

Abstract
Filoviruses are highly infectious, and no FDA-approved drug therapy for filovirus infection is available. Most work to find a treatment has involved only a few strains of Ebola virus and testing of relatively small drug libraries or compounds that have shown efficacy against other virus types. Here we report the findings of a high-throughput screening of 319,855 small molecules from the Molecular Libraries Small Molecule Repository library for their activities against Marburg virus and Ebola virus. Nine of the most potent, novel compounds that blocked infection by both viruses were analyzed in detail for their mechanisms of action. The compounds inhibited known key steps in the Ebola virus infection mechanism by blocking either cell surface attachment, macropinocytosis-mediated uptake, or endosomal trafficking. To date, very few specific inhibitors of macropinocytosis have been reported. The 2 novel macropinocytosis inhibitors are more potent inhibitors of Ebola virus infection and less toxic than ethylisopropylamiloride, one commonly accepted macropinocytosis inhibitor. Each compound blocked infection of primary human macrophages, indicating their potential to be developed as new antifiloviral therapies.
INTRODUCTION
The Filoviridae family includes the highly infectious and pathogenic Ebola virus (EBOV; previously named Zaire Ebola virus) and Marburg virus (MARV) and a recently identified member, Lloviu virus (1). Since the first reported outbreak in 1976, each successive decade has seen increases in the recurrence of filovirus outbreaks, the incidences of infection, as well as the number of infected people. Both Ebola virus and Marburg virus are highly contagious, and average case fatality rates range from 42% to 90% (2, 3). According to WHO, the 2014 Ebola virus outbreak resulted in more than 28,000 infections and 11,000 deaths. Several promising therapeutic candidates are currently being investigated, but none are FDA approved. Strategies such as treatment with antisense oligonucleotides, antibody-based therapies, and treatment with small molecules directed against specific viral as well as cellular factors have been tested, but the outcomes have been mixed (4–17). Additionally, many of these therapies target only closely related strains of Ebola virus, making the development of treatments for other species of Ebola virus and Marburg virus a priority.
Filovirus entry into cells is dependent on the single viral glycoprotein (GP). Though the GPs of Ebola virus and Marburg virus are quite distinct from each other, the entry mechanisms of the two viruses share common traits, offering potential broad-spectrum antifiloviral targets. Filoviral GP binds receptors, and the virus is internalized through macropinocytosis (18, 19) and trafficked through endosomes, which is dependent on the calcium channel TPC2 (14, 20). During trafficking the virus is exposed to an increasingly acidic environment, in which proteases are activated and cleave the GP (21–24), allowing interaction with NPC1, a key protein found in endosomes (25, 26).
The development of small molecules into drugs is inefficient, with only 1 in 100,000 hits from a screen making it to the clinic (27–29). Large screens increase the chances of eventually identifying active and specific small molecules. However, screening is hampered by the dangerous nature of filoviruses, requiring high-containment laboratories for all work. Instead, much of the recent progress in identifying antifilovirus compounds has been made by testing small libraries of medications clinically approved for use for the treatment of other indications or against other virus types (8–10). However, very few specific or potent antifiloviral agents have emerged from these studies.
Here we report a large-scale screen of 319,855 small molecules from the Molecular Libraries Small Molecule Repository (MLSMR) library to identify inhibitors of the GP-mediated entry of EBOV and MARV Musoke. This study used the quantitative high-throughput screening (qHTS) approach to rapidly identify true inhibitors. The results of the screen and the mechanisms of action of nine of the most potent broad-spectrum filovirus inhibitors are presented.
MATERIALS AND METHODS
Cells.
293FT cells (catalog number R700-07; Thermo), Vero cells, HeLa cells, and SW-13 cells were maintained in Dulbecco modified Eagle medium (catalog number MT10017CV; Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; catalog number 100106; Gemini Bio-Products) (referred to here as complete medium). Primary human macrophages were differentiated from the buffy coat fraction of human blood (from the South Texas Blood and Tissue Center) according to previously published protocols (30, 31). Briefly, mononuclear lymphocytes were isolated using LeucoSep tubes (catalog number 19177522; Fisher Scientific), resuspended in Iscove modified Dulbecco medium (IMDM; catalog number SH3022801; Fisher Scientific), and plated in 96-well plates (50,000 cells per well). After the cells were allowed to adhere for 1 h, unattached cells were washed off using IMDM. Attached monocytes were allowed to differentiate into macrophages for 7 to 8 days in IMDM containing 2% heat-inactivated human serum (catalog number 35-060-Cl; Corning Inc.), 100 U/ml penicillin, 100 μg/ml streptomycin, 1× nonessential amino acids (catalog number TMS001C; Fisher Scientific), 50 μM 2-mercaptoethanol (catalog number ICN19470583; Fisher Scientific), and 800 U/ml human macrophage colony-stimulating factor (catalog number 574806; BioLegend). Adherent monocytes were washed, and the medium was replaced on days 2 and 6 while the cells were differentiating. All cells were kept at 37°C in a humidified incubator with 5% CO2.
Wild-type virus production.
For virus naming, we used the current recommendations of ICTV (32). The Zaire Ebola virus (EBOV) Mayinga strain with an insertion of green fluorescent protein (GFP) between the nucleoprotein (NP) and VP35 (33), which was a kind gift of Heinz Feldmann (NIH, Rocky Mountain Laboratory, Hamilton, MT), and the Marburg virus (MARV) Musoke strain were grown in Vero cells. All work was done in a biosafety level 4 (BSL4; protection level 4) lab at the Texas Biomedical Research Institute. Culture supernatants containing virus were concentrated by pelleting through a 20% sucrose cushion. Briefly, culture supernatants from infected Vero cells were collected, and cell debris was removed by centrifugation at 1,800 × g for 15 min. The clarified supernatant was overlaid on a 20% sucrose cushion, and the virus was pelleted by centrifugation at 141,118 × g for 2 h at 4°C. Virus pellets were resuspended in phosphate-buffered saline (PBS) and stored in aliquots at −80°C until they were used.
Production of EBOV VLPs.
Virus-like particles (VLPs) containing EBOV GP were produced in 293FT cells. Plasmid DNA constructs carrying EBOV matrix protein VP40, VP40 fused to GFP (VP40-GFP), NP, and GP were used for VLP production. Briefly, 293FT cells were grown in 10-cm2 tissue culture dishes and transfected with plasmids carrying 3.5 μg of VP40-GFP, 1.5 μg VP40, 5 μg NP, and 1 μg GP. At 24 h posttransfection, the cells were washed once with medium containing no FBS or antibiotics and then overlaid with fresh complete medium. At 2 days posttransfection, the supernatants were collected and cell debris was removed by centrifugation at 1,800 × g for 15 min. The clarified supernatant was overlaid on 20% sucrose cushions, and the VLPs were pelleted by centrifugation at 141,118 × g for 2 h. The pellets containing the VLPs were resuspended in PBS and stored in small aliquots at −80°C until further use.
Effect of compounds on localization of EBOV particles with respect to cells.
HeLa cells were grown in 8-well chamber slides (catalog number 80826; Ibidi) at 20,000 cells per well. All treatments were done in duplicate, with each replicate treatment being performed on a different slide. The cells were treated for 1 h with the compounds of interest at a concentration of 50 μM. At 1 h after incubation with compound, the cells were challenged for 2.5 h with wild-type EBOV. Infected cells were washed 3 times in PBS and fixed by immersing the slides in 4% paraformaldehyde for 24 h at 4°C. The paraformaldehyde from the fixed slides was decanted, and the slides were washed 3 times with PBS. The cells were immunostained without permeabilization with a GP-specific antibody (mouse anti-EBOV GP mAb 4F3, IBT Bioservices, catalog number 0201-020). Anti-mouse immunoglobulin-Alexa Fluor 488 antibody (catalog number A11029; Life Technologies) was used as the secondary antibody. After this, the cells were permeabilized using 0.1% Triton X-100 in PBS and blocked again for 1 h in 3.5% bovine serum albumin. The cells were then immunostained again using anti-GP antibody. Excess antibody was washed off, and the cells were then stained with anti-mouse immunoglobulin Alexa Fluor 546 antibody (catalog number A11030; Life Technologies). After 3 washes to remove any nonspecific antibody, the cell bodies were stained using HCS CellMask blue (catalog number H32720; Life Technologies) per the manufacturer's protocol. The cells were imaged across the z-plane on a Nikon Ti Eclipse automated microscope. Images were deconvolved using AutoQuant X3 software (Media Cybernetics Inc.). The deconvolved images were analyzed using Imaris three-dimensional (3D) image analysis software (Bitplane Inc.). For this 3D analysis, cell bodies were modeled on a CellMask blue outline. Cell surfaces were modeled on the basis of the CellMask blue periphery. Virus particles which were red or green were modeled. Red particles within the cell body were counted as particles inside the cell. Green particles within 1 μm of the cell surface were counted as having a surface interaction.
