

Abstract
In the ‘older’ literature, a definitive renal pathology was described in patients with long-standing hypokalaemia and depletion of the body's potassium reserves. The topic is relevant because possibly a quite cheaply reversible element in the course of chronic kidney disease progression could be addressed. Earlier, pathologists drew attention to vacuolar changes in renal tubular epithelium accompanied by inflammatory interstitial changes in patients with potassium losses. The diagnostic term ‘kaliopenic nephropathy’ was coined to describe such patients. Kaliopenic nephropathy now receives less emphasis than in earlier times. However, with eating disorders, laxative abuse and other potential causes, we suggest that the syndrome should be resurrected.
Keywords: bisacodyl, hypokalaemia, laxatives, melanosis coli, potassium
Introduction
Kaliopenic nephropathy seems largely to have disappeared. Bock et al. [1] described 23 chronically hypokalaemic patients, 21 women, that they followed up for >6 years (not quite) 40 years ago. The causes of hypokalaemia were malnutrition, laxative abuse, self-administered diuretic usage or a combination of these factors. The authors biopsied nine patients and found interstitial nephritis and tubular vacuolization. Gradually over time, renal function deteriorated in many of these patients; they progressed to chronic kidney disease (CKD). Since they were seldom convinced to alter their behaviours, the authors favoured a diagnosis of chronic kaliopenic nephropathy. A similar condition was described in cats; in the felines, dietary rather than behavioural or drug-related problems were more prominent [2]. A recent patient caused us to revisit this syndrome.
The case
A 58-year-old woman was referred to us because of weakness and chronic generalized pain ‘all over’. She lived in a wealthy Middle Eastern country. She claimed chronic weight loss over the past 2 years that she could not explain. Her physician had prescribed methotrexate for ‘rheumatoid factor-negative’ rheumatoid arthritis; however, we could not identify any clinical correlates of this condition. With additional questioning, she admitted to having ingested laxatives (currently bisacodyl) for years because of ‘bloating’, abdominal distension and various discomforts. She chronically ingested acetaminophen and non-steroidal anti-inflammatory drugs for pain, but denied diuretic ingestion. As far as we could discern, she had practically stopped eating altogether. On examination, she appeared in Western dress. The blood pressure was 100/60 mmHg, heart rate 76 bpm, she was afebrile, weighed 44 kg and had a body mass index (weight kg/height m2) of 17.2. She had no evidence of rheumatoid arthritis or any other chronic medical condition aside from cachexia on physical examination. She had no oedema.
Routine laboratory and imaging workup revealed haemoglobin of 8.2 g/dL and haematocrit 25 vol%; the anaemia was normochromic, normocytic (MCV 86 fL, MCH 29.6 pg and MCHC 34.4 g/dL). She had normal platelets (326 Gpt/L) and normal leucocytes (6.4 Gpt/L). The sodium was 138, potassium 2.9, calcium 2.4 and phosphate 0.4 (all mmol/L). The creatinine was 157 µmol/L. We used cystatin C to estimate the glomerular filtration rate (GFR) at 15 mol/min. The 25-OH vitamin D3 was 9.6 µg/L (normal >30 µg/L) and 1,25-OH vitamin D3 was 11.0 ng/L (normal 25–60 ng/L). The parathyroid hormone concentration was 1.68 pmol/L (normal 1.6–6.9 pmol/L). Bone alkaline phosphatase was 62.4 µg/L, which is high, (upper limit of the norm, 24.4 µg/l). Urinalysis showed some white and red blood cells, but no remarkable elements. The protein:creatinine ratio was 436 mg/g, while the albumin:creatinine ratio was 26 mg/g (slightly elevated). A capillary blood sample revealed a pH of 7.29, PaCO2 31, PaO2 75 (both mmHg) and bicarbonate 14 mmol/L. Various antibody tests for chronic systemic illnesses were negative. We collected urine for 24 h. Here, the sodium excretion was 48, potassium 14.4 and phosphate 0.7 (all mmol/L). Ultrasound studies were normal and the gastroscopy showed signs of oesophageal candidiasis. Our gastroenterologist consultant performed a requested colonoscopy. He had not been forewarned but nonetheless thought that the colon showed signs of pseudomelanosis coli and performed a biopsy (Figure 1 ). The specimen confirmed the presence of lipofuscin-laden macrophages.
Fig. 1.
