94D12 and A98M98D18 polymers, respectively.
The micelle formation is further supported by 1H NMR spectroscopic study of a dilute solution of AMD. (Figure S19). In CD2Cl2, which is a good and nonselective solvent for all three blocks in the triblock copolypeptoids, proton signals from the three different blocks (A, M, D) are notably present. In D2O, which is a poor solvent for the D segment and a good solvent for the A and M segments, the proton signals due to the D segment have completely disappeared. This is consistent with micellation where the insoluble D block becomes buried in the core of the micelles. As the temperature is increased to 37 °C, which is above the cloud point of the block copolypeptoids, the proton signals due to the A block was significantly decreased. This indicates the increased dehydration of the A block above the cloud point. The broadening of the proton signals of the M block at elevated temperature was attributed to structural heterogeneity within the gel due to the phase separation during gelation.
To further support the proposed mechanism, a temperature-sweep dynamic light scattering (DLS) measurement within 20–60 °C range was performed on the dilute aqueous solution of A92M94D12 (0.5 wt %) (Figure 7 and S20). At 20 °C, DLS revealed a relatively narrow distribution of micellar sizes with an average 40.6 ± 0.4 nm diameter. As the temperature increases, the average size of the particles also increases (Figure 7). Plot of the derived count rate of the micelle solution versus temperature revealed a sharp increase at 27 °C, indicating the onset of micellar aggregation (Figure S20A). The temperature-dependent DLS measurement suggests the association of micelles at elevated temperature, in agreement with the proposed gelation mechanism (Figure 5).
Figure 7.

Diameter distribution of the A92M94D12 micellar solution (0.5 wt %) at different temperature obtained by DLS measurements.
Tuning the Hydrogel Properties
The gelation temperature and mechanical stiffness of the ABC block copolypeptoid hydrogels can be adjusted by controlling the polymer solution concentration and the polymer composition. For example, the aqueous solutions of A98M98D18 (Entry 1, Table 1) in the 1–5 wt % range all underwent thermoreversible sol–gel transitions (Figure 1). The sol–gel transition temperature can be systematically increased from 26.2 to 33.6 °C as the polymer concentration is decreased from 5 to 1 wt %. The sol–gel transition window also becomes narrower as the polymer concentration increases. The mechanical stiffness of the hydrogel at 37 °C, as indicated by storage modulus (G′), increases from approximately 2 to 251 Pa as the polypeptoid concentration increases from 1 to 5 wt %. It corresponds to an increase of Young’s modulus from 6 to 762 Pa.61 This is attributed to the increased cross-linking density at higher concentrations, resulting in micellar networks with increased stiffness.29 The concentration dependence of the sol–gel transition temperature is consistent with the concentration dependence of the cloud point of block copolymers: the higher the concentration, the lower the cloud point transition.59,62
To investigate the impact of polymer composition on the gelation characteristics, we conducted rheological measurements on the aqueous solutions of triblock copolypeptoids where the chain length of a selective block in the AMD triblock copolypeptoids is systematically varied while the chain length of the remaining two blocks are kept constant. It has been found that the chain length of each block impacts the gelation temperature and gel modulus to different extents. For example, Tgel does not change appreciably when the length of the hydrophobic D end block is increased by 50% from DPn = 12 to 18 (Figure 8C), whereas the G′ at 37 °C decreases from 780 to 251 Pa. By comparison, when the length of the M middle block is increased by 60% from DPn = 98 to 158 while the chain length of the other two blocks are kept constant, Tgel is increased from 26.2 to 30.7 °C and the G′ value at 37 °C was reduced from 251 to 189 Pa (Figure 8B). This is consistent with the general observation that increasing the molar fraction of the terminal hydrophilic moiety in thermoresponsive block copolymers enhances the cloud point.62 By contrast, a dramatic increase of gelation temperature from 26.6 to 40.9 °C occurred (Figure 8A) when the length of A block is decreased by 53% from DPn = 92 to 43. The latter sample did not form a gel at physiological temperature (37 °C). The change in the Tgel is in agreement with the previous report where the cloud point of the A homopolymer exhibits chain length dependence: the shorter chain length of A gives rise to a higher cloud point.59 The gelation temperature (40.9 °C) is much higher than the reported cloud point of the A homopolymer with similar DPn (30 °C). This is expected as the A segment in the triblock copolypeptoids is directly attached to a long hydrophilic M segment, thereby resulting in an increase in cloud point.62 Compared to the ABC hydrogelators, the corresponding ABA triblock copolymer exhibit a very different gelation behavior: no gelation occurs up to 60.0 °C (Figure 8D). It is probably due to the long-range structural arrangement during the simultaneous micellation and gelation process. In addition, the same end block may cause loops or flower-like conformation that suppresses the formation of bridging structure.1,2,29
Figure 8.

Plots of storage (G′, filled symbol) and loss moduli (G″, open symbol) versus temperature for aqueous solutions (5 wt %) of triblock copolypeptoids having varying compositions: A98M98D18 (G′, ●; G″, ○), A92M94D12 (G′, ■; G″, □), A94M158D16 (G′, ●; G″, ○), A43M92D9 (G′, ●; G″, ○), A45M93A45 (G′, ●; G″, ○).
