

Abstract
The current treatment of Chagas disease (CD), based on nifurtimox and benznidazole (Bz), is unsatisfactory. In this context, we performed the phenotypic in vitro screening of novel mono- and diamidines and drug interaction assays with selected compounds. Ten novel amidines were tested for their activities against bloodstream trypomastigote (BT) and amastigote forms of Trypanosoma cruzi (Y and Tulahuen strains) and their toxicities for mammalian host cells (L929 cells and cardiac cells). Seven of 10 molecules were more active than Bz against BT, with the most active compound being the diamidine DB2267 (50% effective concentration [EC50] = 0.23 μM; selectivity index = 417), which was 28-fold more active and about 3 times more selective than the standard drug. Five of the six monoamidines were also more active than Bz. The combination of DB2267 and DB2236 in fixed-ratio proportions showed an additive effect (sum of fractional inhibitory concentrations < 4) on BT. Interestingly, when intracellular forms were exposed to DB2267, its activity was dependent on the parasite strain, being effective (EC50 = 0.87 ± 0.05 μM) against a discrete typing unit (DTU) II strain (strain Y) but not against a representative DTU VI strain (strain Tulahuen) even when different vehicles (β-cyclodextrin and dimethyl sulfoxide) were used. The intrinsic fluorescence of several diamidines allowed their uptake to be studied. Testing of the uptake of DB2236 (inactive) and DB2267 (active) by amastigotes of the Y strain showed that the two compounds were localized intracellularly in different compartments: DB2236 in the cytoplasm and DB2267 in the nucleus. Our present data encourage further studies regarding the activities of amidines and provide information which will help with the identification of novel agents for the treatment of CD.
INTRODUCTION
Chagas disease (CD) is a neglected disorder endemic in 21 countries of Latin America. According to the World Health Organization, 8 million people are infected worldwide and approximately 10 million others are at risk of infection, with 14,000 new cases and 12,000 deaths occurring each year (1). This pathology was discovered by the Brazilian researcher Carlos Chagas (2) and has as the etiological agent the flagellate protozoan Trypanosoma cruzi. There are several transmission routes, including vector transmission by triatomine bugs from the Reduviidae family, blood transfusion or organ transplantation, congenital transmission, and oral contamination (3, 4). The disease is characterized by two distinct phases, the acute and chronic phases. During the acute phase, patients can be asymptomatic or mildly symptomatic, which likely impairs an early and differential diagnosis. After parasitemia is controlled, a chronic phase is established and is represented by subpatent parasitism and positive serology. Approximately 60 to 70% of the patients in this phase present with an indeterminate form, while 30 to 40% develop a cardiac and/or digestive form of the disease years or even decades after the infection (5).
CD therapy is based on two medicines: nifurtimox and benznidazole (Bz). Both require long-term treatment and produce severe adverse effects, while they yield only low cure rates in the late chronic phase (6–8). New studies are essential for the identification of more selective and efficient chemotherapeutic agents with reduced toxic effects, which often result in the avoidance or abandonment of therapy by patients. In this context, it is essential that hit compounds show potent activity and be efficacious against strains representative of the different T. cruzi discrete typing units (DTUs) relevant for human infection to provide new drugs usable in the different areas where CD is endemic (9).
Drug discovery programs are increasingly employing combination strategies, in large part due to the seemingly constant problem of drug resistance but also due to the potential for the discovery of synergism. Use of this approach has recently been successful for the discovery of drugs with activity against several parasitic diseases, including leishmaniasis, malaria, and CD (10–12). It seems important to routinely include this strategy in efforts to discover drugs for the treatment of CD.
Aromatic diamidines, such as pentamidine, are in clinical use for different pathologies, such as leishmaniasis, systemic fungal infection, and stage 1 human African trypanosomiasis, as well as for several veterinary purposes (13). Recently, a new class of diamidines, arylimidamides, has been found to be effective against several pathogens, including T. cruzi. The excellent trypanocidal activity of this new class of amidines in vitro and in vivo encourages further exploration of the amidine family of compounds (14–16).
Thus, the goal of this work is the screening of new heterocyclic amidines with the aim of identifying new compounds with efficacy and selectivity that are equal or superior to those of Bz and that could be considered promising candidates for the treatment of CD.
MATERIALS AND METHODS
Compounds.
