

Abstract
Human African trypanosomiasis (HAT) is a major tropical disease for which few drugs for treatment are available, driving the need for novel active compounds. Recently, morpholino-substituted benzyl amides of the fluoroquinolone-type antibiotics were identified to be compounds highly active against Trypanosoma brucei brucei. Since the lead compound GHQ168 was challenged by poor water solubility in previous trials, the aim of this study was to introduce structural variations to GHQ168 as well as to formulate GHQ168 with the ultimate goal to increase its aqueous solubility while maintaining its in vitro antitrypanosomal activity. The pharmacokinetic parameters of spray-dried GHQ168 and the newly synthesized compounds GHQ242 and GHQ243 in mice were characterized by elimination half-lives ranging from 1.5 to 3.5 h after intraperitoneal administration (4 mice/compound), moderate to strong human serum albumin binding for GHQ168 (80%) and GHQ243 (45%), and very high human serum albumin binding (>99%) for GHQ242. For the lead compound, GHQ168, the apparent clearance was 112 ml/h and the apparent volume of distribution was 14 liters/kg of body weight (BW). Mice infected with T. b. rhodesiense (STIB900) were treated in a stringent study scheme (2 daily applications between days 3 and 6 postinfection). Exposure to spray-dried GHQ168 in contrast to the control treatment resulted in mean survival durations of 17 versus 9 days, respectively, a difference that was statistically significant. Results that were statistically insignificantly different were obtained between the control and the GHQ242 and GHQ243 treatments. Therefore, GHQ168 was further profiled in an early-treatment scheme (2 daily applications at days 1 to 4 postinfection), and the results were compared with those obtained with a control treatment. The result was statistically significant mean survival times exceeding 32 days (end of the observation period) versus 7 days for the GHQ168 and control treatments, respectively. Spray-dried GHQ168 demonstrated exciting antitrypanosomal efficacy.
INTRODUCTION
Human African trypanosomiasis (HAT), also known as sleeping sickness, is caused by Trypanosoma brucei rhodesiense and T. b. gambiense. It is widespread in eastern and southern Africa (T. b. rhodesiense) and in western and central Africa (T. b. gambiense). In spite of currently decreasing numbers of new infections in humans, these regions are threatened by a large reservoir of the parasites in cattle, horses, and other nonmammalian species, creating an imminent risk of the next outbreak of HAT (1). In addition, the rapid development of resistance can be observed (2). Moreover, as the available drugs, such as suramin, pentamidine, melarsoprol, or eflornithine, either are active against only one of the two stages of HAT, the acute or chronic form, or exhibit dangerous adverse events, new anti-HAT drugs are urgently needed. Thus, it is important and perhaps indispensable to rigorously expand the arsenal of potent and safe antiparasitic compounds with demonstrated efficacy against HAT (3). Currently, two new drugs, i.e., the 5-nitroimidazole fexinidazole (phase II/III trials) and the oxaborole SCYX-6759 (phase I trials), are in clinical trials, in which they have shown promising initial results (4; http://www.dndi.org/diseases-projects/diseases/hat.html). However, significant hurdles remain to be overcome before these new drug candidates may become commercially available.
By means of a medium-throughput screening campaign, we recently discovered a new group of antitrypanosomal compounds derived from the fluoroquinolones, the latter of which are active against most Gram-positive and Gram-negative bacterial species but not against T. b. brucei (5–8). The replacement of the carboxylic acid functionality, which is important for the inhibitory activity toward many classes of bacterial topoisomerases, by a benzyl amide group resulted in a library of novel compounds. The newly synthesized compounds are active against T. b. brucei and T. b. rhodesiense at nanomolar concentrations (9) without cell toxicity, as assessed in macrophages. Structure-activity studies revealed that the quinolone carrying a butyl chain in position N-1, an N-benzyl group at the amido function in position 3, and a morpholino ring in position 7 was the most active and is referred to as GHQ168 (Fig. 1).
FIG 1.

Structure and activity/toxicity data for GHQ168, the most active compound from the novel quinolone amide library.
