

Abstract
The preparation of sp3-rich scaffolds to obtain more natural product–like libraries for incorporation into screening decks is challenging. Here, we describe the use of a Diels-Alder reaction between an enone and an azide–containing silyloxydiene to gain efficient access to complex tricyclic amine scaffolds. Derivatization of these scaffolds provided a library of 80 amines, amides, sulfonamides, quinolines and indolenines, all in >20 mg quantities and >90% purities. These library compounds displayed properties more similar to alkaloid natural products than to drugs and commercial drug-like libraries, as shown by a high proportion of sp3 carbon centers.
Keywords: Parallel synthesis, Diels-Alder cycloaddition, polycyclic heterocycles
Graphical Abstract
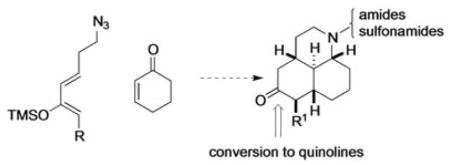
1. Introduction
A growing movement in the drug discovery community has been the introduction of compounds with greater sp3 character into screening collections.1 Such compounds exhibit higher structural complexity than more planar analogs, thereby making them more akin to natural products and better able to probe different regions of chemical space.2 As natural products are extensively employed as drugs, the creation of more drug-like libraries has been proposed to increase the likelihood of finding biological hits.3 In order to create such sp3–rich collections of molecules, methodology needs to be developed to allow the efficient synthesis of these relatively complex scaffolds. Common approaches to this challenge includes the use of natural products themselves as starting points for diversification,4 through diverted total synthesis where advanced intermediates in the synthesis of natural products are repurposed to create analogs of the target molecule,5 and by diversity orientated synthesis.6 We have recently described an approach to diverse scaffolds that contain similar structural features to biologically active alkaloid natural products.7
During this work, we developed a number of enabling technologies to allow access to these complex scaffolds, including a Diels–Alder/Schmidt domino reaction. This combines the most prevalent method for carbocycle formation with a useful method of nitrogen insertion and ring expansion, and allows efficient access to polycyclic amides and lactams.8 This methodology permitted the efficient construction of 5,6,7-tricyclic lactam scaffolds 2 through a Lewis acid-promoted Diels-Alder/Schmidt reaction of azidoenone 1 with a number of silyloxydienes (Scheme 1a).7b By simply tethering the azide to the diene rather than the dienophile, the structurally similar, but 3-dimensionally rather different, lactams 4 were obtained (Scheme 1b).7a,9 In the latter Diels–Alder/Schmidt reaction, an excess (2.5 to 3 equiv) of Lewis acid is required for the lactam formation. However, by reducing the Lewis acid loading to 1 equiv, the domino reaction can be interrupted so that the Diels–Alder cycloaddition step occurs without subsequent alkyl azide Schmidt reaction (Scheme 1c). This was previously employed to synthesize a small (23 member) library of amines and amides, the biological testing of which identified a number of compounds as potent and selective Sigma-1 inhibitors (Ki≥2 nM).7a This promising biological data, as well the as the under-representation of this scaffold in the literature prompted us to revisit this chemistry with the goal of creating a ca. 100 member library containing greater structural diversity. In particular, we wanted to obtain scaffolds with different substituents at C-5, and to prepare library compounds containing functional groups other than amines and amides.
Scheme 1.

Routes to tricyclic N-containing scaffolds
2. Results and Discussion
2. 1 Scaffold preparation
Our synthetic route to tricyclic amine scaffolds 6a–d is outlined in Scheme 2, and represents an improved protocol over the previously reported synthesis of 6b (R = Et).7a Horner–Wadsworth–Emmons reaction of β-ketophosphonates 7a–d with 3-azidopropanal and treatment of the resulting enone with TMSOTf and Et3N afforded silyloxydienes 3a–d. The key Diels–Alder reaction of cyclohexenone with 3a–d was then carried out using 1 equiv of BF3•OEt2 in the presence of flame-dried molecular sieves to afford the cycloadducts 5a–d as a mixture of diastereoisomers. The C-5 chiral center can be epimerized in later transformations, and so this diastereomeric mixture was directly subjected to an aza-Wittig reaction with triphenylphosphine. Reductive amination of the resulting primary amine forms the desired tricyclic scaffold, again as a mixture of diastereoisomers. Under the acid conditions employed, tautomerization of the imine intermediate to its enamine form results in inversion of the C-8a center. Finally, epimerization of the C-5 stereocenter with NaOMe followed by purification by silica gel chromatography allowed the isolation of diastereomerically pure (>9:1 dr) amines 6b–d in multigram (up to 5.0 g) quantities. Compared to the previous route, this method is advantageous as the difficult separation of isomeric azides 5 is avoided, and epimerization of the C-5 center is carried out prior to scaffold diversification. This chemistry can also be used to install alternative R groups at C-5. The sequence where R = H was not as synthetically useful due to consistently low-yielding steps (mainly caused by difficulties in purification/instability to silica gel), nonetheless 800 mg of 6a could be obtained along with a diastereoisomer (see Supporting Information).
Scheme 2.
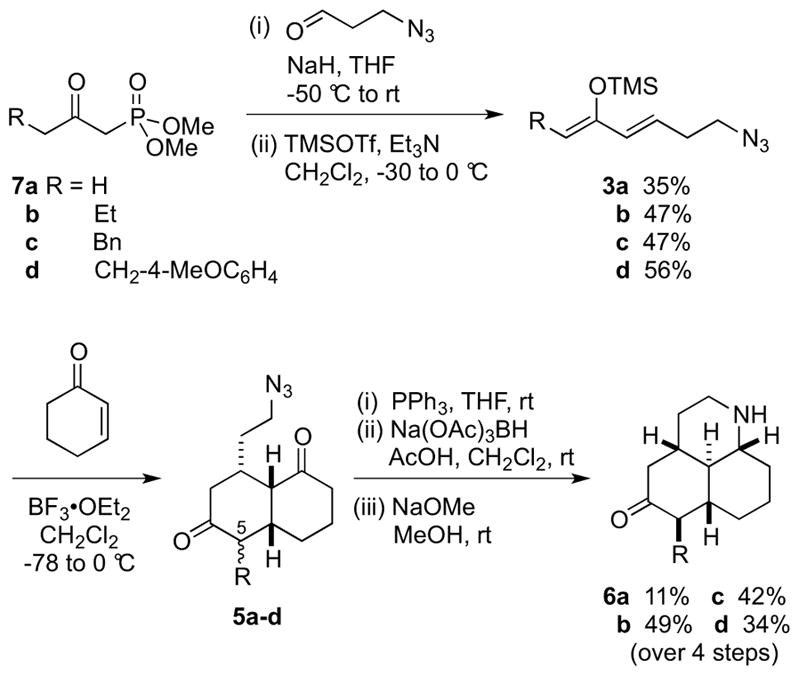
Preparation of scaffolds
2. 2 Library synthesis
During our first foray into tricyclic amine scaffolds 6, we found that only compounds containing a basic amine displayed significant activity against the Sigma receptors, corresponding amides being inactive at these targets. We therefore wished to add further amines to our screening deck to probe the effect of changing the amine substituent and the R1 group on the biological activity (Scheme 3). However, we were also interested in the activity of these scaffolds against other biological targets, and so the secondary amine of 6 was also decorated with different functional groups to provide libraries of amides and sulfonamides, thereby expanding the volume of chemical space covered by this collection. We also wanted to incorporate aromatic heterocycles into these scaffolds as not only are such moieties extensively employed in drug molecules,10 but quinolines derived from related tricyclic scaffolds have been shown to be active at the Sigma-2 receptor.7a
Scheme 3.
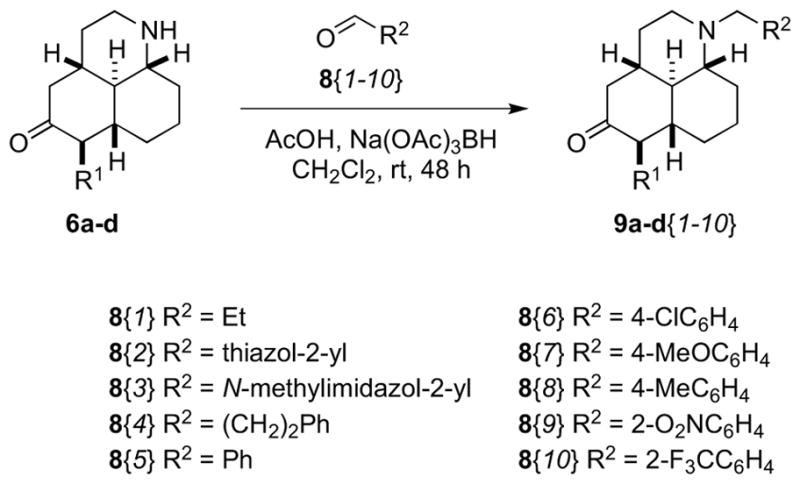
Synthesis of tertiary amine libraries
A library of tertiary amines was prepared by reductive amination with scaffolds 6b–d with ten aldehydes 8 to afford 25 amines 9b–d (Scheme 3). Of these library members, 20 passed our quality hurdles (>20 mg quantities, >90% purity) after mass-directed purification. Scaffold 6a, along with its diastereoisomer (see Supporting Information) was reacted with propionaldehyde to afford two more amines.
A library of amides was prepared by an EDC-promoted coupling of scaffolds 6b–d with 12 carboxylic acids (Scheme 4). We were able to employ acyclic and cyclic aliphatic, and aromatic acids in this reaction, to afford 28 new compounds (25 of which passed our quality controls after mass-directed purification).
Scheme 4.
