

Abstract
The novel ATP synthase inhibitor bedaquiline recently received accelerated approval for treatment of multidrug-resistant tuberculosis and is currently being studied as a component of novel treatment-shortening regimens for drug-susceptible and multidrug-resistant tuberculosis. In a limited number of bedaquiline-treated patients reported to date, ≥4-fold upward shifts in bedaquiline MIC during treatment have been attributed to non-target-based mutations in Rv0678 that putatively increase bedaquiline efflux through the MmpS5-MmpL5 pump. These mutations also confer low-level clofazimine resistance, presumably by a similar mechanism. Here, we describe a new non-target-based determinant of low-level bedaquiline and clofazimine cross-resistance in Mycobacterium tuberculosis: loss-of-function mutations in pepQ (Rv2535c), which corresponds to a putative Xaa-Pro aminopeptidase. pepQ mutants were selected in mice by treatment with clinically relevant doses of bedaquiline, with or without clofazimine, and were shown to have bedaquiline and clofazimine MICs 4 times higher than those for the parental H37Rv strain. Coincubation with efflux inhibitors verapamil and reserpine lowered bedaquiline MICs against both mutant and parent strains to a level below the MIC against H37Rv in the absence of efflux pump inhibitors. However, quantitative PCR (qPCR) revealed no significant differences in expression of Rv0678, mmpS5, or mmpL5 between mutant and parent strains. Complementation of a pepQ mutant with the wild-type gene restored susceptibility, indicating that loss of PepQ function is sufficient for reduced susceptibility both in vitro and in mice. Although the mechanism by which mutations in pepQ confer bedaquiline and clofazimine cross-resistance remains unclear, these results may have clinical implications and warrant further evaluation of clinical isolates with reduced susceptibility to either drug for mutations in this gene.
INTRODUCTION
Multidrug-resistant tuberculosis (MDR-TB) is a major threat to global control of tuberculosis (TB). When multidrug resistance is not diagnosed, patients respond poorly to standardized first-line regimens and additional resistance may develop. When MDR-TB is diagnosed, current second-line regimens require prolonged treatment durations and are less effective, more toxic, and far more expensive than first-line therapy (1).
The diarylquinoline drug bedaquiline (B) received accelerated approval from the U.S. Food and Drug Administration as part of combination therapy for MDR-TB when other alternatives are not available (2). It is now being studied as a component of novel short-course regimens for MDR as well as drug-susceptible TB (ClinicalTrials.gov identifiers NCT02333799, NCT02193776, NCT02589782, NCT02409290, and NCT02454205 [https://clinicaltrials.gov/]). For new drugs such as bedaquiline, it is essential to define and catalog the mechanisms conferring bacterial resistance in order to design appropriate diagnostic tests (including rapid molecular tests), to better manage the treatment of patients who fail therapy or relapse after receiving the drug, and to conduct population level surveillance for changes in drug susceptibility.
The principal mechanism of action of bedaquiline is inhibition of the mycobacterial ATP synthase (3, 4). Strains selected in vitro for resistance to bedaquiline often have mutations in atpE, which encodes the ATP synthase subunit to which bedaquiline binds (5). These target-based mutations cause relatively large (i.e., 10× to 128×) shifts in MIC. However, it was noted that many bedaquiline-resistant isolates selected in vitro, typically with smaller shifts in MIC, do not have mutations in the ATP synthase complex (5). A new non-target-based mechanism conferring low-level bedaquiline resistance and cross-resistance to clofazimine (C) was recently identified among isolates from patients with delayed sputum culture conversion while receiving bedaquiline for MDR-TB, among mice treated with bedaquiline-containing combinations, and in vitro (6, 7). Although both bedaquiline and clofazimine (7) can select for non-target-based mutants in vitro, such mutants have not yet been isolated from mice or patients treated with clofazimine. The responsible mutations were found in Rv0678, which encodes a negative regulator of mmpL5 and mmpS5. Increased transcription of these genes, which comprise a membrane transporter in the resistance-nodulation-division (RND) family, is putatively associated with increased efflux of bedaquiline and clofazimine.
Identification of mutations conferring cross-resistance to clofazimine and bedaquiline is important because clofazimine is a component of some short-course regimens currently under study for treatment of MDR-TB (8–10) and the two drugs are being used together in the ongoing STREAM trial (ClinicalTrials.gov identifier NCT02409290 [https://clinicaltrials.gov/]). Herein, we report a new genetic determinant of low-level bedaquiline and clofazimine cross-resistance in Mycobacterium tuberculosis, conferred by mutations in pepQ.
MATERIALS AND METHODS
Bacterial strains.
M. tuberculosis H37Rv was passaged in mice, subcultured in Middlebrook 7H9 (Fisher Scientific) supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC) complex (Becton-Dickinson) and 0.05% Tween 80 (Sigma-Aldrich), and used for aerosol infection when the optical density at 600 nm (OD600) was approximately 1.0.
Antimicrobials.
Pretomanid (Pa), moxifloxacin (M), bedaquiline, and linezolid (L) were provided by the Global Alliance for Tuberculosis Drug Development (New York, NY), Bayer (Leverkusen, Germany), Janssen (Beerse, Belgium), and Pfizer (Groton, CT), respectively. Rifampin (R), isoniazid (H), pyrazinamide (Z), ethambutol (E), and clofazimine were purchased from Fisher or Sigma. Dosing formulations were prepared and maintained as previously described (11). All drugs were administered once daily by gavage, 5 days per week.
Aerosol infection.
Female BALB/c mice (Charles River, Wilmington, MA) aged 4 to 6 weeks were infected by the aerosol route using an inhalation exposure system (Glas-col Inc., Terre Haute, IN). Mice were randomized to treatment groups (five mice per group per time point) after aerosol infection and were routinely sacrificed (i) on the day after infection to determine the number of CFU implanted in the lungs, (ii) on the day of treatment initiation to determine the pretreatment CFU count, and (iii) at selected time points during and after treatment. Quantitative cultures of lung homogenates were performed in parallel on selective 7H11 agar with and without 0.4% activated charcoal to reduce drug carryover effects, as previously described (11). All procedures involving animals were approved by the Animal Care and Use Committee of Johns Hopkins University.
Efficacy of combinations containing bedaquiline and clofazimine in murine models of TB.
Beginning 14 days after high-dose aerosol infection, as previously described (12), BALB/c mice received no treatment (negative controls) or treatment with the first-line regimen of R-H-Z (positive controls) or one of the following test regimens: bedaquiline (25 mg/kg [of body weight]) alone, the two-drug combination of bedaquiline plus clofazimine (20 mg/kg), or three-drug combinations of bedaquiline plus clofazimine plus one of the following: rifampin (10 mg/kg), isoniazid (10 mg/kg), pyrazinamide (150 mg/kg), ethambutol (100 mg/kg), moxifloxacin (100 mg/kg), pretomanid (50 mg/kg), and linezolid (100 mg/kg). Lung CFU counts were determined for all treatment groups after 4 weeks of treatment and, for mice receiving bedaquiline alone and bedaquiline plus clofazimine, also after 6 and 8 weeks of treatment.