Initial biological studies hinted at the influence of the active compound GHQ168 (compound 29 in reference 9) on the morphology of the mitochondria and its ability to hinder segregation of the kinetoplast (9). In contrast to quinolone carboxylic acids, which are active against the trypanosomal topoisomerase II (from T. b. brucei) (6, 7, 10), we could show that the corresponding amides do not affect the topoisomerase in trypanosomes (9). Taken together, these findings indicate that the mode of action of GHQ168 remains to be elucidated.
Even though GHQ168 has favorable structural properties that predict oral bioavailability (10) (a favorable octanol-water partition coefficient [logP] is ≤5 and the logP for GHQ168 is 3.8, a favorable molecular mass is ≤500 g/mol and the molecular mass of GHQ168 is 437.5 g/mol, a favorable structure has 5 hydrogen bond donors represent and GHQ168 has 1, and a favorable structure has 10 hydrogen bond acceptors and GHQ168 has 7), this compound showed very poor aqueous solubility. Thus, in order to overcome the solubility problem in experiments with mice, the compound was administered intraperitoneally (i.p.) as a lipid formulation. However, these previous experiments had to be stopped due to low tolerance of the vehicle by the test animals (9).
In this study, we aimed to develop by chemical modification fluoroquinolone amide compounds with improved solubility. Furthermore, we developed a formulation of GHQ168 with the same goal of meeting the necessary solubility requirements. The compounds were characterized with regard to their physicochemical properties, in vitro antitrypanosomal activity, and in vitro toxicity. Three of the synthesized compounds were selected and analyzed with regard to intestinal absorption (Caco-2 cell model) for assessment of the feasibility of the development and use of oral dosage forms in the future. Furthermore, the three compounds were profiled for their in vivo pharmacokinetics (PK) and in vivo antitrypanosomal activity in infected mice.
MATERIALS AND METHODS
Materials.
Reference substances (used for the determination of lipophilicity, permeability, in vitro activity, and metabolism), excipients, and reagents were purchased from Sigma-Aldrich, Taufkirchen, Germany, and were of analytical or pharmaceutical grade, unless noted otherwise. Poly(methacrylic acid-comethyl methacrylate) (Eudragit L100; approximate Mw = 125,000 g/mol) for formulation preparation was from Evonik, Essen, Germany. Liquid chromatography (LC) columns were obtained from Phenomenex, Aschaffenburg, Germany (Synergi MAX-RP, Kinetex, Luna) and from MZ-Analytical, Mainz, Germany (Inertsil ODS-2). A PLTK4710 ultrafiltration membrane for albumin binding was purchased from Millipore, Schwalbach, Germany. Caco-2 cells were from ATCC; the polyethylene terephthalate (PET) membrane was from Greiner Bio-One, Frickenhausen, Germany; and Dulbecco's modified Eagle medium (DMEM), high glucose, with l-glutamine, nonessential amino acids, and penicillin-streptomycin solution was from Gibco, Life Technologies, Darmstadt, Germany. For cytotoxicity assays, all cells were purchased from ATCC, Wesel, Germany; RPMI medium was from Gibco, Life Technologies, Darmstadt, Germany; and fetal calf serum (FCS) was from GE Healthcare Europe GmbH, Freiburg, Germany. For metabolism and mutagenesis experiments, NADP sodium salt was obtained from Applichem, Darmstadt, Germany; mass spectrometry (MS)-grade solvents were from VWR, Ismaning, Germany; and all other chemicals, including cell culture medium and supplements, were from Sigma-Aldrich, Taufkirchen, Germany, or Roth, Karlsruhe, Germany, unless specified otherwise. For metabolic stability testing, the S9 fraction was purchased from MP Biomedicals, Illkirch-Graffenstaden, France; the Bradford reagent was from Bio-Rad, Munich, Germany. For mutagenicity testing, fetal calf serum was purchased from Invitrogen, Karlsruhe, Germany, and culture flasks and tissue culture dishes were from Greiner Bio-One, Frickenhausen, Germany. For plasma stability assessment, human plasma was obtained from Bayerisches Rotes Kreuz, Munich, Germany. For the analysis of blood samples, solvents were purchased from Fisher Scientific, Schwerte, Germany.
Synthesis.
The synthesis of GHQ168 was performed as described previously (9). The synthesis of the other related salt compounds presented herein (GHQ237, GHQ242, GHQ243, GHQ250, GHQ232, GHQ215) can be found in the supplemental material. The stoichiometry of the substances and the respective counterion was determined using CHN analysis.