The diamidines (DB2236, DB2259, DB2260, DB2267) were synthesized using a methodology previously reported (17–19), and the synthesis of the monoamidines (DB2261, DB2262, DB2263, DB2266, DB2268, DB2269) has been described previously (20) (Fig. 1). All compounds have been fully characterized by spectral methods (nuclear magnetic resonance, mass spectrometry) and by satisfactory C, H, and N analysis. Stock solutions were prepared in dimethyl sulfoxide (DMSO), whose final concentration never exceeded 0.6% and which did not exert any toxicity toward the parasite or mammalian host cells (data not shown) (21), or β-cyclodextrin in a 1:1 proportion with the compound to be tested and vortexed in dry ice for 10 min. Benznidazole (Bz; N-benzyl-2-nitroimidazole-1-acetamide; Laboratório Farmacêutico do Estado de Pernambuco [LAFEPE], Brazil) was used as the reference drug.
FIG 1.

Structures of the diamidines and monoamidines evaluated in this study.
Parasites.
Bloodstream trypomastigotes (BT) of the T. cruzi Y strain were obtained from Swiss Webster mice at the time of infection with peak parasitemia (14, 22). Trypomastigotes of the T. cruzi Tulahuen strain expressing the β-galactosidase gene from Escherichia coli (23) were collected from the supernatant of infected cell cultures (L929 cell cultures) as reported previously (24).
Mammalian cells.
For the toxicity assays, primary cultures of mammalian cardiac cells (CC) obtained from mouse embryos were plated onto coverslips in 96- or 24-well plates that had previously been coated with 0.01% gelatin (22). For analysis of the effect of the drugs on intracellular parasites, monolayers of mouse L929 fibroblasts were cultivated (4 × 103 cells/well in 96-well microplates) at 37°C in RPMI 1640 medium (pH 7.2 to 7.4) without phenol red (Gibco BRL) supplemented with 10% fetal bovine serum and 2 mM glutamine (RPMI), as reported previously (24).
Trypanocidal activity.
BT of the Y strain (5 × 106 per ml) were incubated for 2 and 24 h at 37°C in RPMI in the presence or absence of serial dilutions of the compounds (0 to 32 μM). After incubation with the compounds, the parasite death rates were determined by light microscopy through direct quantification of the number of live parasites using a Neubauer chamber, and the compound concentration that reduced the number of parasites by 50% (i.e., the 50% effective concentration [EC50]) was then calculated (21). For the assays with the intracellular forms, T. cruzi-infected cell cultures (the Tulahuen strain expressing β-galactosidase) were incubated for 96 h at 37°C with each compound at 10 μM diluted in RPMI. After this period, 500 μM chlorophenol red glycoside in 0.5% Nonidet P-40 was added to each well; the plate was incubated for 18 h at 37°C, and then the absorbance at 570 nm was measured. Controls with uninfected cells treated with the vehicle only and with the reference drug (Bz) were run in parallel. Those compounds with activity similar to that of Bz were further assayed using decreasing concentrations to determine the EC50s, as described above. The results were then expressed as the percentage of T. cruzi growth inhibition by comparing the optical density data obtained for cell cultures treated with the tested compound with those obtained for infected cell cultures treated only with the drug vehicle (24). Next, the most promising compounds (compounds with activity greater than or equal to that of Bz) were then evaluated for 48 h by infection of CC with the Y strain by a standard method described previously (14). After 24 h of infection, the cell cultures were treated with nontoxic concentrations of the compounds, and after 48 h of treatment, the CC were rinsed with saline, fixed with Bouin's fixative (5 min), stained with Giemsa, and examined by light microscopy. The percentage of infected host cells was determined, as was the number of parasites per cell, and the infection index (II), which represents the percentage of infected host cells multiplied by the number of parasites per cell, was calculated. Then, the EC50s were determined as reported previously (14).
Cytotoxicity assays.
CC were incubated for 24 and 48 h at 37°C with different concentrations of each compound serially diluted (1:3) in RPMI, with a concentration range of 0 to 96 µM, and then their morphology and spontaneous contractibility were evaluated by light microscopy and their viability was determined by the PrestoBlue test as reported previously (21). The results were expressed as the percent difference in the reduction of infection between compound-treated and vehicle-treated cells according to the manufacturer's instructions (Thermo Scientific) and the 50% inhibitory concentration (IC50), which corresponds to the concentration that reduces cell viability by 50%. The selectivity index (SI) is expressed as the ratio of the IC50 for host cells and the EC50 for the parasites or infected cell cultures.
Cell internalization of diamidines.
CC were infected with Y strain BT and incubated for 48 h at 37°C. Then, the infected cultures were incubated with each compound (10 μg/ml) for 2 h at 37°C, fixed with paraformaldehyde at 4%, and analyzed by fluorescence microscopy (14).
Determination of drug interactions against BT of T. cruzi.