To elucidate the influence of the total chain length on the gelation characteristics, rheological measurements were also conducted on aqueous solutions of AMD triblock copolypeptoids where the total polymer chain length is varied while the weight fraction of each block is kept constant (Figure 9 and entries 1, 5, and 6 in Table 1). The sol–gel transition temperature was shown to systematically increase from 26.2 to 31.0 to >60.0 °C as the total chain length (DPn) decreases from 214 to 100 to 53. In fact, the shortest chain sample remains a solution at the highest temperature (60.0 °C) that was tested. G′ and G″ appear to crossover at this temperature limit (Figure 9). The chain length dependence of Tgel is consistent with the chain length dependence of the cloud point for A homopolymer: shorter chains exhibit higher cloud points.59 This indicates that tuning the A chain length is the most effective strategy to control the sol–gel transition temperature in the triblock copolypeptoid system. In addition, the hydrogel stiffness at 37 °C, as indicated by the G′ value, is reduced from 251 to 125 Pa with the decrease of total chain length (DPn) from 214 to 53 (Figure 9).
Figure 9.

Plots of storage (G′, filled symbol) and loss moduli (G″, open symbol) versus temperature for aqueous solutions (5 wt %) of triblock copolypeptoids with varying block chain length and same block molar ratio: A98M98D18 (G′, ●; G″, ○), A45M45D10 (G′, ■; G″, □), A23M25D5 (G′, ▲; G″ △).
To elucidate how the hydrophobicity of the core block (i.e., C segment) affect the gelation temperature and mechanical stiffness of the hydrogel, rheological measurements were conducted on the aqueous solutions of triblock copolypeptoids where the hydrophobic D end block was replaced with less hydrophobic O and B (Scheme 1), whereas the length of each block was kept nearly the same. As the hydrophobicity of the core block decreases (D > O > B), the Tgel was slightly increased from around 31.0 to 31.4 to 32.7 °C. The hydrogel at 37 °C shows decreased storage modulus from 125 to 0.2 Pa as the hydrophobic core block is changed from D to B (Figure 10). Thus, the hydrophobicity of the C segment plays an important role in the gelation that increased hydrophobicity lowers the Tgel and raises the hydrogel stiffness.
Figure 10.

Plots of storage modulus (G′, filled symbol) and loss modulus (G″, open symbol) versus temperature for aqueous solutions (5 wt %) of triblock copolypeptoids having varying hydrophobic end block: A45M45D10 (G′, ■; G″, □), A45M47O10 (G′, ●; G″, ○), A41M47B11 (G′, ▲; G″ △).
To further investigate how the hydrophilicity of the B middle block affects the gelation characteristics, rheological measurements were conducted on the aqueous solutions of triblock copolypeptoids where the hydrophilicity of the middle block was altered. As the hydrophilicity of the middle block increases in the following order: M < m < d (Scheme 1), the Tgel was increased from 31.0 to 42.6 °C. The storage modulus at 37 °C is decreased from 125 to 60 Pa with the increase of the middle block hydrophilicity from M to m (Figure 11). In contrast, as the middle block hydrophilicity further increased to d, the polymer solution remains in the sol state at 37 °C. These results strongly indicate that the gelation temperature and stiffness of the gels are highly dependent on the hydrophilicity and hydrophobicity of the constituent block. We are able to adjust the Tgel and G′ by tuning the polymer composition (i.e., chemistry and molar fraction) for targeted applications.
Figure 11.

Plots of storage (G′, filled symbol) and loss moduli (G″, open symbol) versus temperature for aqueous solutions (5 wt %) of triblock copolypeptoids with varying middle block: A45M45D10 (G′, ■; G″, □), A50m55O11 (G′, ●; G″, ○), A46d42D9 (G′, ▲; G″ △).
Protein Encapsulation Study
Thermoreversible hydrogels are useful for encapsulation and delivery of water-soluble therapeutics such as proteins/peptides or cells.10,17 For encapsulation of proteins, it is important that the hydrogels do not adversely affect the protein structure and function. Because the polypeptoids are structurally similar to polypeptides, it is of concern that the hydrogel materials may interact with the proteins and alter the protein functions, even though the polypeptoids only present in low weight fraction in the hydrogel. To assess the suitability of the polypeptoid hydrogel for protein encapsulation, a model water-soluble enzyme (horseradish peroxidases, HRP) was encapsulated in the hydrogel and examined for any functional change over an extended period of time.
HRP is known to catalyze the reaction of guaiacol and H2O2, giving rise to colored product (Scheme S2).57 This allows the enzymatic activity of HRP to be readily quantified by measuring the initial reaction rate using a UV–vis spectrometer (Figure S22). HRP was encapsulated in the A92M94D12 hydrogel (sample 2, Table 1) at 37 °C for up to 7 d, over which period the enzymatic activity was examined and quantified.57 The HRP encapsulated in hydrogels do not exhibit any appreciable changes in the specific enzymatic activity in the first 24 h of encapsulation (Figure 12). The activities are comparable to the control sets (no gel, Figure 12), where the HRP was kept in PBS buffer at 37 °C for the same duration. The encapsulated HRP also shows comparable enzymatic activity to the as-received HRP in PBS buffer at 25 °C without any prolonged incubation (control, Figure 12). Increasing the encapsulation to 7 days resulted in a slight decrease of the enzyme activity by 9.4%. This is ascribed to the prolonged heating as the control sample where the enzyme is incubated in buffer at 37 °C without hydrogel shows the similar percentage reduction of activity. The results indicate that the polypeptoid hydrogel does not adversely affect the HRP enzymatic function.