Formulation.
A solid dispersion was produced using a nanospray dryer (B-90; Büchi, Essen, Germany). GHQ168 (0.2%, wt/vol) and Eudragit L100 (2.0%, wt/vol) were dissolved in methanol, resulting in a 1:10 (wt/wt) spray-dried formulation. Spray drying was performed with the head in the vertical position and with the formulation sprayed through a mesh with a 7-μm mesh size at an airflow set at 150 liters/min, an inlet temperature of 60°C, an outlet temperature of 34°C, a pressure of 5.2 × 103 Pa, and a spray rate of 100%. The resulting white, amorphous nanoparticles were stored in a desiccator over silica gel at room temperature.
Solubility.
A preliminary determination for the fast and approximate assessment of kinetic solubility was achieved by diluting dimethyl sulfoxide (DMSO) stock solutions of the desired compounds (1 mM, 20 mM) in phosphate-buffered saline (PBS) buffer (pH 7.4) in order to give aqueous solutions with concentrations of 10, 100, 500, 1,000, 1,500, 2,000, 2,500, 3,000, 4,000, and 5,000 μM (one experiment was performed with each concentration). Precipitation was detected microscopically (SZ-PT microscope; Olympus, Shinjuku, Japan).
In addition, a more precise determination of kinetic solubility was conducted for those compounds applied in the efficacy studies. “Kinetic solubility” refers to the apparent solubility of a compound in a metastable state, at which, in spite of the fact that the chemical potential of the compound in solution exceeds the chemical potential of the solid compound, precipitation is so slow that a (kinetic) solubility exceeding the equilibrium solubility may be observed.
As described above, dilution series were prepared for each compound but with closer concentration steps (GHQ168, 5, 10, 15, 20, 25, 30, and 35 μg/ml; GHQ242, 150, 200, 250, 300, 350, 400, 450, and 500 μg/ml; GHQ243, 50, 100, 125, 150, 175, 200, 250, and 300 μg/ml; n = 3 each) to end with a maximum of 2.5% residual DMSO content. Detection was accomplished nephelometrically (NEPHEOLOstar BMG, Ortenberg, Germany) using 96-well plates with a flat bottom (Greiner Bio-One, Frickenhausen, Germany). The temperature was set to 37°C, the laser intensity was 80%, and the laser beam focus was 2.20 mm. The gain was adjusted to 60 (GHQ168, GHQ242) and 75 (GHQ243), and the measurement time per well was 0.1 s. The mean result for three dilution series was determined. Two replicate measurements of the same solutions were performed (time frame, 30 min), and the standard deviation (SD) was calculated from the means of three replicate measurements over time.
In contrast to the kinetic solubility, the thermodynamic solubility (also called “equilibrium solubility”) describes a thermodynamically stable state that might take its time to be generated but that is maintained when environmental conditions remain unchanged. The determination of the thermodynamic solubility of GHQ168 was conducted by dosing solid substance (in excess) into 2-ml Eppendorf vials, followed by dissolution in PBS buffer (pH 7.4). Throughout the assay, continuous shaking (800 rpm) and a constant temperature (37°C) were maintained (Eppendorf Thermomixer; Eppendorf AG, Hamburg, Germany). Samples were taken after 10, 30, 60, 120, and 1,200 min, followed by centrifugation (13,000 rpm, 1 min; Micro 2416; VWR International, Darmstadt, Germany) and high-performance liquid chromatography (HPLC)-UV (Jasco, Groß-Umstadt, Germany) analysis of the supernatant (Synergi MAX-RP column; 80 Å; 4 μm; 150 by 4.6 mm; mobile phase, acetonitrile-water [72/28]; temperature, 40°C; flow rate, 1.2 ml/min; injection volume, 20 μl; detection wavelength, 280 nm). Solubility determination was performed in triplicate.
X-ray diffractometry.