MIC determination.
Determination of bedaquiline and clofazimine MICs and the proportion of drug-resistant mutants was performed using the agar proportion method. Serial dilutions of lung homogenates, colony suspensions, or a broth culture were inoculated in 500-μl aliquots onto 7H11 plates with and without the desired drug. MICs were determined using doubling bedaquiline and clofazimine concentrations ranging from 0.003 to 0.25 μg/ml and 0.06 to 2 μg/ml, respectively. The MIC was defined as the lowest drug concentration inhibiting at least 99% of the growth observed on drug-free control plates. To investigate the effect of efflux pump inhibitors on the susceptibility of the strains, the MIC of bedaquiline was also determined using the broth macrodilution method. Doubling concentrations of bedaquiline from 0.007 to 1 μg/ml were tested in the presence or absence of 40 μg/ml of verapamil or 3 μg/ml of reserpine. Briefly, tubes containing in 2.5 ml of 7H9 broth plus OADC with the above-mentioned concentrations of bedaquiline were inoculated with 105 CFU of log-phase culture of H37Rv or the B5 mutant. The MIC was defined as the lowest concentration that prevented visible growth after 14 days of incubation at 37°C. Controls with and without efflux inhibitors and bedaquiline were included for each test. The experiment was performed twice.
DNA sequencing.
Genomic DNA from the parental wild-type strain and resistant mutants was extracted by using the cetyltrimethylammonium bromide (CTAB) protocol (13) and sonicated (Covaris, Inc.). The DNA library was constructed by using a genomic DNA sample preparation kit (Illumina, Inc.). The samples were sequenced on an Illumina Genome Analyzer II, which was operated in paired-end mode, collecting pairs of 51-bp reads from opposite ends of ∼250- to 350-bp fragments. Image analysis and base-calling were done by using the Illumina GA Pipeline software (v0.3). Genome assembly was performed by a comparative assembly method using software developed in-house. Briefly, reads were aligned to the genome of H37Rv as a reference sequence, and then local contig building was used to identify insertions and deletions (14). Mutations in pepQ (Rv2535c) were confirmed by PCR amplification using specific primers (see Table S1 in the supplemental material).
In vivo confirmation of low-level cross-resistance to bedaquiline and clofazimine.
BALB/c mice were infected with either the H37Rv parent strain or an isogenic strain (B5) with a nonsynonymous pepQ mutation and randomized to receive no treatment, isoniazid (10 mg/kg) alone, bedaquiline (12.5, 25, or 50 mg/kg) alone, clofazimine (20 mg/kg) alone, or bedaquiline at 25 mg/kg plus clofazimine. Treatment began 4 days after infection and was administered for 4 weeks for all groups, except the groups receiving bedaquiline at 25 mg/kg, which received 8 weeks of treatment.
Complementation studies to confirm that a pepQ mutation is sufficient for bedaquiline and clofazimine cross-resistance.
The B5 strain was complemented with pDT-Rv2535c (see Fig. S1 in the supplemental material) containing the wild-type pepQ gene from H37Rv. Complementation was confirmed by showing a loss of susceptibility to hygromycin and PCR for presence of the hygromycin resistance gene. Bedaquiline and clofazimine MICs were determined in vitro. Following aerosol infection with the H37Rv parent strain, the B5 mutant, or the B5::pepQ complemented strain, BALB/c mice were randomized to receive no treatment, isoniazid at 10 mg/kg, bedaquiline at 25 mg/kg, or clofazimine at 20 mg/kg. Treatment began 3 days after infection and continued for 4 weeks before mice were sacrificed to determine lung CFU counts.
Expression analysis of mmpS5, mmpL5, and Rv0678.
The H37Rv parent strain and the B5 mutant were grown to mid-log phase (OD600 = 0.6) in 7H9 broth, and cells were pelleted by centrifugation. RNA was extracted by bead beating in TRIzol and column purified according to the manufacturer's instructions (Qiagen, Valencia, CA). RNA was reverse transcribed using the iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA). Real-time PCR was performed with iQ SYBR green supermix on an iCycler (Bio-Rad, Hercules, CA) using primers specific for Rv0676c, Rv0677c, and Rv0678 and 16S rRNA (see Table S1 in the supplemental material). All experiments were performed in biological and technical triplicates. Cycle threshold (CT) values were normalized against 16S rRNA expression, and fold change was calculated by the −2ΔΔCT method (15).
Expression, purification, and activity assays of PepQ.
The entire coding region of Rv2535c was cloned into the pET28b expression vector (Novagen) containing an in-frame N-terminal 6× His tag with the tobacco etch virus (TEV) cleavage site using the NdeI and HindIII restriction sites. The plasmid was transformed into Escherichia coli BL21(DE3) cells for expression of Rv2535c. Cells containing the plasmid were grown at 37°C for 7 h in LB medium with 50 μg/ml of kanamycin followed by induction with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown overnight at 18°C. The cells were lysed via an M-100P microfluidizer (Microfluidics, Worcestershire, United Kingdom) in lysis buffer (50 mM Tris [pH 7.5], 500 mM NaCl, 5% glycerol, DNase, and 2 mM MgCl2) and centrifuged at 15,000 rpm for 1 h. The supernatant was chromatographed over a His tag affinity column (GE Healthcare) charged with Ni. The protein was eluted with 0 to 300 mM imidazole gradient, and the 6× His tag was cleaved using the tobacco etch virus protease. The final purification step was gel filtration chromatography on the standardized s200 Superdex (GE Healthcare) column. The PepQ protein was eluted in two peaks: a large-molecular-mass aggregate that came out near the void volume, followed by a distinct peak with calculated molecular mass corresponding to a tetramer (∼160 kDa). Formation of this tetramer is consistent with other related proteins, e.g., the protein with PDB code 3Q6D. The protein was >95% pure, as observed by SDS-PAGE and was concentrated to 7.5 mg/ml, flash frozen, and stored in dialysis buffer (50 mM Tris [pH 7], 50 mM NaCl, 5% glycerol, 1 mM dithiothreitol [DTT]) at −80°C. The purified recombinant PepQ tetramer in 50 mM Tris (pH 7) was tested for both creatinase activity using the creatinase assay kit (Sigma) and endopeptidase activity using commercially available fluorescent peptides. As the peptides were all labeled with a p-nitroanilide group, the assay monitored for a shift in absorbance at 410 nm, resulting from the cleaved product.