Figure 12.

Specific enzyme activity with different incubation time at 37 °C: incubation in the A92M94D12 hydrogel (sample 2, Table 1) (filled symbol: ■) and incubation without the hydrogel (open symbol: □). Control (circular symbol: ○): the enzymatic activity of as-received HRP was measured in PBS buffer at 25 °C without any treatment.
Cytotoxicity Assessment of the Polypeptoid Solution and Hydrogel
AlamarBlue assay was used to assess the cytotoxicity of ABC triblock copolypeptoids diluted solution to human adipose-derived stem cells (hASCs). The diluted polymer solution (A92M94D12) was shown to be minimally cytotoxic to hASC with concentration up to 20 mg/mL (Figure 13).
Figure 13.

Relative metabolic activity of hASC cultured in dilute solutions of A92M94D12 triblock copolypeptoids (Entry 2, Table 1). The results are normalized to live control.
AlamarBlue assay was further used to investigate the effect of ABC hydrogel on hASCs metabolic activity. After 24 h of culturing in the ABC hydrogel extractives or 3 d of culturing within the hydrogel matrix in direct contacting, hASCs showed a significant decrease (P-value <0.05) of relative metabolic activity compared to the live control (Figure 14A). The corresponding total DNA content was quantified using Quanti-T PicoGreen assay to analyze the hASC proliferation on ABC hydrogel (Figure 14B). No significant inhibition of hASC proliferation compared to the live control was observed when hASCs exposed to the ABC hydrogel extractives for 24 h or cultured within the hydrogel matrix in direct contact for 3 d. The results indicated that the slight decrease of cells proliferation rate does not correlate with a decrease in metabolic activity and may be indicative of stem cells leaving the proliferative cell cycle to differentiate. The differentiation pathway of hASCs happening within the hydrogel matrix was further indicated by the QPCR analysis (Figure 16). Moreover, hASC maintained a healthy spindle shape both when exposed to hydrogel extractives and cultured within hydrogel matrix (Figure 15). Overall, the results demonstrated that ABC hydrogel is a cytocompatible material that can be potentially used as a tissue engineering scaffold.
Figure 14.

(A) Relative metabolic activity of hASC cultured in A92M94D12 hydrogel (Entry 2, Table 1) (5 wt % in PBS). The results are normalized to positive control. (B) Corresponding number of hASC obtained using Quanti-T PicoGreen assay. Star symbol (*) indicates statistical significant difference between two groups.
Figure 16.

QPCR analysis of gene expression within the A92M94D12 hydrogel matrix (Entry 2, Table 1).
Figure 15.

Optical microscopic images of hASC with different treatments.
QPCR Quantification of Chondrogenesis Markers
To further investigate the effect of hydrogel on stem cell differentiation, quantitative real-time polymerase chain reaction (QPCR) analysis were conducted to quantify the expression of two marker genes, Col2a1 and ANGPT1, for chondrogenesis and endotheliogenesis, respectively.64–66
Chondrogenesis is a multistep process characterized cell commitment, expression of chondrogenic markers, condensation, and cellular morphological changes.64 The product of the Col2a1 gene is an early and abundant marker of chondrocytes differentiation pathway.65 The expression of Col2a1 and ANGPT1, a marker of angiogenesis, was assessed by QPCR at the 7 and 21 day time point. The A92M94D12 hydrogel was shown to up-regulate the Col2a1 and down-regulated the ANGPT1 gene expression of hASCs at 7 and 21 d of the chondrogenesis study (Figure 16). Others studies have also shown up-regulation of Col2a1 and down-regulation of ANGPT1 gene expression when hASCs committed to chondrogenesis pathway.64,66 These results indicate that the polypeptoid hydrogel may have potential use as scaffold or graft materials for stem cell based tissue repair.
Several synthetic hydrogels have previously been reported to influence the differentiation of specific cell lines. For example, polypeptide-based hydrogels (PA-PLX-PA67 and PEG-L-PA68) have led to chondrogenesis of chondrocytes and adipose tissue derived cells. Although many factors [e.g., cell morphologies, proliferation rate, cell density, size of cell aggregation, swelling ratio, gel modulus, gel morphology, functional group (e.g., –COO–, –SH, –NH3), and charge state in the hydrogel as well as degradation rate]67–76 have been suggested to contribute to this unique phenomenon, the exact role of each factor and their complex interplays are not well understood. Our future efforts will be directed toward understanding how the structural characteristics of the polypeptoid hydrogels affect the stem cell differentiation by systematically tuning the hydrogel composition and structure.