In vitro drug interactions were assessed using a fixed-ratio method (25) by combining two previously screened diamidines, DB2267 and DB2236, which exhibited the best activity against two different T. cruzi DTUs: the BT (Y strain) and intracellular forms (Tulahuen strain), respectively. In these assays, predetermined EC50s were used to determine the top concentrations of the individual drugs to ensure that the EC50 fell near the midpoint of a six-point 2-fold dilution series. The top concentrations used were 0.96 μM for DB2267 and 16 μM for DB2236 in a 24-h assay. The top concentrations were used to prepare solutions of DB2267 and DB2236 at fixed ratios of 5:0, 4:1, 3:2, 2:3, 1:4, and 0:5, as reported previously (11).
Determination of FIC index, isobologram construction, and classification of the nature of the interaction.
Fractional inhibitory concentrations (FICs) and the sum of the FICs (ΣFICs) were calculated as follows: FIC of DB2267 = EC50 of DB2267 in combination/EC50 of DB2267 alone. The same equation was applied to the partner drug (DB2236). ΣFICs were calculated as the FIC of DB2267 plus the FIC of DB2236. An overall mean ΣFIC was calculated for each combination and used to classify the nature of the interaction. Isobolograms were constructed by plotting the EC50 of DB2267 against the EC50 of DB2236.
Statistical analysis was performed individually for each assay using an analysis of variance program, and the level of significance was set at a P value of ≤0.05. All the assays were conducted at least in duplicate, and results are averages from at least three independent experiments.
RESULTS
The compounds DB2236, DB2259, DB2260, and DB2267 were selected to explore the potency of diamidines against T. cruzi. A small set of monoamidines (DB2261, DB2262, DB2266, DB2268, and DB2269) with tethered potential intercalation units was studied to test the activities of such hybrid molecules against T. cruzi. DB2263 represents a monoamidine control compound without a tethered unit. The biological analysis was started with a time-kill assay of activity against BT of the Y strain, in order to assess the potency of the compounds over a determined period of time. After 2 and 24 h of incubation, most of the tested compounds (7 of 10) showed higher levels of activity than Bz (Table 1). After the shorter period of incubation, DB2266 and DB2269 displayed EC50s of ≤4.2 μM, being at least 24-fold more active than the reference drug. After 24 h, three of the monoamidines (DB2262, DB2266, and DB2269) showed EC50s below 3.0 μM; however, the most effective compound was the diamidine DB2267, which gave an EC50 of 0.23 μM, which made it 28-fold more active than Bz.
TABLE 1.
In vitro trypanocidal activity (EC50 and EC90) and mammalian host cell toxicity (IC50) of the amidines and Bz on BT of the Y strain and CC cultures, respectively, and the corresponding SI
| Compound | Mean ± SD EC50 (μM) |
Mean ± SD EC90 (μM) |
Mean ± SD IC50 (μM) at 24 h | SIa | ||
|---|---|---|---|---|---|---|
| 2 h | 24 h | 2 h | 24 h | |||
| Bz | >100 | 6.65 ± 1.8 | >100 | 18.04 ± 7.6 | 1,000 | >149 |
| DB2236 | >32 | 3.97 ± 1.8 | >32 | 10.64 ± 2.6 | >96 | >24 |
| DB2259 | >32 | 20.00 ± 1.0 | >32 | 32 ± 0 | >96 | >4.8 |
| DB2260 | 24.35 ± 6.8 | 7.26 ± 1.2 | >32 | 10.4 ± 0.7 | >96 | >13 |
| DB2261 | 11.75 ± 4.4 | 3.57 ± 1.2 | >32 | 7.37 ± 7.7 | >96 | >27 |
| DB2262 | 5.57 ± 2.1 | 2.76 ± 0.8 | 14.93 ± 9.5 | 6.80 ± 2.5 | >96 | >34 |
| DB2263 | >32 | 12.34 ± 3.6 | >32 | 28.75 ± 0.2 | 64 | >5 |
| DB2266 | 3.88 ± 1.1 | 2.46 ± 0.9 | 9.42 ± 0.5 | 3.33 ± 0.8 | >96 | >38 |
| DB2267 | 5.37 ± 1.6 | 0.23 ± 0.02 | >32 | 1.00 ± 0.06 | >96 | >417 |
| DB2268 | 5.96 ± 1.9 | 3.07 ± 1.0 | 16.72 ± 5.8 | 7.78 ± 1.9 | >96 | >31 |
| DB2269 | 4.22 ± 1.9 | 2.14 ± 0.1 | 8.62 ± 1.8 | 5.78 ± 2.4 | >96 | >46 |
SIs are based on the EC50.
