

Abstract
Trough gentamicin therapeutic drug monitoring (TDM) is time-consuming, disruptive to neonatal clinical care, and a patient safety issue. Bayesian models could allow TDM to be performed opportunistically at the time of routine blood tests. This study aimed to develop and prospectively evaluate a new gentamicin model and a novel Bayesian computer tool (neoGent) for TDM use in neonatal intensive care. We also evaluated model performance for predicting peak concentrations and the area under the concentration-time curve from time 0 h to time t h (AUC0–t). A pharmacokinetic meta-analysis was performed on pooled data from three studies (1,325 concentrations from 205 patients). A 3-compartment model was used with the following covariates: allometric weight scaling, postmenstrual and postnatal age, and serum creatinine concentration. Final parameter estimates (standard errors) were as follows: clearance, 6.2 (0.3) liters/h/70 kg of body weight; central volume (V), 26.5 (0.6) liters/70 kg; intercompartmental disposition (Q), 2.2 (0.3) liters/h/70 kg; peripheral volume V2, 21.2 (1.5) liters/70 kg; intercompartmental disposition (Q2), 0.3 (0.05) liters/h/70 kg; peripheral volume V3, 148 (52.0) liters/70 kg. The model's ability to predict trough concentrations from an opportunistic sample was evaluated in a prospective observational cohort study that included data from 163 patients and 483 concentrations collected in five hospitals. Unbiased trough predictions were obtained; the median (95% confidence interval [CI]) prediction error was 0.0004 (−1.07, 0.84) mg/liter. Results also showed that peaks and AUC0–t values could be predicted (from one randomly selected sample) with little bias but relative imprecision, with median (95% CI) prediction errors being 0.16 (−4.76, 5.01) mg/liter and 10.8 (−24.9, 62.2) mg · h/liter, respectively. neoGent was implemented in R/NONMEM and in the freely available TDMx software.
INTRODUCTION
The aminoglycoside antibiotic gentamicin is the most commonly used antimicrobial in neonatal units (1, 2) and is effective against Gram-negative bacteria. Gentamicin use is limited by its narrow therapeutic index and risk of toxicity, specifically, nephro- and ototoxicity (3). It is not metabolized in the liver (4) and is almost entirely eliminated by the kidneys; clearance therefore depends on renal function. During the first 2 weeks of life, renal and intrarenal blood flow increase rapidly, causing a steep rise in the glomerular filtration rate (GFR) (5, 6).
Therapeutic drug monitoring (TDM) is required to ensure maximal efficacy and, in particular, minimal toxicity, particularly in the neonatal population, where the variability in pharmacokinetic (PK) parameters is large. Dose individualization approaches focus on toxicity (7, 8) and include single-level methods and nomograms (9, 10), area under the curve (AUC) methods (11), and Bayesian methods (12). The use of nomograms is limited as they cannot readily incorporate covariates affecting PK parameters. AUC methods use a simplified 1-compartment PK model and require at least two gentamicin measurements, which is not appropriate in neonates with limited blood volumes. These drawbacks make Bayesian approaches the most attractive for newborn infants.
Deriving a Bayesian prior for TDM requires a nonlinear mixed-effect PK model, and several such studies of neonatal gentamicin were previously published (13–24). However, those studies were limited by their heterogeneity and use of sparse data (often identifying only a 1-compartment model, whereas gentamicin follows multicompartment kinetics [25, 26]) and failed to account for age-related differences in creatinine levels during the immediate newborn period. Although gentamicin is not a new drug, its dosing and monitoring are still current issues as identified in the United Kingdom National Patient Safety alert (http://www.nrls.npsa.nhs.uk/alerts/?entryid45=66271) and in a recent publication by Valitalo et al. (27), who used simulations to define dosing guidelines.
We aimed to investigate whether opportunistic sampling can predict trough gentamicin concentrations so that standard TDM can be performed using a blood sample taken for other purposes (e.g., routine blood gases). As a secondary aim, we evaluated the model's ability to predict peak gentamicin concentrations and AUC from time 0 h to time t h (AUC0–t) using one randomly selected sample.
MATERIALS AND METHODS
Study population.
This study used two data sets: a model-building data set and a prospectively collected evaluation data set.
To collect data for model development, the electronic bibliographic database PubMed was searched in January 2015 without time limitations. The search strategy included the following: (neonat* OR newborn*) AND (gentamicin) AND (pharmacokinetic* OR PK). Gentamicin samples had to be prospectively collected, and covariates (weight, gestational age [GA], postnatal age [PNA], serum creatinine concentration [SCr]) also had to be reported. We also searched the reference lists in identified papers. The authors of the publications that met the inclusion criteria (n = 8) (11, 15, 21, 22, 28–31) were then invited to contribute their data.
Data for the evaluation of the PK model were collected as a prospective observational cohort study from five United Kingdom hospitals (St George's University Hospitals NHS Foundation Trust, Liverpool Women's NHS Foundation Trust, Oxford University Hospitals, Portsmouth Hospitals NHS Trust, and Coventry & Warwickshire University Hospitals NHS Trust) from July 2012 to November 2013. Infants were eligible for inclusion if the following criteria were met: more than 36 h gentamicin therapy anticipated; postnatal age of less than 90 days; no extracorporeal membrane oxygenation, peritoneal dialysis, or hemofiltration received; and expectation of survival of the study period (as judged by the clinical team). Each patient provided a minimum of two gentamicin concentrations—a trough sample from routine TDM (i.e., a predose sample taken before a noninitial dose) and an additional study sample (taken opportunistically during a course of gentamicin when the infant required blood sampling for clinical care). These samples are referred to as routine (trough) and opportunistic study samples in this article. Exact times of gentamicin dosing and sampling were recorded, along with the patient's weight, age, and serum creatinine concentration (Table 1). Written informed consent was obtained from parents, and the study was approved by the London Central Ethics Committee (reference 12/LO/0455).