CONCLUSIONS
Well-defined amphiphilic ABC triblock copolypeptoids with varying composition and chain length can be synthesized by primary amine-initiated ring-opening polymerization. The polymer aqueous solutions undergo rapid thermoreversible sol–gel transitions. The gelation is attributed to the temperature-induced formation of micellar networks. The hydrogel exhibit shear-thinning behavior and can be injected through 24 gauge syringe needles. The gelation temperature of the hydrogel can be readily adjusted between 26.2 and 60.0 °C, and the mechanical stiffness (G′) at physiological temperature (37 °C) can be tuned from between 0.2 and 780 Pa, corresponding to the Young’s modulus in the 0.5–2346 Pa range. Encapsulation of model proteins (HRP) in the polypeptoid hydrogel for up to 7 d does not adversely affect the enzymatic activity. Furthermore, the ABC hydrogel and hydrogel extractives show minimal cytotoxicity to hASCs as indicated by standard metabolic and proliferation assays. The study of chondrogenic marker expression indicated that the hydrogel may have de novo bioactivity and is capable of modulating the expression of chondrogenic differentiation markers in hASCs. The combination of low cytotoxicity and bioactivity renders the polypeptoid hydrogel a highly promising tissue engineering material. The ABC hydrogel motif is highly versatile and structurally tunable. We envision the further functionalization of the hydrogel by photoinitiated thiol-ene addition chemistry to incorporate various biologically active ligands (e.g., peptides) and further enhancement of the mechanical stiffness of the hydrogels by chemical cross-linking. These studies are currently in progress and will be reported in due course.
Supplementary Material
Acknowledgments
Funding
The work was supported by the National Science Foundation (CHE 0955820 and CBET1403301), NIH (1R01DE024790-01 and R01 CA179902), and the Louisiana State University. The CryoTEM and CryoSEM analysis of the hydrogels was supported by the U.S. Department of Energy under EPSCoR Grant No. DE-SC0012432 with additional support from the Louisiana Board of Regents.
D.H.Z. thanks Dr. Ying Xiao at the LSU Microscopy Facility for assisting the TEM experiments, Dr. Rafael Cueto for assisting the DLS experiments, and Prof. Evgueni Nesterov for providing access to the UV–vis spectrometer and Prof. Qinqlin Wu for access to the rheometer. S.X. also acknowledges the partial financial support from NIH.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemmater. 5b03528.
Procedures for monomer and polymer synthesis and characterization, hydrogel preparation, rheological measurements, DLS measurement of diluted copolymer solutions, regular TEM, SEM and cryo-S/TEM sample preparation and analysis, LCST measurement of ABC triblock copolypetoid solutions, HRP encapsulation study, alamarBlue and PicoGreen assays, and chondrogenesis study, representative 1H NMR spectra of ABC triblock copolypeptoids, SEC chromatograms of ABC triblock copolypeptoids, images showing the sol–gel transitions, temperature dependent DLS and NMR data on the dilute solution of ABC triblock copolypeptoids, and enzymatic activity study data. (PDF)
Video illustrating hydrogels are injectable through a 24 gauge syringe needle and maintain the shape upon contact with 37 °C surfaces. (WMV)
Video illustrating hydrogels are injectable through a 24 gauge syringe needle and maintain the shape upon contact with DI water bath. (WMV)
References
- 1.Li C, Tang Y, Armes SP, Morris CJ, Rose SF, Lloyd AW, Lewis AL. Synthesis and Characterization of Biocompatible Thermo-Responsive Gelators Based on ABA Triblock Copolymers. Biomacromolecules. 2005;6:994–999. doi: 10.1021/bm049331k. [DOI] [PubMed] [Google Scholar]
- 2.Ma Y, Tang Y, Billingham NC, Armes SP, Lewis AL. Synthesis of Biocompatible, Stimuli-Responsive, Physical Gels Based on ABA Triblock Copolymers. Biomacromolecules. 2003;4:864–868. doi: 10.1021/bm034118u. [DOI] [PubMed] [Google Scholar]
- 3.Koonar I, Zhou C, Hillmyer MA, Lodge TP, Siegel RA. ABC Triblock Terpolymers Exhibiting Both Temperature- and pH-Sensitive Micellar Aggregation and Gelation in Aqueous Solution. Langmuir. 2012;28:17785–17794. doi: 10.1021/la303712b. [DOI] [PubMed] [Google Scholar]