Next, we evaluated, using light microscopy and a colorimetric assay with PrestoBlue, the in vitro toxicities of these compounds for mammalian host cells (noninfected primary cultures of cardiac cells) incubated for 24 and 48 h with the different molecules (Fig. 2). After 24 h of incubation, none of the amidines except DB2236 reached an IC50 up to 96 μM, demonstrating their low toxicity profile. DB2236, DB2260, and DB2263 caused only mild toxicity when used at the higher concentration after 48 h of compound exposure. Selectivity determination demonstrated once again that DB2267 was the most promising compound, with an SI value of approximately 417; hence, it was almost 2.8-fold more selective than Bz (Table 1).
FIG 2.

Light microscopy images of untreated (A) and DB2267-treated (B) cardiac cells (1.18 μM for 48 h at 37°C) infected with T. cruzi (Y strain) showing a strong decline (60%) in the number of parasites (arrows) present in the treated cell cultures compared to the number present in the untreated samples.
The third part of the in vitro assays involved evaluation of these compounds using intracellular forms of the parasite. In this case, L929 cell cultures infected with the Tulahuen strain of T. cruzi were treated. In the first step, a fixed concentration of 10 μM, which is equivalent to the EC90 of Bz (21), was used. In this assay, only DB2236 consistently showed activity comparable to that of the reference drug and was then selected to determine its EC50 against these intracellular parasites. Also, as DB2267 was the most active compound against BT forms, it was subjected to this assay (Table 2). The results demonstrated that DB2236 presented trypanocidal activity in the same range as that of Bz, with the EC50 value being 5.4 ± 1.7 μM after 96 h of incubation (Table 2). On the other hand, even though it was the most active compound against the Y strain BT, DB2267 was not effective against the intracellular form of the Tulahuen strain (Table 2). The lack of trypanocidal activity of DB2267 on the intracellular form of this parasite strain persisted even when another vehicle (β-cyclodextrin) was employed (data not shown).
TABLE 2.
Trypanocidal activity of DB2236, DB2267, and Bz against amastigotes and CCa
| Compound | Mean ± SD EC50 (μM) |
IC50 (μM) for CC (48 h) | |
|---|---|---|---|
| Tulahuen (96 h) | Y (48 h) | ||
| Bz | 2.8 ± 1.3 | 3.6 | 1,000 |
| DB2236 | 5.4 ± 1.7 | ND | 45 |
| DB2267 | >32 | 0.87 ± 0.05 | >96 |
L929 cell cultures and CC were infected with the Tulahuen and Y strains of Trypanosoma cruzi, respectively. ND, not determined.
It is unclear if the lack of activity of DB2267 against the intracellular form of the Tulahuen strain is due to differences in the strain (Y versus Tulahuen) or the form (BT versus intracellular form) used. To clarify this matter, we concurrently evaluated the activities of DB2267 and Bz against Y strain amastigotes (Fig. 2 and Table 2). The EC50 for DB2267 against the intracellular form of the Y strain was found to be 0.87 μM, which suggests that the differences in activity noted above are due to differences in the strains and not the form tested.
The intrinsic fluorescent properties of the diamidines allowed cell uptake studies to be undertaken, and we selected two representative diamidines with different molecular sizes, one with a small molecular size (DB2236) and one with a large molecular size (DB2267), which were investigated using Y strain amastigotes. Both compounds were localized intracellularly but in different compartments: DB2267 was found in the DNA-rich nuclear sites (Fig. 3A and B), and DB2236 (Fig. 3C and D) was dispersed in the cytoplasm.
FIG 3.

Fluorescence microscopy images (A and C) and corresponding differential interference contrast images (B and D) of cardiac cells infected with T. cruzi (Y strain) after 2 h of exposure to 10 μg/ml of DB2267 (A and B) and DB2236 (C and D), showing the internalization of both compounds with their intracellular localization in the nuclei (A) and cytoplasm (C) of the mammalian host cells and the parasites (arrows).
Finally, since combination therapeutic strategies are often used in an attempt to reduce toxicity and improve activity, the in vitro interactions of DB2267 and DB2236 at their respective EC50 levels were evaluated. Mean ΣFICs are presented in Table 3, and representative isobolograms are presented in Fig. 4. Interactions were categorized as described by Seifert and Croft (26), in which synergism was classified as a mean ΣFIC of ≤0.5, antagonism was classified as a mean ΣFIC of >4.0, and indifference (or an additive effect) was classified as a mean ΣFIC of between >0.5 and ≤4.0. The interaction of DB2267 and DB2236 was classified as indifferent, as the mean ΣFICs were 1.13 to 1.66 at the EC50 level (Table 3 and Fig. 4).
TABLE 3.