X-ray powder diffractograms were recorded on an X-ray powder diffraction (XRPD) apparatus (D8 Discover; Bruker, Karlsruhe, Germany) using a copper tube operating at 40 kV and 40 mA. A focusing Goebel mirror was installed in the primary beam path (slit, 0.6 mm; axial Soller slit, 2.5°). For the secondary beam path, no slit was applied and the axial Soller slit was set to 2.5°. Detection was done using a LynxEye one-dimensional detector (Bruker AXS). The investigation was performed in coupled two theta/theta mode with a 2-ϴ range of 5 to 45°, a step width of 0.025°, and a 1.0-s measurement time per step. Data collection and processing were conducted with the software packages DIFFRAC.Suite (v2 2.2.690; Bruker AXS 2009-2011) and DIFFRAC.EVA (version 3.0; Bruker AXS 2010-2013). Details on the method used for single crystal structure analysis can be found in the supplemental material.
SEM.
Scanning electron microscopy (SEM) (JSM-7500F SEM microscope; JEOL, Japan) was performed with an accelerating voltage of 2.0 kV and a ×1,000 magnification at a working distance of 8.6 mm. Prior to examination, the samples were sputter coated with gold.
Physicochemical parameters.
LogP data were recorded using HPLC-UV (C18 reversed-phase Inertsil ODS-2 column; 5 μm; 150 by 4.6 mm; mobile phase, phosphate buffer [10 mM; pH 7.4]-methanol [containing 0.02% N,N-dimethylhexylamine], 30/70; temperature, 30°C; flow rate, 1.5 ml/min; injection volume, 40 μl; detection wavelength, 254 nm) as described previously (9). Compounds with known logP values (2-phenylethanol, benzene, N,N-dimethylaniline, toluene, chlorobenzene, ethylbenzene, biphenyl, and anthracene) served as reference substances. The capacity factor (k′) that correlates with logP values can be derived from the retention time (tR) and dead time (t0) of the compounds by the following equation: k′ = (tR – t0)/t0.
Experimental determination of pKa was conducted for GHQ168, GHQ242, and GHQ243 on a Sirius T3 instrument (Sirius, Forest Row, United Kingdom). Due to its low aqueous solubility, determination of the pKa of GHQ168 was conducted from a DMSO stock solution (10 μM) using the assay type fast UV-metric pKa (in which pKa assessment is accomplished by the identification of changes in the absorption profile due to sample ionization). For GHQ242 and GHQ243, the solid compounds (weight, 0.43 mg to 0.56 mg) were dosed into the instrument and the assay type pH-metric pKa (in which the potentiometric pKa is assessed using a pH electrode) was applied. Potassium chloride solution (1.5 ml, 0.15 M) was added to the compounds, and the temperature was maintained at 25°C throughout the assay. The titrations were conducted over the pH range of 2 to 12, starting at low pH values (acidification prior to titration with 0.5 M hydrochloric acid; titration with 0.5 M potassium hydroxide solution).
DSC.
Melting point determinations were performed using a differential scanning calorimetry (DSC) 8000 instrument (PerkinElmer, Waltham, MA, USA). The scanning rate was set to 20°C/min over the range of −50°C to 300°C.
Serum albumin binding.
In principle, albumin binding was determined in vitro by means of the continuous titration methods described herein (11). In brief, the self-made instrument consists of a low-pressure HPLC pump (Bischoff, Leonberg, Germany), an injection valve (Rheodyne, Alsbach, Germany) for injection of the human serum albumin (HSA) solution into the system, an ultrafiltration cell, and a UV detector (Knauer, Berlin, Germany). The ultrafiltration cell contains the PLTK4710 ultrafiltration membrane with a molecular mass cutoff of 30 kDa. HSA and the test compounds were dissolved in 0.03 M phosphate buffer (pH 7.4) containing 0.1 M NaCl for the simulation of physiological conditions. In the case of GHQ168, the substance was dissolved in dimethylformamide (1 mg/ml), and 420 μl of this solution was diluted with 2 mM polysorbate 20 in buffer to 100.0 ml. The concentration of the drug solutions was within the range of 10 to 20 mg/liter. Initially, the drug solution was continuously pumped through the cell and the curves of the measured absorption versus time were plotted. The system was rinsed with buffer, and subsequently, the albumin solution (40 mg/ml) was injected into the system. The ultrafiltration membrane retains the protein in the cell. The drug solution was again pumped through the ultrafiltration cell and the absorbance was measured again. The interaction between drug and HSA present in the cell leads to a shift of the second curve to the right compared to the location of the first recorded curve. The size of the area between the two drug curves is proportional to the amount of drug bound to the HSA.