TABLE 1.
A summary of demographics and dosinga
| Parameter | Model-building data set | Evaluation data set |
|---|---|---|
| No. of subjects | 205 | 163 |
| Wt (kg)b | 2.12 (0.53–5.05) | 2.03 (0.48–5.05) |
| Gestational age (wks)b | 34.0 (23.3–42.1) | 34.3 (23.9–42.3) |
| Postnatal age (days)b | 5.4 (1–66) | 6 (1–78) |
| Postmenstrual age (wks)b | 33.0 (23.3–43.8) | 34.9 (24–43.3) |
| No. (%) of females | 89 (43%) | 68 (41.7%) |
| No. of gentamicin samples per patientc | 6.5 | 3.0 |
| Gentamicin concn (mg/liter)b | 3.4 (0.3–37.6) | 1.0 (0.1–13.2) |
| Time after the dose (h)b | 8.0 (0.02–54.1) | 23.5 (0.08–79.7) |
| No. of occasionsb | 2 (1–22) | 2 (1–7) |
Weight and gestational age data represent values at treatment initiation; the rest of the data represent values at the time of gentamicin sampling/dosing. An occasion was defined as a dose with subsequent gentamicin samples taken; day of birth was defined as day 1.
Data represent medians (ranges).
Data represent means.
Gentamicin dosing and sampling procedure in the prospective evaluation data set.
Gentamicin treatment was initiated at the discretion of the clinical team for possible infection and dosed and monitored using trough concentrations according to the standard practice at each hospital. Gentamicin was administered as a slow (<2-min) bolus via intravenous cannula, percutaneous long line, or umbilical venous catheter.
Bioanalytical techniques.
An enzyme immunoassay (EMIT; Syva) (15), a fluorescence polarization immunoassay (TDx; Abbot) (15, 21), and high-performance liquid chromatography coupled with tandem mass spectrometry (UHPLC-MS/MS) (32) were used to determine gentamicin concentrations in the model-building data set, and the Jaffe reaction (33) was used to determine serum creatinine concentrations. In the prospective evaluation data set, gentamicin serum concentrations were analyzed using immunoassay techniques (see Table S1 in the supplemental material), and creatinine concentrations were determined by either a Jaffe-based method or an enzymatic method (137 neonates and 26 neonates, respectively).
Pharmacokinetic analysis.
The observed concentration-time data from the model-building studies only were pooled and simultaneously analyzed with nonlinear mixed-effects software (NONMEM version 7.3) (34). The first-order conditional estimation method with interaction was used.
Basic model.
One-, 2-, and 3-compartment structural models were considered in defining the basic structural population PK model. The interindividual variability (IIV) was assumed to follow a log-normal distribution and was tested on all parameters. An additive residual error model, a proportional residual error model, and a combination of the two (equation 1) were tested:
| (1) |
where yij is the observed gentamicin concentration at time tij, f is the function that represents the gentamicin model, ϕi is a vector of parameters, and εij is a residual error term.
Interoccasion variability (IOV) was also assumed to be log-normally distributed, and it was tested for all parameters, with an occasion defined as a single dosing interval.
Covariate model.
Allometric scaling was used a priori to standardize all PK parameters to 70 kg (35), and a maturation function, describing the maturation of the GFR with postmenstrual age (PMA) (equation 2) with fixed parameters from a previous study (5), was used to scale clearance. Allometric exponents were fixed to 0.632 for central clearance and 0.75 for intercompartmental clearances. The two different exponents were used because these values were shown to be the best for describing the maturation of renal elimination (5) and tissue blood flows (36), respectively. Allometric exponents for volumes of distribution were fixed to 1. The combination of allometric weight scaling and sigmoidal maturation function was suggested as a standard method for scaling clearance in the pediatric population in a recent comparison of different approaches (37).
| (2) |
where Hill is the sigmoidicity coefficient and PMA50 is the PMA when the maturation of the GFR reaches 50% of adult values.
As it is known that PNA and serum creatinine concentration are important indicators of gentamicin clearance and also based on the post hoc estimates of eta versus covariate plots, they were tested on clearance. These time-varying covariates were considered to significantly improve the fit and were therefore included in the model when the difference in objective function value (ΔOFV) after their inclusion was >3.84 (P < 0.05). Additionally, linear extrapolations between observations were made. To account for endogenous and maternal creatinine concentrations and also for the change in renal function with age, a typical value of serum creatinine concentration for a specific PMA (TSCr), was determined using data from Cuzzolin et al. (38) for preterm (GA, <37 weeks) newborns and data from Rudd et al. (39) for term newborns. A linear decline in TSCr with increasing PMA was found according to equation 3:
| (3) |
A possible influence of serum creatinine on clearance was tested according to equation 4, where the measured serum creatinine concentration (MSCr) was standardized using TSCr and departures from it were estimated as follows:
| (4) |
The effect of PNA was investigated with a logistic function (equation 5) to account for the rapid changes in gentamicin clearance in the first hours of life (the first day of life was defined as day 1) as follows:
| (5) |
where PNA50 is the PNA when clearance has reached 50% of the clearance seen in the typical adult.
After the forward selection (ΔOFV, >3.84) of all covariates (full model), backward elimination was performed, with a P value retention cutoff value of 0.001 (ΔOFV, <10.83).
Evaluation. (i) Internal model evaluation.
Basic goodness-of-fit plots for observations versus population and individual predictions and for conditional weighted residuals versus population predictions and versus time after dose were produced using R statistical software version 3.1.0 (40) and visually examined. The assumptions of normality and homogeneity of the residuals errors were investigated by inspecting a histogram and a q-q plot.
Standard errors from NONMEM covariance step and nonparametric bootstrap analysis performed with 1,000 replicates were used to determine the precision of the final PK parameter estimates.