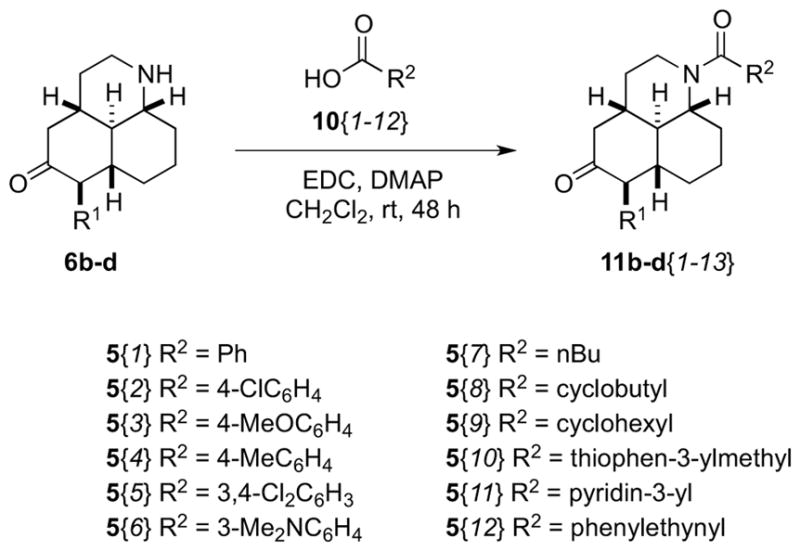
Synthesis of amide libraries
Reaction of scaffolds 6b–d with various sulfonyl chlorides afforded a library of 28 aromatic and heteroaromatic sulfonamides, of which 22 were isolated in >20 mg quantities and >90% purities after mass-directed purification (Scheme 5). We also attempted to incorporate aliphatic sulfonamides into our library by treatment with methylsulfonyl chloride, however this reaction proved unsuccessful, with no desired product isolated. Although we did not carefully examine this, it is possible that mesyl chloride underwent base-mediated elimination before it could react with the relatively hindered amine partner.
Scheme 5.
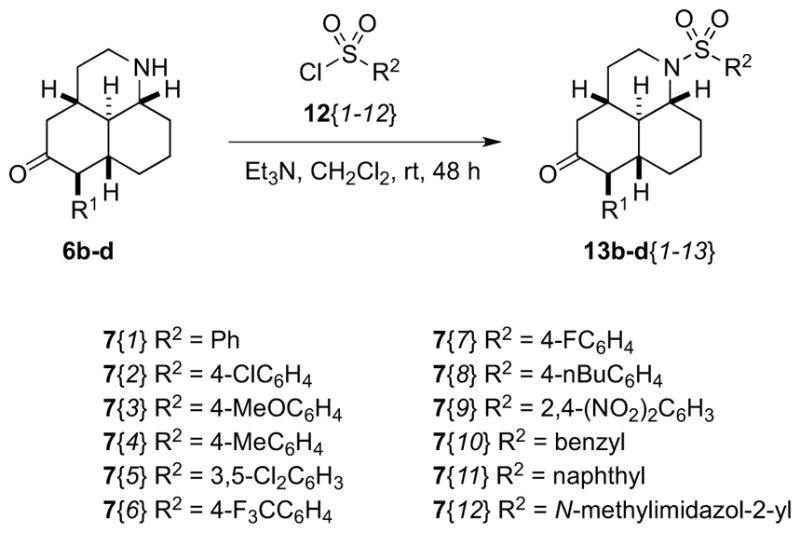
Synthesis of sulfonamide libraries
To prepare compounds containing an indole or quinoline moiety, the synthesis of library member 4b{2} was scaled up to provide 3 g of this tertiary amine. Fisher indole synthesis of 4b{2}with aryl hydrazines and para-toluenesulfonic acid (PTSA) afforded a library of six indolenines 15 as single diastereoisomers (5/6 in >20 mg and >90% purity, Scheme 6), the relative stereochemistry of which was assigned by consideration of the electrocyclization transition state (Figure 1). A small library of 6 quinolines 17 was also prepared using a one-pot modified Friedländer quinoline synthesis protocol.11 Each 2-nitrobenzaldehyde 16 was reduced in situ with Fe0 powder before addition of ketone scaffold 4b{2} and KOH to provide quinolines 17, all in >20 mg quantities and >90% purities after mass-directed purification.
Scheme 6.

Synthesis of indolenine and quinoline libraries
Figure 1.
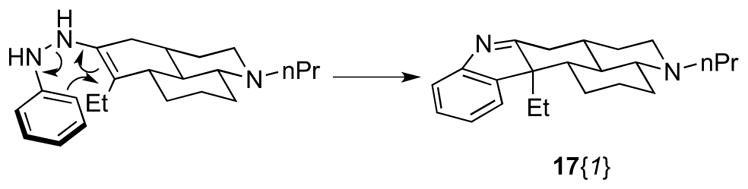
Proposed transition state of electrocyclization leading to indolenine 17{1}
2.3 Cheminformatic analysis
In total, 95 library members were prepared, of which 80 (84%) were isolated in >20 mg quantities and >90% purities. As the compounds are to be included in screening decks, it was desirable to gain an insight as to their expected bioavailability properties. To this effect, the molecular weight (m/w), XlogP, number hydrogen bond donors (HBD) and acceptors (HBA), number of rotatable bonds (rotB) and topological polar surface area (tPSA) were calculated (mean values shown Table 1; see Supporting Information for details). For comparison, the equivalent properties for a set of drugs, natural products (NPs), alkaloid natural products and commercial drug-like libraries are also displayed in Table 1.7b,12 The scaffolds and library compounds were all found to be Veber-compliant, while 59% of library members and 100% of scaffolds met all four of Lipinski’s rules. Non-compliance of Lipinski’s rules for the library compounds was predominantly caused by high XlogP values (average 4.5), presumably arising from the extensive use of aromatic rings used to decorate the scaffolds; the average XlogP of the scaffolds themselves was considerably lower (average 2.5). The XlogP aside, our library compounds were more similar to alkaloid natural products than to the other classes of molecules. In particular they were more rigid and possessed a smaller polar surface area (RotB 4.2, tPSA 43.0) than drugs (6.3, 68.9), and were more akin to alkaloids (2.8, 53.7).
Table 1.
Bioavailability properties and Fsp3 analysis
| Drugs | NPs | Commercial | Alkaloid NPs | Scaffolds | Library Cmpds | |
|---|---|---|---|---|---|---|
| m/w | 361 | 629 | 414 | 319 | 253 | 396 |
| XlogP | 2.7 | 1.5 | 2.4 | 2.0 | 2.5 | 4.5 |
| HBD | 1.5 | 4.9 | 1.5 | 1.3 | 1.0 | 0.0 |
| HBA | 5.4 | 10.8 | 6.8 | 4.4 | 2.3 | 3.1 |
|
| ||||||
| % Pass Lipinski (4/4) | 85 | 42 | 95 | 90 | 100 | 59 |
| % Pass Lipinski (3/4) | 98 | 53 | 100 | 100 | 100 | 100 |
|
| ||||||
| RotB | 6.3 | 9.7 | 5.7 | 2.8 | 1.5 | 4.2 |
| tPSA | 68.9 | 183.2 | 98.3 | 53.7 | 31.4 | 43.0 |
|
| ||||||
| % Pass Veber (2/2) | 88 | 33 | 95 | 95 | 100 | 99 |
| Fsp3 | 0.41 | 0.64 | 0.23 | 0.65 | 0.78 | 0.60 |
The average degree of carbon atom saturation (fraction sp3 (Fsp3))1a for the scaffolds and library compounds was found to be 0.78 and 0.60 respectively. These values are similar to the reference set of natural products (0.64) and alkaloids (0.65) and considerably higher than those of drugs (0.41) and commercial drug-like libraries (0.23).
3. Conclusion
We have prepared a library of 95 previously unreported compounds from four tricyclic amine scaffolds, of which 80 were obtained in useful quantities (>20 mg) and purities (>90%) for incorporation into multiple screening decks. The chemical properties of these compounds were found to be more similar to natural products, in particular alkaloid natural products, than to drugs and commercial drug-like libraries, due to their relatively small polar surface area, rigid structure and high proportion of sp3 carbon centers. One global lesson from our studies and those of similarly minded laboratories4a,13 is that complex-looking molecules, prepared by total synthesis, need not be considered beyond the reach of chemists in studying the biological properties of such molecules. Conversely, we are convinced that the continuing need for precisely targeted compounds represents an important contemporary motivation for continued advances in synthetic methodology.
These compounds will be screened against the Sigma 1 and 2 receptors and/or deposited with the NIH MLSMR (molecular libraries small molecule repository), and their biological activities will be reported in due course.
4. Experimental Section
4.1 General experimental
Unless stated, all solvents and reagents were used as supplied from commercial sources. Dichloromethane and tetrahydrofuran were dried by being passed through two packed columns of neutral ammonia using a commercial solvent purification system prior to use. Parallel library syntheses were performed on a Bohdan Miniblock XT parallel solution phase syntheziser obtained from Mettler-Toledo Auto Chem. Parallel evaporation was performed on a GeneVac EZ-2 plus evaporation system. Purification of the libraries was performed by mass-directed preparative HPLC: the HPLC analysis was carried out using a Waters Acquity system with UV and mass detection with a linear gradient of 5% acetonitrile in pH 9.8 buffered aqueous NH4HCO2 to 100% MeCN at a flow rate of 0.6 mL/min. Preparative reverse-phase purification was carried out using a Waters 2767 preparative system with UV detection (Waters 2996 PAD) and mass detection (Waters Micromass ZQ) with a Waters X-Bridge C18 column (19 × 150 mm, with 19 × 10 mm guard column) eluting with a water (or pH 9.8 aqueous NH4OH)/acetonitrile gradient with a flow rate of 20 mL/min, and sample dilution in DMSO. Flash chromatography was performed using Sorbent Technologies standard grade silica gel (40–63 μm particle size, 230 Å 400 mesh). Automated column chromatography was performed on a Teledyne CombiFlashRf system with pre-packed RediSepRf columns for normal phase, and RediSepRf C18 high performance Gold columns for reverse phase separations. Infrared spectra were acquired on a PerkinElmer Spectrum 100 FT-IR spectrophotometer. NMR spectra were recorded at the frequencies stated using a Bruker AM 400 or Bruker 500 MHz AVIII with 13C-observe cryogenically-cooled probe spectrometer. Chemical shifts were referenced to δ 7.26 and 77.0 ppm from chloroform for 1H and 13C respectively. The multiplicities of 1H signals are designated by the following abbreviations: s = singlet; d = doublet; t = triplet; m = multiplet; br = broad. All 13C NMR spectra were acquired using broadband decoupled mode and assignments were determined using DEPT sequences. Mass spectra were obtained by electrospray ionization in positive ion mode.