Effect of compounds on macropinocytic uptake.
Cells were treated for 1 h with the compounds of interest at a concentration of 50 μM in triplicate. After 1 h, a mixture of 25 μg dextran, Alexa Fluor 488 (molecular weight, 10,000; catalog number D-22910; Life Technologies), and Hoechst stain at a concentration of 1:10,000 was added to the cells. After 30 to 40 min of incubation, images of live cells were captured (minimum, 3 images per well). The number of dextran-containing vesicles in each image was counted using CellProfiler software.
Effect of compound treatment on transferrin uptake.
HeLa cells in 12-well plates were used for binding studies, while cells plated in a 96-well plate were used for uptake studies. Cells were serum starved for 4 h, followed by treatment with the compounds at 50 μM for 1 h in serum-free medium. Treated, serum-starved cells in 12-well plates were then transferred to a 14°C water bath and kept in it for 45 min. The cells were incubated with 25 μg/ml of human transferrin (catalog number T3309; Sigma-Aldrich) for 40 more minutes. Cells were washed twice with PBS at 14°C to wash off unbound transferrin and lysed using cell extraction buffer (300 μl). The amount of transferrin in the lysates (bound to cells) was estimated by enzyme-linked immunosorbent assay (ELISA; human transferrin ELISA kit; SimpleStep; catalog number ab187391; Abcam).
Colocalization of EBOV VLPs with early endosomal marker EEA1 and late endosomal marker LAMP1.
HeLa cells were grown in 8-well chamber slides (catalog number 80826; Ibidi) at 20,000 cells/well. All treatments were done in duplicate for each virus, with each replicate treatment being performed on a different slide. The cells were treated for 1 h with the compounds of interest at a concentration of 50 μM. At 1 h after incubation with compound, the cells were transduced with fluorescent EBOV VLPs for 2 h 30 min at 37°C in a humidified incubator with 5% CO2. After transduction, the cells were washed twice with PBS and fixed in 4% paraformaldehyde (catalog number 15710; Electron Microscopy Science) overnight at 4°C. Fixed cells were washed thrice to remove the paraformaldehyde, and the cells were permeabilized using 0.1% Triton X-100 (catalog number T8787; Sigma) in PBS and blocked for 1 h in 3.5% bovine serum albumin (catalog number BP9704100; Fisher Scientific). The fixed cells were incubated with an anti-EEA1 antibody (catalog number 610457; BD Transduction Laboratories) at a 1:2,000 dilution and anti-LAMP1 antibody raised in a rabbit (catalog number 9091; Cell Signaling Technology) at a 1:200 dilution for 2 h at 37°C. Excess antibody was washed off, and the cells were then stained with anti-mouse immunoglobulin-Alexa Fluor 647 antibody (catalog number A21236; Life Technologies) and anti-rabbit immunoglobulin-Alexa Fluor 546 antibody (catalog number A11035; Life Technologies) at a 1:1,000 dilution. After 3 washes to remove unbound antibody, the cell bodies were stained using HCS CellMask blue (catalog number H32720; Life Technologies) per the manufacturer's protocol. The cells were imaged across the z-plane on a Nikon Ti Eclipse automated microscope. Images were deconvolved using AutoQuant X3 software (Media Cybernetics Inc.). Deconvolved images were analyzed using Imaris 3D image analysis software (Bitplane Inc.). EEA1-positive vesicles, LAMP1-positive vesicles, and VLPs were modeled. VLPs colocalizing with EEA1 and LAMP1 were counted.
Acridine orange assay for analysis of compounds as acidification blockers.
Baby hamster kidney (BHK) fibroblasts were grown in minimal essential medium (MEM) supplemented with 10% FBS at 37°C and 5% CO2. Ten thousand cells in 100 μl medium were added to each well of a 96-well Falcon polystyrene tissue culture plate and grown for 18 to 20 h, at which point the medium was removed and replaced with 50 μl MEM–10% FBS–compound. The compounds were dissolved in dimethyl sulfoxide (DMSO) and diluted in MEM–10% FBS; the initial concentration was 100 μM. Bafilomycin A1 (100 nM) and 1% DMSO in MEM–10% FBS were used as positive and negative controls, respectively. After 4 h at 37°C, the drug-containing medium was removed and replaced with 100 ml of MEM–10% FBS–1 mg/ml acridine orange. The cells were incubated at 37°C for 15 min and washed twice with 100 ml PBS for 5 min. The red and green fluorescence was measured with a BioTek Synergy 4 multimode microplate reader using a 485/20-nm and 665/7.5-nm filter set and a 485/20-nm and 530/30-nm filter set. All compounds were tested in triplicate. The data are presented as the mean ± standard error of the mean ratio of red fluorescence to green fluorescence. Statistical significance was determined by analysis of variance (ANOVA) with posttest comparisons to the results for the DMSO- and bafilomycin-treated controls (GraphPad Prism software).
Cathepsin cleavage assay.
Compounds were tested at 50 μM for their ability to inhibit the cathepsin B cleavage of EBOV GP. A total of 107 transducing units of EBOV GP-pseudotyped vesicular stomatitis virus (VSV) (assessed in Vero cells) was incubated in a low-pH buffer (100 mM sodium acetate [pH 5.0], 1 mM EDTA, 5 mM dithiothreitol) in the presence or absence of 0.5 μg of cathepsin B for 15 min at 37°C. E64 (30 μM), which is a well-established cysteine protease inhibitor, was used as a positive control to block inhibition of protease processing of EBOV GP. Samples were immediately placed on ice following incubation, followed by incubation at 95°C for 5 min to stop the protease reaction, and the proteins were separated on a 4 to 20% SDS-polyacrylamide gel. The proteins were transferred to nitrocellulose and probed with the anti-VSV matrix protein monoclonal antibody 23H12 (34) and rabbit antiserum against EBOV GP residues 83 to 96. Appropriate secondary antibodies that were conjugated to either IRDye 680 or 800, allowing simultaneous imaging of GP and matrix protein, were added. Pixel intensities in the region of full-length EBOV GP (∼140 kDa) and VSV (∼28 kDa) in each lane were determined, and the ratio of GP to matrix protein was determined. This ratio for untreated virions was set equal to 100%, and the other values are expressed relative to this value for the control. All compounds were assessed at least twice, and the means of the ratios are shown.
Effect of compounds on cytotoxicity.
HeLa cells plated in 96-well plates (20,000 cells/well) were treated with the compounds at six concentrations (50, 25, 12.5, 6.25, 3.125, and 1.56 μM) in a 100-μl final volume. After 24 h of incubation with the compounds, cytotoxicity was measured using a CytoTox-Fluor cytotoxicity assay (catalog number G9260; Promega) per the manufacturer's protocol. All compounds were found to be noncytotoxic at 50 μM. Ethylisopropylamiloride (EIPA), on the other hand, was found to have a 50% cytotoxic concentration (CC50) of 37 μM (see Table 1, last column).
TABLE 1.