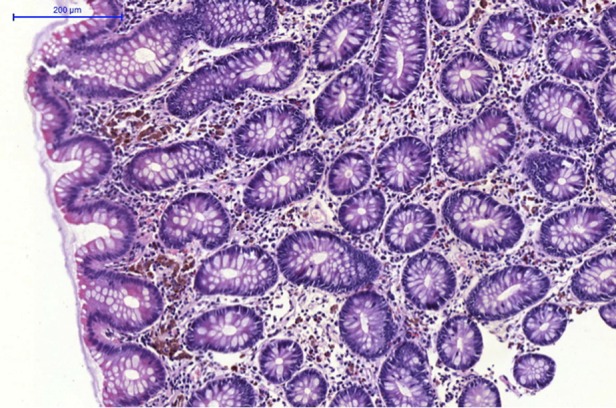
Normal-appearing colonic epithelium is seen with lipofuscin-laden macrophages residing under the mucous layer.
Since the patient had estimated GFR (eGFR) of 15 mL/min, we performed a renal biopsy (Figure 2). The glomeruli were normal (Figure 2A). Interstitial oedema and round-cell infiltrates were present (Figure 2B). Tubules were filled with hyalinaceous casts. Small vessel fibrosis was observed (Figure 2C). However, the invariable and striking feature was vacuolization of the renal tubular epithelium that could be observed in almost every section in both proximal and distal renal tubular epithelium (Figure 2D). We also performed a bone biopsy that disclosed a picture consistent with vitamin D deficiency, namely osteomalacia, and osteoporosis (not shown). We tested for renal tubular acidosis and found that our patient could acidify her urine normally. We did not specifically test her urinary concentrating ability.
Fig. 2.

(A) Normal glomerulus, several proximal tubules with widespread vacuolization and some hyaline casts. (B) Dense lymphocytic interstitial infiltrate, hyaline casts and dilated distal tubules with cellular detritus. (C) Dilated proximal tubules with vacuolization. (D) Normal proximal tubules next to proximal tubules with pronounced vacuolization.
Our treatment programme was dietetic—‘we fed her’. She appeared to cooperate and did not develop symptoms of refeeding [3]. The patient resided in a private, luxurious ward and all her wishes were answered. She was pleasant and gracious towards all clinical questions, although the answers did not invariably satisfy clinical nosiness. Finally, we could not discern how she dressed in her home environment since this state of affairs could clearly have influenced her bone metabolism. Over the ensuing month that she stayed with us, the hypokalaemia, hypophosphataemia and even the metabolic acidosis went away. We stopped the methotrexate. We never found a specific ‘cause’ of our patient's problems. However, under close observation, careful control of surreptitious medications and hospital food, the clinical picture reverted to normal and she gained 7 kg in weight. She travelled back to her country of origin with avuncular advice from us, but with a guarded prognosis nonetheless.
Discussion
We concluded that laxative abuse and self-imposed malnutrition lay at the heart of our patient's problems. Virchow is responsible for the misnomer, melanosis coli [4]. The colonic pigmentation is triggered by deposits of apoptotic cells, which are ingested by adjacent macrophages within the epithelial tissue [5]. Those macrophages migrate in the lamina propria, where lysosomes convert the remains of the cells into lipofuscin pigment. Thus, the term ‘melanosis’ is misleading, since the pigment is not melanin. We used the term ‘pseudomelanosis’ here. In addition to its association with anthraquinones, pseudomelanosis coli may appear during long-term bisacodyl administration.
Our patient did not have classic manifestations of anorexia nervosa, although similar to those patients, her condition was self-inflicted. Anorexia nervosa can affect the kidney in numerous ways, including increased rates of acute kidney injury (AKI) and CKD, electrolyte abnormalities and nephrolithiasis [6]. In anorexia nervosa, there are limitations of serum creatinine level in estimating kidney function, which caused us to rely on cystatin C estimates. Furthermore, the psychosocial challenges inherent with treating systemic manifestations of psychiatric conditions in anorexia nervosa patients are well appreciated. We expect that those in our patient will be little different.
We could identify no renal disease that could explain our patient's symptom constellation, although she clearly had one. An eGFR of 15 mL/min has no trivial connotations. She had interstitial renal disease that could have various explanations. However, the dominant histological finding was the intense vacuolarization of the renal tubular epithelium. Renal tubular vacuoles are nothing new. We were interested to review the report by Jaffé and Sternberg [7], who, to our knowledge, were the first to describe vacuolarization in renal tubular cells after hypokalaemia. Clinicians at that time could not measure potassium. However, they were aware of diarrhoea. Renal biopsies were not done then, so the authors relied on autopsies performed on chronic diarrhoea victims. The disease conditions in their patients lasted between 6 and 21 weeks. The histology shown in their paper, also with haematoxylin and eosin staining, is very similar to the sections we display here. The authors concluded that vacuolar degeneration of renal epithelium could represent an important avenue for investigating renal diseases in general. After all, fatty, hyaline droplets, amyloid degenerations, cloudy swelling, etc. appear in all CKD-related processes.