Additionally, we simulated 1,000 data sets using parameter estimates from the final model and plotted 95% confidence intervals (CI) around the 2.5th, 50th, and 97.5th prediction percentiles of the simulated data. The observations were then overlaid on the plot, also called the visual predictive check (VPC). Perl-speaks-NONMEM (PsN) software (41) was used for the bootstrap analysis and to produce the VPC, which was visualized using R-package Xpose4 (42).
(ii) External model evaluation.
The prospective evaluation data set was used to evaluate the predictive performance of the model. No additional fitting was done, and the diagnostic plots and the VPC were generated as described above.
Bayesian model-predicted trough concentrations were computed using the model as a prior and information from the opportunistic study samples only. These predictions were compared with the observed trough concentrations by calculating the prediction error (PE) (43) and also the mean PE (MPE) (i.e., a measure of bias) and the root mean square error (RMSE), a measure of precision (44) (equations 6):
| (6) |
Also, we counted the number of “correct” predictions that were below or above the currently recommended gentamicin trough concentration threshold of 1 mg/liter or 2 mg/liter (the National Institute for Health and Care Excellence [NICE; http://www.nice.org.uk/guidance/CG149/chapter/1-Guidance#therapeutic-drug-monitoring-for-gentamicin] and British National Formulary for Children [45]).
Further analysis of paired samples (that is, both study and routine samples taken in the same dosing interval) was undertaken for the following scenarios: study samples at concentrations of ≥1, ≥2, and ≥3 mg/liter (compared with unpaired samples only).
Cross-validation.
The subset with the study sample concentration above 3 mg/liter provided the most important comparison, since in this case the study sample concentration was still above the prespecified trough threshold. As there were only 18 pairs with an opportunistic study concentration of ≥3 mg/liter in the evaluation data set, these pairs were merged with paired samples with the same characteristics from the model-building data set. The pooled data set was then randomly split into five subsets, and cross-validation was performed (meaning that 20% of the pairs in each subset were randomly removed and the model was reestimated). The reestimated model was then used as a prior to predict the troughs, and the predicted troughs were compared to the observed trough concentrations as previously described.
Whether the model is able to predict peak concentrations from one randomly selected nonpeak sample was tested essentially as described above, using paired samples from both the model-building data set and the evaluation data set and performing cross-validations. Additionally, in recognition of the fact that a possible pharmacokinetic-pharmacodynamic target for aminoglycosides can also be AUC0–24/MIC (46), the model was also evaluated on how it predicts AUC0–t. Only a subset of the data in which five or more samples were collected after the same dose was used for defining AUC0–t, and the model-predicted versus observed (noncompartmental) AUC0–t values were compared.
Comparison with other models.
To compare our mechanistic model, which scales for size, age, and expected renal function, with previously published models using empirical covariate analysis, predictions of the trough concentrations were generated from the opportunistically collected samples in our prospective data set.
neoGent software.
The model was implemented using R and NONMEM (see the supplemental material). It works as follows: first, an individual's data are read into R; then, Bayesian estimates generated in NONMEM are used to predict outcomes of interest (e.g., the time at which the concentration falls below 2 mg/liter).
RESULTS
Patients.
We contacted eight authors identified in the literature search and obtained two large neonatal gentamicin data sets (15, 21). We received no response from four authors (11, 28–30), and, although an initial response was received from two authors (22, 31), no data were actually shared. Additionally, we obtained some previously unpublished data taken during a PK study of ampicillin and penicillin (32). The data were pooled and comprised 1,325 gentamicin concentrations from 205 neonates (Table 1). This data set was used to derive the model.
For the model evaluation, gentamicin serum concentrations were prospectively collected from a total of 194 neonates. Of the enrolled patients, 163 were included in the PK analysis (Table 1). Reasons for exclusion (31 patients) included inexact sampling times, insufficient samples, or the gentamicin opportunistic study concentration being below the limit of quantification (n = 12). The final evaluation data set comprised 483 gentamicin serum measurements, with 229 study and 254 routinely taken trough concentrations. Median (range) times after dosing were 13.3 (0.08 to 53.3) h and 31.1 (8.0 to 79.7) h for study and routine concentrations, respectively. Patients were on treatment for up to 20 days.
Pharmacokinetic analysis.
Initially, a 2-compartment model provided a better fit to the data (ΔOFV = 7.4 with a 3-compartment model) and was therefore chosen as the basic structural model. But after the addition of the fixed allometric and renal function parameters, covariates, and IOV, a 3-compartment model described the data better (47-unit drop in OFV). The IIV was described with an exponential error structure, and the best residual error model was a combination of a proportional error and an additive error.
Postnatal age and standardized serum creatinine concentration had a significant effect on clearance (ΔOFV = 134.1 and ΔOFV = 17.2, respectively) and were thus included in the final model. Backward elimination (P = 0.001) confirmed that these covariates remained significant with the 3-compartment model. The final gentamicin population PK model is summarized with equations 7:
| (7) |
where CL is gentamicin clearance, V is gentamicin volume of distribution, Q is intercompartmental gentamicin clearance, WT is body weight in kilograms, η is IIV, and κ is IOV.
There was only a small improvement in fit (ΔOFV = 7.6) when the model was parameterized for time-varying covariates (linear extrapolation between observed covariate values), but as this model is more biologically plausible, it was chosen as the final model.
The OFV reduced from 2,305.0 to 1,217.5 between the basic and the final model. The inclusion of the covariates resulted in a reduction of the IIV for the PK parameters: the IIV values for CL and V with the basic model were 71.1% and 62.5%, respectively, and with the final model were 41.8% and 33.5%, respectively. The final PK parameter estimates with uncertainty are reported in Table 2.
TABLE 2.