Statistical analysis.
CFU counts were log10 transformed before analysis. Group means for experimental treatment groups were compared with that of the standard treatment control by one-way analysis of variance (ANOVA) with Dunnett's posttest to adjust for multiple comparisons. All analyses were performed with GraphPad Prism v.4.01 (GraphPad, San Diego, CA).
RESULTS
Bedaquiline and clofazimine have additive activity against M. tuberculosis in mice but select for cross-resistant mutants.
Using a well-established high-dose aerosol model of TB in mice, we evaluated the activity of novel drug combinations containing bedaquiline and clofazimine (Fig. 1). Consistent with prior observations (3, 11, 16), treatment with the standard first-line R-H-Z regimen reduced the mean lung CFU count by 2.46 log10 over the first 4 weeks and treatment with bedaquiline alone reduced the lung CFU counts by an additional 1.08 log10. The addition of pyrazinamide and clofazimine each significantly increased the activity of bedaquiline (P < 0.001), whereas the addition of pretomanid had antagonistic effects (P < 0.05). The addition of rifampin, isoniazid, ethambutol, or moxifloxacin did not significantly affect the activity of the bedaquiline-clofazimine combination, whereas addition of pyrazinamide significantly increased the activity (P < 0.001) and addition of linezolid or pretomanid reduced the activity (P < 0.05). After 8 weeks of treatment, 2 of 5 mice receiving bedaquiline alone had more than 3 log CFU in the lungs, while the mean log CFU count was 0.98 ± 0.53 in the remaining 3 mice. Likewise, 3 of 5 mice receiving bedaquiline plus clofazimine had a mean log CFU count of 2.12 ± 0.69, while the remaining 2 mice were culture negative, as were all 5 mice treated for just 6 weeks (Fig. 1, inset). Unlike those from mice with lower CFU counts at week 8, the isolates from the lungs of mice with higher CFU counts grew equally well on 7H11 agar with and without activated charcoal, indicating that their growth was not impaired by bedaquiline or clofazimine carried over in the lung homogenates.
FIG 1.
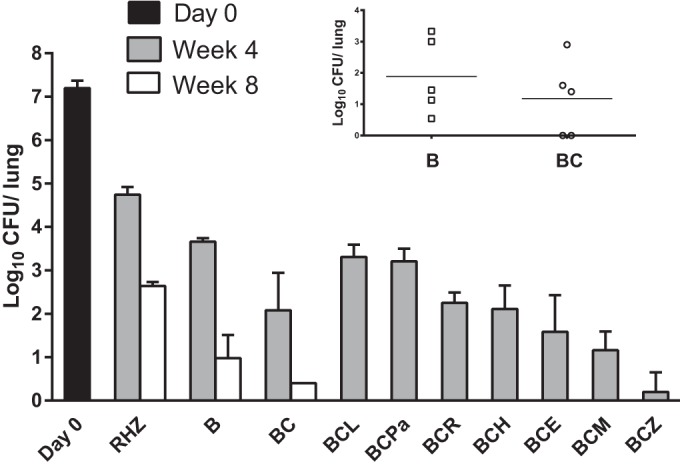
Mean lung log10 CFU counts (±SD) after 4 and 8 weeks of treatment with bedaquiline- and clofazimine-containing regimens. The main graph does not include results from mice in which bedaquiline and clofazimine cross-resistant mutants were selected after 8 weeks of treatment: 2 and 3 mice from the B and BC groups with mean lung CFU counts of 3.22 ± 0.16 and 2.12 ± 0.69 log10, respectively (inset). The CFU counts at the time of treatment initiation are indicated at day 0. Abbreviations: R, rifampin; H, isoniazid; Z, pyrazinamide; B, bedaquiline; C, clofazimine; L, linezolid; Pa, pretomanid; E, ethambutol; M, moxifloxacin.
While bedaquiline and clofazimine MICs for the wild-type H37Rv parent strain were 0.03 and 0.25 μg/ml, respectively, MICs for the isolates from the 2 mice treated with bedaquiline alone for 8 weeks (named B4 and B5) and the 3 mice treated with bedaquiline plus clofazimine for 8 weeks (named BC2, BC3, and BC4) with high, outlying lung CFU counts were 0.12 to 0.25 and 0.5 to 1 μg/ml, respectively, indicating that treatment with bedaquiline, with or without clofazimine, selected for strains with reduced susceptibility to both drugs.
Isolates with bedaquiline and clofazimine cross-resistance harbored mutations in pepQ.
Duplicate samples of genomic DNA from four cross-resistant strains isolated in the first mouse experiment (i.e., B5, BC2, BC3, and BC4) were sequenced on an Illumina GenomeAnalyzer IIx in paired-end mode using a read length of 51 bp. The mean depth of coverage ranged from 118 to 161× (number of reads covering each site, averaged over the whole genome), and the completion was >99% (fraction of sites in 4.4-Mb genome covered by at least 1 read; sites lacking coverage were primarily restricted to PPE and PE_PGRS genes, due to very high GC contents). The parental strain used in these experiments (H37RvJH) was also sequenced, to identify any differences from the public reference genome sequence for H37Rv (17). Compared to the sequence of the parental strain, only one mutation was observed in each strain (Table 1). All 4 strains had a mutation in pepQ (Rv2535c), which encodes a putative cytoplasmic peptidase. The mutations observed included 2 frameshift mutations (−C in Arg271 and +C in Ala14) in mice treated with the bedaquiline-clofazimine combination, as well as a nonsynonymous single nucleotide polymorphism causing a Leu44Pro mutation, obtained from a mouse treated with bedaquiline alone. All mutations were confirmed by PCR amplification and sequencing.
TABLE 1.
Sequencing results for the resistant strainsa
| Mouse no. | Treatment | Coverage | % completion | Mutation |
|---|---|---|---|---|
| B5 | B | 142.1× | 99.25 | pepQ: L44P |
| BC2 | B + C | 118.4× | 99.20 | pepQ: +C in Ala14 |
| BC3 | B + C | 154.3× | 99.43 | pepQ: +C in Ala14 |
| BC4 | B + C | 161.1× | 99.14 | pepQ: −C in Arg271 |
For all strains, the MIC of bedaquiline (B) was 0.12 μg/ml and that of clofazimine (C) was 0.5 to 1 μg/ml.
A pepQ mutant is virulent and less susceptible to bedaquiline and clofazimine in a murine TB model.