- 4.Wang N, Zhang J, Sun L, Wang P, Liu W. Gene-Modified Cell Detachment on Photoresponsive Hydrogels Strengthened through Hydrogen Bonding. Acta Biomater. 2014;10:2529–2538. doi: 10.1016/j.actbio.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 5.Li C, Madsen J, Armes SP, Lewis AL. A New Class of Biochemically Degradable, Stimulus-Responsive Triblock Copolymer Gelators. Angew Chem, Int Ed. 2006;45:3510–3513. doi: 10.1002/anie.200600324. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Ren Y, Sun J, Feng L. Carbon Nanotube-Coated Macroporous Poly(N-isopropylacrylamide) Hydrogel and Its Electrosensitivity. ACS Appl Mater Interfaces. 2013;5:3519–3523. doi: 10.1021/am400927d. [DOI] [PubMed] [Google Scholar]
- 7.Bolisetty S, Vallooran JJ, Adamcik J, Mezzenga R. Magnetic-Responsive Hybrids of Fe3O4 Nanoparticles with β-Lactoglobulin Amyloid Fibrils and Nanoclusters. ACS Nano. 2013;7:6146–6155. doi: 10.1021/nn401988m. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Rodrigues J, Tomas H. Injectable and Biodegradable Hydrogels: Gelation, Biodegradation and Biomedical Applications. Chem Soc Rev. 2012;41:2193–2221. doi: 10.1039/c1cs15203c. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman AS. Hydrogels for Biomedical Applications. Adv Drug Delivery Rev. 2002;54:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 10.Vermonden T, Censi R, Hennink WE. Hydrogels for Protein Delivery. Chem Rev. 2012;112:2853–2888. doi: 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- 11.Orban JM, Wilson LB, Kofroth JA, El-Kurdi MS, Maul TM, Vorp DA. Crosslinking of Collagen Gels by Transglutaminase. J Biomed Mater Res. 2004;68A:756–762. doi: 10.1002/jbm.a.20110. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Yang Y, Yao J, Shao Z, Chen X. Strong Collagen Hydrogels by Oxidized Dextran Modificatio. ACS Sustainable Chem Eng. 2014;2:1318–1324. [Google Scholar]
- 13.LeRoux MA, Guilak F, Setton LA. Compressive and Shear Properties of Alginate Gel: Effects of Sodium Ions and Alginate Concentration. J Biomed Mater Res. 1999;47:46–53. doi: 10.1002/(sici)1097-4636(199910)47:1<46::aid-jbm6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 14.Xu X, Jha AK, Harrington DA, Farach-Carson MC, Jia X. Hyaluronic Acid-based Hydrogels: from A Natural Polysaccharide to Complex Networks. Soft Matter. 2012;8:3280–3294. doi: 10.1039/C2SM06463D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drury JL, Mooney DJ. Hydrogels for Tissue Engineering: Scaffold Design Variables and Applications. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 16.Zhu J. Bioactive Modification of Poly(ethylene glycol) Hydrogels for Tissue Engineering. Biomaterials. 2010;31:4639–4656. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen MK, Lee DS. Injectable Biodegradable Hydrogels. Macromol Biosci. 2010;10:563–579. doi: 10.1002/mabi.200900402. [DOI] [PubMed] [Google Scholar]
- 18.Fujiwara T, Kimura Y. Thermo-Sensitive Gels: Biodegradable Hydrogels from Enantiomeric Copolymers of Poly(lactide) and Poly(ethylene glycol) ACS Symp Ser. 2006;939:216–233. [Google Scholar]
- 19.Altinok H, Yu GE, Nixon SK, Gorry PA, Attwood D, Booth C. Effect of Block Architecture on the Self-Assembly of Copolymers of Ethylene Oxide and Propylene Oxide in Aqueous Solution. Langmuir. 1997;13:5837–5848. [Google Scholar]
- 20.Jeong B, Bae YH, Lee D, Kim S. Biodegradable Block Copolymers as Injectable Drug-delivery Systems. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 21.Tae G, Kornfield JA, Hubbell JA, Johannsmann D, Hogen-Esch TE. Hydrogels with Controlled, Surface Erosion Characteristics from Self-Assembly of Fluoroalkyl-Ended Poly-(ethylene glycol) Macromolecules. 2001;34:6409–6419. [Google Scholar]
- 22.Aamer KA, Sardinha H, Bhatia SR, Tew GN. Rheological Studies of PLLA–PEO–PLLA Triblock Copolymer Hydrogels. Biomaterials. 2004;25:1087–1093. doi: 10.1016/s0142-9612(03)00632-x. [DOI] [PubMed] [Google Scholar]
- 23.Ricardo NMPS, Honorato SB, Yang Z, Castelletto V, Hamley IW, Yuan XF, Attwood D, Booth C. Gelation of Concentrated Micellar Solutions of a Triblock Copolymer of Ethylene Oxide and Styrene Oxide, S5E45S5. Langmuir. 2004;20:4272–4278. doi: 10.1021/la049758c. [DOI] [PubMed] [Google Scholar]
- 24.Bae SJ, Suh JM, Sohn YS, Bae YH, Kim SW, Jeong B. Thermogelling Poly(caprolactone-b-ethylene glycol-b-caprolactone) Aqueous Solutions. Macromolecules. 2005;38:5260–5265. [Google Scholar]
- 25.Ho E, Lowman A, Marcolongo M. Synthesis and Characterization of an Injectable Hydrogel with Tunable Mechanical Properties for Soft Tissue Repair. Biomacromolecules. 2006;7:3223–3228. doi: 10.1021/bm0602536. [DOI] [PubMed] [Google Scholar]