Final parameter estimates with uncertainty from NONMEM output file and from the bootstrap analysisa
| Parameter | Value from the final model |
Value from the bootstrap analysis |
|||||
|---|---|---|---|---|---|---|---|
| Mean | SE | % CV | η shrinkage | Median | 2.5th percentile | 97.5th percentile | |
| CL (liters/h/70 kg) | 6.21 | 0.30 | 6.14 | 5.47 | 6.75 | ||
| θ_SCr | −0.13 | 0.055 | −0.13 | −0.25 | −0.03 | ||
| PNA50 (days) | 1.70 | 0.30 | 1.68 | 1.15 | 2.30 | ||
| V (liters/70 kg) | 26.5 | 1.11 | 26.3 | 23.6 | 28.4 | ||
| Q (liters/h/70 kg) | 2.15 | 0.32 | 2.19 | 1.68 | 3.25 | ||
| V2 (liters/70 kg) | 21.2 | 1.50 | 20.9 | 17.9 | 24.2 | ||
| Q2 (liters/h/70 kg) | 0.27 | 0.047 | 0.28 | 0.19 | 0.38 | ||
| V3 (liters/70 kg) | 148 | 52.0 | 152 | 65.2 | 534 | ||
| IIV on CL | 0.175 | 0.038 | 41.8 | 6.9 | 0.170 | 0.104 | 0.254 |
| IIV on V | 0.112 | 0.032 | 33.5 | 15.2 | 0.113 | 0.057 | 0.190 |
| CL-V covariance | 0.116 | 0.030 | 0.115 | 0.060 | 0.184 | ||
| IIV on V2 | 0.132 | 0.060 | 36.3 | 57.8 | 0.117 | 0.023 | 0.281 |
| IIV on V3 | 0.177 | 0.216 | 42.1 | 85.0 | 0.114 | 0.00002 | 4.18 |
| Interoccasion variability | 0.014 | 0.007 | 11.8 | 0.013 | 0.001 | 0.029 | |
| Residual error (proportional) | 0.036 | 0.006 | 19.0 | 0.036 | 0.025 | 0.049 | |
| Residual error (additive) | 0.016 | 0.007 | 0.015 | 0.000002 | 0.032 | ||
CL, clearance; V, volume of distribution; Q (and Q2), intercompartmental CL; IIV, interindividual variability; SE, standard error obtained with NONMEM 7.3 covariance step; CV, coefficient of variation.
Evaluation. (i) Internal model evaluation.
Figure 1 shows plots assessing goodness of fit by comparing observations and predictions. A VPC of the final model is shown in Fig. 2.
FIG 1.

Observed versus population-predicted gentamicin serum concentrations (top left for the model-building data set and bottom left for the evaluation data set) and conditional weighted residuals versus time after dose (top right for the model-building data set and bottom right for the evaluation data set).
FIG 2.

Visual predictive check of 1,000 simulated concentration-time data sets from the final model, using the model-building data set (left) and the evaluation data set (right). Points represent the observations, black lines represent the 2.5th, 50th, and 97.5th percentiles, and the shaded areas represent the 95% confidence intervals of the corresponding predicted gentamicin concentrations.
(ii) External model evaluation.
The basic diagnostic plots are presented in Fig. 1 and the VPC performed using the evaluation data set and the final parameters from the PK model without additional fitting in Fig. 2.
Table 3 shows the number of correct predictions (for five different data sets from the evaluation data and pooled results from the cross-validation) for gentamicin trough thresholds of 1 and 2 mg/liter together with prediction errors. In the total data set, containing both paired and unpaired samples, the median (95% CI) PE was 0.0004 (−1.1, 0.8) mg/liter. The MPEs from predictions of trough and peak concentrations (using cross-validations) were 0.03 and 0.19 mg/liter and the RMSEs 1.28 and 2.55 mg/liter, respectively (Table 3). When AUC0–t prediction (from one random sample) was evaluated, the MPE was 14.5 mg · h/liter and the RMSE 30.2 mg · h/liter.
TABLE 3.
Summary of external evaluation with the evaluation data seta
| Data set | Limit = 1 mg/liter |
Limit = 2 mg/liter |
PE | MPE | RMSE | ||||
|---|---|---|---|---|---|---|---|---|---|
| No. of correct results/total no. of results (%) | No. of OP | No. of UP | No. of correct results/total no. of results (%) | No. of OP | No. of UP | ||||
| Paired + unpaired | 214/254 (84.3) | 20 | 20 | 242/254 (95.3) | 10 | 2 | 0.0004 mg/liter (−1.07, 0.84) | 0.007 mg/liter | 0.45 mg/liter |
| Paired: study, ≥1 mg/liter | 53/57 (93.0) | 3 | 1 | 56/57 (98.2) | 1 | 0 | −0.04 mg/liter (−0.57, 0.70) | −0.03 mg/liter | 0.32 mg/liter |
| Paired: study ≥ 2 mg/liter | 31/33 (93.9) | 2 | 0 | 33/33 (100) | 0 | 0 | −0.08 mg/liter (−0.50, 0.74) | −0.05 mg/liter | 0.35 mg/liter |
| Paired: study ≥ 3 mg/liter | 19/20 (95.0) | 0 | 1 | 20/20 (100) | 0 | 0 | −0.06 mg/liter (−0.56, 0.82) | −0.02 mg/liter | 0.42 mg/liter |
| Unpaired | 136/161 (84.5) | 14 | 11 | 155/161 (96.3) | 5 | 1 | 0.02 mg/liter (−1.11, 0.70) | −0.001 mg/liter | 0.43 mg/liter |
| XV, paired: study ≥ 3 mg/liter | 478/502 (95.2) | 12 | 12 | 460/502 (91.6) | 21 | 21 | −0.04 mg/liter (−1.77, 3.03) | 0.03 mg/liter | 1.28 mg/liter |
| XV, peaks | 0.16 mg/liter (−4.76, 5.01) | 0.19 mg/liter | 2.55 mg/liter | ||||||
| AUC0–t | 10.8 mg · h/liter (−24.9, 62.2) | 14.5 mg · h/liter | 30.2 mg · h/liter | ||||||
“Correct” indicates that the predicted trough concentration value agrees with the measured concentration value (is above/below the limit). OP, overpredictions; UP, underpredictions; PE, prediction error [median (95% confidence interval]); MPE, mean prediction error; RMSE, root mean square error; XV, cross-validation. Other than the “XV, peaks” data and the AUC0–t data, all results refer to trough prediction evaluations.