Riemenschneider and Bohle [8] also investigated renal morphology in patients with kaliopenic nephropathy. They reviewed renal biopsies from 40 patients with hypokalaemia and hyponatraemia of an average of 10-years duration due to laxative or diuretic abuse, anorexia nervosa or chronic vomiting. Light microscopy revealed juxtaglomerular cells that were sometimes enormously enlarged. In their series, the proximal and distal tubules contained non-specific vacuoles in only 8 of 40 biopsy specimens. In 75% of the cases, the interstitial surface area was increased with predominantly focal lymphocytic cellular infiltration. Interstitial fibrosis was pronounced in emaciated patients. The pathological changes of kaliopenic nephropathy have been modelled in rats. Sarkar and Levine [9] found that the renal tubular epithelial cells showed electron-lucent vacuoles, lysosome-like dense bodies, focal cytoplasmic degradation and dilated intercellular spaces in many proximal tubules. They also described medullary cell droplets with sequestration of cytoplasmic components.
Recently, Reungjui et al. [10] drew attention to the fact that hypokalaemic nephropathy is associated with angiogenesis. They found that Sprague Dawley rats fed low-potassium diets developed peritubular capillary loss. These changes were associated with increased macrophage infiltration, increased expression of both monocyte chemoattractant protein-1 and tumour necrosis factor alpha and a loss of vascular endothelial growth factor (VEGF) and endothelial nitric oxide synthase. Renal markers of oxidative stress were increased late in the disease. The authors concluded that hypokalaemic nephropathy is associated with impaired renal angiogenesis, evidenced by progressive capillary loss, reduced endothelial cell proliferation and loss of VEGF expression. These mechanisms could clearly have played a role in our patient.
Kaliopenic nephropathy could have additional consequences [11]. Resistance to vasopressin has been described by increased prostaglandin production that inhibits adenylate cyclase production in response to vasopressin. In addition to the droplet accumulation, hyperplastic epithelial cells are a feature that could contribute to cyst formation [12]. Torres et al. [13] reported on the occurrence of reversible, medullary renal cysts in patients with chronic potassium depletion. We could identify no such cysts in our patient.
Potassium homeostasis has recently been extensively reviewed [14]. A case example from that review concerns a patient with Gitelman's syndrome, a genetic cause for chronic hypokalaemia that should engender kaliopenic nephropathy. Interestingly, we found few examples of Gitelman's syndrome and hypokalaemic nephropathy. A renal biopsy from an isolated case with renal insufficiency disclosed severe interstitial nephritis, but without the prominent droplet lesions exhibited by our patient [15]. In this condition, total body magnesium depletion may also contribute to nephropathy.
Twenty-four hour urinary sodium and potassium excretion is used to assess intake of these electrolytes. Conceivably, a high-sodium intake and a low potassium intake, perhaps by hypokalaemia-related renal injury, could influence the course of chronic renal disease. Investigators prospectively studied the association of urinary sodium and potassium excretion with CKD progression and all-cause mortality among 3939 patients with CKD in the Chronic Renal Insufficiency Cohort Study. Urinary sodium and potassium excretion was measured using three 24-h urine specimens, and CKD progression was defined as incident end-stage renal disease or halving of eGFR [16]. True to the prediction, the highest quartile of sodium excretion had a relative risk for progression of 1.54. However, similar results were obtained for the quartiles of potassium excretion. The finding that high urinary potassium excretion was significantly and independently associated with an increased risk of CKD progression was unexpected and remains inadequately explained.
An additional issue could be that total body potassium depletion and hypokalaemia could increase the risk of AKI. In isolated case reports, clinicians have postulated a contributory role of hypokalaemia involved in the pathophysiology of renal ischaemia-induced AKI, although mechanisms have not been provided [17]. Here, animal models could be elucidating.
We were surprised that little has been learned mechanistically about hypokalaemia and the increased risk of CKD. Since cats seem particularly subject to CKD when they develop hypokalaemia, perhaps they could become an appropriate animal model. Dow et al. [18] found that within a group of 186 hypokalaemic cats, high serum urea nitrogen concentrations, hyperchloraemia and high serum creatinine concentrations were the most common biochemical abnormalities. When disease diagnosis was compared among cats with severe hypokalaemia (serum potassium concentration <3.0 mmol/L) and those with moderate hypokalaemia, cats with severe hypokalaemia were 3.5 times more likely to have CKD than cats with less severe hypokalaemia. Unfortunately, the authors could provide no biopsy data.