Mean ΣFICs of interaction between DB2267 and DB2236 toward BT of Trypanosoma cruzi Y strain
| Compound ratio | FIC |
ΣFIC | |
|---|---|---|---|
| DB2267 | DB2236 | ||
| 4:1 | 1.05 | 0.08 | 1.13 |
| 3:2 | 1.05 | 0.22 | 1.27 |
| 2:3 | 1.13 | 0.53 | 1.66 |
| 1:4 | 0.64 | 0.80 | 1.44 |
| Mean FIC for combination | 0.97 | 0.41 | 1.37 |
FIG 4.
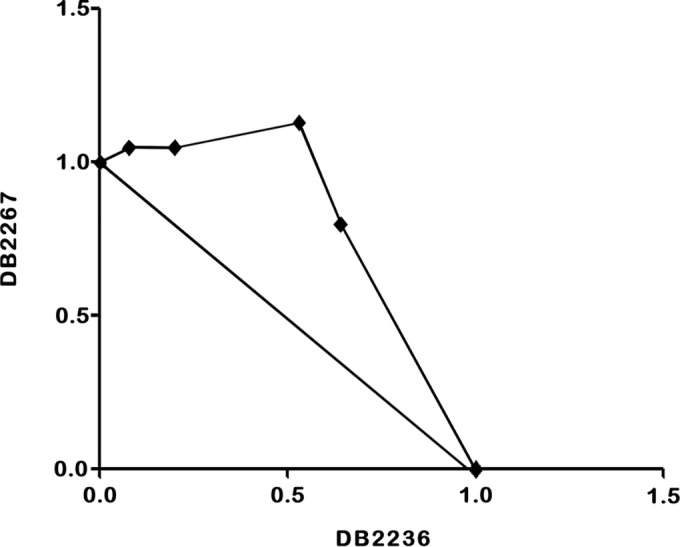
Representative isobologram. In vitro interaction of DB2267 and DB2236 combined against bloodstream trypomastigotes of T. cruzi (Y strain). The EC50 of DB2236 is plotted on the abscissa, and the EC50 of DB2267 is plotted on the ordinate. The plotted points are the EC50 of each fixed ratio of 5:0, 4:1, 3:2, 1:4, and 0:5 of DB2267 and DB2236. ΣFICs of <0.5 indicate synergism. ΣFICs of >0.5 and ≤4.0 indicate additivity, and ΣFICs of >4.0 indicate antagonism.
DISCUSSION
Previous in vitro and in vivo studies demonstrated the excellent activities of many diamidines against T. cruzi: these molecules exhibited dose- and time-dependent trypanocidal effects in the nanomolar range, presented low toxicity for mammalian cells, were able to reduce the cardiac parasitic load in vivo, and resulted in lower levels of tissue damage and higher survival rates for treated animals than for untreated animals (13, 14). Among the diamidines studied, arylimidamides have been shown to be highly effective against T. cruzi and several different species of Leishmania, as well as other intracellular pathogens (13, 16). In this study, 10 amidino molecules were tested in vitro following a well-established work flow and methodology, resulting in the determination of the EC50, IC50, and selectivity index (SI) (24). Among the 10 molecules tested, 7 molecules presented an EC50 lower than 4 μM after 24 h of incubation. In fact, 6 out of 10 amidines presented an EC50 of <6 μM after only 2 h of incubation with BT, being much more effective than the reference drug and corroborating previous data showing the fast action of diamidines, which is a very important characteristic in situations of acute cases of reactivation under conditions of immunosuppression (21). The time-kill assay was performed according to a standardized protocol, in which compound activity was evaluated at two time points: after 2 h of treatment, in order to test the ability of compounds to show rapid action against the parasite, and at 24 h of incubation as the endpoint, since more prolonged incubation times affect BT integrity (8, 13). In addition, evaluation at the early time point (2 h) allows a head-to-head comparison of the test compounds with other fast-acting amidines already assayed by our group and others using protocols similar to those described here (16).
The monoamidines (DB2261, DB2262, DB2266, DB2268, and DB2269) with tethered potential intercalation units gave EC50s of between 2.14 and 3.37 μM against Y strain BT. As this is a new class of potential anti-T. cruzi agents, this constitutes an important finding. The set is too limited to merit discussion of structure-activity relationships, nor does the moderate in vitro activity merit in vivo studies; nevertheless, hit-to-lead investigations of this new class of compounds are merited. DB2263, a control monoamidine of this class but without the tethered unit, exhibited an EC50 of only 12.34 μM, which strongly suggests an important role for the tethered unit in the anti-T. cruzi activity of this series.
