

Abstract
Paclitaxel and rapamycin have been reported to act synergistically to treat breast cancer. Albeit paclitaxel is available for breast cancer treatment, the most commonly used formulation in the clinic presents side effects, limiting its use. Furthermore, both drugs present pharmacokinetics drawbacks limiting their in vivo efficacy and clinic combination. As an alternative, drug delivery systems, particularly liposomes, emerge as an option for drug combination, able to simultaneously deliver co-loaded drugs with improved therapeutic index. Therefore, the purpose of this study is to develop and characterize a co-loaded paclitaxel and rapamycin liposome and evaluate it for breast cancer efficacy both in vitro and in vivo. Results showed that a SPC/Chol/DSPE-PEG (2000) liposome was able to co-encapsulate paclitaxel and rapamycin with suitable encapsulation efficiency values, nanometric particle size, low polydispersity and neutral zeta potential. Taken together, FTIR and thermal analysis evidenced drug conversion to the more bioavailable molecular and amorphous forms, respectively, for paclitaxel and rapamycin. The pegylated liposome exhibited excellent colloidal stability and was able to retain drugs encapsulated, which were released in a slow and sustained fashion. Liposomes were more cytotoxic to 4T1 breast cancer cell line than the free drugs and drugs acted synergistically, particularly when co-loaded. Finally, in vivo therapeutic evaluation carried out in 4T1-tumor-bearing mice confirmed the in vitro results. The co-loaded paclitaxel/rapamycin pegylated liposome better controlled tumor growth compared to the solution. Therefore, we expect that the formulation developed herein might be a contribution for future studies focusing on the clinical combination of paclitaxel and rapamycin.
Keywords: Liposomes, Co-loading, Paclitaxel, Rapamycin, Breast cancer
1. Introduction
Cancer is a complex and highly heterogeneous disease, characterized by indefinite cell growth by different mechanisms, which makes single treatment suboptimal [1]. Among the types of cancer, breast cancer is the most common and lethal in women worldwide [2]. Metastatic breast cancer, developed by 30–40% of patients, represents a great cause of concern, once most patients die from breast cancer metastasis rather than the primary tumor. Therefore, in these cases, the goal of therapy is to delay progression of disease, to improve quality of life and to prolong survival. Among the types of breast cancer, the triple-negative breast cancers, which account for 10 to 17% of all cases and lack expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2), are considered the most aggressive with poor clinical outcome, therefore requiring new therapeutic options [3].
In breast cancer chemotherapy, taxanes represent one of the most effective anticancer drugs available in the clinic. Paclitaxel, the prototype of class, also employed in the treatment of ovarian, head and neck, and non-small cell lung cancers, binds to the β-subunit of tubulin, causing the disruption of the cytoskeleton framework necessary for the tumor cell replication and metastatic spread, thus preventing cell division [4,5]. Paclitaxel faces unfavorable pharmacokinetics. It undergoes extensive tissue distribution, with 90–95% protein binding, has variable systemic clearance, with average clearance ranging from 87 to 503 mL/min/m2 (5.2–30.2 L/h/m2). Paclitaxel is metabolized by the liver to 3 primary metabolites, with involvement of cytochrome P450 [6]. Nonetheless, a major problem of paclitaxel treatment is the development of drug resistance [5].
As an alternative to address this drawback, combination strategies emerge as an approach to enhance the therapeutic efficacy, especially if the combined drugs differ in their mechanism of action, acting synergistically to target multiple key pathways in carcinogenesis and tumor progression [7,8]. Within this context, rapamycin, a macrolide antibiotic used as immunosuppressant, is known to be a potent mTOR (mammalian target of rapamycin) inhibitor and has been reported to enhance the sensitivity to paclitaxel and act as a multidrug resistance reversal agent [9–11]. The mTOR is a serine threonine kinase, downstream mediator in phosphatidylinositol 3-kinase (PI3K) pathway, involved in multiple biologic functions for cell survival, such as transcriptional and translational control [12]. The mTOR pathway is frequently activated in malignant transformation of certain human cancers, such as breast cancer [13]. In addition, rapamycin and its analogs have been extensively evaluated in clinical trials for breast cancer treatment, alone or in combination with paclitaxel and other drugs [14]. In terms of pharmacokinetics, it has poor oral absorption with wide distribution in tissues and has inter- and intrapatient variability in drug clearance. Furthermore, the drug is biotransformed by the cytochrome P450 3A4, resulting in extensive drug-drug interactions. Finally, the drug suffers from long elimination half-life [15].
Despite the advantages for breast cancer treatment, paclitaxel and rapamycin face some problems that limit their efficacy. Because paclitaxel has poor aqueous solubility, the commercially available formulation for intravenous infusion of paclitaxel, Taxol®, employs the surfactant cremophor EL (polyethoxylated castor oil) and dehydrated ethanol. However, this formulation is associated with severe hypersensitivity reactions, including neurotoxicity and neutropenia. Furthermore, the formulation requires non-plasticized containers, due to the leaching of plasticizer diethylhexylphtalate caused by the contact between the formulation with the plastic infusion set [16,17]. Rapamycin is poorly aqueous soluble, has low oral bioavailability, undergoes major sequestration by erythrocytes and is chemically unstable, which altogether limit its employment in cancer therapy [18,19].
The poor clinical outcome associated with conventional chemotherapy has stimulated the development of nanotechnology-based drug delivery systems, such as polymeric micelles, liposomes and dendrimers, with resultant improvement in cancer treatment. These colloidal delivery systems are able to improve unfavorable pharmacokinetics of drugs, with consequent protection against drug degradation, sustained release and reduction of side effects [20]. Furthermore, nanoparticles can be passively accumulated in tumors through their leaky vasculature due to the enhanced permeability and retention (EPR) effect [21]. Of particular interest in drug delivery are liposomes, lipid vesicular systems, extensively studied in clinical trials for anti-cancer, anti-fungal and anti-bacterial drugs, with many products on the market nowadays [22].
Although rapamycin and particularly paclitaxel have been already loaded separately in different delivery systems, such as dendrimers, polymeric nanoparticles, micelles and liposomes, the co-encapsulation of both drugs in liposomes has never been reported [23–27]. Noteworthy, the “same time, same place” delivery of paclitaxel and rapamycin could be an efficient strategy for in vivo synergism [11]. Thus, in this paper, we aim to develop and characterize a novel liposomal formulation for colocalized delivery of paclitaxel and rapamycin, and evaluate it for breast cancer treatment both in vitro, in 4T1 cell line, and in vivo in 4T1-tumor-bearing mice.
2. Material and methods
2.1. Materials
Soy phosphatidylcholine (SPC), cholesterol (Chol), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethyleneglycol)-2000](DSPE-PEG), dipalmitoylphosphatidylcholine (DPPC), and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) were purchased from Avanti polar lipids (Alabaster, AL, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and DMSO were purchased from Sigma Aldrich Co. (St. Louis, MO, USA). Paclitaxel (PAC) was supplied by APIChem (China). Rapamycin (RAP), solvents (chloroform, acetonitrile and DMSO) and the dialysis membrane were obtained from Fisher (Pittsburgh, PA, USA). Sepharose CL-4B was purchased from GE Healthcare (Pittsburgh, PA, USA). Sodium lauryl sulfate was supplied by Henrifarma (Brazil). Antibiotic/antimycotic solutions, fetal bovine serum (FBS), RPMI-1640 medium were obtained from Gibco (Grand Island, NY). 4T1 (ATCC®CRL-2539™) cells were obtained from the American Type Culture Collection (ATCC) and cultivated according to its instructions.
2.2. Development of PAC and RAP-loaded liposomes
Liposomes were prepared by the hydration of the thin lipid film. The composition based on SPC:Chol:DSPE-PEG (2000) was employed for both PAC and RAP-loaded liposomes. RAP and PAC to total lipid ratios were 1:10 and 1:33, respectively. Briefly, lipids and drugs were dissolved in chloroform, which was evaporated in rotary evaporator for 30 min at 65 °C. Then, the lipid film was hydrated with phosphate buffer (pH 7.4) during 30 min at 100 rpm. The formulations were extruded 5 times in 0.2 μm membrane and 3 times in 0.1 μm membranes, filtered (0.45 μm) in PVDF (polyvinylidene difluoride) membranes and purified by gel filtration chromatography in a sepharose CL-4B column.