Despite the fact that the term kaliopenic nephropathy has disappeared from the literature in the last 20 years, we believe the condition still exists. Perhaps we are dealing with a semantic definition. The term hypokalaemic nephropathy with CKD is found more often. A hypokalaemic patient with an aldosterone-producing tumour was reported, but that patient exhibited tubulointerstitial injury [19]. Nevertheless, eating and laxative disorders as exhibited by our patient remain important in the pathogenesis of chronic renal disease [20].
Conflict of interest statement
None declared.
References
- 1.Bock KD, Cremer W, Werner U. Chronic hypokalemic nephropathy: a clinical study. Klin Wochenschr 1978; 56 (Suppl 1): 91–96 [DOI] [PubMed] [Google Scholar]
- 2.Fettman MJ. Feline kaliopenic polymyopathy/nephropathy syndrome. Vet Clin North Am Small Anim Pract 1989; 19: 415–432 [DOI] [PubMed] [Google Scholar]
- 3.O'Connor G, Nicholls D. Refeeding hypophosphatemia in adolescents with anorexia nervosa: a systematic review. Nutr Clin Pract 2013; 28: 358–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wittoesch JH, Jackman RJ, McDonald JR. Melanosis coli: general review and a study of 887 cases. Dis Colon Rectum 1958; 1: 172–180 [DOI] [PubMed] [Google Scholar]
- 5.Mennecier D, Nizou C, Moulin O et al. . Images in medicine. Melanosis coli. Presse Med 1999; 28: 106. [PubMed] [Google Scholar]
- 6.Bouquegneau A, Dubois BE, Krzesinski JM et al. . Anorexia nervosa and the kidney. Am J Kidney Dis 2012; 60: 299–307 [DOI] [PubMed] [Google Scholar]
- 7.Jaffé RH, Sternberg H. Über die vakuoläre nierendegeneration bei chronischer ruhr. Virch Arch 1920; 227: 313–319 [Google Scholar]
- 8.Riemenschneider T, Bohle A. Morphologic aspects of low-potassium and low-sodium nephropathy. Clin Nephrol 1983; 19: 271–279 [PubMed] [Google Scholar]
- 9.Sarkar K, Levine DZ. A correlated study of kidney function and ultrastructure in potassium-depleted rats. Nephron 1975; 14: 347–360 [DOI] [PubMed] [Google Scholar]
- 10.Reungjui S, Roncal CA, Sato W et al. . Hypokalemic nephropathy is associated with impaired angiogenesis. J Am Soc Nephrol 2008; 19: 125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alpern RJ, Toto RD. Hypokalemic nephropathy—a clue to cystogenesis? N Engl J Med 1990; 322: 398–399 [DOI] [PubMed] [Google Scholar]
- 12.Oliver J, Macdowell M, Welt LG et al. . The renal lesions of electrolyte imbalance. I. The structural alterations in potassium-depleted rats. J Exp Med 1957; 106: 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torres VE, Young WF Jr, Offord KP et al. . Association of hypokalemia, aldosteronism, and renal cysts. N Engl J Med 1990; 322: 345–351 [DOI] [PubMed] [Google Scholar]
- 14.Gumz ML, Rabinowitz L, Wingo CS. An integrated view of potassium homeostasis. N Engl J Med 2015; 373: 60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JH, Lee J, Han JS. Gitelman's syndrome with vomiting manifested by severe metabolic alkalosis and progressive renal insufficiency. Tohoku J Exp Med 2013; 231: 165–169 [DOI] [PubMed] [Google Scholar]
- 16.He J, Mills KT, Appel LJ et al. . Urinary sodium and potassium excretion and CKD progression. J Am Soc Nephrol 2015; doi:10.1681/ASN.2015010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee EY, Yoon H, Yi JH et al. . Does hypokalemia contribute to acute kidney injury in chronic laxative abuse? Kidney Res Clin Pract 2015; 34: 109–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dow SW, Fettman MJ, Curtis CR et al. . Hypokalemia in cats: 186 cases (1984–1987). J Am Vet Med Assoc 1989; 194: 1604–1608 [PubMed] [Google Scholar]
- 19.Amoedo ML, Martin ML, Muray S et al. . Hypokaliemic nephropathy as a form of presentation of conn syndrome. Nefrologia 2006; 26: 274–277 [PubMed] [Google Scholar]
- 20.Abdel-Rahman EM, Moorthy AV. End-stage renal disease (ESRD) in patients with eating disorders. Clin Nephrol 1997; 47: 106–111 [PubMed] [Google Scholar]