4.2 Experimental procedures for scaffold synthesis
4.2.1 Amine scaffolds 6a and S1
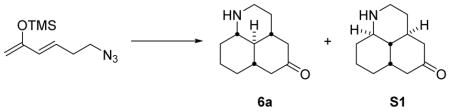
BF3·OEt2 (4.60 mL, 37.1 mmol) was added to a solution of cyclohexenone (3.60 mL, 37.1 mmol) in anhydrous CH2Cl2 (300 mL) with 4Å MS (~8.5 g) at −78 °C under argon. The mixture was stirred at −78 °C for 5 min, then a solution of silyloxydiene 3a 9 (11.8 g, 55.6 mmol) in CH2Cl2 (50 mL) was added over 10 min. The reaction was allowed to warm from −78 °C to rt and stirred for 18 h. The reaction was quenched with H2O (200 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil. The crude product was purified by automated chromatography (2 × 40 g silica columns, 0 to 10% MeOH in CH2Cl2 over 20 min). All of the fractions containing the correct mass were combined to afford an orange oil (4.00 g).
The diastereomeric mixture of above azides was dissolved in anhydrous THF (200 mL) and PPh3 (8.90 g, 34.0 mmol) added. The reaction mixture was stirred at rt for 18 h, then 2M NaOH (50 mL) added and the mixture extracted with EtOAc (3 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil. The residue was dissolved in CH2Cl2 (200 mL) and acetic acid (970 μL, 17.0 mmol) added, followed by sodium triacetoxyborohydride (7.20 g, 34.0 mmol). The reaction was stirred at rt for 18 h, then quenched with 2M NaOH (50 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown solid. The crude product was purified by chromatography (silica gel, 90:9:1 CH2Cl2:MeOH:NH4OH) to afford the title compound (800 mg, 7% from diene 3a, inseparable mixture of diastereoisomers) as a brown oil.
4.2.2 Amines 9a{1} and S2
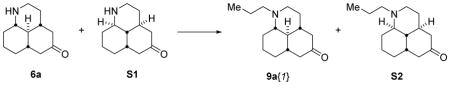
Propionaldehyde (450 μL, 6.21 mmol) was added to a solution of the diastereomeric mixture of amines 6a and S1 (800 mg, 4.14 mmol) in anhydrous CH2Cl2 (30 mL) at rt under argon. Acetic acid (240 μL, 4.14 mmol) was then added, followed by sodium triacetoxyborohydride (1.75 g, 8.28 mmol). The reaction mixture was stirred at rt for 18 h, then quenched with 2 M NaOH (10 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil (850 mg). The crude product was purified by automated reverse-phase chromatography (50 g C18 column, 10% MeCN in basic H2O to 100% over 20 min), to afford 9a{1} (286 mg, 29%) and S2 (152 mg, 16%) as pale yellow solids. 9a{1}: IR 2926, 1711 cm−1; δH (500 MHz, CDCl3) 2.95 – 2.90 (m, 1H), 2.66 – 2.58 (m, 1H), 2.45 – 2.38 (m, 1H), 2.33 – 2.24 (m, 3H), 2.12 – 2.00 (m, 3H), 1.88 – 1.77 (m, 2H), 1.65 – 1.59 (m, 2H), 1.51 – 1.26 (m, 6H), 1.18 – 1.04 (m, 3H), 0.82 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 210.2 (C), 63.8 (CH), 55.1 (CH2), 52.6 (CH2), 50.5 (CH), 48.5 (CH2), 48.4 (CH2), 41.01 (CH), 40.99 (CH), 33.8 (CH2), 33.3 (CH2), 30.0 (CH2), 24.5 (CH2), 17.5 (CH2), 12.0 (CH3); m/z (ESI+) found [M+H]+ 236.2001. C15H26NO+ requires 236.2009. S2: IR 2926, 1710 cm−1; δH (500 MHz, CDCl3) 2.96 – 2.90 (m, 1H), 2.60 – 2.53 (m, 2H), 2.49 – 2.37 (m, 2H), 2.28 – 2.21 (m, 2H), 2.19 – 1.99 (m, 5H), 1.75 – 7.64 (m, 2H), 1.62 – 1.50 (m, 2H), 1.48 – 1.24 (m, 5H), 1.14 (ddd, J = 16.5, 12.5, 4.0 Hz, 1H), 0.85 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 211.5 (C), 59.2 (CH), 54.4 (CH2), 53.1 (CH2), 48.6 (CH2), 47.6 (CH2), 45.1 (CH), 39.1 (CH), 34.9 (CH2), 32.6 (CH), 29.7 (CH2), 28.1 (CH2), 19.8 (CH2), 17.1 (CH2), 12.0 (CH3); m/z (ESI+) found [M+H]+ 236.1999. C15H26NO+ requires 236.2009.
4.2.3 Amine scaffold 6c
BF3·OEt2 (5.20 mL, 42.4 mmol) was added to a solution of cyclohexenone (4.10 mL, 42.4 mmol) in anhydrous CH2Cl2 (350 mL) with 4Å MS (~20 g) at −78 °C under argon. The mixture was stirred at −78 °C for 5 min, then a solution of silyloxydiene 3c 9 (17.9 g, 59.4 mmol) in CH2Cl2 (50 mL) was added over 10 min. The reaction was allowed to warm from −78 °C to rt and stirred for 18 h. The reaction was quenched with H2O (200 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil (19.5 g). The crude product was purified by automated chromatography (40 g silica column, 0 to 10% MeOH in CH2Cl2 over 20 min). All of the fractions containing the correct mass were combined to afford an orange oil (6.60 g).
The diastereomeric mixture of above azides was dissolved in anhydrous THF (400 mL) and PPh3 (10.6 g, 40.6 mmol) added. The reaction mixture was stirred at rt for 18 h, then 2M NaOH (60 mL) added and the mixture extracted with EtOAc (3 × 200 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil. The residue was dissolved in CH2Cl2 (350 mL) and acetic acid (1.20 mL, 20.3 mmol) added, followed by sodium triacetoxyborohydride (8.60 g, 40.6 mmol). The reaction was stirred at rt for 18 h, then quenched with 2M NaOH (70 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil. The crude product was purified by chromatography (silica gel, 90:9:1 CH2Cl2:MeOH:NH4OH) to afford the title compound (3.30 g, 20% from diene 3c, mixture of diastereoisomers) as a brown oil.
The diastereomeric mixture was dissolved in MeOH (150 mL) and K2CO3 (7.20 g, 52.4 mmol) added. The reaction was stirred at rt for 18 h, then concentrated to a brown oil, which was dissolved in CH2Cl2 (100 mL) and H2O (30 mL). The aqueous layer was extracted with CH2Cl2 (4 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford the title compound (2.44 g, 74%, >9:1 dr) as a yellow solid foam. δH (500 MHz, CDCl3) 7.26 – 7.12 (m, 5H), 3.11 – 3.05 (m, 2H), 2.79 (dd, J = 14.5, 2.6 Hz, 1H), 2.72 (ddd, J = 12.2, 12.2, 2.7 Hz, 1H), 2.43 (ddd, J = 10.8, 8.1, 1.9 Hz, 1H), 2.37 (dd, J = 12.8, 3.8 Hz, 1H), 2.31 – 2.22 (m, 1H), 2.18 – 2.12 (m, 2H), 1.89 – 1.83 (m, 2H), 1.81 – 1.76 (m, 1H), 1.66 (ddd, J = 12.9, 5.9, 2.5 Hz, 1H), 1.58 – 1.49 (m, 1H), 1.42 – 1.23 (m, 4H), 1.19 – 1.05 (m, 2H); δC (125 MHz, CDCl3) 209.9 (C), 141.5 (C), 129.3 (CH), 128.2 (CH), 125.7 (CH), 59.9 (CH), 58.0 (CH), 52.3 (CH), 48.9 (CH2), 46.2 (CH2), 46.0 (CH), 41.5 (CH), 34.3 (CH2), 33.2 (CH2), 31.9 (CH2), 31.2 (CH2), 24.3 (CH2); m/z (ESI+) found [M+H]+ 284.1991. C19H26NO+ requires 284.2009.
4.2.4 Dimethyl (4-(4-methoxyphenyl)-2-oxobutyl)phosphonate 7d
n-Butyllithium (2.5 M in hexanes, 22.0 mL, 55.1 mmol) was added slowly to a solution of dimethyl methylphosphonate (6.00 mL, 55.1 mmol) in anhydrous THF (100 mL) at −78 °C under argon. The reaction mixture was stirred at −78 °C for 1 h, then a solution methyl 3-(4-methoxyphenyl)propanoate14 (10.7 g, 55.1 mmol) in THF (50 mL) was added dropwise. The mixture was stirred at −78 °C for 5 min, then at rt for 30 min, after which the reaction was quenched with saturated aqueous NH4Cl (100 mL) and extracted with Et2O (3 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford a pale yellow oil (15.8 g). The crude product was purified by chromatography (silica gel, 100% EtOAc) to afford the title compound (9.00 g, 57%) as a colourless oil. IR 2956, 1712 cm−1; δH (400 MHz, CDCl3) 7.23 – 7.06 (m, 2H), 6.93 – 6.76 (m, 2H), 3.80 (s, 3H), 3.79 (s, 3H), 3.76 (s, 3H), 3.09 (d, J = 22.7 Hz, 2H), 2.98 – 2.84 (m, 4H); δC (100 MHz, CDCl3) 201.1 (C, d, J = 6.1 Hz), 158.0 (C), 132.6 (C), 129.3 (CH), 113.9 (CH), 55.3 (CH3), 53.1 (CH3), 53.0 (CH3), 45.8 (CH2), 41.5 (CH2, d, J = 128 Hz), 28.6 (CH2); m/z (ESI+) found [M+H]+ 287.1049. C13H20O5P+ requires 287.1043.