Recent findings for aromatic diamidines, especially for the arylimidamide class, showed that many of them are highly active against different strains of the parasite, including those naturally resistant to Bz, and also against both parasite forms relevant to human infection: BT and amastigotes (14). DB2236 was moderately active against both the Y strain (BT) and the Tulahuen strain (intracellular form), showing EC50s of 3.97 and 5.4 μM, respectively. However, the most active compound against Y strain BT, DB2267, was inactive against intracellular forms of the Tulahuen strain (even when different types of drug vehicles, such as DMSO and β-cyclodextrin, were used) but retained efficacy against amastigotes of the Y strain. Hence, our data show that the activity of DB2267 is strain dependent, which is not a desirable feature for novel drug candidates for the treatment of CD (12). It can be concluded that structural modifications of the diamidines may impact their mechanisms of action and/or cellular targets and that compound metabolization and/or uptake/extrusion with different parasite strains deserves to be further investigated.
Fluorescence microscopy studies conducted with molecules such as furamidine took advantage of the intrinsic fluorescent properties of diamidines to identify intracellular targets (such as kinetoplast DNA [kDNA]) of trypanosomatids. For T. cruzi, previous studies identified diamidines located in different intracellular compartments, including the nucleus, kinetoplast, and acidocalcisomes, suggesting the site(s) of cellular insult (primary and secondary) and/or the site of compound storage (27–31). Therefore, we performed fluorescent microscopy analysis to ascertain the uptake of two representative molecules with different sizes: a small diamidine (DB2236) and a larger one (DB2267). Both were internalized by the parasite; DB2236 was found to be diffuse in the host cell and parasite cytoplasm, whereas the larger diamidine was localized in DNA-rich compartments, such as the nucleus and kDNA, as previously reported for other diamidines (13). The different localizations may reflect distinct cellular targets that merit further studies. Several classical aromatic amidines selectively target AT-rich DNA to form a complex in the minor groove of the double helix. Both the mitochondrial and nuclear DNAs are well established as dication targets, and strong evidence indicates that the functional destruction of DNA is related to their antiparasitic action (7, 8). In trypanosomes, leishmania, and related organisms, the mitochondrial kDNA network typically contains DNA that, like the genomic DNA of Plasmodium, exhibits a high AT content (>75%), extensive closely spaced sequences that act as strong binding sites, and selective therapeutic target sites for these dication compounds, conferring higher selectivity than host cell DNA (13).
The strategy of targeting different molecular sites has recently been evaluated by several groups (32–34) in order to try to avoid strain resistance and enhance biological activity during drug therapy for several neglected diseases, including CD. In this work, when the two diamidines DB2236 and DB2267 were combined in a serially diluted fashion (25) and evaluated against the Y strain BT form, the combination showed no difference in activity compared to that of monotherapy.
These results corroborate the general anti-T. cruzi activity of aromatic diamidines; however, the discovery of strain dependency for DB2267 underscores the importance of complete screening of strains and forms. The discovery of a new class of amidines (monoamidines with tethered units) with in vitro activity worthy of future hit-to-lead studies is a significant finding. Despite the lack of improvement of activity by use of a combination of compounds with the combination studied, it remains prudent to include such studies with future analogues. Thus, taken as a whole, our data encourage further analysis of novel amidino analogues to find ones which display safer and more potent antiparasitic phenotypic effects that may be helpful for the future identification of novel promising agents for the therapy of Chagas disease.
ACKNOWLEDGMENTS
The present study was financially supported by the Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Oswaldo Cruz, PAEF/CNPq/Fiocruz, PDTIS, and CAPES. This work was also supported, in part, by the U.S. National Institutes of Health (grant no. RO1AI64200 to D.W.B.). M.N.C.S. is a research fellow of CNPq and CNE research.
Funding Statement
The present study was financially supported by Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Oswaldo Cruz, PROEP/CNPq/Fiocruz, PDTIS, and CAPES. This work was also supported, in part, by U.S. National Institutes of Health grant no. R01AI64200 (D.W.B.). M.N.C.S. is a research fellow of CNPq and CNE Research.