Activities of selected compounds against MARV and EBOV and cytotoxicities for HeLa cellsa
| Compound | MARV |
EBOV |
Activity in macrophages IC50 (μM) | CC50 (μM) | ||
|---|---|---|---|---|---|---|
| Curve class | IC50 (μM) in HeLa cells | Curve class | IC50 (μM) in HeLa cells | |||
| MLS000078751 | 1.5 | 6.1 ± 0.75 | 1.3 | 25.6 ± 1.5 | ∼25 ± 3.5 | >50 |
| MLS000394177 | 1.3 | 12.9 ± 1 | 1.6 | 1.9 ± 0.25 | 8.6 ± 2 | >50 |
| MLS000534476 | 1.3 | 6.2 ± 0.5 | 1.6 | 24.4 ± 2 | 3.6 ± 0.8 | >50 |
| MLS000554255 | 1.3 | 6.4 ± 0.5 | 1.5 | 6.8 ± 0.75 | 6.0 ± 1.4 | >50 |
| MLS000555232 | 1.3 | 6.3 ± 0.6 | 1.6 | 1.7 ± 0.8 | 9.4 ± 1 | >50 |
| MLS000730532 | 1.3 | 1.9 ± 0.5 | 1.1 | 1.6 ± 0.2 | 9.5 ± 1 | >50 |
| MLS000733230 | 1.3 | 11.7 ± 1.2 | 1.3 | 6.7 ± 0.5 | 4.4 ± 0.3 | >50 |
| MLS000762907 | 1.3 | 12.5 ± 0.8 | 1.3 | 3.2 ± 3 | 23.4 ± 3 | >50 |
| MLS001101371 | 1.3 | 13.0 ± 2 | 1.3 | 12.9 ± 2 | 24.6 ± 2.5 | >50 |
Nine of the 17 compounds confirmed to be hits from the counterscreen were obtained from independent sources, and their activities against both EBOV and MARV were confirmed on HeLa cells. Both the quality of the dose-response curve (curve class, as defined in Fig. S3 in the supplemental material) and inhibitory potency (IC50) are shown. Activity against EBOV was also tested in primary monocyte-derived macrophages (last column). The cytotoxicities of the compounds were measured using a CytoTox-Fluor cytotoxicity assay (Promega). All compounds were found to be noncytotoxic at 50 μM when the viabilities of treated cells were compared to those of untreated cells. The IC50 values shown for MARV and EBOV in HeLa cells are averages ± standard deviations from 3 independent experiments, while the activity in human macrophages is the average ± standard deviations from 2 independent experiments.
RESULTS
Optimization of a high-throughput assay for identifying entry inhibitors of MARV.
GP mediates the earliest steps of virus infection and is an attractive target for therapeutic intervention. By blocking virus entry, infection and spread can be treated before the virus has an opportunity to impact cell health. To specifically detect inhibitors of GP function, virus pseudotypes were used for primary screening (see Fig. S1A in the supplemental material). Pseudotyped viruses bear the glycoproteins of one virus on the core of another virus type (35). We were successful in producing VSV pseudotyped with MARV GP and encoding firefly luciferase as a reporter (MARVGP-VSVluc) with a titer of 1011 relative light units/ml, corresponding to 109 PFU/ml. The MARVGP-VSVluc pseudotype yielded a signal-to-noise ratio of more than 30 (see Fig. S2A in the supplemental material) and a Z′-factor above 0.55 (see Fig. S2B in the supplemental material) in a 1,536-well format, indicating its suitability for qHTS. Extensive attempts to produce a pseudotype using EBOV GP with a similarly high titer were not successful.
Identification of 7,200 compounds as inhibitors of infection by MARVGP-VSVluc.
Compounds in the MLSMR library were screened at 6 concentrations each using an automated robotic assay platform at the National Center for Advancing Translational Sciences (NCATS). Compounds were prioritized on the basis of their curve class (see Fig. S3 in the supplemental material) and the efficacy of reporter expression inhibition. To eliminate compounds impacting luciferase reporter function, the hits were first filtered by removing compounds that showed activity in multiple unrelated assays performed at NCATS. Using this approach, 7,200 compounds (2.3% of the library) were identified to be inhibitors of MARVGP-VSVluc infection (PubChem assay identifiers 540276 and 720532).
Counterscreening using retrovirus-based pseudotype.
Counterscreening was performed to eliminate inhibitors of VSV genome replication and firefly luciferase (used as the reporter of infection) in the primary screen. A second pseudotype in which the VSV core was replaced with that from Moloney murine leukemia virus (MLV) and the luciferase reporter was replaced with a red fluorescent protein, mStrawberry, was used (see Fig. S1B in the supplemental material). MLV shares little similarity with VSV in its replication strategy, and luciferase and the fluorescent proteins have different mechanisms for producing signals. Even though the MLV pseudotype had a 100-fold lower titer (107 focus-forming units/ml) than the VSV pseudotype, the MLV pseudotype infection assay yielded a Z′ value of >0.7 in 384-well plates. All 7,200 compounds were tested at a single concentration of 50 μM to prioritize compounds of moderate potency or better. Cells were imaged by microscopy, and the infection rate was calculated. At the same time, those compounds reducing the total cell nucleus count by >20% were deprioritized as they potentially impacted cell viability, the cell cycle, or adherence to the well. Of the 7,200 hits, 554 compounds (7.7%) inhibited the MLV pseudotype infection by >90% without affecting the cell count. This observation suggested that >90% of the primary screen hits were either of low potency or impacted the VSV pseudotype and its luciferase reporter rather than MARV GP function. Chemical fingerprinting to determine the structural relationship of the 554 compounds was done using the Tanimoto criteria (36), and the results are graphically represented in Fig. 1A. More than half of the compounds shared similarity with at least one other compound, forming 20 distinct groups of structurally related molecules. The remaining compounds appeared to be structurally unrelated to each other.
FIG 1.

Structural analysis of active compounds identified after counterscreens. (A) The relatedness of compounds that inhibited MARV GP pseudotype infection by >80% at 50 μM was evaluated by determining the structural similarity using PubChem criteria and the relationship stringency based upon Tanimoto coefficients. Compounds with Tanimoto scores of >0.8 (high stringency) were considered related and are shown as nodes connected in each network. Seventeen compounds (black nodes) inhibited infection of both wild-type MARV and wild-type EBOV at a 50% effective concentration of less than 30 μM. Of the 17 compounds, 9 were available for more detailed study and are numbered. (B) Structures of the 9 compounds indicated by their PubChem identification numbers.
Identification of 17 novel inhibitors of both wild-type MARV and wild-type EBOV infection.
Compounds identified after counterscreening with the MLV pseudotype were tested for inhibition of wild-type MARV and then wild-type EBOV. Each compound was tested at 8 doses ranging from 0.39 μM up to 50 μM in HeLa cells (a human cell line susceptible to infection by both viruses). Of the 554 compounds tested, 61 (11%) inhibited MARV infection by more than 80% at a minimum of one of the test concentrations (see Table S1 in the supplemental material) and did not cause a more than 20% reduction in cell number. Interestingly, 42 of these 61 compounds also inhibited EBOV infection and in most cases were about 4-fold more potent than that required to inhibit MARV infection. Of these, 17 yielded a 50% inhibitory concentration (IC50) of less than 30 μM against both viruses. Nine of the compounds were readily available from an independent source, and so their activities could be verified (Table 1 and Fig. 1B). These 9 compounds were determined to be nontoxic at the highest concentration tested (50 μM; Table 1). In each case, the independently synthesized compound had activity within 1 μM of that seen when the MLSMR library was used as the source of the compound (see Fig. S4 to S6 in the supplemental material). It is likely that these pan-filovirus inhibitors were affecting cellular pathways shared by both viruses for entry. Additionally, all nine compounds inhibited EBOV infection in primary human macrophages, which are a primary target of infection in vivo (Table 1, last column; see also Fig. S8 in the supplemental material), suggesting their potential for use for the treatment of human infection.
Entry was the most likely point at which the compounds affected infection by the two pseudotypes and wild-type viruses, as they all shared the MARV or EBOV GPs and were affected at similar potencies. So, known steps of the virus entry pathway were examined using quantitative assays to understand the mechanism of inhibition. The microscopic analysis approach used to analyze virus uptake relies heavily on sensitive antibodies against virus structural proteins with high signal-to-noise ratios. Unfortunately, such antibodies against MARV were not commercially available, and the polyclonal antibody used for primary hit confirmation produced a diffuse background staining that was not amenable for quantitative analysis. In contrast, high-affinity antibodies against EBOV with low background staining are available, and so the assays were focused on EBOV.