- 26.Park SH, Choi BG, Joo MK, Han DK, Sohn YS, Jeong B. Temperature-Sensitive Poly(caprolactone-co-trimethylene carbonate)-Poly(ethylene glycol)-Poly(caprolactone-co-trimethylene carbonate) as in Situ Gel-Forming Biomaterial. Macromolecules. 2008;41:6486–6492. [Google Scholar]
- 27.O’Lenick TG, Jin N, Woodcock JW, Zhao B. Rheological Properties of Aqueous Micellar Gels of a Thermo- and pH-Sensitive ABA Triblock Copolymer. J Phys Chem B. 2011;115:2870–2881. doi: 10.1021/jp2001332. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Yu L, Ding J. Roles of Hydrophilic Homopolymers on the Hydrophobic-Association-Induced Physical Gelling of Amphiphilic Block Copolymers in Water. Macromolecules. 2008;41:6493–6499. [Google Scholar]
- 29.Zhou C, Hillmyer MA, Lodge TP. Efficient Formation of Multicompartment Hydrogels by Stepwise Self-Assembly of Thermoresponsive ABC Triblock Terpolymers. J Am Chem Soc. 2012;134:10365–10368. doi: 10.1021/ja303841f. [DOI] [PubMed] [Google Scholar]
- 30.Yuan Xiong X, Chiu Tam K, Huat Gan L. Synthesis and Thermally Responsive Properties of Novel Pluronic F87/Polycaprolactone (PCL) Block Copolymers with Short PCL Blocks. J Appl Polym Sci. 2006;100:4163–4172. [Google Scholar]
- 31.Zhou C, Toombes GES, Wasbrough MJ, Hillmyer MA, Lodge TP. Structure of Two-Compartment Hydrogels from Thermoresponsive ABC Triblock Terpolymers. Macromolecules. 2015;48:5934–5943. [Google Scholar]
- 32.Ruel-Gariépy E, Leroux JC. In Situ-forming Hydrogels—Review of Temperature-sensitive Systems. Eur J Pharm Biopharm. 2004;58:409–426. doi: 10.1016/j.ejpb.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 33.Gupta MK, Martin JR, Werfel TA, Shen T, Page JM, Duvall CL. Cell Protective, ABC Triblock Polymer-Based Thermoresponsive Hydrogels with ROS-Triggered Degradation and Drug Release. J Am Chem Soc. 2014;136:14896–14902. doi: 10.1021/ja507626y. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Buurma NJ, Haq I, Turner C, Armes SP, Castelletto V, Hamley IW, Lewis AL. Synthesis and Characterization of Biocompatible, Thermoresponsive ABC and ABA Triblock Copolymer. Langmuir. 2005;21:11026–11033. doi: 10.1021/la0515672. [DOI] [PubMed] [Google Scholar]
- 35.Badylak SF, Tullius R, Kokini K, Shelbourne KD, Klootwyk T, Voytik SL, Kraine MR, Simmons C. The Use of Xenogeneic Small Intestinal Submucosa as a Biomaterial for Achille’s Tendon Repair in a Dog Model. J Biomed Mater Res. 1995;29:977–985. doi: 10.1002/jbm.820290809. [DOI] [PubMed] [Google Scholar]
- 36.Badylak S, Kokini K, Tullius B, Whitson B. Strength over Time of a Resorbable Bioscaffold for Body Wall Repair in a Dog Model. J Surg Res. 2001;99:282–287. doi: 10.1006/jsre.2001.6176. [DOI] [PubMed] [Google Scholar]
- 37.Park JS, Yang HJ, Woo DG, Yang HN, Na K, Park KH. Chondrogenic Differentiation of Mesenchymal Stem Cells Embedded in a Scaffold by Long-term Release of TGF-β3 Complexed with Chondroitin Sulfate. J Biomed Mater Res, Part A. 2009;92:806–816. doi: 10.1002/jbm.a.32388. [DOI] [PubMed] [Google Scholar]
- 38.Hwang NS, Varghese S, Theprungsirikul P, Canver A, Elisseeff J. Enhanced Chondrogenic Differentiation of Murine Embryonic Stem Cells in Hydrogels with Glucosamine. Biomaterials. 2006;27:6015–6023. doi: 10.1016/j.biomaterials.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 39.Nuttelman CR, Tripodi MC, Anseth KS. Dexamethasone-functionalized Gels Induce Osteogenic Differentiation of Encapsulated hMSCs. J Biomed Mater Res, Part A. 2006;76A:183–195. doi: 10.1002/jbm.a.30537. [DOI] [PubMed] [Google Scholar]
- 40.Liu SQ, Tian Q, Wang L, Hedrick JL, Hui JHP, Yang YY, Ee PLR. Injectable Biodegradable Poly(ethylene glycol)/RGD Peptide Hybrid Hydrogels for in vitro Chondrogenesis of Human Mesenchymal Stem Cells. Macromol Rapid Commun. 2010;31:1148–1154. doi: 10.1002/marc.200900818. [DOI] [PubMed] [Google Scholar]
- 41.Jha AK, Xu X, Duncan RL, Jia X. Controlling the Adhesion and Differentiation of Mesenchymal Stem Cells Using Hyaluronic Acid-based, Doubly Crosslinked Networks. Biomaterials. 2011;32:2466–2478. doi: 10.1016/j.biomaterials.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park H, Guo X, Temenoff JS, Tabata Y, Caplan AI, Kasper FK, Mikos AG. Effect of Swelling Ratio of Injectable Hydrogel Composites on Chondrogenic Differentiation of Encapsulated Rabbit Marrow Mesenchymal Stem Cells In Vitro. Biomacromolecules. 2009;10:541–546. doi: 10.1021/bm801197m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lampe KJ, Bjugstad KB, Mahoney MJ. Impact of Degradable Macromer Content in a Poly(Ethylene Glycol) Hydrogel on Neural Cell Metabolic Activity, Redox State, Proliferation, and Differentiation. Tissue Eng, Part A. 2010;16:1857–1866. doi: 10.1089/ten.tea.2009.0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo L, Zhang D. Cyclic Poly(r-peptoid)s and Their Block Copolymers from N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-Substituted N-Carboxylanhydrides. J Am Chem Soc. 2009;131:18072–18074. doi: 10.1021/ja907380d. [DOI] [PubMed] [Google Scholar]