2.3. Physicochemical characterization
2.3.1. Determination of encapsulation efficiency of PAC and RAP
First, the encapsulated drug was separated from the free drug by filtration in PVDF membranes and further purification was done in sepharose CL-4B column. The total liposome fraction (obtained previously to purification) and the purified liposomal fraction (fractions 4–6) were solubilized in DMSO for liposomal rupture and drug solubilization. The samples were diluted with acetonitrile and finally sonicated for 10 min, filtered (0.22 μm) in PTFE (polytetrafluorethylene) membranes and then analyzed in triplicate by spectrophotometry (RAP) at 277 nm and HPLC (PAC). The HPLC method for PAC quantification employed mobile phase composed of water:acetonitrile (50:50, v/v), at a flow rate of 1 mL/min, wavelength at 227 nm and injection volume of 20 μL, using a C18 Lichrospher® column (Merck). The methods were validated according to the ICH (Harmonized Tripartite Guidelines) for selectivity, linearity, precision, accuracy, detection and quantification limits [28]. The encapsulation efficiency (EE) was calculated according to the equation:
2.3.2. Particle size and polydispersity index analyses
Dynamic light scattering was employed for particle size and polydispersity index determination at 25 °C, using the Zetasizer 3000HSa (Malvern Instruments) under a HeNe laser operated at 4 mV and wavelength at 633 nm.
2.3.3. Zeta potential determination
Zeta potential was determined by electrophorectic mobility of dispersed drops under an electrical field, using Zetasizer 3000HSa equipment.
2.3.4. Transmission Electron Microscopy (TEM)
Samples were placed on a copper grid, allowed to absorb and then excess sample was removed. Subsequently, a drop of 1% (w/v) aqueous solution of uranyl acetate was added for negative staining. After 2 min, excess liquid was removed and the sample was dried at room temperature. The analyses were carried out on a JEOL JEM-100CX2 transmission electron microscope with an acceleration of 100 kv [29].
2.3.5. Fourier Infrared Spectroscopy (FTIR) analyses
A Shimadsu IRPrestige-21 equipment was employed. For the analyses, samples were lyophilized with sucrose as cryoprotectant (5/1, molar ratio of sucrose per total lipid) and ground and mixed with potassium bromide, by compressing the powders in a hydraulic press. Scans were obtained at a resolution of 2 cm−1 from 4000 to 400 cm−1. In the experiment, it was ensured that the method had enough sensitivity to detect the amount of loaded drugs.
2.3.6. Differential Scanning Calorimetry (DSC) analyses
A Jade Perkin Elmer equipment was used. The samples, previously lyophilized were placed in aluminum pans and heated from 15 °C to 250 °C at a rate of 10 °C/min, under nitrogen pressure of 3 kgf/cm2. Calibration was performed using indium and n-octadecane as reference materials. In the experiment, it was ensured that the method had enough sensitivity to detect the amount of loaded drugs.
2.3.7. Thermogravimetry analysis (TGA)
TGA was carried out with a PerkinElmer 4000 instrument calibrated with standard weights. The lyophilized samples were weighted and heated from 15 °C to 400 °C at a rate of 10 °C/min, under nitrogen pressure of 2 kgf/cm2.
2.3.8. Colloidal stability
Co-loaded PAC and RAP liposome suspension stability was assessed for percentage of the initial drug loading, particle size and polydispersity index for 30 days of storage at 4 °C
2.3.9. In vitro drug release
PAC and RAP solutions (formulated in cremophor EL40 and dehydrated ethanol) and lyophilized liposomes containing PAC and RAP, co-encapsulated or not, evaluated for release in 50 mL 7.4 phosphate buffer containing 1% sodium lauryl sulfate with agitation speed at 150 rpm. In this study, samples were dispersed in 1 mL of PBS buffer, pH 7.4, and placed inside PVC tubes wrapped with 12–14 kDa MWCO (molecular weight cut-off) cellulose dialysis membranes (Fisherbrand) and connected to the dissolution shafts of the apparatus 1 (SR8 Plus, Hanson Coorporation) [30]. Samples were collected until 72 h, diluted with acetronitrile, filtered, and analyzed by the HPLC method described previously described for PAC. For RAP quantification, an HPLC method was employed with mobile phase composed of water:acetonitrile (25:75, v/v), at a flow rate of 1.6 mL/min, at 40 °C, detected at 277 nm wavelength, with and injection volume of 20 μL in a C18 Lichrospher® column (Merck).
2.4. Cell culture
2.4.1. Cytotoxicity evaluation in 4T1 cell line
Cytotoxicity was evaluated in 4T1 cell line obtained from ATCC (CRL 2539TM) cultivated in RPMI-1640 supplemented with 10% FBS and 1% antibiotic/antimycotic solution. Cells were trypsinized and then seeded onto 96-well flat-bottom tissue-culture plates (10.000 cells/well) and incubated in 5% CO2 incubator for 24 h. After complete culture medium removal, the wells were washed with PBS pH 7.4, the diluted experimental groups were added and the plates were incubated at 37 °C for 72 h. After incubation, the wells were washed with PBS and incomplete fresh medium with MTT solution (2,5 mg mL−1), followed by incubation for 4 h at 37 °C. Then, the medium containing MTT was replaced for DMSO to dissolve the MTT formazan crystals. The absorbance was read at 570 nm. The concentration resulting in 50% cell death (IC50) was calculated from concentration-effect curves, considering the optical density of the control well (non-treated cells) as 100%.
2.4.2. Combination index determination in 4T1 cell line
The combination index (CI) of PAC and RAP in solution or liposome was determined according to method proposed by Chou and Talalay [31] and calculated as follows:
In this equation, (D)1 and (D)2 represent the doses of drug 1 and drug 2 necessary to produce IC50 effect in combination, whereas (Dx)1 and (Dx)2 are the doses of drug 1 and drug 2 required for the IC50 effect individually. An average CI < 1 indicates synergism, CI > 1 indicates antagonism and an average CI of 1 indicates additivity.
2.5. In vivo studies
2.5.1. Animals
BALB/c mice (6–8 weeks old) were employed in the experiments and all animal procedures were performed according to an animal care protocol approved by the University of Sao Paulo Institutional Animal Care and Use committee in accordance with the “Principles of Laboratory Animal Care” (protocol # 14.1.883.53.6). Animals were housed in groups of 5 at 25 °C with a 12 h light–dark cycle and allowed to food and water ad libitum.
2.5.2. Anti-tumor efficacy of PAC and RAP solutions, PAC-loaded, RAP-loaded liposomes and PAC/RAP co-loaded liposome
To investigate whether liposomes were better than the solutions and if liposomal co-loaded PAC and RAP had superior therapeutic efficacy, intramammary tumors were developed by inoculating 4T1 murine breast cancer cells (1 × 105) in the left and right mammary fat fads of BALB/c mice. After injection, the time for tumor appearance was 10–14 days, when the treatment was started [32]. Lyophilized liposomes and negative and positive controls were diluted in PBS buffer, pH 7.4, prior to administration. The theoretical dose of PAC and RAP administered were 5 mg/kg and 15 mg/kg, twice a week, totalizing 5 administrations [11]. Groups received intraperitoneal injection as follows: (I) saline (negative control); (II) PAC solution; (III) PAC liposome; (IV) RAP solution; (V) RAP liposome; (VI) PAC RAP solution; (VII) PAC RAP liposome (n = 5 in each group). The tumor volume and body weight were recorded for all tumor-bearing mice until the end of the study, after which animals were euthanized with overdose of anesthetics. The length and width of the tumors were measured every 2 days using a caliper and the tumor volume was calculated with the equation:(width2×length)/2. To compare groups, relative tumor volume was calculated at each measurement point. The values were calculated through the tumor volume at a given time point divided by the tumor volume prior to first treatment, in percentage, according to Lu et al., [33]. At the end of the experiment, the tumors had an average volume of 485 mm3, and the maximum tumor volume allowed was 1100 mm3. After completion of the study, the tumors were removed, photographed and submitted to histological evaluation by anti-Ki67 antibody staining.