MLS000078751 and MLS000534476 inhibit virus binding to cells.
The first step of virus entry into cells is binding to the cell surface. Virus particle binding was analyzed by counting the number of particles bound per cell as well as the number of cells that had no virus particles attached. Only compounds MLS000078751 and MLS000534476 significantly decreased the total number of viral particles bound by 53% and 55%, respectively (Fig. 2A and B). Both compounds, as well as MLS000554255, increased the proportion of cells without any bound virus. In contrast, MLS000394177 and MLS000555232 enhanced virus-cell binding by 44% and 89%, respectively, suggesting that each may cause virus to accumulate on the cell surface by preventing internalization.
FIG 2.
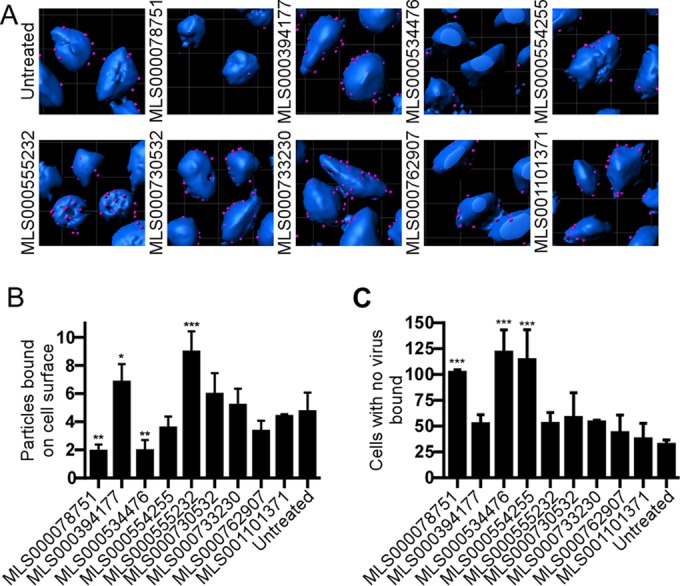
Effect of compound treatment on binding of EBOV to the cell surface. (A) HeLa cells were pretreated with 50 μM of the indicated compound for 1 h and were then incubated with wild-type EBOV for 2.5 h. The cells were then fixed and stained without permeabilization using an EBOV GP-specific antibody followed by an Alexa Fluor 546-labeled secondary antibody (red). Cell bodies were stained using CellMask blue. 3D modeling of deconvolved image z-stacks was done using Imaris software. (B) The number of virus particles present on the cell surface was counted. (C) A second analysis of the number of cells that had no virus bound was also performed. Data are the averages ± SDs for at least 100 cells. All assays were performed 3 times with similar outcomes each time. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To determine if the inhibition of virus binding was specific for EBOV, binding of transferrin to cells was measured (see Fig. S7 in the supplemental material). Of the two compounds that blocked EBOV binding to cells, MLS000534476 reduced transferrin binding to an extent similar to that seen for EBOV binding. In contrast, MLS000078751 reduced transferrin binding by only 35%, which suggests greater specificity against EBOV. The remaining 7 compounds were investigated further for their ability to cause defects in other steps of virus entry.
Inhibition of macropinocytic uptake.
EBOV predominantly uses macropinocytosis for productive uptake into cells. High-molecular-weight dextran is commonly used as a marker of macropinocytic uptake and is strongly associated with EBOV particles after uptake (18, 19, 37). We found that compounds MLS000394177, MLS000730532, and MLS000733230 reduced the uptake of fluorescently labeled dextran (Fig. 3A) by approximately 91%, 74%, and 91%, respectively (Fig. 3B). As a control, EIPA, a commonly accepted inhibitor of macropinocytosis, was used and inhibited dextran uptake (95%) at a level similar to MLS000394177, MLS000730532, and MLS000733230. MLS000394177, MLS000730532, and MLS000733230 also reduced the number of viral particles inside the cell body by 62%, 89%, and 91%, respectively (Fig. 3E and F).
FIG 3.

Effect of compound treatment on macropinocytosis and uptake of EBOV into cells. (A) HeLa cells pretreated with 50 μM of the indicated compounds for 1 h, followed by incubation with fluorescently labeled dextran (molecular weight, 10,000; green) as a marker of macropinocytic uptake. Images of cells were captured, and the number of dextran-positive vesicles was counted. EIPA, a known inhibitor of macropinocytosis, was used to block uptake. The cell nucleus (intense blue) and cytoplasm (weak blue) were stained with CellMask blue. (B) The average number of vesicles per cell ± SD was determined for images of at least 50 cells. (C) Measurement of transferrin uptake. HeLa cells were serum starved for 4 h, followed by treatment with the indicated compounds for 1 h in serum-free medium. Treated cells were incubated with 25 μg/ml of transferrin conjugated to Alexa Fluor 488, unbound transferrin was washed off, and the cells were fixed in formalin. Fixed cells were imaged. (D) The fluorescence intensity of the cells after excitation at 488 nm was measured. The fluorescence of cells without transferrin treatment was used to determine the background signal. The percentage of transferrin-positive cells for each treatment was plotted. EIPA (25 μM), a specific inhibitor of macropinocytosis, and chlorpromazine (25 μM), an inhibitor of clathrin-mediated endocytosis and a known inhibitor of transferrin uptake, were used as controls. All data are represented as the means ± SDs for 3 replicates. (E) Cells were incubated with EBOV for 2.5 h in the presence of each of the indicated compounds, and then nonpermeabilized cells were stained for EBOV GP followed by staining with an Alexa Fluor 546-labeled secondary antibody (red). Cells were then permeabilized and the staining was repeated but an Alexa Fluor 488-labeled secondary antibody was used. Cell bodies (blue) were stained with CellMask blue. (F) Deconvolved image stacks were used to generate 3D models of cells with bound virus particles using Imaris software. Internalized virus particles (green, not red) and cell numbers were counted. The number of cells with 15 ± 5 virus particles (mean ± SD for the control) inside the cell cytoplasm was calculated as a measure of virus uptake efficiency. The average ± SD for more than 200 cells is shown. All assays were performed 3 times with similar outcomes. **, P < 0.01; ***, P < 0.001.
As a measure of specificity and to control for a generalized disruption of membrane function and endocytic uptake, transferrin uptake, a well-characterized marker of clathrin-mediated endocytosis, was measured. Neither MLS000394177 nor MLS000733230 had an effect on transferrin uptake. In contrast, MLS000730532 impaired uptake of this marker, a result comparable to that obtained with chlorpromazine, a known inhibitor of clathrin-mediated endosome trafficking (Fig. 3C and D). Similarly, MLS000762907 also inhibited transferrin uptake.
Since MLS000394177 and MLS000733230 inhibited EBOV and dextran uptake but did not affect virus binding to cells or transferrin uptake, each appeared to specifically inhibit filovirus entry by affecting macropinocytosis similarly to EIPA, while MLS000730532 appeared to cause a more generalized disruption of endocytosis.
Effect of compounds on colocalization of EBOV VLPs to EEA1- and LAMP1-positive compartments.
After macropinocytic uptake, virus is trafficked through the endosomal network, during which the GP is cleaved by endosomal proteases. The remaining 4 compounds that did not affect macropinocytosis (including MLS000762907) were then tested for their impact on trafficking of EBOV VLPs beyond the macropinosome to early endosomes (colocalized with EEA1) and then late endosomes (colocalized with LAMP1). VLPs tagged with GFP-VP40 were used to follow trafficking. Since the GFP is encapsulated by the viral membrane, it should resist degradation by the endosomal proteases. After treatment with MLS000762907 and MLS001101371, VLPs colocalized with EEA1-positive vesicles normally. In contrast, MLS000555232 reduced the colocalization of EBOV VLPs with EEA1-positive vesicles by 77%, while MLS000554255 increased the colocalization of VLPs with EEA1-positive vesicles by 83% (Fig. 4A). When the association with LAMP1 was tested, MLS000554255, MLS000555232, and MLS001101371 significantly reduced the number of VLPs present in LAMP1-positive vesicles (P < 0.05) by 33%, 89%, and 41%, respectively (Fig. 4B). Taken together, MLS000555232 appeared to block colocalization to EEA1-positive and LAMP1-positive vesicles, whereas MLS000554255 and MLS001101371 impaired colocalization with LAMP1-positive vesicles only. Of these compounds, MLS000555232 also produced a nearly 2-fold increase in virus particle binding to the cell surface (Fig. 2B), suggesting that this compound may alter receptor and virus trafficking from the cell surface. Only MLS000762907 had no effect on VLP colocalization with EEA1- and LAMP1-positive vesicles. As described above, MLS000762907 reduced the uptake of transferrin but not that of dextran (Fig. 3C and D) or virus. Together, these observations suggest that MLS000762907 may interfere with trafficking (via clathrin-mediated uptake) of an unidentified cofactor that interacts with virus after uptake into cells.