Figure 3 shows the median and the range of PE for this model and previously published gentamicin population PK models.
FIG 3.
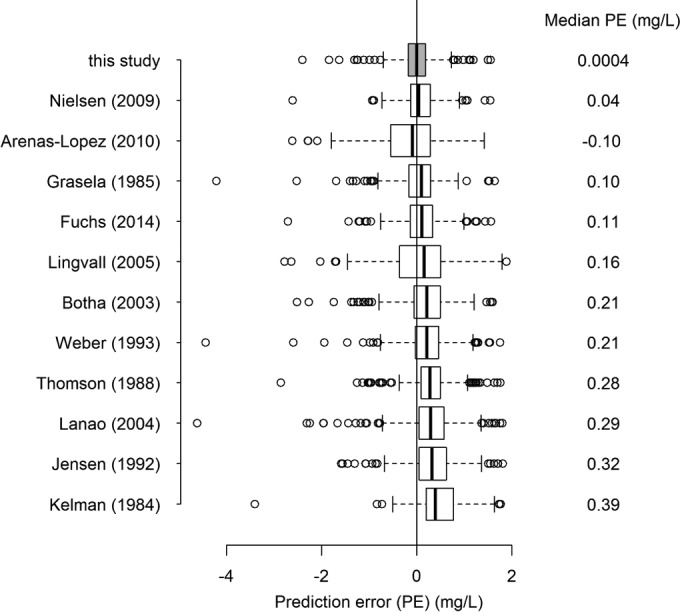
Comparison of predictive performances of the developed model (shaded box plot) and previously published neonatal gentamicin PK models.
neoGent.
Figure S1 in the supplemental material shows an example of an output from neoGent.
DISCUSSION
A PK model for gentamicin in neonates was developed and evaluated with prospectively collected data. Through its use of mechanistic covariates, the model gave unbiased predictions of trough concentrations from an opportunistic sample. Using this model, concentrations from samples taken at any time can be used to generate informative TDM, potentially eliminating the need for specifically timed trough gentamicin samples and the safety concerns and inconvenience associated with them. An exploratory analysis to evaluate whether such an approach could be used for predicting individual peak concentration and AUC0–t showed that, while the predictions were unbiased, they were relatively imprecise (Table 3).
The small median PE (0.0004 mg/liter) for trough concentrations suggests that the model implemented in neoGent performs well, although some outliers were not captured (range, −2.4 to 1.6 mg/liter). The median prediction errors were in most cases negative (Table 3), indicating that the model slightly overpredicts the trough concentrations (i.e., predicts them to be higher than they are), which might be (from a safety perspective) preferable to underpredicting. Cross-validations confirmed that samples do not need to be taken at a specific time when using this model for TDM, as the predictions of trough concentrations (as determined using an opportunistic sample) were unbiased, with a median PE of −0.04 mg/liter (Table 3). Although we did not test the effect of the sampling time on model predictions, the samples were collected from a wide range of times (0.1 to 53.3 h after the dose), as they would be in routine hospital tests.
Comparison of the developed model with the existing published models showed that the predicted trough concentrations were the least biased (i.e., the median prediction error was the smallest) when our model was used (Fig. 3). However, due to unavailability of some covariates in our data set, three models were used without all of the covariates (Apgar score [15, 19], sepsis [19], and comedication with dopamine [23]) included, which could explain their worse predictive performance.
The rich data in our model-building data set (6.5 samples per patient) supported a 3-compartment model, where the final estimates for the third compartment were as follows: intercompartmental clearance, 0.3 liters/h/70 kg; peripheral volume of distribution, 148 liters/70 kg. Additionally, the terminal half-life for a typical subject from the prospective evaluation data set (weight, 2.0 kg; PMA, 34.9 weeks; PNA, 6 days; MSCr, 47.0 μmol/liter; TSCr, 66.4 μmol/liter) was 189.7 h. This could indicate uptake of gentamicin into the renal cortex, and slow excretion from it (47), and is in agreement with previously found evidence of deep tissue accumulation of gentamicin (26, 48).
Unfortunately, many authors were unwilling or unable to share their data, and we managed to obtain data from only two (15, 21) of eight identified studies for our model-building data set. We did obtain one further subsequent data set corresponding to results of assays performed in another pharmacokinetic study in neonates also receiving gentamicin (32). Due to differences with respect to model structure and parameterization, it was not possible to extract relevant information for model building from the published reports. However, thanks in part to the fact that the data obtained from Nielsen et al. (21) were of such high quality, with multiple samples assayed per patient, our final model described both the model-building data set and the evaluation data set well, as shown in Fig. 1 and 2. The histogram and the q-q plot of the conditional weighted residuals (data not shown) confirmed that they follow a normal distribution pattern. The mean (standard error) final estimates for clearance (CL) and volume of distribution (V) were 6.21 (0.30) liters/h/70 kg and 26.5 (1.11) liters/70 kg, respectively (Table 2). The values of the PK parameters for a typical infant from the model-building data set (weight, 2.12 kg; PMA, 33.0 weeks; PNA, 5.4 days; MSCr, 78 μmol/liter; TSCr, 71.4 μmol/liter) were 0.077 liters/h and 0.80 liters (and 0.10 liters/h and 0.78 liters for a neonate from the evaluation data set) for CL and V, respectively. These values are in agreement with clearance estimates from previous neonatal studies of gentamicin pharmacokinetics (13, 14, 18, 22–24). The reported value (0.026 liters/h) for CL from Nielsen et al. (21) may appear to be lower, but using our median demographic values in their model, the CL value becomes similar to our estimates (0.095 liters/h). The final estimate for volume of distribution is consistent with the estimates from Fuchs et al. (23) and Botha et al. (24), but it is not in accordance with what was found by García et al. (20) (0.252 liters). The probable reason for this is the use of a different studied population, because when the median weight from our data set was used in their model, the resulting V was 0.968 liters, in agreement with our estimate.