2.5.3. Statistical analysis
The results were presented as mean ± standard deviation (SD). Student t test was used and statistical significance was set at P < 0.05.
3. Results and discussion
3.1. Formulation development
For the preparation of liposomes, different formulation variables were considered, such as the type of lipid employed for drug encapsulation, the drug to lipid ratio, the effect of Chol and pegylation, because of their influence on drug encapsulation and delivery [20]. For PAC-loaded liposome preparation, the drug to lipid ratio employed was fixed at 1:33, which has been previously reported, considering that higher PAC molar concentrations could result in precipitation because of drug tendency to undergo concentration-dependent aggregation in hydrophobic environments [34,35]. In the case of RAP encapsulation, the first step of the study corresponded to the optimization of the drug to lipid ratio, aiming at the balance of reduced particle size and high encapsulation efficiency (unpublished results). The best results were achieved with the ratio at 1:10, with small particle, higher values of RAP encapsulation efficiency and negative zeta potential.
Among other lipids initially evaluated, SPC resulted in a better lipid for RAP encapsulation, because it forms less rigid lipid bilayer, enabling higher drug encapsulation. For PAC encapsulation, SPC was also employed, considering the many reports on PAC encapsulation employing natural PC [26,34,36]. Another component employed in the formulations is Chol, a lipid used for better in vitro and in vivo liposome stability [37]. On the other hand, our unpublished results showed that the presence of Chol reduced both RAP and PAC encapsulation, in accordance with other reports stating that Chol is more rigid than SPC and thus restricts the lipid bilayer flexibility, preventing the drug from accommodating within the vesicle [4,35,38]. Noteworthy, Haeri and collaborators defined as 25 mol% the maximum amount of Chol that can be added to liposomes containing RAP, once higher ratios of Chol may induce rapid vesicle aggregation [27].
Finally, the third lipid employed in the formulations is DSPE-PEG 2000, with the purpose of longer blood circulation times, due to the steric hindrance effect of polyethylene glycol (PEG), resulting in reduced uptake by the mononuclear phagocytic system (MPS). Regarding the pegylated lipid MW, it has been previously reported that DSPE-PEG (2000) is able to better prolong blood circulation than DSPE-PEGs with higher MWs [39]. When DSPE-PEG 2000 was added to the PAC and RAP-loaded SPC:Chol liposomes, particle size, polydispersity index and encapsulation efficiency were not compromised, while the zeta potential was neutral due to the nature of the pegylated lipid. Because SPC:Chol:DSPE-PEG (10:2:0.5) proved to be a suitable colloidal carrier for both PAC and RAP when loaded separately, we decided to prepare a formulation with both drugs co-encapsulated, expecting that this strategy of drug combination could result in improved in vivo anticancer effect. Our results in Table 1 shows that the characteristics of the liposomes were preserved after the co-encapsulation, with 73.31% and 56.32% RAP and PAC encapsulation efficiency values, respectively, exhibiting 136.95 nm particle size with 0.27 polydispersity and neutral zeta potential. Furthermore, considering the values of EE for PAC and RAP in the co-encapsulated liposome, the actual ratio of encapsulated PAC/RAP is 0.56/2.20. Finally, the spherical unilamellar vesicular structure of PAC/RAP co-loaded pegylated liposomes were confirmed by TEM (Fig. 1), in accordance with a previous observation of PAC-loaded liposomes [29].
Table 1.
Characteristics of liposomes: particle size, polydispersity, zeta potential and encapsulation efficiency.
| Formulation | Encapsulation efficiency (%) | Total drug (mg)/100 mg liposome | Particle Size (nm) | Polydispersity | Zeta potential (mV) |
|---|---|---|---|---|---|
| Blank SPC:Chol:DSPE-PEG2000 | – | – | 127.59 ± 5.98 | 0.24 ± 0.01 | −1.12 ± 0.03 |
| SPC:Chol:DSPE-PEG2000:PAC (10:2:0.5) | 65.03 ± 9,93 | 2.01 ± 0.31 | 124.1 ± 4.67 | 0.48 ± 0.02 | −1.26 ± 0.11 |
| SPC:Chol:DSPE-PEG2000:RAP (10:2:0.5:1) | 59.58 ± 4.97 | 5.87 ± 0.48 | 133.84 ± 8.35 | 0.26 ± 0.01 | −3.51 ± 0.24 |
| SPC:Chol:DSPE-PEG2000:RAP:PAC (10:2:0.5:1:0.33) | 73.31 ± 4.53 (RAP)/56.32 ± 6.32 (PAC) | 7.22 ± 0.45 (RAP)/1.75 ± 0.20 (PAC) | 136.95 ± 7.28 | 0.27 ± 0.01 | 0.02 ± 0.19 |
Fig. 1.
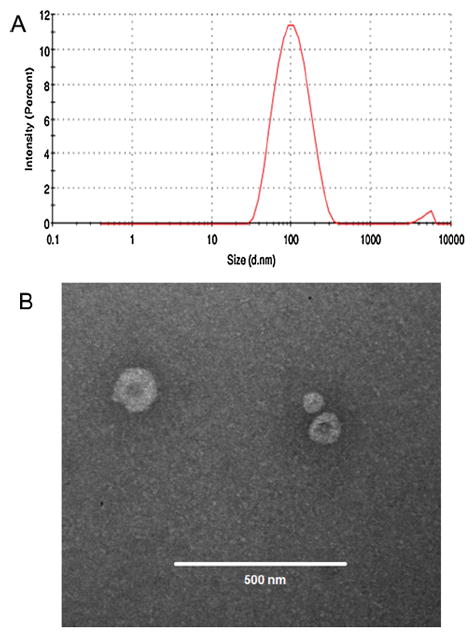
Particle size distribution by dynamic light scattering (A) and TEM micrographs of PAC/RAP loaded liposomes (B).
Compared with the blank liposome (Table 1), the characteristics are similar to the drug-loaded liposomes, especially in terms of zeta potential, considering that the molar percentage of drugs is low and the majority of the liposome is composed by SPC and Chol, which do not bear charge [26]. It can be noted, however, that the particle size increased when RAP was added, because it is present in 10mol% in the lipid bilayer.
Regarding the PdI of formulations, the co-loaded liposome had a PdI of 0.27, which is comparable to a previous report on liposomes [40]. However, a high PdI value and a heterogeneous liposomal suspension could represent a scale up challenge. An alternative for this drawback could be the increase of the number of extrusion cycles and the use of other serial extrusion membranes with smaller porosity. Furthermore, many other methods have been proposed as alternatives to produce homogeneous liposomes, besides the extrusion method, employed herein. Some examples are the homogenization techniques, such as the microfluidization and ethanol injection method, both known for the easy scale up [41].
3.2. FTIR characterization
The occurrence of interaction between drugs and excipients and also the the nature of the drugs after encapsulation, i.e crystalline, molecular or amorphous, were investigated by infrared spectroscopy and thermal analysis. FTIR analysis is a useful tool to investigate intermolecular interactions, through shift or broadening of the bands in the spectra [42,43]. In Fig. 2, FTIR spectra corresponding to the drugs, blank liposome, PAC-loaded, RAP-loaded and PAC RAP-loaded liposomes are represented. PAC exhibited main infrared peaks as follows: N-H stretching vibrations at 3479–3300 cm−1, CH2 asymmetric and symmetric stretching vibrations at 2976–2885 cm−1; C O stretching vibration from the ester groups at 1725 cm−1; amide bound at around 1647 cm−1 and C N stretching vibrations around 1240 cm−1 [44]. Regarding the PAC-loaded liposomes, the characteristic peaks of PAC drug were not evident, which could be explained by drug presence as a molecular dispersion within the liposome matrix, similarly to the findings by Martins et al. who studied PAC-loaded polymeric microspheres [44]. The spectrum of RAP shows peaks at 3400, 2934, 1738, 1633 and 1444 cm−1 which can be assigned to the stretching vibrations of O H, C H, C O, C C and C H adjacent to carbonyl, respectively [45]. These bands appeared in reduced intensity in liposomes, because of due of the low percentage of drug loaded, but no other change was identified.