- 45.Zhang D, Lahasky SH, Guo L, Lee CU, Lavan M. Polypeptoid Materials: Current Status and Future Perspectives. Macromolecules. 2012;45:5833–5841. [Google Scholar]
- 46.Fetsch C, Luxenhofer R. Highly Defined Multiblock Copolypeptoids: Pushing the Limits of Living Nucleophilic Ring-Opening Polymerization. Macromol Rapid Commun. 2012;33:1708–1713. doi: 10.1002/marc.201200189. [DOI] [PubMed] [Google Scholar]
- 47.Lahasky SH, Hu X, Zhang D. Thermoresponsive Poly(α-peptoid)s: Tuning the Cloud Point Temperatures by Composition and Architecture. ACS Macro Lett. 2012;1:580–584. doi: 10.1021/mz300017y. [DOI] [PubMed] [Google Scholar]
- 48.Fetsch C, Flecks S, Gieseler D, Marschelke C, Ulbricht J, van Pée K, Luxenhofer R. Self-Assembly of Amphiphilic Block Copolypeptoids with C2-C5 Side Chains in Aqueous Solution. Macromol Chem Phys. 2015;216:547–560. [Google Scholar]
- 49.Sun J, Zuckermann RN. Peptoid Polymers: A Highly Designable Bioinspired Material. ACS Nano. 2013;7:4715–4732. doi: 10.1021/nn4015714. [DOI] [PubMed] [Google Scholar]
- 50.Ulbricht J, Jordan R, Luxenhofer R. On the Biodegradability of Polyethylene Glycol, Polypeptoids and Poly(2-oxazoline)s. Biomaterials. 2014;35:4848–4861. doi: 10.1016/j.biomaterials.2014.02.029. [DOI] [PubMed] [Google Scholar]
- 51.Luxenhofer R, Fetsch C, Grossmann A. Polypeptoids: A Perfect Match for Molecular Definition and Macromolecular Engineering? J Polym Sci, Part A: Polym Chem. 2013;51:2731–2752. [Google Scholar]
- 52.Zuckermann RN. Peptoid Origins. Biopolymers. 2011;96:545–555. doi: 10.1002/bip.21573. [DOI] [PubMed] [Google Scholar]
- 53.Secker C, Brosnan SM, Luxenhofer R, Schlaad H. Macromol Biosci. 2015;15:881–891. doi: 10.1002/mabi.201500023. [DOI] [PubMed] [Google Scholar]
- 54.Lee CU, Smart TP, Guo L, Epps TH, III, Zhang D. Synthesis and Characterization of Amphiphilic Cyclic Diblock Copolypeptoids from N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-Carboxyanhyrdride. Macromolecules. 2011;44:9574–9585. doi: 10.1021/ma2020936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Birke A, Huesmann D, Kelsch A, Weilbacher M, Xie J, Bros M, Bopp T, Becker C, Landfester K, Barz M. Polypeptoid-block-polypeptide Copolymers: Synthesis, Characterization, and Application of Amphiphilic Block Copolypept(o)ides in Drug Formulations and Miniemulsion Techniques. Biomacromolecules. 2014;15:548–557. doi: 10.1021/bm401542z. [DOI] [PubMed] [Google Scholar]
- 56.Makino a, Kizaka-Kondoh s, Yamahara R, Hara I, Kanzaki T, Ozeki E, Hiraoka M, Kimura S. Near-infrared fluorescence tumor imaging using nanocarrier composed of poly(l-lactic acid)-block-poly(sarcosine) amphiphilic polydepsipeptide. Biomaterials. 2009;30:5156–5160. doi: 10.1016/j.biomaterials.2009.05.046. [DOI] [PubMed] [Google Scholar]
- 57.Yamamoto F, Yamahara R, Makino A, Kurihara K, Tsukada H, Hara E, Hara I, Kizaka-Kondoh S, Ohkubo Y, Ozeki E, Kimura S. Radiosynthesis and initial evaluation of 18F labeled nanocarrier composed of poly(L-lactic acid)-block-poly-(sarcosine) amphiphilic polydepsipeptide. Nucl Med Biol. 2013;40:387–394. doi: 10.1016/j.nucmedbio.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 58.Makino A, Hara E, Hara I, Yamahara R, Kurihara K, Ozeki E, Yamamoto F, Kimura S. Control of in vivo blood clearance time of polymeric micelle by stereochemistry of amphiphilic polydepsipeptides. J Controlled Release. 2012;161:821–825. doi: 10.1016/j.jconrel.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 59.Robinson JW, Secker C, Weidner S, Schlaad H. Thermoresponsive Poly(N-C3 glycine)s. Macromolecules. 2013;46:580–587. [Google Scholar]
- 60.Robinson JW, Schlaad H. A versatile polypeptoid platform based on N-allyl glycine. Chem Commun. 2012;48:7835–7837. doi: 10.1039/c2cc33881e. [DOI] [PubMed] [Google Scholar]