4.2.5 (E)-7-Azido-1-(4-methoxyphenyl)hept-4-en-3-one S3
Sodium hydride (60% dispersion in oil, 1.54 g, 38.6 mmol) was added portionwise to a solution of 7d (8.50 g, 29.7 mmol) in anhydrous THF (100 mL) at −50 °C under argon. The resulting suspension was warmed from −78 °C to 0 °C over 2.5 h, then cooled to −10 °C and a solution of 3-azidopropanal15 (3.82 g, 38.6 mmol) in THF (20 mL) was added over 5 min. The reaction mixture was stirred between 0 and −10 °C for 1 h, then at rt for 45 min. Water (50 mL) was then added and extracted with Et2O (2 × 50 mL). The combined organics were washed with brine (50 mL), dried (Na2SO4) and concentrated to afford a brown oil (10.1 g). The crude product was purified by automated chromatography (2 × 40 g columns, 0 to 25% EtOAc in hexanes over 15 min) to afford the title compound (4.5 g, 58%) as a yellow oil. IR 2935, 2094, 1511 cm−1; δH (400 MHz, CDCl3) 7.17 – 7.05 (m, 2H), 6.86 – 6.81 (m, 2H), 6.76 (dt, J = 15.9, 6.9 Hz, 1H), 6.19 (dt, J = 15.9, 1.5 Hz, 1H), 3.79 (s, 3H), 3.42 (t, J = 6.7 Hz, 2H), 3.04 – 2.80 (m, 4H), 2.49 (dtd, J = 6.9, 6.9, 1.5 Hz, 2H); δC (100 MHz, CDCl3) 199.1 (C), 158.0 (C), 142.0 (CH), 133.1 (C), 132.3 (CH), 129.3 (CH), 113.9 (CH), 55.3 (CH3), 49.7 (CH2), 42.2 (CH2), 31.8 (CH2), 29.1 (CH2); m/z (ESI+) found [M+H]+ 260.1421. C14H18N3O2+ requires 260.1394.
4.2.6 (((2Z,4E)-7-azido-1-(4-methoxyphenyl)hepta-2,4-dien-3-yl)oxy)trimethylsilane 3d
TMSOTf (4.65 mL, 25.7 mmol) was added dropwise to a solution of azide S3 (4.45 g, 17.2 mmol) and Et3N (7.18 mL, 51.5 mmol) in anhydrous CH2Cl2 (150 mL) at −30 °C under argon. The resulting yellow solution was stirred at −30 °C for 15 min then at 0 °C for 15 min, then quenched with sat. aq. NaHCO3 (50 mL) and extracted with CH2Cl2 (2 × 50 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil, which was dissolved in 4:1 hexanes: EtOAc and passed through a Florisil plug with 4:1 hexanes: EtOAc washings. The filtrate was concentrated to afford the title compound (5.50 g, 96%) as a yellow oil that was used immediately in the next step. δH (400 MHz, CDCl3) 6.93 – 6.88 (m, 2H), 6.65 – 6.60 (m, 2H), 5.80 (d, J = 15.4 Hz, 1H), 5.55 (dt, J = 15.0, 7.1 Hz, 1H), 4.71 (t, J = 7.1 Hz, 1H), 3.58 (s, 3H), 3.20 (d, J = 7.1 Hz, 2H), 3.12 (t, J = 7.1 Hz, 2H), 2.19 (dt, J = 7.1, 7.1 Hz, 2H), 0.05 (s, 9H).
4.2.7 Amine scaffold 6d
BF3·OEt2 (1.47 mL, 11.9 mmol) was added to a solution of cyclohexenone (1.15 mL, 11.9 mmol) in anhydrous CH2Cl2 (100 mL) with 4Å MS (~5 g) at −78 °C under argon. The mixture was stirred at −78 °C for 5 min, then a solution of silyloxydiene 3d (5.50 g, 16.6 mmol) in CH2Cl2 (20 mL) was added over 10 min. The reaction was allowed to warm from −78 °C to rt and stirred for 18 h. The reaction was quenched with H2O (50 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oily solid (5.72 g). The crude product was purified by automated chromatography (40 g silica column, 0 to 10% MeOH in CH2Cl2 over 20 min). All of the fractions containing the correct mass were combined to afford a brown oil (4.85 g).
The diastereomeric mixture of above azides was dissolved in anhydrous THF (200 mL) and PPh3 (7.16 g, 27.3 mmol) added. The reaction mixture was stirred at rt for 18 h, then 2M NaOH (50 mL) added and the mixture extracted with EtOAc (3 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil. The residue was dissolved in CH2Cl2 (200 mL) and acetic acid (780 μL, 13.6 mmol) added, followed by sodium triacetoxyborohydride (5.79 g, 27.3 mmol). The reaction was stirred at rt for 18 h, then quenched with 2M NaOH (50 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organics were dried (Na2SO4) and concentrated to afford a brown oil (15 g). The crude product was purified by chromatography (silica gel, 90:9:1 CH2Cl2:MeOH:NH4OH) to afford the title compound (1.70 g, 46% from silyloxydiene 3d, 4:1 dr) as a brown oil.
The diastereomeric mixture was dissolved in MeOH (50 mL) and K2CO3 (3.37 g, 24.4 mmol) added. The reaction was stirred at rt for 18 h, then concentrated to a brown oil, which was dissolved in CH2Cl2 (100 mL) and H2O (30 mL). The aqueous layer was extracted with CH2Cl2 (4 × 100 mL). The combined organics were dried (Na2SO4) and concentrated to afford the title compound (1.65 g, 97%, 9:1 dr) as a brown solid foam. IR2925, 1707, 1511 cm−1; δH (400 MHz, CDCl3) 7.12 – 7.07 (m, 2H), 6.81 – 6.75 (m, 2H), 3.77 (s, 3H), 3.32 (d, J = 12.4 Hz, 1H), 2.96 (dd, J = 14.3, 7.9 Hz, 1H), 2.88 – 2.78 (m, 2H), 2.65 – 2.58 (m, 1H), 2.49 – 2.39 (m, 2H), 2.25 – 2.07 (m, 3H), 1.94 – 1.87 (m, 1H), 1.80 – 1.52 (m, 5H), 1.42 – 1.26 (m, 2H), 1.24 – 1.13 (m, 1H); δC (100 MHz, CDCl3) 208.4 (C), 157.7 (C), 132.6 (C), 130.2 (CH), 113.7 (CH), 59.0 (CH), 57.4 (CH), 55.2 (CH3), 48.0 (CH), 47.7 (CH2), 45.2 (CH), 43.7 (CH2), 39.5 (CH), 30.9 (CH2), 30.1 (CH2), 30.1 (CH2), 29.7 (CH2), 23.8 (CH2); m/z (ESI+) found [M+H]+ 314.2137. C20H28NO2+ requires 314.2115.
4.2.8 Amine 9b{1}
Propionaldehyde (1.22 mL, 16.8 mmol) was added to a solution of amine 6b (2.48 g, 11.2 mmol) in anhydrous CH2Cl2 (100 mL) at rt under argon. Acetic acid (640 μL, 11.2 mmol) was then added, followed by sodium triacetoxyborohydride (4.75 g, 22.4 mmol). The reaction mixture was stirred at rt for 48 h, then quenched with 2 M NaOH (50 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organics were dried (Na2SO4) and concentrated to afford the title compound (2.94 g, 100%) as a yellow oil, which was used without further purification. IR 2929, 1711 cm−1; δH (400 MHz, CDCl3) 3.04-2.99 (1H, m), 2.74-2.66 (1H, m), 2.58-2.51 (1H, m), 2.40-2.32 (2H, m), 2.17-1.87 (6H, m), 1.70-1.41 (7H, m), 1.36-1.19 (4H, m), 1.10-1.00 (1H, m), 0.87 (3H, t, J 7.2), 0.85 (3H, t, J 7.6); δC (100 MHz, CDCl3) 210.8 (C), 63.9 (CH), 56.1 (CH), 54.8 (CH2), 52.4 (CH2), 50.4 (CH), 48.8 (CH2), 44.7 (CH), 41.4 (CH), 32.9 (CH2), 31.2 (CH2), 29.5 (CH2), 24.6 (CH2), 17.9 (CH2), 17.1 (CH2), 12.0 (CH3), 11.2 (CH3); ); m/z (ESI+) found [M+H]+ 264.2314. C17H30NO+ requires 264.2322.