Permeation through Caco-2 cell monolayers.
The evaluation of permeation was performed in principle as reported previously (12). In brief, Caco-2 cells at passage number 51 were thawed, passaged four times, and seeded at 1.3 × 105 cells/cm2 on 24-well cell culture inserts with a PET membrane (pore size, 0.4 μm). The cells were cultivated in DMEM, high glucose, with l-glutamine, nonessential amino acids, and penicillin-streptomycin solution for 23 days at 37°C in 5% CO2. Transepithelial electric resistance (TEER) in DMEM was monitored three times a week during the medium change (EVOM2 electrode; World Precision Instruments, USA). Prior to the permeability experiment, the cells were washed twice with Hanks balanced salt solution (HBSS), 25 mM HEPES, pH 7.4, and TEER was measured in order to assess the integrity of the monolayer (cells with TEER values of <200 Ω · cm2 were excluded from the experiment). For permeation testing, 150 μl of 20 μM GHQ168, GHQ242, and GHQ243 in HBSS–0.5% DMSO was added to the apical compartment and 600 μl of HBSS was added to the basolateral compartment, followed by gentle shaking for 1 h at 37°C in 5% CO2. The cells were washed with HBSS, and the integrity of the monolayer was reconfirmed by TEER measurement. Fluorescein isothiocyanate-dextran was used as a nonpermeant negative control, and propranolol HCl was used as a positive control with high levels of permeation. For each compound, at least three independent Caco-2 cell assays were conducted within 1 day. The concentration of fluorescein isothiocyanate-dextran was determined fluorimetrically (excitation wavelength, 490 nm; emission wavelength, 514 nm; LS50B luminescence spectrometer; PerkinElmer). All other compounds were analyzed by LC/MS/MS using the analytical method described in “Pharmacokinetics” below.
In vitro activity.
The methods described by Räz et al. (13), Baltz et al. (29), Papadopoulou et al. (14), Larson et al. (15), and Muth et al. (16) were applied for determination of in vitro activity.
Cytotoxicity.
The methods used to determine cytotoxicity are described in the supplemental material.
Metabolism and mutagenesis.
Phase I metabolism of GHQ168 by cytochrome P450-dependent monooxygenase was investigated in rat liver microsomes. Microsomes were prepared by ultracentrifugation (100,000 × g, 60 min, 4°C) from a commercially available S9 fraction from Aroclor 1254-treated male Sprague-Dawley rats. After resuspension in HEPES buffer (25 mM HEPES, 100 mM NaCl, 1.5 mM disodium EDTA, 1 mM dithiothreitol, 10% glycerol, pH 7.4), the protein concentration was determined with the Bradford reagent, yielding 7 mg/ml, and the microsomes were stored at −80°C until use.
Microsomal incubation mixtures (final volume, 500 μl) contained GHQ168 (100 μM, 1% DMSO), microsomal protein (1 mg/ml), 0.1 M phosphate buffer (pH 7.4), and an NADPH-generating system and were incubated at 37°C for 30, 60, and 90 min. The NADPH-generating system was prepared from isocitrate (10 mM), isocitrate dehydrogenase (0.05 U), MgCl2 (4 mM), and NADP (1 mM) and was preincubated for 5 min at 37°C prior to addition to the incubation system. Control incubations were carried out under the same conditions with heat-deactivated microsomes.
After the incubation period, the reaction was stopped by addition of ethyl acetate. Then, 7-(4-acetylpiperazin-1-yl)-N-(2,4-dichlorobenzyl)-6-fluoro-1-(2-fluorophenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide, which is structurally similar to GHQ168, was added as an internal standard and the reaction mixture was extracted three times with ethyl acetate. After evaporation of the solvent, the residues were dissolved in 100 μl acetonitrile and subjected to HPLC with photodiode array detection (LaChrome; Merck Hitachi, Darmstadt, Germany) to determine the rate of GHQ168 decrease (Kinetex column; 2.6 μm; C18; 100 by 3 mm; mobile phase A, water, 0.5% formic acid; mobile phase B, acetonitrile, 0.5% formic acid; gradient, 38% mobile phase B for 6 min, gradually increasing to 60% mobile phase B within 44 min; room temperature; flow rate, 0.2 ml/min; injection volume, 10 μl; detection wavelength, 282 nm). The m/z ratios of the metabolites were obtained by HPLC-mass spectrometry (Agilent LC/MSD G1946D single quadrupole mass spectrometer; Agilent Technologies, Waldbronn, Germany) in full scan mode using positive ionization, a fragmentor voltage of 70 V, a capillary voltage of 4,000 V, and full scan range of m/z 100 to 500.