Because transposon mutagenesis experiments have suggested that inactivation of pepQ may reduce the fitness of M. tuberculosis (18), and because the pepQ mutants displayed only low-level resistance to bedaquiline and clofazimine in vitro, we sought to confirm their virulence and reduced susceptibility in vivo. We compared the parental H37Rv strain and the B5 mutant on the basis of their abilities to multiply in untreated BALB/c mice and their susceptibilities to treatment with isoniazid (positive control), bedaquiline, and/or clofazimine. The aerosol infectious dose was approximately 1 log10 higher for the parent strain than for the B5 mutant (Fig. 2). By 4 weeks postinfection, untreated (UT) control mice infected with H37Rv had died, and therefore, none were available for CFU counts (although lung CFU counts are typically >8 log10, or approximately 4 log10 higher than baseline, at the time of death). Untreated mice infected with B5 did not die within 4 weeks of infection because the infectious dose was not high enough. However, the approximately 4-log10 increase in lung CFU counts over this period is evidence that the strain multiplies more or less normally in mice. The treatment with isoniazid (positive control) was at least as bactericidal against the B5 strain as against H37Rv, reducing the lung CFU counts by more than 1 log10 in both strains. Dose-dependent activity of bedaquiline was observed against both strains. However, while bedaquiline at 12.5 mg/kg was bacteriostatic and 50 mg/kg reduced the lung CFU counts by more than 2 log10 against H37Rv, even 50 mg/kg of bedaquiline did not fully inhibit multiplication of the B5 mutant but rather allowed a nearly 2-log10 increase in CFU counts. Clofazimine enabled growth of both strains in this acute infection model, as previously observed (19), but was more effective against the H37Rv strain than against the B5 mutant.
FIG 2.

Mean lung log10 CFU counts (±SD) (A and B) and change in CFU counts (week 4 − day 0) (C and D) after 4 weeks of treatment in mice infected with the parental H37Rv strain (A and C) or the B5 mutant (B and D). CFU counts were not determined for untreated mice infected with the parental strain, which required euthanasia prior to the predetermined endpoint. Abbreviations: UT, untreated; H, isoniazid (10 mg/kg); C, clofazimine (20 mg/kg); B, bedaquiline (12.5, 25, or 50 mg/kg).
Complementation of pepQ restores susceptibility to bedaquiline and clofazimine.
Complementation of the B5 and BC2 strains with the wild-type pepQ gene from H37Rv restored the susceptibility of the strains to bedaquiline, as confirmed by the MIC of 0.03 μg/ml, which was no different from that for the parental H37Rv control. Following this, 3 groups of mice were infected with H37Rv, the B5 strain, or the B5::pepQ strain and initiated on treatment with isoniazid at 10 mg/kg, bedaquiline at 25 mg/kg, or clofazimine at 20 mg/kg 4 days later. In untreated mice, lung CFU counts increased by approximately 4 log10 over the subsequent 4 weeks (Fig. 3) and were accompanied by visible lung lesions (see Fig. S2 in the supplemental material). Isoniazid was bactericidal, with the greatest effect against the B5 strain, and prevented the formation of lung lesions. Like isoniazid, bedaquiline reduced the mean lung CFU counts by 0.5 to 1 log10 CFU against H37Rv and the B5::pepQ strain, and it did prevent formation of macroscopic lung lesions in mice infected with the B5 mutant (see Fig. S2 in the supplemental material). However, bedaquiline allowed a >1-log10 increase in mean CFU counts of the B5 mutant. Clofazimine, which has a relatively poor activity in this acute infection model when given alone (19), allowed multiplication of all strains but significantly reduced the multiplication of the H37Rv (P < 0.05) and B5::pepQ (P < 0.05) strains but not the B5 strain (P > 0.05) compared to findings with no treatment. In addition, clofazimine prevented formation of lung lesions in mice infected with H37Rv and the B5::pepQ complemented strain but not the B5 strain (see Fig. S2 in the supplemental material). These results clearly demonstrate that complementation with the wild-type pepQ gene was sufficient to restore susceptibility to bedaquiline and clofazimine in the B5 mutant and thus confirm that the loss of PepQ function is sufficient for low-level resistance to bedaquiline and clofazimine.
FIG 3.

Mean lung log10 CFU counts (±SD) (A to C) and change in lung CFU counts (week 4 − day 0) (D to F) after 4 weeks of treatment in mice infected with the parental H37Rv strain (A and D), the B5 mutant (B and E), or the complemented B5::pepQ strain (C and F). Abbreviations: UT, untreated; H, isoniazid (10 mg/kg); C, clofazimine (20 mg/kg); B, bedaquiline (25 mg/kg).
Structural analysis of M. tuberculosis pepQ product.
pepQ encodes a 372-amino-acid (aa) protein that has two domains: an ∼100-aa N-terminal alpha/beta domain and an ∼250-aa C-terminal peptidase domain (Fig. 4). Based on homology to Ypdf in E. coli (e.g., 37% amino acid identity over 363 residues), M. tuberculosis pepQ is predicted to encode a proline-specific aminopeptidase (prolidase), active on substrates with Xaa1-Pro2 at the amino terminus. The C-terminal domain of YpdF houses the catalytic activity and is homologous to the E. coli methionine aminopeptidase (MetAP; 29% amino acid identity) (20). However, MetAP lacks the N-terminal domain. In enzyme assays, YpdF was found to have weak activity on substrates with an N-terminal methionine (Met-Xaa) if the second amino acid is alanine, proline, or serine and higher activity for other substrates with a proline in the second position (Xaa-Pro) (21). In E. coli, YpdF is encoded in an operon with YpdE, which is a methionine aminopeptidase. YpdF and YpdE are proposed to work in concert to degrade proteins, with YpdE removing amino acids from the N terminus until a proline is encountered in the second position and YpdF removing the block (21). The M. tuberculosis pepQ product also has homology (∼30% amino acid identity) to other E. coli proline aminopeptidases, the pepP and pepQ products, which have both the catalytic C-terminal domain and a noncatalytic N-terminal domain that plays a role in oligomerization and substrate specificity. The pepP product and similar aminopeptidase P enzymes (EC 3.4.11.9) may preferentially hydrolyze the Xaa-Pro bond at the terminus of larger oligopeptides (but also dipeptides) and can discriminate the 3rd and 4th residues in their catalytic specificity.
FIG 4.

Locations of mutations and domains in pepQ.