Fig. 2.
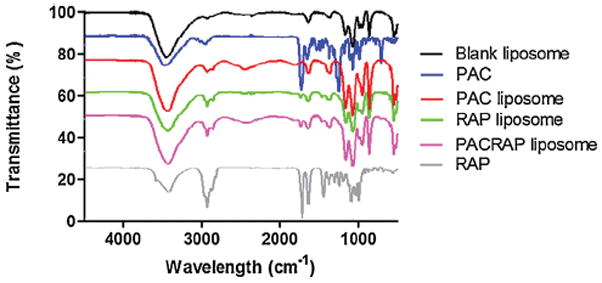
FTIR spectra from 4550 to 450 cm−1 of blank liposome, PAC solution, RAP solution, PAC-loaded liposome, RAP-loaded liposome and PAC/RAP-loaded liposome.
3.3. Thermal analysis characterization
Herein, in Fig. 3A, PAC presents an endotherm band of vitreous transition at 167 °C and an exotherm band at 234 °C, in accordance with the observations by Liggins et al. who attributed this behavior to PAC in the amorphous state [46]. These two bands disappeared in the PAC-loaded liposomes spectra, which could indicate that PAC is molecularly dispersed within the liposomes, in accordance with the FTIR data. RAP spectrum exhibited two endothermic bands at 187 and 196 °C, attributed to the crystal melting (Fig. 3A) [47]. It is important to note that RAP bands in the liposomes were more broadened, appearing at a higher temperature, suggesting that RAP was at least partially converted from the crystalline to the amorphous state in liposomes. An important consequence is that amorphous drugs are more soluble and thus could present enhanced bioavailability [43]. In some spectra in Fig. 3A we can also observe wide endotherm bands around 50 °C, attributed to water loss. Finally, TGA (Fig. 3B) evidenced that the blank liposome is thermally stable until 400 °C, whereas both PAC and RAP degrade after 200 °C. This characteristic was preserved in the liposomes, however the degradation was less pronounced due to the small amount of drugs added to the formulations.
Fig. 3.
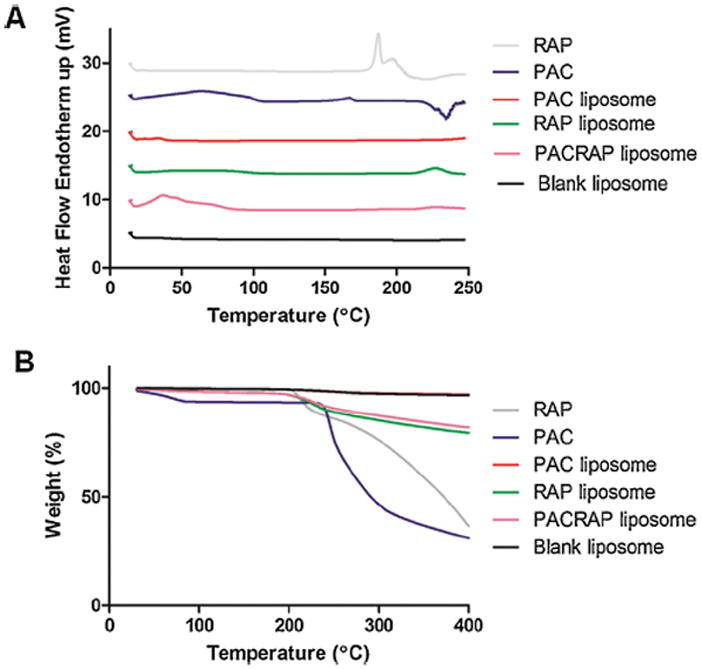
DSC analysis (A) and TGA (B) of blank liposome, PAC, RAP, PAC-loaded liposome, RAP-loaded liposome and PAC/RAP-loaded liposome. Scanning was done from 15 to 250 °C for DSC and from 15 to 400 °C for TGA, at a rate of 10 °C/min.
3.4. In vitro release and colloidal stability characterization
In vitro release studies are an essential step during drug development because not only it serves for quality control of formulations, but can also reflect the in vivo performance of the product [30]. This analysis is particularly well described for PAC, considering the publications available, reporting the sustained release of PAC from nanoparticles evaluated through the dialysis method [26,36]. PEGy-lated liposomes containing both PAC and RAP, co-encapsulated or not, presented very slow release, less than 20% of total drug after 72 h (Fig. 4A), indicating that the drugs remained encapsulated within the liposomes, which agreed with other reports on PAC and RAP slow and incomplete release from liposomes [35,48]. PEGylated liposomes were able to retain drugs encapsulated, creating a depot effect, similarly to the observations by Yang et al. who studied PAC-loaded liposomes [26]. They argued that within liposomes the drug could possibly be stable in the blood circulation and would be slowly released at the tumor site, where the liposome would accumulate through the EPR effect, associated to an intravenous administration. Conversely, PAC and RAP formulated in cremophor and dehydrated ethanol solutions exhibited higher release, corresponding to approximately 80 and 50% release, respectively. Colloidal stability of the PEGlyated liposomes containing PAC and RAP was also investigated (Fig. 4B).
Fig. 4.
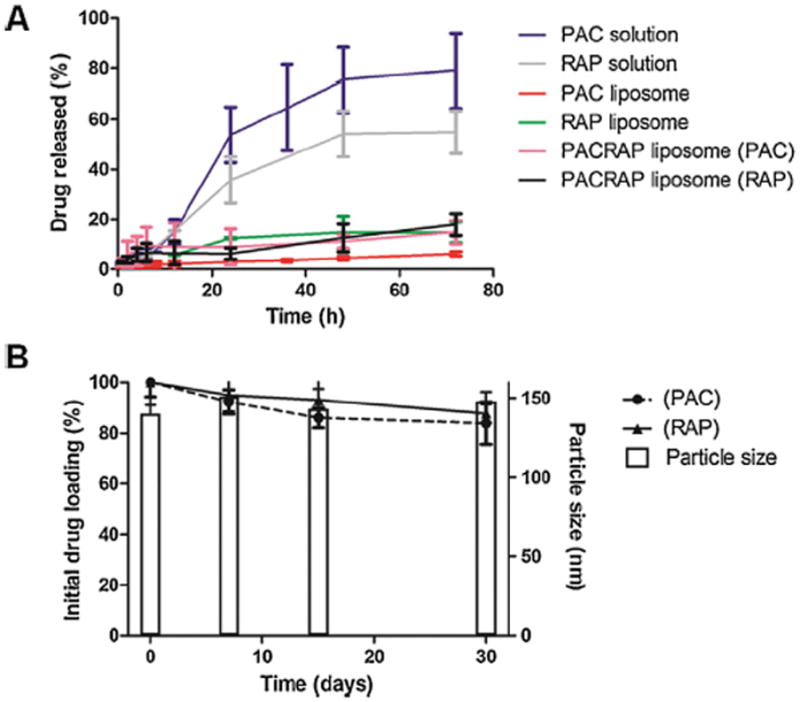
A represents the in vitro drug release studies done in 50 mL phosphate buffer medium supplemented with 1% sodium lauryl sulfate using dialysis membrane (12–14 kDa MWCO) under 150 rpm agitation at 37 °C, for PAC solution, RAP solution, PAC-loaded liposome, RAP-loaded liposome and PAC/RAP-loaded liposome. B represents the colloidal stability evaluation (percentage drug loading and particle size) for 30 days at 4 °C of the PAC/RAP-loaded liposome.
The maintenance of particle size, polydispersity and encapsulation efficiency after 1 month at 4 °C proved the stability of the formulations during the period evaluated. The stability of the neutral liposomes was attributed to the presence of cholesterol. Kirby et al. investigated the in vitro and in vivo stability of neutral, negatively and positively charged unilamellar liposomes composed of egg phosphatidylcholine and cholesterol and concluded that the stability was dependent on the cholesterol content, regardless of the zeta potential. Furthermore, the authors showed that the stability of neutral liposomes at 4 °C was maintained for several days [37].