FIG 4.
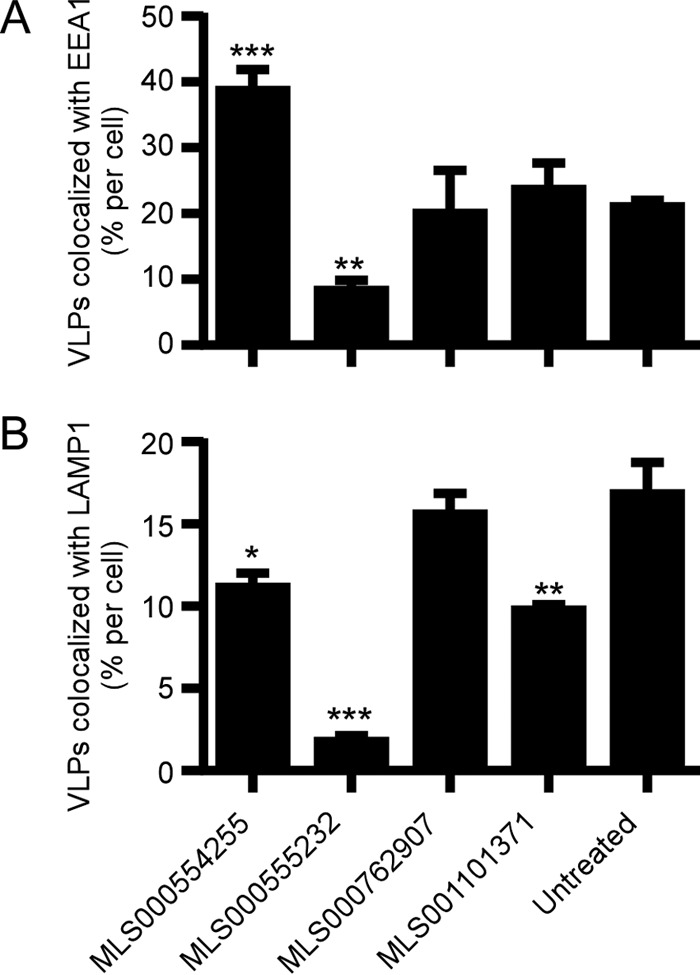
Effect of compound treatment on VLP trafficking. HeLa cells treated with 50 μM of the indicated compounds for 1 h were incubated with EBOV VLPs containing VP40-GFP for 2.5 h. Fixed cells were permeabilized and stained for EEA1 (an early endosome marker) and LAMP1 (a lysosome marker). 3D modeling of deconvolved image stacks was done using Imaris software. The portion of VLPs colocalized with EEA1 (A) or LAMP1 (B) is shown as the average ± SD for more than 200 cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Inhibition of endosomal acidification and cleavage of EBOV GP by endosomal proteases.
Filoviruses are pH dependent, requiring acidification of endosomes for productive infection. Both the cleavage of GP by endosomal proteases, such as cathepsins, and the structural rearrangement of GP and its interaction with cellular factors, leading to fusion of the virus and cell membranes, are pH dependent (21, 38). Compounds were therefore checked for their inhibition of endosomal acidification and GP cleavage. Acridine orange, a pH-sensitive dye that accumulates in endosomes, was incubated with cells, and its fluorescence was measured. Treatment of cells with each of MLS000394177, MLS000534476, MLS000730532, MLS000733230, MLS000762907, and MLS001101371 reduced endosomal acidification by between 30 and 60% (Fig. 5A), but none was as potent as bafilomycin A1, a specific inhibitor of the vacuolar ATPase, the primary endosomal proton pump. Three of these compounds also inhibited macropinocytosis (Fig. 3A and B). The partial endosomal acidification block likely contributes to the antifilovirus action of these compounds but does not appear to account for most of their activity. When tested for inhibition of GP cleavage by cathepsin B, none of the compounds showed a statistically significant effect compared to that of no treatment as a control (Fig. 5B; see also Fig. S9 in the supplemental material).
FIG 5.

Effect of compound treatment on interaction with endosomal acidification and cathepsin protease activity. (A) HeLa cells were pretreated with the indicated compounds at 50 μM for 1 h and then incubated for 1 h with acridine orange, a pH-sensitive fluorescent dye that accumulates in endosomes and which has a pH-sensitive fluorescent emission peak measured at 665 nm and a pH-independent emission peak measured at 530 nm. The ratio of the fluorescent emission at 665 nm to that at 530 nm was used to measure endosomal acidification relative to that for untreated cells and is given as the average ± SD for 3 independent measurements. (B) EBOV GP-pseudotyped VSV pseudovirions were incubated with cathepsin B in the presence or absence of 50 μM each inhibitor. E64 served as a positive control that blocked cathepsin B activity. Proteins from treated virions were separated and immunoblotted for the GP and VSV matrix protein. Ratios of quantitation of full-length GP (uncleaved) to matrix protein were generated. Shown are the mean values ± SDs relative to the values for pseudovirions not treated with cathepsin B.
In summary, of the 9 compounds evaluated, 2 appeared to block virus binding to cells, 3 blocked macropinocytic uptake, and 3 altered endocytic trafficking of virus particles in cells; for 1 compound, the mechanism by which virus entry was blocked could not be determined (Table 2). Each compound inhibited both EBOV and MARV infection of cells, and none of the compounds was toxic at a concentration of up to 50 μM, so these are promising candidates for development as therapeutic agents.
TABLE 2.
Summary of assay outcomes used to determine the mechanism of action of the indicated compounds against EBOV infection
| Compound | Effecta |
Mechanism of action | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Surface interaction | Uninfected cell no. | Dextran uptake | Transferrin uptake | Effect on EEA1 | Effect on LAMP1 | Acidification block | Cathepsin cleavage | ||
| MLS000078751 | − | + | 0 | 0 | ND | ND | − | 0 | Surface interaction block |
| MLS000394177 | + | 0 | − | 0 | ND | ND | + | 0 | Macropinocytosis block |
| MLS000534476 | − | + | 0 | − | ND | ND | + | 0 | Surface interaction block |
| MLS000554255 | 0 | + | 0 | 0 | + | − | − | 0 | Late endocytic trafficking block |
| MLS000555232 | + | 0 | 0 | 0 | − | − | − | 0 | Early endocytic trafficking block |
| MLS000730532 | 0 | 0 | − | − | ND | ND | + | 0 | Uptake block |
| MLS000733230 | 0 | 0 | − | 0 | ND | ND | + | 0 | Macropinocytosis block |
| MLS000762907 | 0 | 0 | 0 | − | 0 | 0 | + | 0 | Trafficking block |
| MLS001101371 | 0 | 0 | 0 | 0 | 0 | − | + | 0 | Late endocytic trafficking block |
0, no effect; +, increase in assay activity; −, decrease in assay activity; ND, not determined.
DISCUSSION
EBOV and MARV have high case fatality rates averaging above 45% (2, 3). Successful palliative treatment of patients from the latest EBOV outbreak demonstrated the importance of early medical intervention. At the same time, it emphasized the lack of availability of specific antiviral therapies and prompted the more thorough testing of promising vaccine and therapy candidates. However, most of these efforts targeted only one virus species (16, 39). Like any RNA virus, filoviruses can easily mutate, and so the development of resistance to any one antiviral compound is inevitable. Therefore, a robust therapy will require a cocktail of inhibitors targeting different steps of the viral replication cycle. Screening of hundreds of thousands of compounds for activity increases the likelihood of successfully identifying disease-specific, drug-like small molecules and bringing them to the clinic. To date, this is the first report of a large-scale screen for antifiloviral compounds.