4.2.9 Alcohol S4
L-Selectride (1 M in THF, 8.10 mL) was added dropwise to a solution of 9b{1} (850 mg, 3.23 mmol) in anhydrous THF (20 mL) at −78 °C under argon. The reaction mixture was allowed to warm slowly to rt and stirred for 18 h, then quenched with 30% aq H2O2 (5.1 mL) and 2 M NaOH (6.8 mL). The mixture was stirred at rt for 1 h, then extracted with CH2Cl2 (3 × 20 mL). The combined organics were dried (Na2SO4) and concentrated to afford a yellow oil. The crude product was purified by chromatography (silica gel, 90:9:1 CH2Cl2:MeOH:NH4OH) to afford the title compound (825 mg, 96%) as a yellow oil. δH (500 MHz, CDCl3) 4.03 (br s, 1H), 2.93 (ddd, J = 11.3, 3.8, 3.8 Hz, 1H), 2.70 – 2.56 (m, 1H), 2.55 – 2.40 (m, 1H), 2.32 (ddd, J = 12.0, 12.0, 2.5 Hz, 1H), 2.14 – 2.00 (m, 1H), 1.96 – 1.76 (m, 4H), 1.68 – 1.60 (m, 1H), 1.53 (ddd, J = 12.6, 2.7, 2.7 Hz, 1H), 1.50 – 1.40 (m, 3H), 1.41 – 0.94 (m, 7H), 0.94 – 0.87 (m, 3H), 0.84 (t, J = 7.4 Hz, 3H), 0.81 – 0.71 (m, 2H); δC (125 MHz, CDCl3) 66.5 (CH), 63.5 (CH), 55.3 (CH2), 52.7 (CH2), 51.4 (CH), 47.6 (CH), 40.3 (CH2), 38.6 (CH), 33.3 (CH), 32.8 (CH2), 29.9 (CH2), 29.8 (CH2), 24.5 (CH2), 21.4 (CH2), 17.1 (CH2), 12.0 (CH3), 11.7 (CH3); m/z (ESI+) found [M+H]+ 266.2480. C17H32NO+ requires 266.2478
4.3. Experimental procedures for library synthesis
4.3.1 General procedure for the preparation of amines
Each reaction tube of a 24-position Bohdan Miniblock XT was flushed with argon, then a solution of the amine scaffold 6b–d (70 mg) in anhydrous dichloromethane (2 mL) was added, followed by the appropriate aldehyde (1.5 equiv), acetic acid (1 equiv) and Na(OAc)3BH (2 equiv). The reactions were shaken at 500 rpm at rt for 48 h, then 2 M NaOH (1 mL) added. The reactions were passed through Isolute® hydrophobic phase separator tubes, which allowed the halogenated solvent layer to pass through. The aqueous layers were extracted with dichloromethane (2 × 2 mL). The combined organics were evaporated in a Genevac EZ-2 Plus parallel evaporator and subjected to mass-directed preparative HPLC purification to afford amines 9b–d{1–10} in average yields of 43% (range 4–76%) and purities of 97.9% (range 94.1–100%), excluding three compounds that did not meet quantity or purity requirements (9b{9}, 9d{2}, and 9d{5}). Full data are provided in Supporting Information.
4.3.2 General procedure for the preparation of amides
Each reaction tube of a 24-position Bohdan Miniblock XT was flushed with argon, then a solution of the amine scaffold 6b–d (70 mg) in anhydrous dichloromethane (2 mL) was added, followed by the appropriate acid (1.2 equiv), EDC (1.2 equiv) and DMAP (1.2 equiv). The reactions were shaken at 500 rpm at rt for 48 h, then water (2 mL) added. The reactions were passed through Isolute® hydrophobic phase separator tubes, which allowed the halogenated solvent layer to pass through. The aqueous layers were extracted with dichloromethane (2 × 2 mL). The combined organics were evaporated in a Genevac EZ-2 Plus parallel evaporator and subjected to mass-directed preparative HPLC purification to afford pure amides 11b–d{1–13} in average yields of 39% (range 20–57%) and purities of 97.3% (range 85.9–100%), excluding two compounds that did not meet quantity or purity requirements (11b{9} and 11b{11}). Full data are provided in Supporting Information.
4.3.3 General procedure for the preparation of sulfonamides
Each reaction tube of a 24-position Bohdan Miniblock XT was flushed with argon, then a solution of the amine scaffold 6b–d (70 mg) in anhydrous dichloromethane (2 mL) was added, followed by the appropriate sulfonyl chloride (1.5 equiv) and Et3N (1.5 equiv). The reactions were shaken at 500 rpm at rt for 48 h, then water (2 mL) added. The reactions were passed through Isolute® hydrophobic phase separator tubes, which allowed the halogenated solvent layer to pass through. The aqueous layers were extracted with dichloromethane (2 × 2 mL). The combined organics were evaporated in a Genevac EZ-2 Plus parallel evaporator and subjected to mass-directed preparative HPLC purification to afford pure sulfonamides 13b–d{1–13} in average yields of 39% (range 6–63%) and purities of 98.0% (range 87.8–100%), excluding three compounds that did not meet quantity or purity requirements (13b{9}, 13c{8}, and 13c{9}). Full data are provided in Supporting Information.
4.3.4 General procedure for the preparation of 3H-indoles
PTSA (510 mg, 2.3 mmol) was added to a solution of amine scaffold 4b{2} (60 mg, 0.23 mmol) in EtOH (1.5 mL) in a microwave vial, followed by the appropriate hydrazine (hydrochloride) (2.5 equiv). The vial was sealed then heated in an oil bath at 90 °C for 4 h, then cooled to rt, 2 M NaOH (2 mL) added and the mixture extracted with CH2Cl2 (3 × 5 mL). The combined organics were dried (Na2SO4) and concentrated under reduced pressure. The residues were subjected to mass-directed preparative HPLC purification to afford pure 3H-indoles 15{1–6} in average yields of 40% (range 27–57%) and purities of 99.4% (range 87.8–100%), excluding compound 15{4}, which was not obtained in sufficient purity for screening. Full data are provided in Supporting Information.
4.3.5 General procedure for the preparation of quinolines
Fe0 powder (77 mg, 1.4 mmol) was added to a solution of nitrobenzaldehyde 16{1–6} (0.35 mmol) in ethanol (1 mL) in a microwave vial, followed by 0.1 M HCl (180 μL). The vial was sealed, then heated in an oil bath at 85 °C until complete by TLC (~2 h). The mixture was cooled to rt, then a solution of ketone scaffold 4b{2} (0.27 mmol) in EtOH (2 mL) added, followed by powdered KOH (18 mg, 0.32 mmol). The mixture was heated at 85 °C for 18 h, then cooled to rt, passed through a celite plug and eluted with dichloromethane, and concentrated under reduced pressure. The residues were subjected to mass-directed preparative HPLC purification to afford pure quinolines 17{1–6} in average yields of 42% (range 26–62%) and purities of 97.0% (range 91.7–100%). Full data are provided in Supporting Information.
4.4. Characterization for representative library examples
4.4.1 Amine 9b{2}
δH (500 MHz, CDCl3) 7.72 (d, J = 3.3 Hz, 1H), 7.26 (d, J = 3.3 Hz, 1H), 4.23 (d, J = 15.8 Hz, 1H), 3.94 (d, J = 15.8 Hz, 1H), 3.05 – 2.93 (m, 1H), 2.53 – 2.40 (m, 1H), 2.34 (dd, J = 13.1, 3.3 Hz, 1H), 2.30 – 2.20 (m, 1H), 2.20 – 2.10 (m, 1H), 2.10 – 1.99 (m, 2H), 1.99 – 1.91 (m, 1H), 1.91 – 1.82 (m, 1H), 1.70 – 1.45 (m, 5H), 1.40 – 1.14 (m, 4H), 1.05 (qd, J = 13.0, 3.6 Hz, 1H), 0.84 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 210.7 (C), 170.7 (C), 142.4 (CH), 119.1 (CH), 64.3 (CH), 56.1 (CH), 54.3 (CH2), 53.9 (CH2), 50.9 (CH), 48.7 (CH2), 44.4 (CH), 41.3 (CH), 33.2 (CH2), 31.4 (CH2), 30.3 (CH2), 24.4 (CH2), 17.9 (CH2), 11.1 (CH3); m/z (ESI+) found [M+H]+ 319.1847. C18H27N2OS+ requires 319.1839.
4.4.2 Amine 9c{1}
δH (500 MHz, CDCl3) 7.26 – 7.11 (m, 5H), 3.08 (dd, J = 14.2, 8.1 Hz, 1H), 2.97 – 2.92 (m, 1H), 2.80 (dd, J = 14.2, 2.6 Hz, 1H), 2.69 – 2.61 (m, 1H), 2.52 – 2.45 (m, 1H), 2.45 – 2.38 (m, 1H), 2.38 – 2.28 (m, 2H), 2.17 – 2.10 (m, 3H), 1.92 – 1.86 (m, 2H), 1.66 – 1.61 (m, 1H), 1.52 – 1.41 (m, 4H), 1.37 – 1.07 (m, 5H), 0.85 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 210.1 (C), 141.6 (C), 129.3 (CH), 128.2 (CH), 125.6 (CH), 63.8 (CH), 57.8 (CH), 55.2 (CH2), 52.5 (CH2), 51.2 (CH), 48.9 (CH2), 46.5 (CH), 41.6 (CH), 33.4 (CH2), 32.0 (CH2), 31.2 (CH2), 30.0 (CH2), 24.6 (CH2), 17.4 (CH2), 12.0 (CH3); m/z (ESI+) found [M+H]+ 326.2516. C22H32NO+ requires 326.2478.
4.4.3 Amine 9c{2}
δH (400 MHz, CDCl3) 7.72 (d, J = 3.3 Hz, 1H), 7.28 – 7.12 (m, 6H), 4.23 (d, J = 15.8 Hz, 1H), 3.94 (d, J = 15.4 Hz, 1H), 3.08 (dd, J = 14.2, 8.1 Hz, 1H), 3.03 – 2.95 (m, 1H), 2.82 (dd, J = 14.2, 2.6 Hz, 1H), 2.51 – 2.40 (m, 2H), 2.37 (dd, J = 12.9, 3.2 Hz, 1H), 2.29 – 2.23 (m, 1H), 2.20 – 2.01 (m, 3H), 1.94 – 1.87 (m, 1H), 1.67 – 1.61 (m, 1H), 1.57 – 1.42 (m, 2H), 1.40 – 1.20 (m, 4H), 1.19 – 1.08 (m, 1H); δC (100 MHz, CDCl3) 209.8 (C), 170.9 (C), 142.5 (CH), 141.4 (C), 129.3 (CH), 128.2 (CH), 125.7 (CH), 119.1 (CH), 64.2 (CH), 57.7 (CH), 54.4 (CH2), 53.9 (CH2), 51.2 (CH), 48.7 (CH2), 46.2 (CH), 41.3 (CH), 33.3 (CH2), 32.0 (CH2), 31.2 (CH2), 30.4 (CH2), 24.4 (CH2); m/z (ESI+) found [M+H]+ 381.2039. C23H29N2OS+ requires 381.1995.