Three independent microsomal incubations were performed (with a new vial of microsomes and a fresh solution of test compound for each test).
The mutagenicity of GHQ168 was assessed in a hypoxanthine-guanine phosphoribosyltransferase (HPRT) assay in cells of the V79 Chinese hamster fibroblast cell line (17). V79 cells were cultured in DMEM supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FCS, referred to as DMEM complete. The HPRT assay was performed as described previously (18). Briefly, 1.5 × 106 V79 cells were seeded in 175-cm2 cell culture flasks containing 20 ml DMEM complete. After 24 h, the medium was changed (day 0) and the cells were treated with different concentrations of GHQ168, 1 μM 4-nitroquinoline-N-oxide (NQO; positive control), or 1% DMSO (negative control) for 24 h. A total of 1 × 106 cells were subcultured in fresh medium directly after treatment (day 1) and again on day 4.
On days 1, 4, and 6, the numbers of viable cells and cells with a disintegrated cell membrane were counted with a CASY model DT electronic cell counter (Schaerfe, Reutlingen, Germany) as a reference point for cytotoxicity and proliferation.
On day 6, cells with mutations at the Hprt gene locus were selected by growing cells in DMEM complete and 7 μg 6-thioguanine (6-TG)/ml using three tissue culture-treated dishes (diameter, 145 mm) with 1 × 106 cells per dish. To determine the plating efficiency (PE) on days 1 and 6, cells were grown in the absence of 6-TG (500 cells per 100-mm dish, three dishes). After 1 week, the cells were fixed with ethanol and stained with methylene blue (0.5% in methanol). The colonies were counted, and the PE, i.e., the number of colonies per number of seeded cells, and the mutant frequency, i.e., the number of colonies/(number of seeded cells × PE on day 6), were calculated. Three independent HPRT tests were performed (with a new batch of cells and a fresh solution of test compound for each test).
Plasma stability.
A plasma stability assay was performed as described by Di et al. (19). In brief, a 1 mM DMSO stock solution of the selected compound was added to human plasma, and this mixture was diluted 1:2 with PBS buffer (pH 7.4) to achieve a final compound concentration of 10 μM. The mixture was incubated at 37°C for 2 h. Within that time, aliquots of 100 μl were taken at time zero (reference), 30, 60, and 120 min. All samples were diluted with 300 μl acetonitrile and centrifuged (3,000 rpm for 15 min), and the supernatant (200 μl) was analyzed by means of HPLC (Synergi MAX-RP column; 80 Å; 4 μm; 150 by 2 mm; mobile phase, acetonitrile [ACN], 10 mM ammonium acetate buffer [pH 4]; gradient, 10% ACN, increasing to 90% in 7 min, 90% ACN for 2 min, 90% ACN decreasing to 10% ACN in 2 min, 10% ACN for 2 min; temperature, 40°C; flow rate, 0.4 ml/min; injection volume, 30 μl; detection wavelength, 281 nm [GHQ242] or 278 nm [GHQ243]). The resulting peak areas of each aliquot were divided by the peak area of the reference sample (time zero) to calculate the percent decrease. The stability of each compound was measured in duplicate.
PBPK modeling and pharmacokinetics.
Prior to the in vivo efficacy studies, in silico physiologically based pharmacokinetic (PBPK) modeling (20) was conducted using the Simcyp software package (Simcyp, Sheffield, United Kingdom) in order to identify a suitable dose and study design. The study design (e.g., duration, dosing interval) and the prediction of the plasma levels were performed with a mouse model. The parameters calculated experimentally to enable determination of a relevant PK study design took into account molar mass, pKa, logP, and the fraction unbound in plasma. The volume of distribution (V) was calculated by the software according to the corrected Poulin and Theil method (21–24). Clearance (CL) was calculated from the predicted volume of distribution normalized per kilogram of body weight (V) according to the formula CL = V × kel × weight of the mouse, where kel is the elimination rate constant derived experimentally from in vitro metabolic stability assays. The weight of a mouse was set to be 25 g, and simulations were conducted assuming first-order elimination kinetics.