We did not attempt to estimate the allometric power exponents and constants of the maturation function, as the PMA in the studied neonates (23.3 to 43.8 weeks) was insufficient to capture the age when maturation was complete (PMA50 = 55.4 weeks [5]); instead, these constants were fixed to the values from another study in which the main focus was renal maturation (5). This type of scaling was used to improve the model usefulness by allowing it to be extrapolated to different subpopulations (for example, neonates with a different weight or a different PMA). In addition to capturing changes in clearance due to the long-term maturation that extends throughout gestation and into the first 2 years of life, we attempted to capture the short-term changes in clearance that occur after birth regardless of gestational age. A benefit of fixing the long-term maturation based on known relationships between PMA and renal function was that this short-term maturation was apparent with our estimate of PNA50 of 40.8 h, indicating that clearance rapidly increases over the first few days of life. In the first day of life, the clearance was at 37% of the value for a typical adult, and it reached 95% by the end of the first month of infancy.
The typical serum creatinine concentration (used in the model) was determined using SCr concentrations from the Jaffe assay, because the same method was used to determine SCr concentrations in the model-building data set. However, for the evaluation data set, assays based on both the Jaffe and enzymatic methods were used to determine SCr concentrations. However, the goodness of fit to the evaluation data set and the predictive performance of the model were good; therefore, no correction factor was included. Also, the enzymatic assay was used in only 16% of patients. Due to the range of the data that were used to determine typical-for-PMA SCr concentrations, the model can be used for a neonate with a PMA of <44 weeks or for a term neonate of <4 weeks of age. The power exponent on the creatinine function was estimated to be −0.13, meaning that, if observed SCr and typical SCr concentrations were 70 μmol/liter and 60 μmol/liter, respectively, clearance would be 2% lower.
Large η shrinkage values indicate that the data did not contain enough information to make a reliable individual estimation. And while the level of shrinkage corresponding to the peripheral volumes of distribution (V2 and V3) was high, that corresponding to clearance was relatively low (6.9%) (Table 2), which is important for making predictions of trough gentamicin concentrations and AUC0–t. The η shrinkage corresponding to the central volume of distribution was also relatively low (15%) (Table 2).
Although the main aim of the present study was to evaluate whether the model can predict trough concentrations, the ability of the model to predict the peak gentamicin concentration (from a randomly selected nonpeak sample) was also examined. Cross-validations showed that the median prediction error (95% CI) in predicting peaks was 0.16 (−4.76, 5.01) mg/liter, indicating unbiased but not very precise predictions. This is perhaps not surprising, given that the concentrations determined at a median time after dosing of 19.3 h were used to predict concentrations at a median of 1 h postdose. The prediction of AUC0–t (also from one sample) was similarly unbiased (median prediction error, 10.8 mg · h/liter) but imprecise (95% CI, −24.9 to 62.2 mg · h/liter) (Table 3). However, normalized RMSEs (to the range of observed data) for peak and AUC0–t prediction were 7.0% and 17.6%, respectively, indicating that, considering the range of possible values, the precision is perhaps more acceptable. Target AUC0–24 or peak values have not been defined in neonates, and slow clearance and a narrow therapeutic index mean that adjusting doses to target efficacy in this population may not be realistic. However, our model does now give unbiased predictions of both metrics from an opportunistically collected single sample, which should prove useful in future clinical research to define efficacy targets in this age group. At present, due to their imprecision, these predictions (for peak concentration and AUC0–t) should currently be used only for research purposes and not for dose adjustment.
Conclusion.
A new gentamicin model has been developed and evaluated with prospectively collected data. We used mechanistic covariate parameterization informed by principles of allometric size scaling, known scaling of glomerular filtration maturation, and standardization for age-expected serum creatinine concentration. This “biological prior” information gave a model with better predictive performance for prospectively collected external data than any previously published gentamicin model. Using this, we developed a software tool, neoGent (see the supplemental material for the provisional stand-alone version and the version implemented in the Web TDM application TDMx [http://www.tdmx.eu/] [49]), which can be used to predict when the trough concentration falls below 2 mg/liter and thus to guide the dosing interval. Furthermore, the peak concentration or AUC0–24 from any postdose sample can also be predicted with little bias.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alison Thomson for making the data from reference 15 freely available online (NIH/NIBIB grant P41-EB01975) and all patients, families, and staff members of the hospitals participating in the neoGent study. We also thank the members of the neoGent collaboration, Mark Anthony, Tim Scorrer, Prakash Satodia, and Nasreen Aziz, and their research teams. We acknowledge the contribution of the United Kingdom National Institute for Health Research Clinical Research Network.
We declare that we have no conflicts of interest.
Funding Statement
This project was funded by Action Medical Research (grant code SP4650, GN1834). E.G. received funding from the NeoMero study, part of the European Union Seventh Framework Programme for research, technological development and demonstration (grant agreement number 242146). J.F.S. received funding from a United Kingdom Medical Research Council Fellowship (grant number G1002305).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00577-16.