3.5. In vitro efficacy evaluation in 4T1 breast cancer cells
The first step of the efficacy evaluation of the formulations for breast cancer was the cytotoxicity assay in 4T1, a highly metastatic mammary carcinoma cell line. Results in Fig. 5 showed that PAC is more cytotoxic than RAP and the liposomes were more cytotoxic to 4T1 cells. However, the effect of encapsulation was more evident for RAP than PAC, considering that the IC50 of the solution decreased from 1228.07 nM to 278.19 nM for RAP-loaded liposome and from 66.69 nM (PAC solution) to 46.48 nm (PAC-loaded liposome). The better cytotoxicity of liposomes was also found in another study, which reported that RAP encapsulation in both the conventional and pH sensitive liposomes enabled superior delivery and cytotoxicity to HT-29 cancer cells compared to drug in solution [49]. The better cytotoxicity achieved with liposomes might result from the enhanced cellular uptake, as already shown for doxorubicin-loaded liposomes aimed for 4T1 cell delivery [50]. Furthermore, when the two drugs were combined, it was found a potent synergism for both the solution and particularly with the co-loaded liposome (CI values corresponded to 0.73 and 0.39, respectively). Our findings agree with the potent synergism of PAC and RAP both in solution and co-loaded polymeric micelle, previously reported [9,51]. Regarding the mechanism of the synergism between PAC and RAP, Mondesire et al. (2004) proposed that RAP treatment may enhance PAC induced apoptosis involving caspase activation, by allowing cells to progress to G2-M arrest, through the downregulation of the mTOR pathway [9].
Fig. 5.
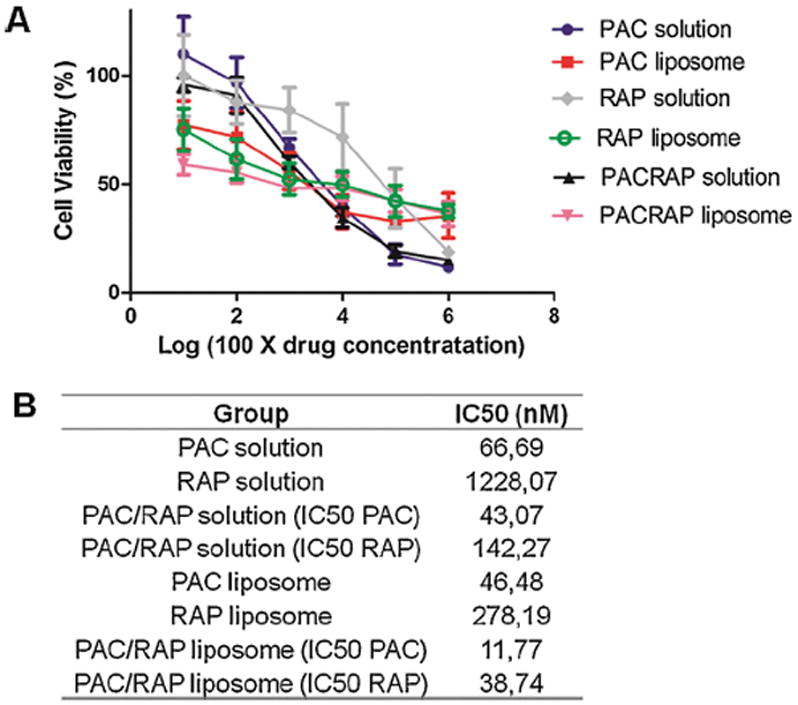
Cytotoxicity evaluation in 4T1 breast cancer cell lines of PAC solution, RAP solution, PAC-loaded liposome, RAP-loaded liposome and PAC/RAP-loaded liposome employing the MTT assay. A represents the dose-response curves and B represents the IC50 calculated for each group.
3.6. In vivo efficacy evaluation in 4T1-tumor-bearing mice
For the in vivo study, 4T1 mammary carcinoma cells were used for animal transplantation. These cells are highly tumorigenic, invasive, and can spontaneously metastize to distant sites, including lymph nodes, blood, liver, lung, brain and bone. Therefore, 4T1 tumor has many characteristics that make it an adequate experimental model for human mammary breast cancer and has been employed for the therapeutic efficacy evaluation of drugs and nanoparticles in breast cancer [5,32,52].
To determine whether the liposomal encapsulation and also the combination therapy with PAC and RAP are able to improve the therapeutic efficacy on breast carcinoma in vivo, we evaluated PAC and RAP solutions administered separately and together, PAC-loaded liposome, RAP-loaded liposome and PAC/RAP co-loaded liposome. Results in Fig. 6 shows that PAC solution had little effect on inhibiting tumor growth and was not statistically different from the negative control, confirming the results obtained by Luo et al. [5]. Conversely, 4T1-bearing tumors were more sensitive to RAP solution administration at the dose employed (15 mg/kg every 3 days), which caused a statistically significant tumor reduction compared to the negative group (Fig. 6A), also evident from tumors visualization after biopsy (Fig. 6B). The effect of the liposomal encapsulation as a strategy to improve the in vivo anticancer efficacy was evident for PAC, considering that the solution administration caused a 307% increase in the tumor volume at the final of the experiment compared to a 173% increase for PAC-loaded liposome, whereas in the case of RAP there was no statistical difference between the solution and the liposome group. It should be considered, on the other hand, that other benefits must be taken into account in the case of RAP encapsulation, especially the potential to prevent it from erythrocyte sequestration and chemical degradation, which altogether limit its employment in cancer therapy [18,19].
Fig. 6.
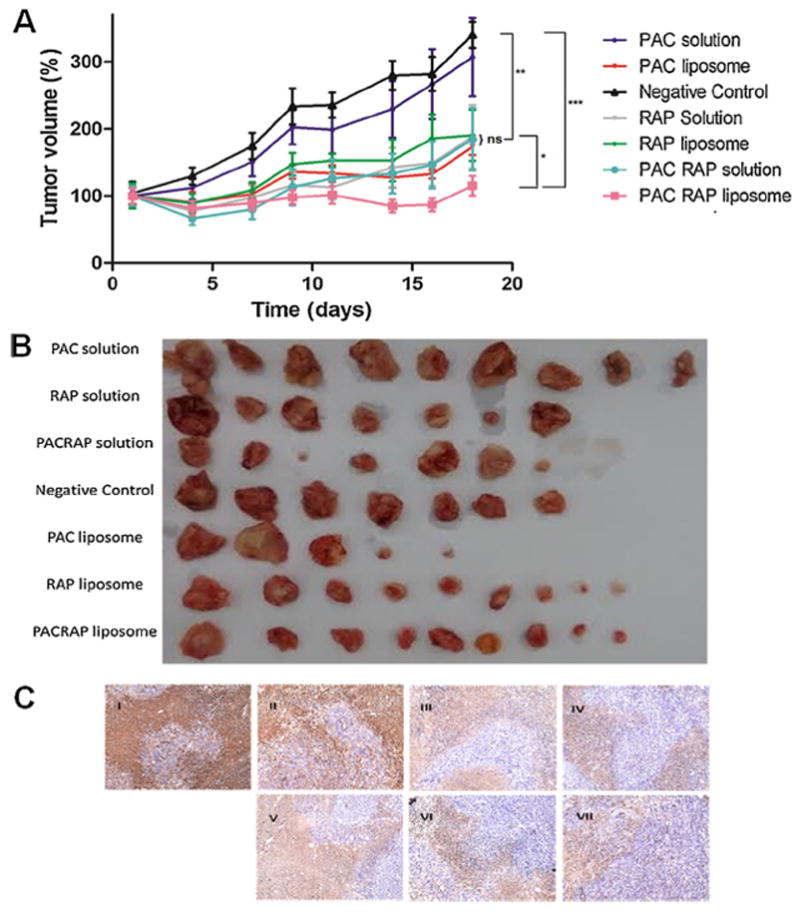
Therapeutic evaluation of PAC solution, RAP solution, PAC/RAP solution, PAC-loaded liposome, RAP-loaded liposome and PAC/RAP-loaded liposome in 4T1 tumor-bearing BALB/C mice. A represents the percentage tumor volume increase thought out the study, in which PAC and RAP were administered at theoretical doses of 5 mg/kg and 15 mg/kg twice a week, 5 times in total. B represents the photograph of the tumors after biopsy. C represents anti-Ki67 antibody staining of tumors (I—negative control; II—PAC solution; III—RAP solution; IV—PAC/RAP solution; V—PAC-loaded liposome; VI—RAP-loaded liposome and VII—PAC/RAP-loaded liposome).