Analysis of mouse blood samples was performed using a Triple Quad 5500 LC/MS/MS system from AB Sciex (Darmstadt, Germany) [Luna column; 3 μm; C18(2); 100 Å; 75- by 3-mm reversed-phase column; mobile phase, methanol (800 ml), LC/MS-grade water (200 ml), ammonium formate (0.636 g), and concentrated formic acid (1.25 ml) (pH 4.5); room temperature; injection volume, 3 μl; detection masses, 438.1 Da to 331.0 Da (GHQ168), 439.1 Da to 331.0 Da (GHQ242), 453.1 Da to 72.0 Da (GHQ243), and 494.2 Da to 368.9 Da (glibenclamide; internal standard); turbospray ionization in the positive mode]. The dwell time was set to 150 ms. Nitrogen was used both as the nebulizing and the drying gas (validation data on the analytical method are provided in Table S3A to F and Fig. S6A to C in the supplemental material).
In vivo efficacy studies.
The T. b. rhodesiense STIB900 strain is a derivative of the STIB704 strain isolated from a patient in Ifakara, Tanzania, in 1982 (25). The mouse model of acute STIB900 infection mimics the first stage of the disease, in which the trypanosomes are localized in the hemolymphatic system. Prior to the efficacy studies, a donor female NMRI mouse was inoculated intraperitoneally with 1 × 104 bloodstream forms of STIB900. In the efficacy studies, four female NMRI mice were used per experimental group. Each mouse was inoculated i.p. with 104 bloodstream forms of STIB900. For that, heparinized blood from the donor mouse with a level of parasitemia of approximately 5 × 106/ml was suspended in phosphate-saline-glucose (PSG) to obtain a trypanosome suspension of 4 × 104 trypanosomes/ml. Each mouse used for the experiments was injected with 0.25 ml trypanosome suspension. Two efficacy studies were conducted: in the first study, compound administration began on day 3 postinfection (stringent model; test compounds, GHQ168, GHQ242, and GHQ243), and in the second study, compound administration began 1 day postinfection (early-treatment model; test compound, GHQ168). Other than this difference, both studies were designed equally (route of administration, i.p.; dosing interval, 12 h; study duration, 4 days; single doses of GHQ168 at 3.5 mg/kg of body weight, GHQ242 at 22.9 mg/kg, or GHQ243 at 21.9 mg/kg; fluid intake with dose, 0.1 ml/10 g body weight).
For in vivo administration, GHQ168 spray-dried particles and crystalline GHQ242 and GHQ243 were dissolved in the following delivery vehicles prior to i.p. application: for GHQ168, 5% (wt/vol), glucose, 1% (wt/vol) polysorbate 80, and PBS buffer (pH 7.4); for GHQ242 and GHQ243, 5% (wt/vol) glucose, 1% (wt/vol) polysorbate 80, 10% (vol/vol) DMSO, and PBS buffer (pH 7.4).
Four mice served as infected, untreated controls. They were not injected with the vehicle alone, since it was established in the lab that these vehicles do not affect parasitemia or the mice (data not shown). All mice were monitored for parasitemia by microscopic examination of tail blood twice a week until day 32 postinfection and afterwards once per week until day 60. The time to parasite relapse was recorded to calculate the mean relapse duration (MRD; in days) after infection. Mice were euthanized after detection of a relapse of parasitemia (at least 2 trypanosomes per field of view). The mean survival duration (MSD; in days) of the treated groups and the MSD of the control group were compared using an unpaired t test; the level of significance was defined at an α level of 0.05 (Minitab software, version 17.2.1). Data for mice that died during the treatment but without parasites in the blood were excluded from the calculation of MSD and MRD, as the cause of death was not related to parasitemia. Mice that survived to and that were aparasitemic at day 60 (stringent model) or at day 32 (early-treatment model) were considered cured (which was endpoint of the model) and euthanized.