REFERENCES
- 1.World Health Organization. 2015. Chagas disease. World Health Organization, Geneva, Switzerland: http://www.who.int/topics/chagas_disease/en/. [Google Scholar]
- 2.Chagas CJ. 1909. Nova tripanosomíase humana: estudos sobre a morfologia e o ciclo evolutivo do Schizotrypanum cruzi, n. gen., n. sp., agente etiológico de nova entidade mórbida do homem. Mem Inst Oswaldo Cruz 1:159–218. [Google Scholar]
- 3.Coura JR, Viñas PA. 2010. Chagas disease: a new worldwide challenge. Nature 465:S6–S7. doi: 10.1038/nature09221. [DOI] [PubMed] [Google Scholar]
- 4.Bern C. 2011. Antitrypanosomal therapy for chronic Chagas' disease. N Engl J Med 364:2527–2534. doi: 10.1056/NEJMct1014204. [DOI] [PubMed] [Google Scholar]
- 5.Coura JR, Dias JCP. 2009. Epidemiology, control and surveillance of Chagas disease—100 years after its discovery. Mem Inst Oswaldo Cruz 104:31–40. [DOI] [PubMed] [Google Scholar]
- 6.Rodrigues Coura J, De Castro SL. 2002. A critical review on Chagas disease chemotherapy. Mem Inst Oswaldo Cruz 97:3–24. doi: 10.1590/S0074-02762002000100001. [DOI] [PubMed] [Google Scholar]
- 7.Soeiro MNC, De Castro SL. 2009. Trypanosoma cruzi targets for new chemotherapeutic approaches. Expert Opin Ther Targets 13:105–121. doi: 10.1517/14728220802623881. [DOI] [PubMed] [Google Scholar]
- 8.Soeiro MNC, De Castro SL. 2011. Screening of potential anti-Trypanosoma cruzi candidates: in vitro and in vivo studies. Open Med Chem J 5:21–30. doi: 10.2174/1874104501105010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zingales B, Miles MA, Moraes CB, Luquetti A, Guhl F, Schijman AG, Ribeiro I. 2014. Drug discovery for Chagas disease should consider Trypanosoma cruzi strain diversity. Drugs for Neglected Disease Initiative; Chagas Clinical Research Platform Meeting. Mem Inst Oswaldo Cruz 109:828–833. doi: 10.1590/0074-0276140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coura JR. 2013. Chagas disease: control, elimination and eradication. Is it possible? Mem Inst Oswaldo Cruz 108:962–967. doi: 10.1590/0074-0276130565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diniz LDF, Urbina JA, de Andrade IM, Mazzeti AL, Martins TA, Caldas IS, Talvani A, Ribeiro I, Bahia MT. 2013. Benznidazole and posaconazole in experimental Chagas disease: positive interaction in concomitant and sequential treatments. PLoS Negl Trop Dis 7:e2367. doi: 10.1371/journal.pntd.0002367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chatelain E. 2015. Chagas disease drug discovery: toward a new era. J Biomol Screen 20:22–35. doi: 10.1177/1087057114550585. [DOI] [PubMed] [Google Scholar]
- 13.Soeiro MNC, Werbovetz K, Boykin DW, Wilson WD, Wang MZ, Hemphill A. 2013. Novel amidines and analogues as promising agents against intracellular parasites: a systematic review. Parasitology 140:929–951. doi: 10.1017/S0031182013000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batista DG, Batista MM, Oliveira GM, Amaral PB, Lannes-Vieira J, Britto CM, Junqueira A, Lima MM, Romanha AJ, Sales PA Jr, Stephens CE, Boykin DW, Soeiro MNC. 2010. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas' disease treatment. Antimicrob Agents Chemother 54:2940–2952. doi: 10.1128/AAC.01617-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Da Silva CF, Batista DGJ, Oliveira GM, De Souza EM, Hammer ER. 2012. In vitro and in vivo investigation of the efficacy of arylimidamide DB1831 and its mesylated salt form—DB1965—against Trypanosoma cruzi infection. PLoS One 7:e30356. doi: 10.1371/journal.pone.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Araújo JS, Da Silva CF, Batista DGJ, Da Silva PB, Meuser MB, Aiub CAF, Da Silva MFV, Araújo-Lima CF, Banerjee M, Farahat AA, Stephens CE, Kumar A, Boykin DW, Soeiro MNC. 2014. In vitro and in vivo studies of the biological activity of novel arylimidamides against Trypanosoma cruzi. Antimicrob Agents Chemother 58:4191–4195. doi: 10.1128/AAC.01403-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ismail MA, Brun R, Wenzler T, Miao Y, Wilson WD, Boykin DW. 2004. Dicationic biphenyl benzimidazole derivatives as antiprotozoal agents. Bioorg Med Chem 12:5405–5413. doi: 10.1016/j.bmc.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 18.Farahat AA, Paliakov E, Kumar A, Barghash A-EM, Goda FE, Eisa HM, Wenzler T, Brun R, Liu Y, Wilson WD, Boykin DW. 2011. Exploration of larger central ring linkers in furamidine analogues: synthesis and evaluation of their DNA binding, antiparasitic and florescence properties. Bioorg Med Chem 19:2156–2167. doi: 10.1016/j.bmc.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 19.Hu L, Patel A, Bondada L, Yang S, Wang MZ, Munde M, Wilson WD, Wenzler T, Brun R, Boykin DW. 2013. Synthesis and antiprotozoal activity of dicationic 2, 6-diphenylpyrazines and aza-analogues. Bioorg Med Chem 21:6732–6741. doi: 10.1016/j.bmc.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green J. 2014. Synthesis, of aza-heterocyclic monoamidines as potential DNA minor groove binders, anti-trypanosomals, and boron neutron capture therapy agents. Dissertation. Georgia State University, Atlanta, GA. [Google Scholar]