REFERENCES
- 1.Turner MA, Lewis S, Hawcutt DB, Field D. 2009. Prioritising neonatal medicines research: UK Medicines for Children Research Network scoping survey. BMC Pediatr 9:50. doi: 10.1186/1471-2431-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cantey JB, Wozniak PS, Sanchez PJ. 2014. Prospective surveillance of antibiotic use in the neonatal intensive care unit: results from the SCOUT Study. Pediatr Infect Dis J doi: 10.1097/INF.0000000000000542. [DOI] [PubMed] [Google Scholar]
- 3.Pacifici GM. 2009. Clinical pharmacokinetics of aminoglycosides in the neonate: a review. Eur J Clin Pharmacol 65:419–427. doi: 10.1007/s00228-008-0599-y. [DOI] [PubMed] [Google Scholar]
- 4.Ramirez MS, Tolmasky ME. 2010. Aminoglycoside modifying enzymes. Drug Resist Updat 13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhodin MM, Anderson BJ, Peters AM, Coulthard MG, Wilkins B, Cole M, Chatelut E, Grubb A, Veal GJ, Keir MJ, Holford NH. 2009. Human renal function maturation: a quantitative description using weight and postmenstrual age. Pediatr Nephrol 24:67–76. doi: 10.1007/s00467-008-0997-5. [DOI] [PubMed] [Google Scholar]
- 6.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. 2003. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med 349:1157–1167. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 7.Turnidge J. 2003. Pharmacodynamics and dosing of aminoglycosides. Infect Dis Clin North Am 17:503–528. doi: 10.1016/S0891-5520(03)00057-6. [DOI] [PubMed] [Google Scholar]
- 8.Begg EJ, Barclay ML, Kirkpatrick CM. 2001. The therapeutic monitoring of antimicrobial agents. Br J Clin Pharmacol 52(Suppl 1):35S–43S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dersch-Mills D, Akierman A, Alshaikh B, Yusuf K. 2012. Validation of a dosage individualization table for extended-interval gentamicin in neonates. Ann Pharmacother 46:935–942. doi: 10.1345/aph.1R029. [DOI] [PubMed] [Google Scholar]
- 10.Boyle EM, Brookes I, Nye K, Watkinson M, Riordan FA. 2006. “Random” gentamicin concentrations do not predict trough levels in neonates receiving once daily fixed dose regimens. BMC Pediatr 6:8. doi: 10.1186/1471-2431-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stickland MD, Kirkpatrick CM, Begg EJ, Duffull SB, Oddie SJ, Darlow BA. 2001. An extended interval dosing method for gentamicin in neonates. J Antimicrob Chemother 48:887–893. doi: 10.1093/jac/48.6.887. [DOI] [PubMed] [Google Scholar]
- 12.Burton ME, Brater DC, Chen PS, Day RB, Huber PJ, Vasko MR. 1985. A Bayesian feedback method of aminoglycoside dosing. Clin Pharmacol Ther 37:349–357. doi: 10.1038/clpt.1985.51. [DOI] [PubMed] [Google Scholar]
- 13.Kelman AW, Thomson AH, Whiting B, Bryson SM, Steedman DA, Mawer GE, Samba-Donga LA. 1984. Estimation of gentamicin clearance and volume of distribution in neonates and young children. Br J Clin Pharmacol 18:685–692. doi: 10.1111/j.1365-2125.1984.tb02530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grasela TH, Ott R, Faix RG. 1985. Population pharmacokinetics of gentamicin in neonates using routine clinical data, abstr B23. Abstr Am So Clin Pharmacol Ther, 86th Annu Meet, San Antonio, Texas, USA. [Google Scholar]
- 15.Thomson AH, Way S, Bryson SM, McGovern EM, Kelman AW, Whiting B. 1988. Population pharmacokinetics of gentamicin in neonates. Dev Pharmacol Ther 11:173–179. [DOI] [PubMed] [Google Scholar]
- 16.Jensen PD, Edgren BE, Brundage RC. 1992. Population pharmacokinetics of gentamicin in neonates using a nonlinear, mixed-effects model. Pharmacotherapy 12:178–182. [PubMed] [Google Scholar]
- 17.Weber W, Kewitz G, Rost KL, Looby M, Nitz M, Harnisch L. 1993. Population kinetics of gentamicin in neonates. Eur J Clin Pharmacol 44(Suppl 1):S23–S25. doi: 10.1007/BF01428387. [DOI] [PubMed] [Google Scholar]
- 18.Lanao JM, Calvo MV, Mesa JA, Martín-Suárez A, Carbajosa MT, Miguelez F, Domínguez-Gil A. 2004. Pharmacokinetic basis for the use of extended interval dosage regimens of gentamicin in neonates. J Antimicrob Chemother 54:193–198. doi: 10.1093/jac/dkh261. [DOI] [PubMed] [Google Scholar]
- 19.Lingvall M, Reith D, Broadbent R. 2005. The effect of sepsis upon gentamicin pharmacokinetics in neonates. Br J Clin Pharmacol 59:54–61. doi: 10.1111/j.1365-2125.2005.02260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.García B, Barcia E, Pérez F, Molina IT. 2006. Population pharmacokinetics of gentamicin in premature newborns. J Antimicrob Chemother 58:372–379. doi: 10.1093/jac/dkl244. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen EI, Sandstrom M, Honore PH, Ewald U, Friberg LE. 2009. Developmental pharmacokinetics of gentamicin in preterm and term neonates: population modelling of a prospective study. Clin Pharmacokinet 48:253–263. doi: 10.2165/00003088-200948040-00003. [DOI] [PubMed] [Google Scholar]
- 22.Arenas-Lopez S, Mulla H, Durward A, Tibby SM. 2010. Extended-interval gentamicin: population pharmacokinetics in pediatric critical illness. Pediatr Crit Care Med 11:267–274. doi: 10.1097/PCC.0b013e3181b80693. [DOI] [PubMed] [Google Scholar]