It is important to point out that in vitro the encapsulation in liposome enhanced the efficacy particularly for RAP, probably because the liposome improved the uptake to the cell, although PAC-loaded liposomes remained more toxic than RAP-loaded liposomes (about 6 times higher). On the other hand, in vivo, PAC-loaded liposomes better benefited from encapsulation compared to RAP-loaded liposome. The in vivo environment is more complex than the cell environment, which might sometimes be difficult to correlate. As discussed by Poondru et al. [53], in solid tumors there might be a lack of in vitro-in vivo correlation of anticancer drugs, because drug activity is rather complex and it involves the permeability barrier to tumor cells, interstitial hypertension and the enzymatic barrier, involving metabolic degradation. In other words, we hypothesize that encapsulating PAC might better improve its pharmacokinetics compared to RAP, which could have higher relevance in terms of enhancing efficacy.
Considering that the combination therapy of two drugs with different mechanisms of action is an interesting approach to synergistically target multiple key pathways in cancer progression and also motivated by our promising in vitro results, we tested the in vivo efficacy of the drugs combined in solution and in a liposomal formulation [7,8]. Regarding the employed ratio of PAC to RAP, it was used the theoretical ratio of 1:3 (or 0.56/2.2, considering the values of EE), resulting in the administered doses of 5 and 15 mg/kg (or 2.8 mg/kg and 11 mg/kg, considering the EE values for each drug), respectively for PAC and RAP, for each chemotherapy cycle, the same theoretical doses employed in a previous report [11]. It should be considered, however, that the volume administered in vivo was added with more formulation in order to make up for the loss of drug due to EE, resulting in the actual doses of 3.8 mg/kg and 15 mg/kg, respectively for PAC and RAP. Noteworthy, in the paper by Blanco et al., 2014 the antitumor efficacy evaluated in mice bearing MCF-7 breast tumors was superior for the 1:3 ratio compared to the 2:1 ratio of PAC:RAP, which evidences the proper selection of PAC:RAP ratio for synergism [11]. From Fig. 6, confirming our in vitro results, it is evident that the drug combination in the liposome was better than the solution, resulting in 115% and 185% tumor increase, respectively, compared to 340% tumor increase in the negative control group at the final of the experiment. Furthermore, Fig. 6C shows decreased tumor proliferation, evaluated by anti-Ki67 antibody brown staining, for RAP and PAC/RAP solutions and liposomes, compared to the negative control.
The better results achieved of drug combination in liposome could be attributed to the enhancement of the pharmacokinetics hurdles faced by both PAC and RAP, which could be anticipated from the conversion of drugs to more soluble forms, i.e the molecular state for PAC and to the amorphous form for RAP, revealed by the FTIR and thermal analysis. Nanoparticles are also able to provide a sustained release of the entrapped drug, resulting in multiple benefits, such as the decrease of the duration/number of administrations and also continuously exposure of drug to the tumor, with positive influence on the in vivo anticancer effect [54]. Moreover, as discussed by Blanco et al. the synergistic antitumor effects of PAC and RAP could be significantly enhanced when both drugs are delivered site specifically and simultaneously to tumors, through co-delivery from nanoparticles, which supports the results obtained herein [11]. Using this combination approach, it possible to take the advantages of both PAC and RAP and also make up for the deficiencies of each other. Thus, an enhanced synergism emerges, considering that RAP is known to enhance the sensitivity to PAC and overcome its resistance [9–11]. Noteworthy, it is know that PAC suffers from efflux form the cancer cells by the P-glycoprotein pump, expressed in normal and malignant tissue [55], which can be benefited from the property of RAP to overcome multidrug resistance by P-glycoprotein inhibition [56]. Furthermore, very advantageously, liposomal drugs have decreased susceptibility than free drugs to the efflux activity by P-glycoprotein [57].
Finally, mice body weight and survival were monitored in order to evaluate liposome toxicity. Fig. 7 shows that there were no significant changes in mice body weight of the groups tested and although there were deaths in some groups, all the animals survived until the completion of the study in the co-loaded liposome group, suggesting minor acute toxicity. Indeed, this result was expected, considering that liposomes are regarded as one of the most biocompatible and biosafe delivery systems, which has enabled their enormous clinical success, making liposomes the most successful nanoparticles on the market nowadays for the treatment of many diseases, particularly cancer [22,58].
Fig. 7.
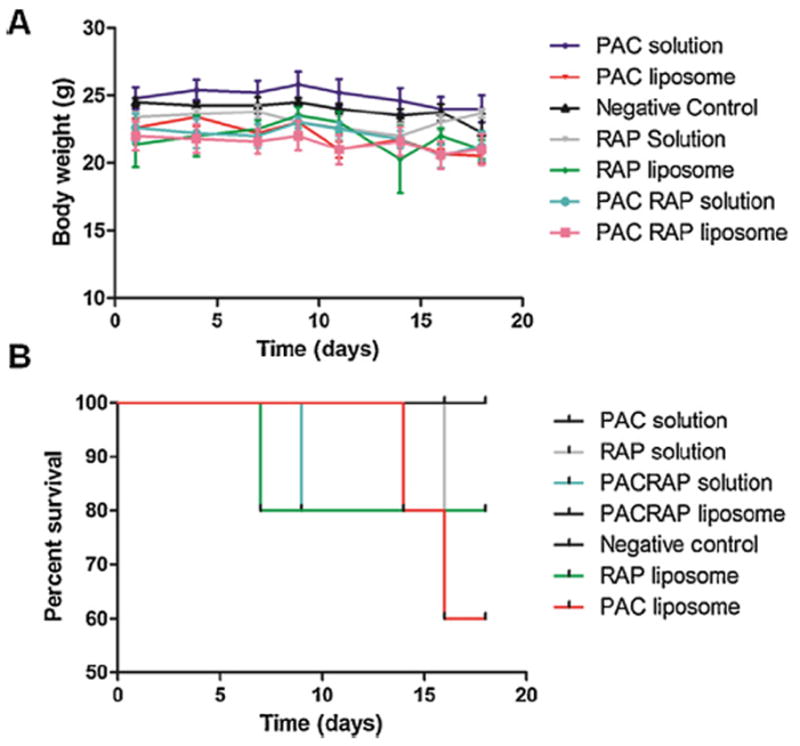
Therapeutic evaluation of PAC solution, RAP solution, PAC/RAP solution, PAC-loaded liposome, RAP-loaded liposome and PAC/RAP-loaded liposome in 4T1 tumor-bearing BALB/C mice. A represents the body weights recorded every 3 days and B represents the percent survival of groups until the completion of the study.
4. Conclusion
In this paper, it was reported for the first time the co-encapsulation of paclitaxel and rapamycin in a liposomal formulation. Our studies proved that a composition based on SPC/Chol/DSPE-PEG (2000) enabled a satisfactory encapsulation of both drugs, with suitable particle size for parenteral drug delivery. FTIR and thermal analysis evidenced drug conversion to the more soluble molecular and amorphous forms, for paclitaxel and rapamycin, respectively. Moreover, the formulation showed excellent colloidal stability and retained drugs encapsulated, with sustained release. From our results, it was evident that both drugs acted synergistically both in vitro, in 4T1 breast cancer cell lines, and in vivo, in 4T1 tumor-bearing mice, with promising tumor growth control. We expect that the formulation reported herein provides insight in future studies aiming for the clinical use of paclitaxel and rapamycin.