Although significant progress in performing high-throughput screening (HTS) in a BSL4 facility has been made, it is still cumbersome and the throughput is lower than that needed to evaluate a large number of compounds in a reasonable amount of time. Here, we optimized virus pseudotypes of MARV and EBOV to allow identification of inhibitors of virus entry into cells. The replication-defective pseudotyped viruses allowed testing at a lower level of biological containment and enabled the use of a state-of-the-art robotics platform to assess the efficacy of multiple concentrations of each compound during the primary screening (qHTS). This approach greatly reduces false-positive and -negative hits by examining the dose-response curves of each compound. However, since qHTS increased the number of assay points required by 6-fold, a high plating density (1,536-well format) was needed. This was a significant challenge to implementation of the screening with a cell-based assay since as few as 1,000 cells were present in each well and high virus titers were required for the small volume of the inoculum. Unfortunately, EBOV GP pseudotypes of sufficient activity could not be obtained due to the toxicity of the GP in cells used to make the pseudotype. In contrast, MARV GP was not as toxic and a sufficient virus titer was achieved.
A second attribute that aided the qHTS approach was the choice of the infection reporter, firefly luciferase. Firefly luciferase provided a high signal-to-noise ratio (>100) and a broad signal linearity (6 orders of magnitude) that could be sensitively detected. However, while the use of the pseudotype and luciferase reporter aided the primary screening, counterscreening was needed to distinguish compounds that impacted the virus GP function from those that interfered with VSV replication and luciferase activity. Indeed, of the 7,200 compounds that were identified from the primary screen, only 554 were effective in the retrovirus pseudotype-based counterscreen. All of the 7,200 compounds are reported in PubChem (identifiers 540276 and 720532) and may be useful as inhibitors of VSV or similar rhabdoviruses. Importantly, the approach of using the two pseudotypes to screen and counterscreen the library was validated when wild-type viruses were used to check compound inhibition. A significant enrichment was seen for active compounds, with 10% of the 554 compounds being active at <50 μM against either wild-type MARV or wild-type EBOV. This represents a 100-fold enrichment over that seen when screening a random library of compounds for antivirals (40, 41).
Of the most potent 61 novel compounds that inhibited MARV infection, 42 were also inhibitors of EBOV. This suggests that even though MARV and EBOV are distantly related and their GPs share less than 35% identity (2, 42), there are conserved features that can be targeted by small molecules. Conversely, the fact that the other 19 compounds affected only MARV indicates that specific differences in cell entry may be exploited in studying infection mechanisms unique for each virus. Indeed, this is supported by work showing that each virus has different dependencies on cellular proteases for activation of the GP for membrane fusion (22, 23) and is potentially the reason that none of the broadly active compounds were found to be cathepsin protease inhibitors.
One of the objectives after identifying inhibitory compounds was to deduce their mechanism of action. Quantitative high-resolution microscopy-based image analysis approaches were developed to identify the site of inhibitor activity. This was achieved using the CellProfiler platform (43, 44), a freely available software image analysis tool, as well as 3D image reconstruction of images of labeled virus interacting with labeled cells and organelles. This approach used robust assays, and the results were quantitated using automated computer-based image analysis to evaluate hundreds of cells and thousands of virus particle-cell interactions. The analysis allowed unbiased segregation of compounds into those impacting binding to cells (MLS000078751 and MLS000534476), early uptake (MLS000394177, MLS000730532, MLS000733230), early endocytic trafficking (MLS000555232), and late endosome trafficking (MLS000554255 and MLS001101371). We expect that the same approach can be applied to similar antiviral screens in the future and will enable a better understanding of the compound mechanism of action to be obtained. MLS000762907 did not block any of these steps and is likely a general inhibitor of endocytosis that targets steps other than the known markers of filovirus cell entry that we tested.
One of the common features of filovirus entry into cells is the macropinocytic uptake of the large filamentous virus particle (up to 2 μm in length). The long filamentous form of the virus means that virus particles are sterically precluded from classical endocytic uptake routes, such as clathrin- and caveola-mediated endocytosis. Instead, macropinocytosis appears to play a dominant role in productive infection (18, 19). However, in general, as an endocytic uptake route, it remains poorly defined. Few macropinocytosis-specific inhibitors with which to study the pathway are available, and only a small number of cellular proteins that distinguish it from other endocytic uptake mechanisms have been identified. Amilorides, such as EIPA, which inhibit an endosomal Na+/H+ pump, have been extremely valuable as well-accepted inhibitors of macropinocytosis (45) and were helpful in defining the EBOV uptake mechanism (19). Aside from amilorides, few other specific inhibitors of macropinocytosis are available. The finding that MLS000394177 and MLS000733230 appear to specifically block EBOV and dextran uptake but not transferrin uptake indicates that they are novel macropinocytosis inhibitors. We expect that further work to identify the cellular targets of each compound will reveal new components involved in macropinocytosis as well as novel filovirus entry inhibitors.
A second common feature of filovirus cell entry is the requirement for a low pH to promote cleavage of the GP during endocytic trafficking as well as the eventual virus-to-cell membrane fusion mediated by the cleaved GP. The known proteases that mediate cleavage reside in endosomes and are activated by low pH (46, 47). Therefore, inhibitors of vesicle proton pumps are a potential class of broad-spectrum antiviral therapies. However, attempts to use bafilomycin A1 or chloroquine have not been successful due to toxicity or low potency, respectively (48). MLS000394177, MLS000733230, MLS000730532, and MLS000534476 each affected endosomal acidification but not as dramatically as bafilomycin A did. Interestingly, three of these compounds also inhibited macropinocytosis. The change in endosomal acidification is not unusual, as many compounds that alter vesicle trafficking also affect endosome acidification and vice versa, as each is closely coupled (49, 50). Indeed, EIPA, the inhibitor of macropinocytosis, also decreases endosomal acidification to an extent similar to that seen for the compounds identified here (45).
A third class of inhibitors blocked passage of virus particles from the macropinosome to the early and late endosomes. MLS000555232 appeared to block movement to early endosomes. Two other inhibitors impacted passage from the early endosome to the late endosome (MLS000554255 and MLS001101371). In the case of MLS000554255, particles accumulated in EEA1-positive vesicles (an 83% increase over that with no treatment) and did not advance to later compartments. While each of these compounds inhibited the trafficking of EBOV, which happens through macropinocytosis, they did not inhibit transferrin uptake, suggesting that they target a specific component of endosomal trafficking specific for EBOV. Another compound, MLS000762907, did not affect any of the steps of viral entry tested here. However, it blocked the uptake of transferrin into cells, suggesting a role for a factor that is trafficked through a pathway involved in transferrin uptake and is necessary for virus to escape from the endosome.
In screening the MLSMR small-molecule library, we identified 9 novel broadly acting filovirus entry inhibitors and the steps of entry at which each acts. We are currently working on determining the specificity and molecular targets of these compounds. Using already annotated data in PubChem, we found that these compounds have been screened in more than 400 other assays targeting cellular processes or specific protein-ligand interactions (Table 3). All the compounds, except MLS000554255, were reported to be active in, on average, 1.5% of these assays. This low rate suggests that the compounds are acting specifically and may provide insight into their cellular targets. MLS000394177, the macropinocytosis inhibitor, was active in assays for inhibition of Janus kinase 2 (JNK2), Synuclein activity, and Shiga toxins A and B. Shiga toxins are known to be taken into cells by macropinocytosis (51, 52). However, a role for JNK signaling or Synuclein in macropinocytosis has not been reported, but each is associated with cytoskeletal rearrangements that could promote macropinocytosis. MLS000394177, MLS000730532, MLS000733230, and MLS000078751 were found to inhibit the secretion of IL-1β, which is known to involve exocytosis (53). Exocytosis is also important in initiating macropinocytosis by delivering to the cell surface lipids that form the macropinosome, and inhibition of exocytosis inhibits EBOV infection (54). The remaining majority of assays involved inhibition of various ion channels. As discussed above, such channels are known to play roles in macropinocytosis (45), endocytosis, and EBOV infection (14); however, none of the compounds were structurally related to previously reported channel inhibitors. The compound MLS001101371 was an exception in being identified in only two other assays, despite being tested in the most assays (>800). The two assays tested for antagonists of the DNA repair gene TDP1 and acetylcholine receptor M1. A role for a DNA repair inhibitor is unclear, but known antagonists of G protein-coupled receptors (GPCRs), including acetylcholine receptors, was recently reported to inhibit EBOV and MARV infection during endocytosis (5). MLS001101371 shares no similarity to the reported GPCR inhibitors that prevent EBOV infection and so may also represent a novel GPCR antagonist. Future work will involve a detailed structure-activity analysis to determine important features of the molecules and improve their potency and medicinal chemistry toward development of a therapeutic. The derivatives will also be used to label their protein targets. We expect the further study of each compound will provide a deeper understanding of the filovirus infection mechanism as well as the cellular processes involved in driving virus infection.