4.4.4 Amine 9d{1}
δH (500 MHz, CDCl3) 7.15 – 7.10 (m, 2H), 6.80 – 6.76 (m, 2H), 3.77 (s, 3H), 3.01 – 2.91 (m, 2H), 2.78 (dd, J = 14.2, 2.5 Hz, 1H), 2.70 – 2.61 (m, 1H), 2.54 – 2.44 (m, 1H), 2.39 – 2.27 (m, 3H), 2.17 – 2.08 (m, 3H), 1.93 – 1.85 (m, 2H), 1.67 – 1.60 (m, 1H), 1.54 – 1.38 (m, 4H), 1.36 – 1.05 (m, 5H), 0.85 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 210.2 (C), 157.6 (C), 133.4 (C), 130.3 (CH), 113.5 (CH), 63.8 (CH), 57.9 (CH), 55.21 (CH3), 55.18 (CH2), 52.5 (CH2), 51.1 (CH), 48.9 (CH2), 46.2 (CH), 41.5 (CH), 33.3 (CH2), 31.9 (CH2), 30.2 (CH2), 30.0 (CH2), 24.6 (CH2), 17.4 (CH2), 12.0 (CH3); m/z (ESI+) found [M+H]+ 356.2596. C23H34NO2+ requires 356.2584.
4.4.5 Amine 9d{4}
δH (500 MHz, CDCl3) 7.32 – 7.28 (m, 2H), 7.23 – 7.18 (m, 3H), 7.15 – 7.12 (m, 2H), 6.81 – 6.77 (m, 2H), 3.78 (s, 3H), 3.01 – 2.95 (m, 2H), 2.84 – 2.77 (m, 2H), 2.65 – 2.53 (m, 3H), 2.40 – 2.32 (m, 3H), 2.18 – 2.04 (m, 3H), 1.97 – 1.74 (m, 4H), 1.67 – 1.62 (m, 1H), 1.61 – 1.41 (m, 2H), 1.39 – 1.08 (m, 5H); δC (125 MHz, CDCl3) 210.1 (C), 157.6 (C), 141.9 (C), 133.3 (C), 130.3 (CH), 128.4 (CH), 128.3 (CH), 125.9 (CH), 113.5 (CH), 63.9 (CH), 57.8 (CH), 55.2 (CH3), 52.5 (CH2), 52.4 (CH2), 50.8 (CH), 48.7 (CH2), 46.1 (CH), 41.3 (CH), 33.8 (CH2), 33.0 (CH2), 31.8 (CH2), 30.2 (CH2), 29.7 (CH2), 26.0 (CH2), 24.5 (CH2); m/z (ESI+) found [M+H]+ 432.2902. C29H38NO2+ requires 432.2897.
4.4.6 Amine 9d{6}
δH (500 MHz, CDCl3) 7.29 – 7.23 (m, 4H), 7.15 – 7.12 (m, 2H), 6.81 – 6.77 (m, 2H), 4.09 (d, J = 13.7 Hz, 1H), 3.78 (s, 3H), 3.16 (d, J = 13.5 Hz, 1H), 2.98 (dd, J = 14.2, 8.1 Hz, 1H), 2.84 – 2.77 (m, 2H), 2.41 – 2.31 (m, 2H), 2.29 – 2.23 (m, 1H), 2.19 – 2.10 (m, 2H), 2.06 – 2.00 (m, 1H), 1.95 – 1.87 (m, 2H), 1.57 – 1.52 (m, 1H), 1.50 – 1.10 (m, 7H); δC (125 MHz, CDCl3) 210.1 (C), 157.6 (C), 138.1 (C), 132.4 (C), 133.4 (C), 130.3 (CH), 130.2 (CH), 128.3 (CH), 113.5 (CH), 65.2 (CH), 57.9 (CH), 56.7 (CH2), 55.2 (CH3), 52.9 (CH2), 51.1 (CH), 48.8 (CH2), 46.1 (CH), 41.5 (CH), 33.0 (CH2), 32.0 (CH2), 30.6 (CH2), 30.3 (CH2), 24.5 (CH2); m/z (ESI+) found [M+H]+ 438.2191. C27H33ClNO2+ requires 438.2194.
4.4.7 Amide 11b{1}
δH (500 MHz, CDCl3) 7.43 – 7.35 (m, 5H), 3.56 – 3.46 (m, 2H), 3.41 (ddd, J = 13.9, 6.1, 4.1 Hz, 1H), 2.46 (dd, J = 13.1, 4.4 Hz, 1H), 2.31 – 2.24 (m, 1H), 2.17 – 2.06 (m, 2H), 2.03 – 1.98 (m, 1H), 1.95 – 1.86 (m, 2H), 1.82 – 1.52 (m, 5H), 1.51 – 1.33 (m, 2H), 1.26 – 1.19 (m, 1H), 1.18 – 1.08 (m, 1H), 0.86 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 210.0 (C), 171.7 (C), 137.2 (C), 129.6 (CH), 128.5 (CH), 126.9 (CH), 60.2 (CH), 55.9 (CH), 49.0 (CH2), 46.4 (CH), 45.7 (CH), 43.0 (CH2), 37.0 (CH), 32.1 (CH2), 31.4 (CH2), 29.7 (CH2), 24.7 (CH2), 17.9 (CH2), 11.3 (CH3); m/z (ESI+) found [M+H]+ 326.2130. C21H28NO2+ requires 326.2115.
4.4.8 Amide 11b{4}
δH (500 MHz, CDCl3) 7.30 (d, J = 7.8 Hz, 2H), 7.18 (d, J = 7.8 Hz, 2H), 3.54 – 3.46 (m, 2H), 3.43 (ddd, J = 13.8, 6.1, 4.3 Hz, 1H), 2.45 (dd, J = 13.1, 4.4 Hz, 1H), 2.36 (s, 3H), 2.28 – 2.23 (m, 1H), 2.17 – 2.06 (m, 2H), 2.03 – 1.97 (m, 1H), 1.94 – 1.85 (m, 2H), 1.81 – 1.51 (m, 5H), 1.50 – 1.33 (m, 2H), 1.25 – 1.18 (m, 1H), 1.17 – 1.08 (m, 1H), 0.86 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 210.1 (C), 172.0 (C), 139.7 (C), 134.3 (C), 129.0 (CH), 127.0 (CH), 60.2 (CH), 55.9 (CH), 49.0 (CH2), 46.4 (CH), 45.7 (CH), 43.3 (CH2), 37.1 (CH), 32.2 (CH2), 31.4 (CH2), 29.7 (CH2), 24.7 (CH2), 21.4 (CH3), 17.9 (CH2), 11.3 (CH3); m/z (ESI+) found [M+H]+ 340.2284. C22H30NO2+ requires 340.2271.
4.4.9 Amide 11c{8}
δH (500 MHz, CDCl3) 7.26 – 7.12 (m, 5H), 3.56 – 3.38 (m, 2H), 3.30 – 3.16 (m, 2H), 3.05 (dd, J = 14.2, 8.5 Hz, 1H), 2.76 (dd, J = 14.2, 2.6 Hz, 1H), 2.51 – 2.44 (m, 2H), 2.38 – 2.27 (m, 2H), 2.22 – 2.07 (m, 5H), 2.06 – 1.80 (m, 4H), 1.75 – 1.64 (m, 1H), 1.61 – 1.33 (m, 4H), 1.29 – 1.13 (m, 2H); δC (125 MHz, CDCl3) 209.3 (C), 173.8 (C), 141.2 (C), 129.2 (CH), 128.2 (CH), 125.8 (CH), 59.3 (CH), 57.3 (CH), 49.1 (CH2), 47.4 (CH), 46.4 (CH), 37.8 (CH), 35.9 (CH), 32.3 (CH2), 31.7 (CH2), 31.1 (CH2), 30.2 (CH2), 25.3 (CH2), 25.1 (CH2), 24.5 (CH2), 18.0 (CH2); m/z (ESI+) found [M+H]+ 366.2454. C24H32NO2+ requires 366.2428.
4.4.10 Amide 11c{10}
δH (500 MHz, CDCl3) 7.29 – 7.27 (m, 1H), 7.25 – 7.21 (m, 2H), 7.19 – 7.12 (m, 3H), 7.05 – 7.03 (m, 1H), 6.99 (dd, J = 4.9, 1.3 Hz, 1H), 3.69 (s, 1H), 3.64 (br s, 1H), 3.52 – 3.45 (m, 1H), 3.30 – 3.21 (m, 1H), 3.04 (dd, J = 14.2, 8.5 Hz, 1H), 2.75 (dd, J = 14.2, 2.6 Hz, 1H), 2.49 – 2.41 (m, 2H), 2.22 – 2.15 (m, 2H), 2.08 (t, J = 12.3 Hz, 2H), 1.93 – 1.86 (m, 1H), 1.84 – 1.73 (m, 1H), 1.67 – 1.31 (m, 5H), 1.22 – 1.12 (m, 2H); δC (125 MHz, CDCl3) 209.2 (C), 169.8 (C), 141.2 (C), 135.1 (C), 129.2 (CH), 128.2 (CH), 128.0 (CH), 126.0 (CH), 125.8 (CH), 121.7 (CH), 59.4 (CH), 57.1 (CH), 48.9 (CH2), 47.4 (CH), 46.3 (CH), 39.0 (CH2), 36.7 (CH2), 35.7 (CH), 32.2 (CH2), 31.2 (CH2), 31.1 (CH2), 30.1 (CH2), 24.5 (CH2); m/z (ESI+) found [M+H]+ 408.2026. C25H30NO2S+ requires 408.1992.
4.4.11 Amide 11d{7}
δH (500 MHz, CDCl3) 7.13 – 7.09 (m, 2H), 6.80 – 6.76 (m, 2H), 3.76 (s, 3H), 3.76 – 3.67 (m, 1H), 3.44 (dt, J = 10.7, 5.5 Hz, 1H), 3.31 – 3.21 (m, 1H), 2.96 (dd, J = 14.2, 8.5 Hz, 1H), 2.73 (dd, J = 14.2, 2.5 Hz, 1H), 2.48 (dd, J = 12.8, 4.6 Hz, 1H), 2.42 (ddd, J = 11.0, 8.6, 2.0 Hz, 1H), 2.34 – 2.16 (m, 3H), 2.15 – 2.03 (m, 3H), 1.91 – 1.85 (m, 1H), 1.75 – 1.65 (m, 1H), 1.63 – 1.55 (m, 3H), 1.54 – 1.24 (m, 6H), 1.23 – 1.13 (m, 1H), 0.92 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 209.5 (C), 172.4 (C), 157.7 (C), 133.1 (C), 130.2 (CH), 113.6 (CH), 59.3 (CH), 57.4 (CH), 55.2 (CH3), 49.1 (CH2), 47.2 (CH), 46.4 (CH), 38.1 (CH2), 35.7 (CH), 33.6 (CH2), 32.3 (CH2), 31.5 (CH2), 30.4 (CH2), 30.1 (CH2), 27.6 (CH2), 24.5 (CH2), 22.6 (CH2), 14.0 (CH3); m/z (ESI+) found [M+H]+ 398.2692. C25H36NO3+ requires 398.2690.