- 23.Fuchs A, Guidi M, Giannoni E, Werner D, Buclin T, Widmer N, Csajka C. 2014. Population pharmacokinetic study of gentamicin in a large cohort of premature and term neonates. Br J Clin Pharmacol doi: 10.1111/bcp.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botha JH, du Preez MJ, Adhikari M. 2003. Population pharmacokinetics of gentamicin in South African newborns. Eur J Clin Pharmacol 59:755–759. doi: 10.1007/s00228-003-0663-6. [DOI] [PubMed] [Google Scholar]
- 25.Heimann G. 1983. Renal toxicity of aminoglycosides in the neonatal period. Pediatr Pharmacol (New York) 3:251–257. [PubMed] [Google Scholar]
- 26.Laskin OL, Longstreth JA, Smith CR, Lietman PS. 1983. Netilmicin and gentamicin multidose kinetics in normal subjects. Clin Pharmacol Ther 34:644–650. doi: 10.1038/clpt.1983.227. [DOI] [PubMed] [Google Scholar]
- 27.Valitalo PA, van den Anker JN, Allegaert K, de Cock RF, de Hoog M, Simons SH, Mouton JW, Knibbe CA. 12 March 2015. Novel model-based dosing guidelines for gentamicin and tobramycin in preterm and term neonates. J Antimicrob Chemother doi: 10.1093/jac/dkv052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakae S, Yamada M, Ito T, Chiba Y, Sasaki E, Sakamoto M, Tada K, Yamada T, Mori S. 1988. Gentamicin dosing and pharmacokinetics in low birth weight infants. Tohoku J Exp Med 155:213–223. doi: 10.1620/tjem.155.213. [DOI] [PubMed] [Google Scholar]
- 29.Ali AS, Farouq MF, Al-Faify KA. 2012. Pharmacokinetic approach for optimizing gentamicin use in neonates during the first week of life. Indian J Pharmacol 44:36–40. doi: 10.4103/0253-7613.91864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lannigan R, Thomson AH. 2001. Evaluation of 22 neonatal gentamicin dosage protocols using a Bayesian approach. Paediatr Perinat Drug Ther 4:92–100. [Google Scholar]
- 31.Rastogi A, Agarwal G, Pyati S, Pildes RS. 2002. Comparison of two gentamicin dosing schedules in very low birth weight infants. Pediatr Infect Dis J 21:234–240. doi: 10.1097/00006454-200203000-00014. [DOI] [PubMed] [Google Scholar]
- 32.Metsvaht T, Tasa T, Kipper K, Padari H, Ilmoja M, Lutsar I. 2015. Population pharmacokinetics (PK) of gentamicin in term and near-term neonates, abstr WSPID-0499. Abstr World Soc Pediatr Infect Dis (WSPID), 9th World Congr, Rio de Janeiro, Brazil. [Google Scholar]
- 33.Jaffe M. 1886. Ueber den Niederschlag, welchen Pikrinsäure in normalem Harn erzeugt und über eine neue Reaction des Kreatinins. Z Physiol Chem 10:391–400. [Google Scholar]
- 34.Boeckmann AJ, Beal SL, Sheiner LB. 1999. NONMEM users guide. University of California at San Francisco, San Francisco, CA. [Google Scholar]
- 35.Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 36.West GB, Brown JH, Enquist BJ. 1997. A general model for the origin of allometric scaling laws in biology. Science 276:122–126. doi: 10.1126/science.276.5309.122. [DOI] [PubMed] [Google Scholar]
- 37.Germovsek E, Barker CI, Standing JF. 2015. An argument for standardised scaling: comparison of methods for scaling clearance in children, p 24, abstr 3635. Abstr Annu Meet Popul Approach Group Eur, Crete, Greece: www.page-meeting.org/?abstr=3635. [Google Scholar]
- 38.Cuzzolin L, Fanos V, Pinna B, di Marzio M, Perin M, Tramontozzi P, Tonetto P, Cataldi L. 2006. Postnatal renal function in preterm newborns: a role of diseases, drugs and therapeutic interventions. Pediatr Nephrol 21:931–938. doi: 10.1007/s00467-006-0118-2. [DOI] [PubMed] [Google Scholar]
- 39.Rudd PT, Hughes EA, Placzek MM, Hodes DT. 1983. Reference ranges for plasma creatinine during the first month of life. Arch Dis Child 58:212–215. doi: 10.1136/adc.58.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 41.Lindbom L, Pihlgren P, Jonsson EN. 2005. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 79:241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Jonsson EN, Karlsson MO. 1999. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58:51–64. [DOI] [PubMed] [Google Scholar]
- 43.Brendel K, Dartois C, Comets E, Lemenuel-Diot A, Laveille C, Tranchand B, Girard P, Laffont CM, Mentre F. 2007. Are population pharmacokinetic and/or pharmacodynamic models adequately evaluated? A survey of the literature from 2002 to 2004. Clin Pharmacokinet 46:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheiner LB, Beal SL. 1981. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 9:503–512. doi: 10.1007/BF01060893. [DOI] [PubMed] [Google Scholar]
- 45.Paediatric Formulary Committee. 2016. BNF for children. Pharmaceutical Press, London, United Kingdom. [Google Scholar]
- 46.Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, Drusano GL. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis 44:79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- 47.Prayle A, Watson A, Fortnum H, Smyth A. 2010. Side effects of aminoglycosides on the kidney, ear and balance in cystic fibrosis. Thorax 65:654–658. doi: 10.1136/thx.2009.131532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schentag JJ, Jusko WJ. 1977. Renal clearance and tissue accumulation of gentamicin. Clin Pharmacol Ther 22:364–370. doi: 10.1002/cpt1977223364. [DOI] [PubMed] [Google Scholar]
- 49.Wicha SG, Kees MG, Solms A, Minichmayr IK, Kratzer A, Kloft C. 2015. TDMx: a novel Web-based open-access support tool for optimising antimicrobial dosing regimens in clinical routine. Int J Antimicrob Agents 45:442–444. doi: 10.1016/j.ijantimicag.2014.12.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.