Acknowledgments
The authors would like to acknowledge FAPESP for grants 2012/10388-3, 2013/05362-8, 2012/21513-3 and 2014/08462-6 and Dr. Yasuro Sugimoto and Dr. Robert Brueggemeier for their kind assistance.
References
- 1.Yin T, Wang L, Yin L, Zhou J, Huo M. Co-delivery of hydrophobic paclitaxel and hydrophilic AURKA specific siRNA by redox-sensitive micelles for effective treatment of breast cancer. Biomaterials. 2015;61:10–25. doi: 10.1016/j.biomaterials.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 2.Ramadass SK, Anantharaman NV, Subramanian S, Sivasubramanian S, Madhan B. Paclitaxel/epigallocatechin gallate coloaded liposome: a synergistic delivery to control the invasiveness of MDA-MB-231 breast cancer cells. Colloids Surf B: Biointerfaces. 2015;125:65–72. doi: 10.1016/j.colsurfb.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H, Cohen AL, Krishnakumar S, Wapnir IL, Veeriah S, Deng G, Coram MA, Piskun CM, Longacre TA, Herrler M, Frimannsson DO, Telli ML, Dirbas FM, Matin AC, Dairkee SH, Larijani B, Glinsky GV, Bild AH, Jeffrey SS. Patient derived xenografts of triple negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014;16:1–16. doi: 10.1186/bcr3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kouldelka S, Turánek J. Liposomal paclitaxel formulations. J Controlled Release. 2012;163:322–334. doi: 10.1016/j.jconrel.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Luo T, Wang J, Yin Y, Hua H, Jing J, Sun X, Li M, Zhang Y, Jiang Y. (-)-Epigallocatechin gallate sensitizes breast cancer cells to paclitaxel in a murine model of breast carcinoma. Breast Cancer Res. 2010;12:1–10. doi: 10.1186/bcr2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonnichsen DS, Relling MV. Clinical pharmacokinetics of paclitaxel. Clin Pharmacokinet. 1994;27:256–269. doi: 10.2165/00003088-199427040-00002. [DOI] [PubMed] [Google Scholar]
- 7.He X, Wang Y, Zhu J, Orlaff M, Eng C. Resveratrol enhances the anti-tumor activity of the mTOR inhibitor rapamycin in multiple breast cancer cell lines mainly by suppressing rapamycin-induced AKT signaling. Cancer Lett. 2011;301:168–176. doi: 10.1016/j.canlet.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Hasenstein JR, Shin HC, Kasmerchak K, Buehler D, Kwon GS, Kozak KR. Antitumor activity of triolimus: a novel multidrug-loaded micelle containing paclitaxel, rapamycin, and 17-AAG. Mol Cancer Ther. 2012;11:2233–2242. doi: 10.1158/1535-7163.MCT-11-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mondesire WH, Jian W, Zhang H, Ensor J, Hung M-C, Mills GB, Meric-Bernstam F. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10:7031–7042. doi: 10.1158/1078-0432.CCR-04-0361. [DOI] [PubMed] [Google Scholar]
- 10.VanderWeele DJ, Zhou R, Rudin CM. Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther. 2004;3(12):1605–1613. [PubMed] [Google Scholar]
- 11.Blanco E, Sangai T, Wu S, Hsiao A, Ruiz-Esparza GU, Gonzalez-Delgado CA, Cara FE, Granados-Principal S, Evans KW, Akcakanat A, Wang Y, Do KA, Meric-Bernstam F, Ferrari M. Colocalized delivery of rapamycin and paclitaxel to tumors enhances synergistic targeting of the PI3K/Akt/mTORpathway. Mol Ther. 2014;22:1310–1319. doi: 10.1038/mt.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acevedo-Gadea C, Hatzis C, Chung G, Fishbach N, Lezon-Geyda K, Zelterman D, DiGiovanna MP, Harris L, Abu-Khalaf MM. Sirolimus and trastuzumab combination therapy for HER2 positive metastatic breast cancer after progression onprior trastuzumab therapy. Breast Cancer Res Treat. 2015;150:157–167. doi: 10.1007/s10549-015-3292-8. [DOI] [PubMed] [Google Scholar]
- 13.LoRusso PM. Mammalian target of rapamycin as a rational therapeutic target for breast cancer treatment. Oncology. 2013;84:43–56. doi: 10.1159/000343063. [DOI] [PubMed] [Google Scholar]
- 14.Vicier C, Dieci MV, Arnedos M, Delaloge S, Viens P, Andre F. Clinical development of mTOR inhibitors in breast cancer. Breast Cancer Res. 2014;16:1–9. doi: 10.1186/bcr3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malahati K, Kahan BD. Clinical pharmacokinetics of sirolimus. Clin Pharmacokinet. 2001;40:573–585. doi: 10.2165/00003088-200140080-00002. [DOI] [PubMed] [Google Scholar]
- 16.Yoshizawa Y, Kono Y, Ogawara KI, Kimura T, Higaki K. PEG liposomalization of paclitaxel improved its in vivo disposition and anti-tumor efficacy. Int J Pharm. 2011;412:132–141. doi: 10.1016/j.ijpharm.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, Jr, Baker JR, Van Echo DA, Von Hoff DD, Leyland-Jones B. Hypersensitivity reactions from taxol. J Clin Oncol. 2016;8(1190):1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 18.Mita MM, Mita A, Rowinsky EK. Mammalian target of rapamycin: a new molecular target for breast cancer. Clin Breast Cancer. 2003;4:126–137. doi: 10.3816/cbc.2003.n.018. [DOI] [PubMed] [Google Scholar]
- 19.Yatscoff RW. Pharmacokinetics of rapamycin. Transpl Proc. 1996;28:970–973. [PubMed] [Google Scholar]
- 20.Eloy JO, de Souza MC, Petrilli R, Barcellos JPA, Lee RJ, Marchetti JM. Liposomes as carriers of hydrophilic small molecule drugs: strategies to enhance encapsulation and delivery. Colloids Surf B: Biointerfaces. 2014;123:345–363. doi: 10.1016/j.colsurfb.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 21.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics incancer chemotherapy; mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 22.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Delivery Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 23.Teow HM, Zhou ZM, Najilah SR, Yusof NJ, Abbott, D’Emanuele A. Delivery of paclitaxel across cellular barriers using a dendrimer-based nanocarrier. Int J Pharm. 2013;441:701–711. doi: 10.1016/j.ijpharm.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 24.Emami J, Rezazadeh M, Hasanzadeh F, Sadeghi H, Mostafavi HA, Minaiyan M, Rostami M, Davies N. Development and in vitro/in vivo evaluation of a novel targeted polymeric micelle for delivery of paclitaxel. Int J Biol Macromol. 2015;80:29–40. doi: 10.1016/j.ijbiomac.2015.05.062. [DOI] [PubMed] [Google Scholar]
- 25.Le Broc-Ryckewaert D, Carpentier R, Lipka E, Daher S, Vaccher C, Betbeder D, Furman C. Development of innovative paclitaxel-loaded small PLGA. nanoparticles: study of their antiproliferative activity and their molecular interactions on prostatic cancer cells. Int J Pharm. 2013;454:712–719. doi: 10.1016/j.ijpharm.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Yang T, Cui FD, Choi MK, Cho JW, Chung SJ, Shim CK, Kim DD. Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm. 2007;338:317–326. doi: 10.1016/j.ijpharm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Haeri A, Sadeghian S, Rabbani S, Anvari MS, Boroumand MA, Dadashzadeh S. Use of remote film loading methodology to entrap sirolimus into liposomes: preparation characterization and in vivo efficacy for treatment of restenosis. Int J Pharm. 2011;414:16–27. doi: 10.1016/j.ijpharm.2011.04.055. [DOI] [PubMed] [Google Scholar]
- 28.I.C.H. ICH Harmonised Tripartite Guideline Validation of Analytical Procedures: Text and Methodology Q2(R1) I.C.H; 2005. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. [Google Scholar]