TABLE 3.
Assays in the PubChem database reporting the compounds to be active
| Compound | Total no. of reported assays | % unique assays reporting compound to be activea |
|
|---|---|---|---|
| Cellular assays | In vitro assays | ||
| MLS000555232 | 731 | 0.69 | 0.00 |
| MLS000762907 | 639 | 1.59 | 0.48 |
| MLS001101371 | 535 | 0.56 | 0.00 |
| MLS000394177 | 715 | 2.69 | 0.29 |
| MLS000733230 | 705 | 1.57 | 0.14 |
| MLS000730532 | 713 | 2.39 | 0.29 |
| MLS000078751 | 906 | 1.34 | 0.34 |
| MLS000534476 | 773 | 1.83 | 0.27 |
| MLS000554255 | 808 | 0.14 | 4.40 |
Assays were grouped on the basis of whether they were identified as hits in cell-based or in vitro-only systems.
Supplementary Material
ACKNOWLEDGMENTS
It would have not been possible to carry out this work without the constructive criticism and help of lab members Ann Reyes, Maria Villa, Olena Shtanko, and Yasuteru Sakurai, who were immensely useful in shaping the direction of work and interpretation of data.
This work was supported by NIH grants R21 5R21AI115082-02 (to R.A.D.), RO3 5R03MH086850-02 (to R.A.D.), and RO1 AI077519 (to W.M.), DOD grant HDTRA1-12-1-0002 (to R.A.D.), a Southwest Foundation Forum grant (to M.A.), the Cowles Fellowship Program at the Texas Biomedical Research Institute (to M.A.), and the Ewing Halsell Foundation (to R.A.D.).
M.A. performed screening with MLV pseudotypes, screened compounds against wild-type viruses, did viral particle distribution and colocalization experiments, performed dextran and transferrin uptake experiments, designed experiments, analyzed the data, and wrote and edited the manuscript. J.K., H.W., R.H., and R.G. performed the VSV-based primary screen, analyzed the data, and provided a description of the methods. A.R.L. performed the acridine orange experiments, analyzed the data, and wrote the corresponding text. O.S. provided human monocytes isolated from blood and helped with design of the experiment. D.J.L. designed the acridine orange experiment, wrote the corresponding text, and edited the manuscript. A.S., D.J.M., and A.J. coordinated the procurement of compounds, the design of the experiments, the collection of data, prioritization of the compounds, and communication with the corresponding author. W.M. performed the cathepsin cleavage experiments, analyzed the data, wrote the corresponding text, and edited the manuscript. R.A.D. procured funding, coordinated the experiments, designed the experiments, helped to set up experiments, and wrote and edited the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00543-16.
REFERENCES
- 1.Bukreyev AA, Chandran K, Dolnik O, Dye JM, Ebihara H, Leroy EM, Muhlberger E, Netesov SV, Patterson JL, Paweska JT, Saphire EO, Smither SJ, Takada A, Towner JS, Volchkov VE, Warren TK, Kuhn JH. 2014. Discussions and decisions of the 2012-2014 International Committee on Taxonomy of Viruses (ICTV) Filoviridae Study Group, January 2012-June 2013. Arch Virol 159:821–830. doi: 10.1007/s00705-013-1846-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartman AL, Towner JS, Nichol ST. 2010. Ebola and Marburg hemorrhagic fever. Clin Lab Med 30:161–177. doi: 10.1016/j.cll.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Rougeron V, Feldmann H, Grard G, Becker S, Leroy EM. 2015. Ebola and Marburg haemorrhagic fever. J Clin Virol 64:111–119. doi: 10.1016/j.jcv.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu A, Li B, Mills DM, Panchal RG, Cardinale SC, Butler MM, Peet NP, Majgier-Baranowska H, Williams JD, Patel I, Moir DT, Bavari S, Ray R, Farzan MR, Rong L, Bowlin TL. 2011. Identification of a small-molecule entry inhibitor for filoviruses. J Virol 85:3106–3119. doi: 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H, Lear-Rooney CM, Johansen L, Varhegyi E, Chen ZW, Olinger GG, Rong L. 2015. Inhibition of Ebola and Marburg viral entry by G protein-coupled receptor antagonists. J Virol 89:9932–9938. doi: 10.1128/JVI.01337-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garrison AR, Giomarelli BG, Lear-Rooney CM, Saucedo CJ, Yellayi S, Krumpe LR, Rose M, Paragas J, Bray M, Olinger GG Jr, McMahon JB, Huggins J, O'Keefe BR. 2014. The cyanobacterial lectin scytovirin displays potent in vitro and in vivo activity against Zaire Ebola virus. Antiviral Res 112:1–7. doi: 10.1016/j.antiviral.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heald AE, Iversen PL, Saoud JB, Sazani P, Charleston JS, Axtelle T, Wong M, Smith WB, Vutikullird A, Kaye E. 2014. Safety and pharmacokinetic profiles of phosphorodiamidate morpholino oligomers with activity against Ebola virus and Marburg virus: results of two single-ascending-dose studies. Antimicrob Agents Chemother 58:6639–6647. doi: 10.1128/AAC.03442-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kouznetsova J, Sun W, Martínez-Romero C, Tawa G, Shinn P, Chen CZ, Schimmer A, Sanderson P, McKew JC, Zheng W, García-Sastre A. 2014. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg Microbes Infect 3:e84. doi: 10.1038/emi.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, Hoffstrom BG, Dewald LE, Schornberg KL, Scully C, Lehar J, Hensley LE, White JM, Olinger GG. 2013. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med 5:190ra179. doi: 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansen LM, DeWald LE, Shoemaker CJ, Hoffstrom BG, Lear-Rooney CM, Stossel A, Nelson E, Delos SE, Simmons JA, Grenier JM, Pierce LT, Pajouhesh H, Lehar J, Hensley LE, Glass PJ, White JM, Olinger GG. 2015. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci Transl Med 7:290ra289. doi: 10.1126/scitranslmed.aaa5597. [DOI] [PubMed] [Google Scholar]
- 11.Johnson JC, Martinez O, Honko AN, Hensley LE, Olinger GG, Basler CF. 2014. Pyridinyl imidazole inhibitors of p38 MAP kinase impair viral entry and reduce cytokine induction by Zaire ebolavirus in human dendritic cells. Antiviral Res 107:102–109. doi: 10.1016/j.antiviral.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mudhasani R, Kota KP, Retterer C, Tran JP, Whitehouse CA, Bavari S. 2014. High content image-based screening of a protease inhibitor library reveals compounds broadly active against Rift Valley fever virus and other highly pathogenic RNA viruses. PLoS Negl Trop Dis 8:e3095. doi: 10.1371/journal.pntd.0003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, Johnson A, Morton J, Swope K, Bohorov O, Bohorova N, Goodman C, Kim D, Pauly MH, Velasco J, Pettitt J, Olinger GG, Whaley K, Xu B, Strong JE, Zeitlin L, Kobinger GP. 2014. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 514:47–53. doi: 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakurai Y, Kolokoltsov AA, Chen CC, Tidwell MW, Bauta WE, Klugbauer N, Grimm C, Wahl-Schott C, Biel M, Davey RA. 2015. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347:995–998. doi: 10.1126/science.1258758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salata C, Baritussio A, Munegato D, Calistri A, Ha HR, Bigler L, Fabris F, Parolin C, Palu G, Mirazimi A. 2015. Amiodarone and metabolite MDEA inhibit Ebola virus infection by interfering with the viral entry process. Pathog Dis 73:ffv032. doi: 10.1093/femspd/ftv032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thi EP, Mire CE, Lee AC, Geisbert JB, Zhou JZ, Agans KN, Snead NM, Deer DJ, Barnard TR, Fenton KA, MacLachlan I, Geisbert TW. 2015. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature 521:362–365. doi: 10.1038/nature14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warren TK, Wells J, Panchal RG, Stuthman KS, Garza NL, Van Tongeren SA, Dong L, Retterer CJ, Eaton BP, Pegoraro G, Honnold S, Bantia S, Kotian P, Chen X, Taubenheim BR, Welch LS, Minning DM, Babu YS, Sheridan WP, Bavari S. 2014. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508:402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. 