- 21.Timm BL, Da Silva PB, Batista MM, Farahat AA, Kumar A, Boykin DW, Soeiro MNC. 2014. In vitro investigation of the efficacy of novel diamidines against Trypanosoma cruzi. Parasitology 141:1272–1276. doi: 10.1017/S0031182014000407. [DOI] [PubMed] [Google Scholar]
- 22.Meirelles MN, de Araujo-Jorge TC, Miranda CF, de Souza W, Barbosa HS. 1986. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur J Cell Biol 41:198–206. [PubMed] [Google Scholar]
- 23.Buckner FS, Verlinde CL, La Flamme AC, Van Voorhis WC. 1996. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrob Agents Chemother 40:2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romanha AJ, De Castro SL, Soeiro MNC, Lannes-Vieira J, Ribeiro I, Talvani A, Bourdin B, Blum B, Olivieri B, Zani C, Spadafora C, Chiari E, Chatelain E, Chaves G, Calzada JE, Bustamante JM, Freitas-Junior LH, Romero LI, Bahia MT, Lotrowska M, Soares M, Andrade SG, Armstrong T, Degrave W, Andrade ZA. 2010. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz 105:233–238. doi: 10.1590/S0074-02762010000200022. [DOI] [PubMed] [Google Scholar]
- 25.Fivelman QL, Adagu IS, Warhust DC. 2004. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother 48:4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seifert K, Croft SL. 2006. In vivo and in vitro interactions between miltefosine and other antileishmanial drugs. Antimicrob Agents Chemother 50:73–79. doi: 10.1128/AAC.50.1.73-79.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Souza EM, Lansiaux A, Bailly C, Wilson WD, Hu Q, Boykin DW, Batista MM, Araújo-Jorge TC, Soeiro MNC. 2004. Phenyl substitution of furamidine markedly potentiates its antiparasitic activity against Trypanosoma cruzi and Leishmania amazonensis. Biochem Pharmacol 68:593–600. doi: 10.1016/j.bcp.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 28.De Souza EM, Menna-Barreto R, Araújo-Jorge TC, Kumar A, Hu Q, Boykin DW, Soeiro MNC. 2006. Antiparasitic activity of aromatic diamidines is related to apoptosis-like death in Trypanosoma cruzi. Parasitology 133:75–79. doi: 10.1017/S0031182006000084. [DOI] [PubMed] [Google Scholar]
- 29.Silva CF, Batista MM, De Souza EM, Meirelles MNL, Stephens CE, Som P, Boykin DW, Soeiro MNC. 2007. Cellular effects of reversed amidines on Trypanosoma cruzi. Antimicrob Agents Chemother 51:3803–3809. doi: 10.1128/AAC.00047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daliry A, Da Silva PB, Da Silva CF, Batista MM, De Castro SL, Tidwell RR, Soeiro MNC. 2009. In vitro analyses of the effect of aromatic diamidines upon Trypanosoma cruzi. J Antimicrob Chemother 64:747–750. doi: 10.1093/jac/dkp290. [DOI] [PubMed] [Google Scholar]
- 31.Da Silva EN Jr, Cavalcanti BC, Guimarães TT, Pinto MDC, Cabral IO, Pessoa C, Costa-Lotufo LV, De Moraes MO, De Andrade CK, Dos Santos MR, De Simone CA, Goulart MO, Pinto AV. 2011. Synthesis and evaluation of quinonoid compounds against tumor cell lines. Eur J Med Chem 46:399–410. doi: 10.1016/j.ejmech.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 32.Cencig S, Coltel N, Truyens C, Carlier Y. 2012. Evaluation of benznidazole treatment combined with nifurtimox, posaconazole or AmBisome® in mice infected with Trypanosoma cruzi strains. Int J Antimicrob Agents 40:527–532. doi: 10.1016/j.ijantimicag.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Moreira da Silva R, Oliveira LT, Silva Barcellos NM, de Souza J, de Lana M. 2012. Preclinical monitoring of drug association in experimental chemotherapy of Chagas' disease by a new HPLC-UV method. Antimicrob Agents Chemother 56:3344–3348. doi: 10.1128/AAC.05785-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bustamante JM, Craft JM, Crowe BD, Ketchie SA, Tarleton RL. 2014. New, combined, and reduced dosing treatment protocols cure Trypanosoma cruzi in mice. J Infect Dis 209:150–162. doi: 10.1093/infdis/jit420. [DOI] [PMC free article] [PubMed] [Google Scholar]