- 29.Chen MX, Li BK, Yin DK, Liang J, Li SS, Peng DY. Layer-by-layer assembly of chitosan stabilized multilayered liposomes for paclitaxel delivery. Carbohydr Polym. 2014;111:298–304. doi: 10.1016/j.carbpol.2014.04.038. [DOI] [PubMed] [Google Scholar]
- 30.de Souza MC, Marotta-Oliveira SS, Rocha NHS, Eloy JO, Marchetti JM. Development of a method to evaluate the release profile of tamoxifen from pegylated hybrid micelles. J Liquid Chromatogr Related Technol. 2015;38:1223–1229. [Google Scholar]
- 31.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 32.Meraz IM, Hearnden CH, Liu X, Yang M, Williams L, Savage DJ, Gu J, Rhudy JR, Yokoi K, Lavelle EC, Serda RE. Multivalent presentation of MPL by porous silicon microparticles favors T helper 1 polarization enhancing theanti-tumor efficacy of doxorubicin nanoliposomes. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu J, Huang Y, Zhao W, Chen Y, Li J, Gao X, Venkataramanan R, Li S. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol Pharm. 2013;10:2880–2890. doi: 10.1021/mp300729y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crosasso P, Ceruti M, Brusa P, Arpicco S, Dosio F, Cattel L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J Controlled Release. 2000;63:19–30. doi: 10.1016/s0168-3659(99)00166-2. [DOI] [PubMed] [Google Scholar]
- 35.Zhang JA, Anyarambhatla G, Ma L, Ugwu S, Xuan T, Sardone T, Ahmad I. Development and characterization of a novel cremophorw EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm. 2005;59:177–187. doi: 10.1016/j.ejpb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Yang T, Choi MK, Cui FD, Kim JS, Chung SJ, Shim CK, Kim DD. Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Release. 2007;120:169–177. doi: 10.1016/j.jconrel.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 37.Kirby C, Clarke J, Gregoriadis G. Effect of the cholesterol content of small unilamellar liposomes on their stability in vivo and in vitro. Biochem J. 1980;186:591–598. doi: 10.1042/bj1860591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Senior J, Gregoriadis G. Stability of small unilamella liposomes in serum and clearance from the circulation: the effect of the phospholipid and cholesterol components. Life Sci. 1982;30:2123–2136. doi: 10.1016/0024-3205(82)90455-6. [DOI] [PubMed] [Google Scholar]
- 39.Maruyama K, Yuda T, Okamoto A, Ishikura C, Kojima S, Iwatsuru M. Effect of molecular weight in amphipathic polyethyleneglycol on prolonging the circulation time of large unilamellar liposomes. Chem Pharm Bull. 1991;39:1620–1622. doi: 10.1248/cpb.39.1620. [DOI] [PubMed] [Google Scholar]
- 40.Paini M, Daly SR, Aliakbarian B, Fathi A, Tehrany EA, Perego P, Dehghani F, Valtchev P. An efficient liposome based method for antioxidants encapsulation. Colloids Surf B: Biointerfaces. 2015;136:1067–1072. doi: 10.1016/j.colsurfb.2015.10.038. [DOI] [PubMed] [Google Scholar]
- 41.Wagner A, Vorauer-Uhl K. Liposome technology for industrial purposes. J Drug Delivery. 2011(2011) doi: 10.1155/2011/591325. Article ID 591325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eloy JO, Marchetti JM. Solid dispersions containing ursolic acid in poloxamer 407 and PEG 6000: a comparative study of fusion and solvent methods. Powder Technol. 2014;253:98–106. [Google Scholar]
- 43.Eloy JO, Saraiva J, Albuquerque S, Marchetti JM. Solid dispersion of ursolic acid in gelucire 50/13: a strategy to enhance drug release and trypanocidal activity. AAPS PharmSciTech. 2012;13:1436–1445. doi: 10.1208/s12249-012-9868-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martins KF, Messias AD, Leite FL, Duek EAR. Preparation and characterization of paclitaxel-loaded PLDLA microspheres. Mater Res. 2014;17:650–656. [Google Scholar]
- 45.Bian H, Zhou S, Liang X, Li Q, Han W. In vitro study of poly(ethylene carbonate) as a drug-eluting stent coating. Prog Nat Sci: Mater Int. 2012;22:295–302. [Google Scholar]
- 46.Liggins RT, Hunter WL, Burt HM. Solid-state characterization of paclitaxel. J Pharm Sci. 1997;86:1458–1463. doi: 10.1021/js9605226. [DOI] [PubMed] [Google Scholar]
- 47.Rouf MA, Vural I, Billensoy E, Hincal A, Erol DD. Rapamycin-cyclodextrin complexation: improved solubility and dissolution rate. J Inclusion Phenom Macrocyclic Chem. 2011;70:167–175. [Google Scholar]
- 48.Rouf MA, Vural I, Renoir JM, Rincal AA. Development and characterization of liposomal formulations for rapamycin delivery and investigation of their antiproliferative effect on MCF7 cells. J Liposome Res. 2009;19:322–331. doi: 10.3109/08982100902963043. [DOI] [PubMed] [Google Scholar]
- 49.Ghanbarzadeh S, Arami S, Pourmoazzen Z, Khorrani A. Improvement of the antiproliferative effect of rapamycin on tumorcell lines by poly (monomethylitaconate)-based pH-sensitive, plasmastable liposomes. Colloids Surf B: Biointerfaces. 2014;115:323–330. doi: 10.1016/j.colsurfb.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y, Ji M, Wong MK, Joo KI, Wang P. Enhanced therapeutic efficacy of irgd-conjugated crosslinked multilayer liposomes for drug delivery. BioMed Res Int. 2013;2013 doi: 10.1155/2013/378380. Article ID 378380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shin HC, Alani AWG, Cho H, Bae Y, Kolesar JM, Kwon GS. A 3-in-1 polymeric micelle nanocontainer for poorly water-soluble drugs. Mol Pharm. 2011;8:1257–1265. doi: 10.1021/mp2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]
- 53.Poondru S, Parchment RE, Purohit V, LoRusso P, Horwitz JP, Hazeldine ST, Polin L, Corbett T, Jasti BR. Lack of in vitro-in vivo correlation of a novel investigational anticancer agent, SH30. Invest New Drugs. 2002;20:23–33. doi: 10.1023/a:1014457510073. [DOI] [PubMed] [Google Scholar]
- 54.Palma G, Conte CA, Barbieri S, Bimonte, Luciano A, Rea D, Ungaro F, Tirino P, Quaglia FF, Arra C. Antitumor activity of PEGylated biodegradable nanoparticles for sustained. release of docetaxel in triple-negative breast cancer. Int J Pharm. 2014;473:55–63. doi: 10.1016/j.ijpharm.2014.06.058. [DOI] [PubMed] [Google Scholar]
- 55.Yoshizawa Y, Ogawara K-I, Kimura T, Higaki K. A novel approach to overcome multidrug resistance: utilization of P-gp mediated efflux of paclitaxel to attack neighboring vascular endothelial cells in tumors. Eur J Pharm Sci. 2014;62:274–280. doi: 10.1016/j.ejps.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 56.Pawarode A, Minderman H, O’loughilin KL, Greco WR, Baer MR. Rapamycin overcomes multidrug resistance (MDR) mediated by P-glycoprotein (Pgp), multidrug resistance protein-1 (MRP-1), breast cancer resistance protein (BCRP) and lung resistance protein (LRP) and synergizes with substrate drugs in MDR cells. Cancer Res. 2006;66:1270. [Google Scholar]
- 57.Kopecka J, Salzano G, Campia I, Lusa S, Ghigo D, De Rosa G, Riganti C. Insights in the chemical components of liposomes responsible for P-glycoprotein inhibition, nanomedicine: nanotechnology. Biol Med. 2014;10:77–87. doi: 10.1016/j.nano.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 58.Mallick S, Choi JS. Liposomes: versatile and biocompatible nanovesicles for efficient biomolecules delivery. J Nanosci Nanotechnol. 2014;14:755–765. doi: 10.1166/jnn.2014.9080. [DOI] [PubMed] [Google Scholar]