Proline aminopeptidases have a 2-domain architecture similar to that of creatinases (which catalyze a similar reaction, hydrolysis of creatine into sarcosine and urea), although the peptidase activity of the former is metal ion dependent and they bind cations in the active site (20), whereas creatinase activity does not require metal ions (22, 23). The mutations selected by treatment with bedaquiline with or without clofazimine in mice occurred in both domains. The frameshift mutation in Ala14 and the nonsynonymous mutation L44P occur in the N-terminal domain, and the frameshift mutation in Arg271 occurs in the catalytic domain. Figure 4 illustrates where these mutations fall in the domain structure of the pepQ product. To better understand the role of these mutations, we used Phyre2 (24) to build a homology model of the three-dimensional (3D) atomic structure of the pepQ product. Phyre2 detected the greatest homology to a proline aminopeptidase from Bacillus anthracis (PDB code 3Q6D; 39% amino acid identity; deposited by Midwest Center for Structural Genomics). Phyre2 created a hidden Markov model based on a PSI-BLAST alignment of closely related sequences and used it to optimally thread the M. tuberculosis pepQ product amino acid sequence onto the 3Q6D structure as a template, followed by loop optimization and energy minimization (24). The active site can be inferred to be in a pocket in the C-terminal domain containing a Ca2+ metal ion, in the same position as the dinuclear divalent cation sites in pepP (1a16) and MetAP (3mat), which is involved in catalyzing the cleavage of the scissile peptide bond. The cations are coordinated by D221, D232, E339, E325, and His296 (M. tuberculosis pepQ), all perfectly conserved in all of these structures. Based on the homology model, Leu44 of M. tuberculosis pepQ appears to be buried in the core of the N-terminal domain (on a beta strand that shows a high level of conservation with E. coli pepP and both prokaryotic and eukaryotic prolidases [25]), not in close proximity to the active site. Thus, L44P is likely a structure-destabilizing mutation, as is the frameshift mutation in Ala14. A frameshift in Arg271 would truncate half the C-terminal domain (splitting the pseudosymmetric alpha/beta “pita bread” fold in half and destroying the active site).
The PepQ model can be superimposed with the crystal structure of the E. coli MetAP protein (3mat), which was complexed with a peptide inhibitor (bestatin analog). The mechanism and specificity determinants of E. coli MetAP have been well characterized (20). MetAP has a dinuclear cation site (2 Co2+) in the same location as in 3Q6D. The N-terminal residue of the peptide inhibitor of MetAP binds in a P1 pocket, and the second residue binds in a P2 pocket, with the polar backbone atoms of the scissile bond between them coordinating the cations. The residues of M. tuberculosis PepQ lining the P1 part of the pocket are considerably different than MetAP. For example, F190 is replaced with Cys in MetAP, and I302 is replaced with F. This could potentially explain differences in specificity for the N-terminal residue between prolidases and methionine aminopeptidases (26). While the inhibitor in the 3mat structure mimics Met1-Ala2, YpdF would be expected to bind Xaa1-Pro2 as a prolidase. The putative position of the P2 pocket can also be inferred by superimposing the crystal structure of E. coli PepP in complex with the inhibitor Pro-Leu (PDB code 1a16), which putatively mimics the 2nd and 3rd residues of a peptide substrate. In both MetAP and the pepP product, the P2 pocket is adjacent to residue His-204. While the homology between M. tuberculosis pepQ and E. coli pepP is relatively low (31% amino acid identity), nearly every residue in the vicinity of the active site was either identical or highly conserved. The residues lining the P1 pocket are F190 (Y), I302 (V), H303, T341 (D), T232, and V398 (L), with residues of the E. coli pepP product shown in parentheses. The residues lining P2 are T234, I193, R337, H204, P203 (L), and H292 (Fig. 5). Thus, we expect M. tuberculosis PepQ to be able to coordinate metal ions similarly to E. coli PepP and the specificity of M. tuberculosis PepQ to be very similar to that of E. coli PepP, i.e., to correspond to a proline aminopeptidase (Xaa-Pro).
FIG 5.

Multiple amino acid sequence alignment for the products of M. tuberculosis pepQ, Lactococcus lactis pepP, and E. coli pepP, constructed using ClustalW. Residues coordinating the Mn2+ ions are shown in red. Asterisks indicate positions which have a single, fully conserved residue. Colons indicate conservation between groups of strongly similar properties, scoring >0.5 in the Gonnet PAM 250 matrix. Periods indicate conservation between groups of weakly similar properties, scoring ≤0.5 in the Gonnet PAM 250 matrix.
Enzymatic characterization of M. tuberculosis PepQ.
Recombinant purified M. tuberculosis pepQ product exhibited no creatinase activity (data not shown). Of the 6 proline-containing peptide substrates tested, only Arg-Pro-p-nitroanilide showed modest cleavage activity. Several substrates with an N-terminal methionine were also tested. However, none of these showed cleavage, suggesting that the pepQ product is not a methionine aminopeptidase.
Mass spectrometry showed that recombinant M. tuberculosis pepQ product does not bind to or modify the structure of bedaquiline or clofazimine. Overexpression of M. tuberculosis pepQ in Mycobacterium smegmatis did not significantly alter the MIC of bedaquiline or clofazimine. This is not surprising, as the mutations presumably result in loss of function. Taken together, these results suggest that PepQ is not a target or activating enzyme for either drug.
Resistance mediated by pepQ mutation may be associated with increased drug efflux, but this is not due to upregulation of mmpL5 and mmpS5 expression.
Addition of verapamil and reserpine reduced the MIC against the H37Rv parent strain from 0.03 to 0.007 μg/ml and 0.015 to 0.03 μg/ml, respectively. Both efflux inhibitors reduced the MIC of both the B5 mutant and the H37Rv parent to the same level that was below the MIC against the parent strain in the absence of the efflux inhibitor, indicating a possible role of one or more efflux pumps in the mechanism of low-level bedaquiline resistance mediated by loss-of-function mutations in pepQ (see Table S2 in the supplemental material). Further, quantitative PCR (qPCR) for the genes involved in the efflux-mediated resistance reported in previous studies did not show any significant fold change in transcription levels (see Table S3 in the supplemental material).
DISCUSSION
Bedaquiline is a promising new drug for the treatment of TB. Its limited clinical usage to date has provided little opportunity to select for drug-resistant mutants or to assess the impact of such mutants on treatment outcomes. In vitro-selected target-based mutations in atpE confer high-level resistance to bedaquiline (e.g., 16 to 128× increase in MIC) (5, 6). However, to our knowledge, no atpE mutant has been isolated from a patient treated with bedaquiline. On the other hand, nontarget mutations in Rv0678 conferring low-level resistance to bedaquiline and clofazimine were recently described for sputum isolates with at least 4-fold increases in bedaquiline MIC after treatment including bedaquiline (6). More recently, Rv0678 mutants were identified in the sputa of both MDR-TB and drug-susceptible TB patients that had not received bedaquiline or clofazimine (27). Although these Rv0678 mutations cause relatively small (2× to 8×) increases in bedaquiline and clofazimine MICs compared to mutations in atpE, their selection during combination therapy in TB patients and in mice is a cause of concern (6). In mice, Rv0678 resistant mutants may be selected with 8 weeks of bedaquiline monotherapy (D. Almeida, unpublished observation) but emerge only late in the course of combination therapy and are eventually cleared, presumably by the action of companion agents (6). Clinically, these mutants emerged almost entirely among patients with pre-XDR- and XDR-TB (6), but their isolation has not clearly been associated with poorer clinical outcomes among patients receiving bedaquiline with more effective companion agents (28). These results underscore the importance of appropriate combination therapy and adherence and the risk of premature discontinuation of therapy.