4.4.12 Sulfonamide 13b{12}
δH (500 MHz, CDCl3) 7.46 (d, J = 1.2 Hz, 1H), 7.39 (d, J = 1.2 Hz, 1H), 4.15 (dt, J = 13.1, 3.9 Hz, 1H), 3.75 (s, 3H), 2.97 – 2.91 (m, 1H), 2.70 (ddd, J = 11.8, 9.5, 3.8 Hz, 1H), 2.47 – 2.42 (m, 1H), 2.33 (dd, J = 13.4, 3.1 Hz, 1H), 2.17 – 2.10 (m, 1H), 2.07 – 2.02 (m, 1H), 1.90 – 1.82 (m, 2H), 1.80 – 1.61 (m, 3H), 1.57 – 1.42 (m, 4H), 1.31 – 1.14 (m, 2H), 1.07 – 0.98 (m, 1H), 0.80 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 210.0 (C), 140.9 (C), 138.8 (CH), 123.6 (CH), 63.6 (CH), 55.8 (CH), 49.8 (CH), 48.6 (CH2), 48.5 (CH2), 44.8 (CH), 40.9 (CH), 34.1 (CH3), 32.8 (CH2), 31.0 (CH2), 30.6 (CH2), 24.8 (CH2), 17.8 (CH2), 10.9 (CH3); m/z (ESI+) found [M+H]+ 366.1856. C18H28N3O3S+ requires 366.1846.
4.4.13 Sulfonamide 13b{11}
δH (500 MHz, CDCl3) 8.38 – 8.36 (m, 1H), 7.99 – 7.95 (m, 2H), 7.92 (d, J = 8.1 Hz, 1H), 7.77 (dd, J = 8.7, 1.9 Hz, 1H), 7.67 – 7.60 (m, 2H), 4.35 (dt, J = 13.0, 3.7 Hz, 1H), 2.96 – 2.89 (m, 1H), 2.73 – 2.67 (m, 1H), 2.36 (dd, J = 13.5, 3.4 Hz, 1H), 2.20 – 2.13 (m, 2H), 2.09 – 2.03 (m, 1H), 1.88 – 1.73 (m, 4H), 1.70 – 1.46 (m, 5H), 1.29 – 1.22 (m, 1H), 1.17 – 0.98 (m, 2H), 0.81 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 209.6 (C), 138.4 (C), 134.7 (C), 132.2 (C), 129.3 (CH), 129.2 (CH), 128.7 (CH), 128.0 (CH), 127.9 (CH), 127.5 (CH), 122.4 (CH), 63.8 (CH), 55.9 (CH), 49.8 (CH), 48.7 (CH2), 48.5 (CH2), 44.8 (CH), 41.2 (CH), 33.2 (CH2), 30.7 (CH2), 30.5 (CH2), 24.8 (CH2), 17.8 (CH2), 10.9 (CH3); m/z (ESI+) found [M+H]+ 412.1971. C24H30NO3S+ requires 412.1941.
4.4.14 Sulfonamide 13c{7}
δH (500 MHz, CDCl3) 7.73 – 7.69 (m, 2H), 7.24 – 7.20 (m, 2H), 7.18 – 7.11 (m, 3H), 6.98 – 6.94 (m, 2H), 4.20 (dt, J = 12.9, 3.8 Hz, 1H), 3.87 (s, 3H), 3.01 (dd, J = 14.3, 7.9 Hz, 1H), 2.84 – 2.76 (m, 2H), 2.57 (ddd, J = 11.9, 9.7, 3.7 Hz, 1H), 2.45 (ddd, J = 10.8, 8.0, 2.3 Hz, 1H), 2.37 (dd, J = 13.3, 3.4 Hz, 1H), 2.20 – 2.13 (m, 2H), 2.07 – 2.03 (m, 1H), 1.86 – 1.81 (m, 1H), 1.75 – 1.66 (m, 2H), 1.57 – 1.43 (m, 3H), 1.29 – 1.21 (m, 1H), 1.18 – 1.05 (m, 2H); δC (125 MHz, CDCl3) 208.9 (C), 162.6 (C), 141.0 (C), 132.5 (C), 129.2 (CH), 129.1 (CH), 128.2 (CH), 125.8 (CH), 114.2 (CH), 63.4 (CH), 57.4 (CH), 55.6 (CH3), 49.9 (CH), 48.4 (CH2), 48.2 (CH2), 46.4 (CH), 41.0 (CH), 32.9 (CH2), 31.1 (CH2), 31.1 (CH2), 30.9 (CH2), 24.7 (CH2); m/z (ESI+) found [M+H]+ 454.2046. C26H32NO4S+ requires 454.2047.
4.4.15 Sulfonamide 13c{5}
δH (500 MHz, CDCl3) 7.65 (d, J = 1.8 Hz, 2H), 7.54 (t, J = 1.8 Hz, 1H), 7.26 – 7.22 (m, 2H), 7.19 – 7.13 (m, 3H), 4.24 (dt, J = 13.2, 3.6 Hz, 1H), 3.03 (dd, J = 14.3, 7.9 Hz, 1H), 2.94 – 2.83 (m, 2H), 2.76 (ddd, J = 13.0, 9.4, 3.7 Hz, 1H), 2.48 (ddd, J = 10.9, 7.9, 2.4 Hz, 1H), 2.42 (dd, J = 13.4, 2.7 Hz, 1H), 2.25 – 2.17 (m, 1H), 2.11 – 2.05 (m, 1H), 1.98 – 1.92 (m, 1H), 1.90 – 1.75 (m, 3H), 1.65 – 1.52 (m, 3H), 1.33 – 1.25 (m, 1H), 1.21 – 1.07 (m, 2H); δC (125 MHz, CDCl3) 208.5 (C), 144.7 (C), 140.9 (C), 136.0 (C), 132.4 (CH), 129.2 (CH), 128.2 (CH), 125.8 (CH), 125.2 (CH), 63.8 (CH), 57.3 (CH), 49.8 (CH), 48.8 (CH2), 48.3 (CH2), 46.3 (CH), 41.0 (CH), 33.2 (CH2), 31.1 (CH2), 30.9 (CH2), 30.4 (CH2), 24.7 (CH2); m/z (ESI+) found [M+H]+ 492.1176. C25H28Cl2NO3S+ requires 492.1161.
4.4.16 Sulfonamide 13d{2}
δH (500 MHz, CDCl3) 7.74 – 7.70 (m, 2H), 7.49 – 7.46 (m, 2H), 7.11 – 7.07 (m, 2H), 6.79 – 6.75 (m, 2H), 4.23 (dt, J = 13.0, 3.6 Hz, 1H), 3.76 (s, 3H), 2.92 (dd, J = 14.3, 7.8 Hz, 1H), 2.86 – 2.79 (m, 2H), 2.67 – 2.60 (m, 1H), 2.43 – 2.36 (m, 2H), 2.20 – 2.14 (m, 1H), 2.08 – 2.02 (m, 2H), 1.88 – 1.79 (m, 1H), 1.78 – 1.69 (m, 2H), 1.59 – 1.45 (m, 3H), 1.28 – 1.20 (m, 1H), 1.18 – 1.04 (m, 2H); δC (125 MHz, CDCl3) 208.9 (C), 157.7 (C), 139.9 (C), 138.9 (C), 132.8 (C), 130.2 (CH), 129.4 (CH), 128.4 (CH), 113.6 (CH), 63.6 (CH), 57.5 (CH), 55.2 (CH3), 49.8 (CH), 48.5 (CH2), 48.4 (CH2), 46.0 (CH), 40.9 (CH), 33.0 (CH2), 31.0 (CH2), 30.7 (CH2), 30.1 (CH2), 24.7 (CH2); m/z (ESI+) found [M+H]+ 488.1643. C26H31ClNO4S+ requires 488.1657.
4.4.18 Indolenine 15{1}
δH (500 MHz, CDCl3) 7.64 (d, J = 7.7 Hz, 1H), 7.32 (td, J = 7.6, 1.4 Hz, 1H), 7.25 – 7.16 (m, 2H), 3.37 (d, J = 11.4 Hz, 1H), 2.98 – 2.63 (m, 4H), 2.36 (td, J = 11.5, 3.2 Hz, 1H), 2.30 – 2.14 (m, 3H), 2.11 – 1.94 (m, 2H), 1.94 – 1.74 (m, 4H), 1.73 – 1.35 (m, 5H), 1.26 (h, J = 14.3, 13.2 Hz, 1H), 1.11 – 1.00 (m, 1H), 0.95 (t, J = 7.3 Hz, 3H), 0.37 (q, J = 8.2, 7.8 Hz, 3H); δC (125 MHz, CDCl3) 188.5 (C), 155.0 (C), 144.0 (C), 127.8 (CH), 125.1 (CH), 121.4 (CH), 120.5 (CH), 64.5 (CH), 58.3 (C), 53.3 (CH2, br), 51.9 (CH2, br), 51.5 (CH, br), 42.7 (CH2), 41.6 (CH), 35.0 (CH), 29.6 (CH2, br), 28.1 (CH2), 27.9 (CH2), 26.1 (CH2), 23.6 (CH2), 16.3 (CH2), 11.5 (CH3), 8.0 (CH3); m/z (ESI+) found [M+H]+ 337.2645. C23H33N2+ requires 337.2638.