2010. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog 6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. 2010. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog 6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simmons JA, D'Souza RS, Ruas M, Galione A, Casanova JE, White JM. 2016. Ebolavirus glycoprotein directs fusion through NPC1+ endolysosomes. J Virol 90:605–610. doi: 10.1128/JVI.01828-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gnirss K, Kuhl A, Karsten C, Glowacka I, Bertram S, Kaup F, Hofmann H, Pohlmann S. 2012. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology 424:3–10. doi: 10.1016/j.virol.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misasi J, Chandran K, Yang JY, Considine B, Filone CM, Cote M, Sullivan N, Fabozzi G, Hensley L, Cunningham J. 2012. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J Virol 86:3284–3292. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. 2006. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol 80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cote M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bleicher KH, Bohm HJ, Muller K, Alanine AI. 2003. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discov 2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 28.Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DV, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam GS. 2011. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov 10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 29.Pereira DA, Williams JA. 2007. Origin and evolution of high throughput screening. Br J Pharmacol 152:53–61. doi: 10.1038/sj.bjp.0707373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuss IJ, Kanof ME, Smith PD, Zola H. 2009. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr Protoc Immunol Chapter 7:Unit 7.1. doi: 10.1002/0471142735.im0701s85. [DOI] [PubMed] [Google Scholar]
- 31.Nair S, Archer GE, Tedder TF. 2012. Isolation and generation of human dendritic cells. Curr Protoc Immunol Chapter 7:Unit 7.32. doi: 10.1002/0471142735.im0732s99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhn JH, Bao Y, Bavari S, Becker S, Bradfute S, Brauburger K, Rodney Brister J, Bukreyev AA, Cai Y, Chandran K, Davey RA, Dolnik O, Dye JM, Enterlein S, Gonzalez JP, Formenty P, Freiberg AN, Hensley LE, Hoenen T, Honko AN, Ignatyev GM, Jahrling PB, Johnson KM, Klenk HD, Kobinger G, Lackemeyer MG, Leroy EM, Lever MS, Muhlberger E, Netesov SV, Olinger GG, Palacios G, Patterson JL, Paweska JT, Pitt L, Radoshitzky SR, Ryabchikova EI, Saphire EO, Shestopalov AM, Smither SJ, Sullivan NJ, Swanepoel R, Takada A, Towner JS, van der Groen G, Volchkov VE, Volchkova VA, Wahl-Jensen V, Warren TK, Warfield KL, Weidmann M, Nichol ST. 2014. Virus nomenclature below the species level: a standardized nomenclature for filovirus strains and variants rescued from cDNA. Arch Virol 159:1229–1237. doi: 10.1007/s00705-013-1877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebihara H, Theriault S, Neumann G, Alimonti JB, Geisbert JB, Hensley LE, Groseth A, Jones SM, Geisbert TW, Kawaoka Y, Feldmann H. 2007. In vitro and in vivo characterization of recombinant Ebola viruses expressing enhanced green fluorescent protein. J Infect Dis 196(Suppl 2):S313–S322. doi: 10.1086/520590. [DOI] [PubMed] [Google Scholar]
- 34.Lyles DS, Puddington L, McCreedy BJ Jr. 1988. Vesicular stomatitis virus M protein in the nuclei of infected cells. J Virol 62:4387–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basu A, Mills DM, Bowlin TL. 2010. High-throughput screening of viral entry inhibitors using pseudotyped virus. Curr Protoc Pharmacol Chapter 13:Unit 13B.3. doi: 10.1002/0471141755.ph13b03s51. [DOI] [PubMed] [Google Scholar]
- 36.Bajusz D, Racz A, Heberger K. 2015. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Cheminform 7:20. doi: 10.1186/s13321-015-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aleksandrowicz P, Marzi A, Biedenkopf N, Beimforde N, Becker S, Hoenen T, Feldmann H, Schnittler HJ. 2011. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J Infect Dis 204(Suppl 3):S957–S967. doi: 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A 94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marzi A, Robertson SJ, Haddock E, Feldmann F, Hanley PW, Scott DP, Strong JE, Kobinger G, Best SM, Feldmann H. 2015. Ebola vaccine. VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science 349:739–742. doi: 10.1126/science.aab3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung DH, Jonsson CB, Tower NA, Chu YK, Sahin E, Golden JE, Noah JW, Schroeder CE, Sotsky JB, Sosa MI, Cramer DE, McKellip SN, Rasmussen L, White EL, Schmaljohn CS, Julander JG, Smith JM, Filone CM, Connor JH, Sakurai Y, Davey RA. 2014. Discovery of a novel compound with anti-Venezuelan equine encephalitis virus activity that targets the nonstructural protein 2. PLoS Pathog 10:e1004213. doi: 10.1371/journal.ppat.1004213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malecka KA, Fera D, Schultz DC, Hodawadekar S, Reichman M, Donover PS, Murphy ME, Marmorstein R. 2014. Identification and characterization of small molecule human papillomavirus E6 inhibitors. ACS Chem Biol 9:1603–1612. doi: 10.1021/cb500229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrette RW, Xu L, Rowland JM, McIntosh MT. 2011. Current perspectives on the phylogeny of Filoviridae. Infect Genet Evol 11:1514–1519. doi: 10.1016/j.meegid.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. 2011. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. 2010. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol 188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dube D, Brecher MB, Delos SE, Rose SC, Park EW, Schornberg KL, Kuhn JH, White JM. 2009. The primed ebolavirus glycoprotein (19-kilodalton GP1,2): sequence and residues critical for host cell binding. J Virol 83:2883–2891. doi: 10.1128/JVI.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunt CL, Lennemann NJ, Maury W. 2012. Filovirus entry: a novelty in the viral fusion world. Viruses 4:258–275. doi: 10.3390/v4020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller EH, Harrison JS, Radoshitzky SR, Higgins CD, Chi X, Dong L, Kuhn JH, Bavari S, Lai JR, Chandran K. 2011. Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J Biol Chem 286:15854–15861. doi: 10.1074/jbc.M110.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu YB, Dammer EB, Ren RJ, Wang G. 2015. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener 4:18. doi: 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scott CC, Gruenberg J. 2011. Ion flux and the function of endosomes and lysosomes: pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays 33:103–110. doi: 10.1002/bies.201000108. [DOI] [PubMed] [Google Scholar]
- 51.In J, Lukyanenko V, Foulke-Abel J, Hubbard AL, Delannoy M, Hansen AM, Kaper JB, Boisen N, Nataro JP, Zhu C, Boedeker EC, Giron JA, Kovbasnjuk O. 2013. Serine protease EspP from enterohemorrhagic Escherichia coli is sufficient to induce Shiga toxin macropinocytosis in intestinal epithelium. PLoS One 8:e69196. doi: 10.1371/journal.pone.0069196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malyukova I, Murray KF, Zhu C, Boedeker E, Kane A, Patterson K, Peterson JR, Donowitz M, Kovbasnjuk O. 2009. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am J Physiol Gastrointest Liver Physiol 296:G78–G92. doi: 10.1152/ajpgi.90347.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lopez-Castejon G, Brough D. 2011. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev 22:189–195. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. 2012. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol 86:7473–7483. doi: 10.1128/JVI.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.