The present study provides the first evidence of nontarget mutations in pepQ conferring low-level resistance to bedaquiline and cross-resistance to clofazimine. Zhang et al. (29) recently reported in vitro selection of a pepQ mutant in M. tuberculosis with clofazimine. However, they did not report the results of direct susceptibility testing with clofazimine or bedaquiline or confirm the causative role of pepQ mutation in clofazimine resistance. Like mutations in Rv0678 (6), the observed pepQ mutations produce modest increases (up to 4-fold) in bedaquiline and clofazimine MICs and reduce the efficacy of bedaquiline and clofazimine in vivo. However, these mutations did not result in complete resistance to these drugs, as increasing doses of bedaquiline and, to a lesser extent, combining bedaquiline and clofazimine were associated with a greater antituberculosis effect. These data and the additive bactericidal and sterilizing effects of bedaquiline and clofazimine against drug-susceptible bacilli (30) indicate that despite the presence of two shared resistance mechanisms, the combined use of clofazimine and bedaquiline may be advantageous. However, these two drugs will not protect each other against the emergence of resistance, and it is therefore important to use them in combination only if they are protected from emergence of resistance by additional antibiotics that are not substrates of the same efflux pumps. Both Rv0678 and pepQ mutants are selected and able to grow in mice treated with bedaquiline, despite plasma bedaquiline concentrations exceeding the MIC against these resistant organisms (11, 31). This may be due to the high protein binding of bedaquiline, which results in free drug concentrations below the MIC at the site of infection. Rv0678 mutants and pepQ mutants may be selected preferentially over atpE mutants in vivo during bedaquiline treatment because they strike the right balance between reduced susceptibility to bedaquiline and maintenance of fitness.
Our findings may have implications for breakpoint selection for bedaquiline susceptibility testing. Although the bedaquiline MIC against the pepQ mutants is below the provisional susceptibility breakpoint of 0.5 μg/ml proposed by EUCAST (32), monotherapy with clinically relevant bedaquiline doses in mice infected with a pepQ mutant had limited efficacy. These findings and the limited clinical data available to date warrant careful monitoring of bedaquiline MICs and treatment outcomes and further consideration whether a lower breakpoint would better predict patient response to bedaquiline treatment. Polymorphisms in Rv0678 have recently been observed among isolates from patients that have not received bedaquiline or clofazimine and are not always associated with bedaquiline MICs that exceed the provisional breakpoint (27), and it remains unclear whether such Rv0678 mutations are associated with poorer clinical outcomes among patients receiving bedaquiline-containing regimens (6, 28). Thus, the clinical impact of the small shift in MIC conferred by pepQ mutations remains to be determined. Further surveillance is required to identify Rv0678 and pepQ mutants and to correlate their presence with MICs and clinical outcomes among patients receiving therapy that includes one or both drugs.
It is interesting that single nucleotide polymorphisms in pepQ were occasionally observed in clinical isolates that we have sequenced. In particular, pepQ::Ser66Pro was observed in two XDR-TB isolates (X16 and X23; Beijing strain family) and an MDR-TB isolate (R1792; LCC strain family) from the Western Cape of South Africa, all isolated prior to the introduction of bedaquiline (33). No other strains were observed to have mutations in pepQ among over 50 clinical isolates from South Africa and South America. It is possible that use of clofazimine to treat MDR-TB in this region could have contributed to selective amplification of these mutants. However, this seems unlikely given the limited usage of clofazimine in South Africa during the period in which such selection would have occurred. The clinical isolates with polymorphisms in pepQ were not available for drug susceptibility testing. However, the presence of Rv0678 and pepQ mutations in isolates from patients naive to bedaquiline and clofazimine raises the possibility that they may also be selected by other drugs used to treat TB or other infections.
What is the function of PepQ in M. tuberculosis, and how might loss of function result in reduced susceptibility to bedaquiline and clofazimine? While further characterization of the enzymatic activity of PepQ is clearly required before drawing conclusions, the genomic and structural characterization described here suggests that the pepQ product is a proline-specific aminopeptidase and ortholog of the pepP product in E. coli and Lactococcus lactis and PapA (YqhT) in Bacillus subtilis (34). This is supported by homology in both genetic sequence and predicted protein structure. Although the DNA sequence homology is not particularly high, there is a high degree of conservation of amino acids in the predicted active site, including strict conservation of metal-binding residues between PepQ and PepP from both E. coli and L. lactis. Moreover, each of these orthologous genes is situated next to and coexpressed with elongation factor P (EF-P; Rv2534c), one of the few universally conserved elongation factors (34). Conserved homologs also exist in other pathogenic mycobacteria (M. bovis, M. leprae, and M. paratuberculosis) (35).
Proline-specific aminopeptidases are among only 80 enzymes conserved in the 3 major kingdoms of life (36). Such high conservation may stem from the exceptional conformational rigidity that proline residues introduce into peptide chains, which presents challenges to proteolytic enzyme systems. For example, in E. coli, the essential methionine aminopeptidase (PepM) responsible for cotranslational N-terminal methionine excision readily liberates the N-terminal methionine when proline or an amino acid with a similarly small side chain occupies the penultimate position. However, a proline in the third position inhibits PepM activity (37, 38), making PepP required for efficient N-terminal methionine excision in such cases (39).
The conformational constraints of polyproline motifs also present challenges during ribosomal peptide synthesis by stalling translation. As EF-P is required to prevent such stalling and promote efficient translation of polyproline-containing peptides (40, 41), the genomic organization of a putative proline-specific aminopeptidase like the pepQ product with EF-P is likely no coincidence. Work with Gram-negative enteric pathogens has revealed that proteins with EF-P target motifs (and hence dependent on EF-P for optimal translation) are enriched for metabolic enzymes, membrane-associated proteins, transporters, and two-component regulatory systems (42). The same appears to be true for M. tuberculosis. This system may have evolved for rapid remodeling of the cellular proteome in response to changing environmental conditions, such as carbon source (43, 44). The highly conserved organization of genes for proline-specific aminopeptidases and EF-P in bacteria thus supports a potential role for pepQ in regulating maturation and/or turnover of specific proteins in a manner that is sensitive to the metabolic status of M. tuberculosis.