4.4.19 Quinoline 17{6}
δH (500 MHz, CDCl3) 8.31 (s, 1H), 7.94 (s, 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.61 (dd, J = 8.5, 1.5 Hz, 1H), 3.23 – 3.17 (m, 1H), 2.91 – 2.86 (m, 1H), 2.73 (dt, J = 13.4, 7.8 Hz, 1H), 2.60 – 2.47 (m, 4H), 2.34 – 2.26 (d, 1H), 2.20 – 2.11 (m, 2H), 2.09 – 2.03 (m, 1H), 1.95 – 1.82 (m, 2H), 1.75 – 1.59 (m, 2H), 1.56 – 1.43 (m, 3H), 1.29 – 1.11 (m, 3H), 0.89 (t, J = 7.3 Hz, 3H), 0.73 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 163.3 (C), 145.6 (C), 136.9 (C), 130.6 (CH), 130.2 (C, q, J = 32 Hz), 128.4 (CH), 128.2 (C), 126.4 (CH, q, J = 4.2 Hz), 124.2 (C, q, J = 270 Hz), 121.1 (CH, q, J = 3.1 Hz), 65.4 (CH), 54.8 (CH2), 52.3 (CH2), 48.9 (CH), 47.0 (CH), 40.3 (CH), 39.4 (CH), 33.3 (CH2), 29.8 (CH2), 29.7 (CH2), 27.0 (CH2), 24.8 (CH2), 17.5 (CH2), 12.0 (CH3), 9.7 (CH3); m/z (ESI+) found [M+H]+ 417.2518. C25H32F3N2+ requires 417.2512.
4.4.20 Quinoline 17{5}
δH (500 MHz, CDCl3) 7.85 (s, 1H), 7.37 – 7.30 (m, 2H), 6.96 (d, J = 7.4 Hz, 1H), 4.05 (s, 3H), 3.21 (d, J = 11.3 Hz, 1H), 3.08 – 3.03 (m, 1H), 2.77 – 2.70 (m, 1H), 2.62 – 2.45 (m, 4H), 2.27 – 2.13 (m, 3H), 2.05 – 1.99 (m, 1H), 1.94 – 1.81 (m, 2H), 1.74 – 1.42 (m, 5H), 1.30 – 1.12 (m, 3H), 0.90 (t, J = 7.3 Hz, 3H), 0.73 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 160.6 (C), 154.9 (C), 138.8 (C), 135.1 (C), 130.7 (CH), 127.9 (C), 125.5 (CH), 119.3 (CH), 106.9 (CH), 65.5 (CH), 56.1 (CH3), 54.7 (CH2), 52.3 (CH2), 48.5 (CH), 47.1 (CH), 40.7 (CH), 39.1 (CH), 33.4 (CH2), 29.7 (CH2), 28.1 (CH2), 25.0 (CH2), 17.5 (CH2), 12.0 (CH3), 9.8 (CH3); m/z (ESI+) found [M+H]+ 379.2762. C25H35N2O+ requires 379.2744.
4.4.21 Quinoline 17{4}
δH (500 MHz, CDCl3) 7.91 (d, J = 9.0 Hz, 1H), 7.80 (s, 1H), 7.72 (s, 1H), 7.55 (d, J = 9.0, 1.5 Hz, 1H), 3.30 (d, J = 11.4 Hz, 1H), 2.90 – 2.85 (m, 1H), 2.84 – 2.73 (m, 1H), 2.71 – 2.61 (m, 2H), 2.57 – 2.44 (m, 2H), 2.31 – 2.22 (m, 2H), 2.20 – 2.14 (m, 1H), 2.05 (d, J = 12.9 Hz, 1H), 1.97 – 1.91 (m, 1H), 1.89 – 1.72 (m, 2H), 1.65 – 1.43 (m, 4H), 1.38 – 1.12 (m, 3H), 0.92 (t, J = 7.3 Hz, 3H), 0.72 (t, J = 7.4 Hz, 3H); δC (125 MHz, CDCl3) 161.7 (C), 145.2 (C), 135.2 (C), 131.1 (C), 130.1 (CH), 130.0 (CH), 129.5 (CH), 127.3 (C), 125.9 (CH), 65.3 (CH), 54.2 (CH2), 51.9 (CH2), 48.7 (CH), 46.3 (CH), 40.5 (CH), 39.1 (CH), 33.1 (CH2), 29.2 (CH2), 29.1 (CH2), 27.1 (CH2), 24.8 (CH2), 17.1 (CH2), 11.9 (CH3), 9.7 (CH3); m/z (ESI+) found [M+H]+ 383.2255. C24H32ClN2+ requires 383.22
Supplementary Material
Acknowledgments
We thank Kevin Frankowski for helpful discussions and comments on the manuscript, and Patrick Porubsky for mass-directed purification of the library compounds. This work was supported by the National Institutes of General Medical Sciences (P41GM089164 and 1R24GM111385).
ABBREVIATIONS
- PTSA
para-toluenesulfonic acid
- NP
natural product
- Fsp3
fraction sp3
- HBD
hydrogen bond donors
- HBA
hydrogen bond acceptors
- tPSA
topological polar surface area
- rotB
rotatable bonds
Footnotes
Supporting Information. Experimental procedures for the synthesis of scaffolds, tabulated results for all libraries, characterization data and 1H and 13C NMR spectra for scaffolds and representative library compounds.
Author Contributions
M.C.M. and J.A. conceived and designed the experiments, M.C.M. carried out the experiments, and M.C.M and J.A. co-wrote the manuscript and Supporting Information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Lovering F, Bikker J, Humblet C. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; (b) Lovering F. Med Chem Comm. 2013;4:515–519. [Google Scholar]
- 2.(a) Shelat AA, Guy RK. Nat Chem Biol. 2007;3:442–446. doi: 10.1038/nchembio0807-442. [DOI] [PubMed] [Google Scholar]; (b) Clemons PA, Wilson JA, Dančík V, Muller S, Carrinski HA, Wagner BK, Koehler AN, Schreiber SL. Proc Natl Acad Sci USA. 2011;108:6817–6822. doi: 10.1073/pnas.1015024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; (b) Butler MS. Nat Prod Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- 4.(a) Huigens RW, III , Morrison KC, Hicklin RW, Flood TA, Jr, Richter MF, Hergenrother PJ. Nat Chem. 2013;5:195–202. doi: 10.1038/nchem.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hanessian S, Kothakonda KK. J Comb Chem. 2005;7:837–842. doi: 10.1021/cc0500351. [DOI] [PubMed] [Google Scholar]
- 5.(a) Njardarson JT, Gaul C, Shan D, Huang XY, Danishefsky SJ. J Am Chem Soc. 2004;126:1038–1040. doi: 10.1021/ja039714a. [DOI] [PubMed] [Google Scholar]; (b) Szpilman AM, Carreira EM. Angew Chem, Int Ed. 2010;49:9592–9628. doi: 10.1002/anie.200904761. [DOI] [PubMed] [Google Scholar]
- 6.(a) Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; (b) Galloway WRJD, Isidro-Llobet A, Spring DR. Nat Commun. 2010;1:80. doi: 10.1038/ncomms1081. [DOI] [PubMed] [Google Scholar]
- 7.(a) Frankowski KJ, Setola V, Evans JM, Neuenswander B, Roth BL, Aube J. Proc Natl Acad Sci USA. 2011;108:6727–6732. doi: 10.1073/pnas.1016558108. S6727/6721-S6727/6106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McLeod MC, Singh G, Plampin JN, 3rd, Rane D, Wang JL, Day VW, Aubé J. Nat Chem. 2014;6:133–140. doi: 10.1038/nchem.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng Y, Reddy DS, Hirt E, Aubé J. Org Lett. 2004;6:4993–4995. doi: 10.1021/ol047809r. [DOI] [PubMed] [Google Scholar]
- 9.Frankowski KJ, Neuenswander B, Aube J. J Comb Chem. 2008;10:721–725. doi: 10.1021/cc800078h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Of the 145 best selling small molecule pharmaceutical products for 2012, 81 (56%) contained at least one aromatic heterocycle. http://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top200%20Pharmacetical%20Products%20by%20US%20Retail%20Sales%20in%202012_0.pdf
- 11.Li AH, Beard DJ, Coate H, Honda A, Kadalbajoo M, Kleinberg A, Laufer R, Mulvihill KM, Nigro A, Rastogi P, Sherman D, Siu KW, Steinig AG, Wang T, Werner D, Crew AP, Mulvihill MJ. Synthesis. 2010;1678–1686 [Google Scholar]
- 12.Kopp F, Stratton CF, Akella LB, Tan DS. Nat Chem Biol. 2012;8:358–365. doi: 10.1038/nchembio.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Marcaurelle LA, Comer E, Dandapani S, Duvall JR, Gerard B, Kesavan S, Lee MDt, Liu H, Lowe JT, Marie JC, Mulrooney CA, Pandya BA, Rowley A, Ryba TD, Suh BC, Wei J, Young DW, Akella LB, Ross NT, Zhang YL, Fass DM, Reis SA, Zhao WN, Haggarty SJ, Palmer M, Foley MA. J Am Chem Soc. 2010;132:16962–16976. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nielsen TE, Schreiber SL. Angew Chem, Int Ed. 2008;47:48–56. doi: 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Breinbauer R, Vetter IR, Waldmann H. Angew Chem, Int Ed. 2002;41:2878–2890. doi: 10.1002/1521-3773(20020816)41:16<2878::AID-ANIE2878>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Mayhugh D, Saeed A, Wang X, Thompson RC, Dominianni SJ, Kauffman RF, Singh J, Bean JS, Bensch WR, Barr RJ, Osborne J, Montrose-Rafizadeh C, Zink RW, Yumibe NP, Huang N, Luffer-Atlas D, Rungta D, Maise DE, Mantlo NB. J Med Chem. 2003;46:5121–5124. doi: 10.1021/jm034173l. [DOI] [PubMed] [Google Scholar]
- 15.Boyer JH. J Am Chem Soc. 1951;73:5248–5252. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.