- 61.Chaudhuri O, Koshy ST, Branco da Cunha C, Shin JW, Verbeke CS, Allison KH, Mooney DJ. Extracellular Matrix Stiffness and Composition Jointly Regulate the Induction of Malignant Phenotypes in Mammary Epithelium. Nat Mater. 2014;13:970–978. doi: 10.1038/nmat4009. [DOI] [PubMed] [Google Scholar]
- 62.Chung JE, Yokoyama M, Aoyagi T, Sakurai Y, Okano T. Effect of Molecular Architecture of Hydrophobically Modified Poly(N-isopropylacrylamide) on the Formation of Thermoresponsive Coreshell Micellar Drug Carriers. J Controlled Release. 1998;53:119–130. doi: 10.1016/s0168-3659(97)00244-7. [DOI] [PubMed] [Google Scholar]
- 63.Hidalgo-Cuadrado N, Pérez-Galende P, Manzano T, De Maria CG, Shnyrov VL, Roig MG. Screening of Postharvest Agricultural Wastes as Alternative Sources of Peroxidases: Characterization and Kinetics of a Novel Peroxidase from Lentil (Lens culinaris L.) Stubble. J Agric Food Chem. 2012;60:4765–4772. doi: 10.1021/jf300245z. [DOI] [PubMed] [Google Scholar]
- 64.Zhao Q, Eberspaecher H, Lefebvre V, de Crombrugghe B. Parallel Expression of Sox9 and Col2a1 in Cells Undergoing Chondrogenesis. Dev Dyn. 1997;209:377–386. doi: 10.1002/(SICI)1097-0177(199708)209:4<377::AID-AJA5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 65.Estes BT, Wu AW, Guilak F. Potent Induction of Chondrocytic Differentiation of Human Adipose-derived Adult Stem Cells by Bone Morphogenetic Protein 6. Arthritis Rheum. 2006;54:1222–1232. doi: 10.1002/art.21779. [DOI] [PubMed] [Google Scholar]
- 66.Kishiya M, Sawada T, Kanemaru K, Kudo H, Numasawa T, Yokoyama T, Tanaka S, Motomura S, Ueyama K, Harata S, et al. A Functional RNAi Screen for Runx2-regulated Genes Associated with Ectopic Bone Formation in Human Spinal Ligaments. J Pharmacol Sci. 2008;106:404–414. doi: 10.1254/jphs.fp0072043. [DOI] [PubMed] [Google Scholar]
- 67.Choi BG, Park MH, Cho SH, Joo MK, Oh HJ, Kim EH, Park K, Han DK, Jeong B. In Situ Thermal Gelling Polypeptide for Chondrocytes 3D Culture. Biomaterials. 2010;31:9266–9272. doi: 10.1016/j.biomaterials.2010.08.067. [DOI] [PubMed] [Google Scholar]
- 68.Yeon B, Park MH, Moon HJ, Kim SJ, Cheon YW, Jeong B. 3D Culture of Adipose-Tissue-Derived Stem Cells Mainly Leads to Chondrogenesis in Poly(ethylene glycol)-Poly(L-alanine) Diblock Copolymer Thermogel. Biomacromolecules. 2013;14:3256–3266. doi: 10.1021/bm400868j. [DOI] [PubMed] [Google Scholar]
- 69.Ko CY, Yang CY, Yang SR, Ku KL, Tsao CK, Chwei-Chin Chuang D, Chu IM, Cheng MH. Cartilage Formation Through Alterations of Amphiphilicity of Poly(ethylene glycol)-poly(caprolactone) Copolymer Hydrogels. RSC Adv. 2013;3:25769–25779. [Google Scholar]
- 70.Kye EJ, Kim SJ, Park MH, Moon HJ, Ryu KH, Jeong B. Differentiation of Tonsil-Tissue-Derived Mesenchymal Stem Cells Controlled by Surface-Functionalized Microspheres in PEG-Polypeptide Thermogels. Biomacromolecules. 2014;15(6):2180–2187. doi: 10.1021/bm500342r. [DOI] [PubMed] [Google Scholar]
- 71.Tan F, Xu X, Deng T, Yin M, Zhang X, Wang J. Fabrication of Positively Charged Poly(ethylene glycol)-diacrylate Hydrogel as a Bone Tissue Engineering Scaffold. Biomed Mater. 2012;7:055009. doi: 10.1088/1748-6041/7/5/055009. [DOI] [PubMed] [Google Scholar]
- 72.Bryant SJ, Durand KL, Anseth KS. Manipulations in Hydrogel Chemistry Control Photoencapsulated Chondrocyte Behavior and Their Extracellular Matrix Production. J Biomed Mater Res. 2003;67A:1430–1436. doi: 10.1002/jbm.a.20003. [DOI] [PubMed] [Google Scholar]
- 73.Tang J, Peng R, Ding J. The Regulation of Stem Cell Differentiation by Cell-cell Contact on Micropatterned Material Surfaces. Biomaterials. 2010;31:2470–2476. doi: 10.1016/j.biomaterials.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 74.Zhang L, Yuan T, Guo L, Zhang X. An in Vitro Study of Collagen Hydrogel to Induce the Chondrogenic Differentiation of Mesenchymal Stem Cells. J Biomed Mater Res, Part A. 2012;100A:2717–2725. doi: 10.1002/jbm.a.34194. [DOI] [PubMed] [Google Scholar]
- 75.Wolf F, Candrian C, Wendt D, Farhadi J, Heberer M, Martin I, Barbero A. Cartilage Tissue Engineering Using Pre-aggregated Human Articular Chondrocytes. Eur Cell Mater. 2008;16:92–99. doi: 10.22203/ecm.v016a10. [DOI] [PubMed] [Google Scholar]
- 76.Moreira Teixeira LS, Leijten JC, Sobral J, Jin R, Van Apeldoom AA, Feijen J, Van Blitterswijk C, Dijkstra PJ, Karperien M. High Throughput Generated Micro-aggregates of Chondrocytes Stimulate Cartilage Formation in Vitro and in Vivo. Eur Cell Mater. 2012;5:387–399. doi: 10.22203/ecm.v023a30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.