Proline-specific aminopeptidases also have important roles in the utilization of exogenous and endogenous proteins as sources of essential amino acids useful for protein synthesis, energy production, osmoprotection, and recycling of reduced cofactors. Proline itself is increasingly recognized as having a critical role in energy metabolism, redox control, and bacterial virulence (45). In early transposon mutagenesis studies (18), disruptions of pepQ were observed to cause a growth defect (thus pepQ is neither absolutely essential nor nonessential in vitro). More recent high-resolution transposon mutagenesis studies suggest that pepQ is conditionally essential when grown on glycerol as a carbon source but not essential when grown on cholesterol (P value < 0.01 for comparison of transposon insertion counts) (46). This is interesting given recent evidence linking mycobacterial proline catabolism to protection from the toxic effects of methylglyoxal produced during growth on glycerol (47). If mutation of pepQ reduces intracellular availability of proline, this might increase susceptibility to methylglyoxal and reduce growth on glycerol.
The mechanism by which pepQ mutations result in reduced susceptibility to bedaquiline and clofazimine is unclear. Rv0678 mutations appear to confer cross-resistance to bedaquiline and clofazimine via derepression of mmpL5-mmpS5 (Rv0676c-Rv0677c), which encode an RND transporter capable of exporting bedaquiline and clofazimine (6, 7). Reversion of bedaquiline susceptibility of a pepQ mutant to that of its wild-type parent in the presence of efflux pump inhibitors suggests that drug efflux is involved in the mechanism of resistance. However, unlike the mutations in Rv0678, the mutation in pepQ is not associated with overexpression of mmpL5 or mmpS5. One possibility is that pepQ mutations increase efflux through this transporter by a different means, such as preventing degradation of MmpL5, which contains a Val-Pro-Pro stretch near the amino terminus.
In conclusion, we demonstrate that loss-of-function mutations in pepQ confer reduced susceptibility to bedaquiline and clofazimine. While the shift in susceptibility is relatively small, it is of a magnitude similar to those of mutations in Rv0678 that have been selected in patients and mice receiving combination therapy including bedaquiline. Therefore, these results should provide the impetus to include pepQ in the genetic analysis of bedaquiline- and clofazimine-resistant strains identified during clinical usage. Further study of the biological function of pepQ in M. tuberculosis and the mechanism by which pepQ mutations confer reduced susceptibility is clearly warranted.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by the Global Alliance for TB Drug Development and the National Institutes of Health (R01-AI111992 to E.N.).
K.A. is employed by, and holds stock in, Janssen Pharmaceutica. None of the other authors has a financial relationship with a commercial entity that has an interest in the subject of this article.
Funding Statement
This research was funded by the Global Alliance for TB Drug Development and the National Institutes of Health (R01-AI111992 to E.N.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00753-16.
REFERENCES
- 1.Matteelli A, Migliori GB, Cirillo D, Centis R, Girard E, Raviglione M. 2007. Multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis: epidemiology and control. Expert Rev Anti Infect Ther 5:857–871. doi: 10.1586/14787210.5.5.857. [DOI] [PubMed] [Google Scholar]
- 2.Cohen J. 2013. Infectious disease. Approval of novel TB drug celebrated—with restraint. Science 339:130. doi: 10.1126/science.339.6116.130. [DOI] [PubMed] [Google Scholar]
- 3.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 4.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. 2007. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 5.Huitric E, Verhasselt P, Koul A, Andries K, Hoffner S, Andersson DI. 2010. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob Agents Chemother 54:1022–1028. doi: 10.1128/AAC.01611-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andries K, Villellas C, Coeck N, Thys K, Gevers T, Vranckx L, Lounis N, de Jong BC, Koul A. 2014. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9:e102135. doi: 10.1371/journal.pone.0102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartkoorn RC, Upekar S, Cole ST. 3 March 2014. Cross-resistance between clofazimine and bedaquiline through up-regulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob Agents Chemother doi: 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Deun A, Maug AK, Salim MA, Das PK, Sarker MR, Daru P, Rieder HL. 2010. Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med 182:684–692. doi: 10.1164/rccm.201001-0077OC. [DOI] [PubMed] [Google Scholar]
- 9.Piubello A, Harouna SH, Souleymane MB, Boukary I, Morou S, Daouda M, Hanki Y, Van Deun A. 2014. High cure rate with standardised short-course multidrug-resistant tuberculosis treatment in Niger: no relapses. Int J Tuberc Lung Dis 18:1188–1194. doi: 10.5588/ijtld.13.0075. [DOI] [PubMed] [Google Scholar]
- 10.Aung KJ, Van Deun A, Declercq E, Sarker MR, Das PK, Hossain MA, Rieder HL. 2014. Successful ‘9-month Bangladesh regimen’ for multidrug-resistant tuberculosis among over 500 consecutive patients. Int J Tuberc Lung Dis 18:1180–1187. doi: 10.5588/ijtld.14.0100. [DOI] [PubMed] [Google Scholar]
- 11.Tasneen R, Li SY, Peloquin CA, Taylor D, Williams KN, Andries K, Mdluli KE, Nuermberger EL. 2011. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 55:5485–5492. doi: 10.1128/AAC.05293-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nuermberger EL, Yoshimatsu T, Tyagi S, O'Brien RJ, Vernon AN, Chaisson RE, Bishai WR, Grosset JH. 2004. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am J Respir Crit Care Med 169:421–426. doi: 10.1164/rccm.200310-1380OC. [DOI] [PubMed] [Google Scholar]
- 13.Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR Jr. 2007. Genetic manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol Chapter 10:Unit 10A.2. doi: 10.1002/9780471729259.mc10a02s6. [DOI] [PubMed] [Google Scholar]
- 14.Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, Jacobs WR Jr, Mizrahi V, Parish T, Rubin E, Sassetti C, Sacchettini JC. 2010. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J Bacteriol 192:3645–3653. doi: 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Ibrahim M, Andries K, Lounis N, Chauffour A, Truffot-Pernot C, Jarlier V, Veziris N. 2007. Synergistic activity of R207910 combined with pyrazinamide against murine tuberculosis. Antimicrob Agents Chemother 51:1011–1015. doi: 10.1128/AAC.00898-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 18.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 19.Lu Y, Zheng M, Wang B, Fu L, Zhao W, Li P, Xu J, Zhu H, Jin H, Yin D, Huang H, Upton AM, Ma Z. 2011. Clofazimine analogs with efficacy against experimental tuberculosis and reduced potential for accumulation. Antimicrob Agents Chemother 55:5185–5193. doi: 10.1128/AAC.00699-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowther WT, McMillen DA, Orville AM, Matthews BW. 1998. The anti-angiogenic agent fumagillin covalently modifies a conserved active-site histidine in the Escherichia coli methionine aminopeptidase. Proc Natl Acad Sci U S A 95:12153–12157. doi: 10.1073/pnas.95.21.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Roberts RJ, Kasif S, Guan C. 2005. Characterization of two new aminopeptidases in Escherichia coli. J Bacteriol 187:3671–3677. doi: 10.1128/JB.187.11.3671-3677.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coll M, Knof SH, Ohga Y, Messerschmidt A, Huber R, Moellering H, Russmann L, Schumacher G. 1990. Enzymatic mechanism of creatine amidinohydrolase as deduced from crystal structures. J Mol Biol 214:597–610. doi: 10.1016/0022-2836(90)90201-V. [DOI] [PubMed] [Google Scholar]
- 23.Bazan JF, Weaver LH, Roderick SL, Huber R, Matthews BW. 1994. Sequence and structure comparison suggest that methionine aminopeptidase, prolidase, aminopeptidase P, and creatinase share a common fold. Proc Natl Acad Sci U S A 91:2473–2477. doi: 10.1073/pnas.91.7.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 25.Weaver J, Watts T, Li P, Rye HS. 2014. Structural basis of substrate selectivity of E. coli prolidase. PLoS One 9:e111531. doi: 10.1371/journal.pone.0111531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilce MC, Bond CS, Dixon NE, Freeman HC, Guss JM, Lilley PE, Wilce JA. 1998. Structure and mechanism of a proline-specific aminopeptidase from Escherichia coli. Proc Natl Acad Sci U S A 95:3472–3477. doi: 10.1073/pnas.95.7.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coeck N, Villelas C, Meehan CJ, Lounis N, Niemann S, Rigouts L, de Jong B, Andries K. 2015. Unexpected high frequency of Rv0678 mutations in MDR-TB patients without documented prior use of clofazimine or bedaquiline. Int J Tuberc Lung Dis 19:S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pym AS, Diacon AH, Tang SJ, Conradie F, Danilovits M, Chuchottaworn C, Vasilyeva I, Andries K, Bakare N, De Marez T, Haxaire-Theeuwes M, Lounis N, Meyvisch P, Van Baelen B, van Heeswijk RP, Dannemann B, TMC207-C209 Study Group. 2016. Bedaquiline in the treatment of multidrug- and extensively drug-resistant tuberculosis. Eur Respir J 47:564–574. doi: 10.1183/13993003.00724-2015. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Chen J, Cui P, Shi W, Zhang W, Zhang Y. 2015. Identification of novel mutations associated with clofazimine resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 70:2507–2510. doi: 10.1093/jac/dkv150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams K, Minkowski A, Amoabeng O, Peloquin CA, Taylor D, Andries K, Wallis RS, Mdluli KE, Nuermberger EL. 2012. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob Agents Chemother 56:3114–3120. doi: 10.1128/AAC.00384-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rouan MC, Lounis N, Gevers T, Dillen L, Gilissen R, Raoof A, Andries K. 2012. Pharmacokinetics and pharmacodynamics of TMC207 and its N-desmethyl metabolite in a murine model of tuberculosis. Antimicrob Agents Chemother 56:1444–1451. doi: 10.1128/AAC.00720-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.European Committee on Antimicrobial Susceptibility Testing. 2014. Workshop on recommendations for pharmaceutical companies regarding data required for new antituberculous drugs, 11–12 November 2014, Basel, Switzerland. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Mycobacteria/Mycobacterial_workshop_Basel_11-12Nov2014.pdf.
- 33.Ioerger TR, Feng Y, Chen X, Dobos KM, Victor TC, Streicher EM, Warren RM, Gey van Pittius NC, Van Helden PD, Sacchettini JC. 2010. The non-clonality of drug resistance in Beijing-genotype isolates of Mycobacterium tuberculosis from the Western Cape of South Africa. BMC Genomics 11:670. doi: 10.1186/1471-2164-11-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matos J, Nardi M, Kumura H, Monnet V. 1998. Genetic characterization of pepP, which encodes an aminopeptidase P whose deficiency does not affect Lactococcus lactis growth in milk, unlike deficiency of the X-prolyl dipeptidyl aminopeptidase. Appl Environ Microbiol 64:4591–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribeiro-Guimaraes ML, Pessolani MC. 2007. Comparative genomics of mycobacterial proteases. Microb Pathog 43:173–178. doi: 10.1016/j.micpath.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Harris JK, Kelley ST, Spiegelman GB, Pace NR. 2003. The genetic core of the universal ancestor. Genome Res 13:407–412. doi: 10.1101/gr.652803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Bassat A, Bauer K, Chang SY, Myambo K, Boosman A, Chang S. 1987. Processing of the initiation methionine from proteins: properties of the Escherichia coli methionine aminopeptidase and its gene structure. J Bacteriol 169:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirel PH, Schmitter MJ, Dessen P, Fayat G, Blanquet S. 1989. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc Natl Acad Sci U S A 86:8247–8251. doi: 10.1073/pnas.86.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasueda H, Kikuchi Y, Kojima H, Nagase K. 1991. In-vivo processing of the initiator methionine from recombinant methionyl human interleukin-6 synthesized in Escherichia coli overproducing aminopeptidase-P. Appl Microbiol Biotechnol 36:211–215. [DOI] [PubMed] [Google Scholar]
- 40.Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. 2013. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339:85–88. doi: 10.1126/science.1229017. [DOI] [PubMed] [Google Scholar]
- 41.Zou SB, Hersch SJ, Roy H, Wiggers JB, Leung AS, Buranyi S, Xie JL, Dare K, Ibba M, Navarre WW. 2012. Loss of elongation factor P disrupts bacterial outer membrane integrity. J Bacteriol 194:413–425. doi: 10.1128/JB.05864-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hersch SJ, Wang M, Zou SB, Moon KM, Foster LJ, Ibba M, Navarre WW. 2013. Divergent protein motifs direct elongation factor P-mediated translational regulation in Salmonella enterica and Escherichia coli. mBio 4:e00180-13. doi: 10.1128/mBio.00180-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guédon E, Renault P, Ehrlich SD, Delorme C. 2001. Transcriptional pattern of genes coding for the proteolytic system of Lactococcus lactis and evidence for coordinated regulation of key enzymes by peptide supply. J Bacteriol 183:3614–3622. doi: 10.1128/JB.183.12.3614-3622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doerfel LK, Rodnina MV. 2013. Elongation factor P: function and effects on bacterial fitness. Biopolymers 99:837–845. doi: 10.1002/bip.22341. [DOI] [PubMed] [Google Scholar]
- 45.Tanner JJ. 2008. Structural biology of proline catabolism. Amino Acids 35:719–730. doi: 10.1007/s00726-008-0062-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berney M, Weimar MR, Heikal A, Cook GM. 2012. Regulation of proline metabolism in mycobacteria and its role in carbon metabolism under hypoxia. Mol Microbiol 84:664–681. doi: 10.1111/j.1365-2958.2012.08053.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.