During the efficacy study with the stringent mouse model, blood samples were collected from tail blood for subsequent LC/MS/MS analysis at predetermined time points (1 h and 4 h after the 7th treatment and 16 h after the 8th treatment) in order to assess pharmacokinetics (see Table S4 in the supplemental material). In vivo efficacy studies in mice were conducted at the Swiss Tropical and Public Health Institute (Basel, Switzerland) according to the rules and regulations for the protection of animal rights (Tierschutzverordnung) of the Swiss Bundesamt für Veterinärwesen. They were approved by the veterinary office of the Canton Basel-Stadt, Switzerland.
Statistics.
Statistical tests (in metabolism and mutagenesis studies) were performed using OriginPro software (version 9.1; OriginLab Corp., Northampton, MA, USA). Multiple data sets were analyzed by analysis of variance with post hoc comparison by the Scheffé test. Pairwise comparisons were performed by Student's t test. The evidence was postulated to have statistical significance if the P value was <0.05.
RESULTS
Solubility in water.
The thermodynamic solubility of GHQ168 was 0.005 ± 0.001 μg/ml, as assessed after equilibration for 20 h in PBS buffer and under the conditions described above in Methods and Materials. The kinetic solubility was 15 ± 1 μg/ml; i.e., the kinetic solubility exceeded the thermodynamic solubility by a factor of 3,000. Therefore, GHQ168 was practically insoluble in water (22), driving the need for solubility improvement before in vivo efficacy studies commenced. Solubility improvement was addressed by (i) formulation approaches and (ii) structural modification.
The formulation strategy was fueled by the coplanar stacking of GHQ168 within its crystal (Fig. 2), with the crystalline nature being corroborated by XRPD (see Fig. S1A in the supplemental material). These highly ordered structures may allow a low free enthalpy state, consequently demanding high levels of work for molecular escape from the crystal. The resulting high melting point of the crystal (171°C) reflects these patterns and favors the low solubility of GHQ168. Consequently, a formulation strategy pushing GHQ168 to free enthalpy states higher than the state of the crystal was deployed. For that, GHQ168 was spray dried into a polymer, thereby embedding the drug in an amorphous state (see Fig. S1B in the supplemental material) within microparticles (see the scanning electron micrographs in Fig. S2 in the supplemental material). In addition to the amorphous presentation, the spray-dried formulation massively increased the surface area of the compound compared to the compact arrangement within the crystal, typically increasing the dissolution rate on the basis of simple Fickian diffusion considerations. The methacrylic acid polymer used for embedding the compound was selected to readily dissolve at a pH exceeding approximately 5.5, thereby gradually releasing the molecularly dispersed drug when its exposed to a pH that is at or that exceeds this pH threshold (26). Stability studies demonstrated the physical stability of this formulation for 1 year of storage by the absence of (re-)crystallization (the formulation was not assessed at later time points; see Fig. S1B in the supplemental material). Spray drying of GHQ168 resulted in white nanoparticles of a size ranging below 350 nm, which was confirmed by subsequently performed dynamic light scattering (DLS) experiments (see Fig. S3 in the supplemental material) in order to monitor the pattern in solution over time. Furthermore, a smooth surface, spherical form, and narrow size distribution were illustrated with the help of scanning electron micrographs (see Fig. S2 in the supplemental material). The overall spray-drying process yielded an efficacy of about 90%. The maximum solubility (supersaturated state) in PBS buffer was determined to be 32.9 ± 1.6 μg/ml after 30 min, and this decreased to 7.3 ± 0.7 μg/ml after 6 h (Fig. 3), thereby transiently exceeding the thermodynamic solubility of GHQ168 by a factor >6,000. Physical mixtures of GHQ168 with the methacrylic acid polymer did not impact the solubility profile, indicating that amorphization is essential to improve the solubility and corroborating the formulation hypothesis, in that breaking the crystalline forces by amorphization is instrumental to increasing the pharmaceutical properties of GHQ168 (Fig. 3).
FIG 2.

Crystal structure of GHQ168. (Left) View of the unit cell with four molecules of GHQ168; (right) view from another direction with a restricted depiction of the quinolone backbone of the molecule only, illustrating the arrangement of GHQ168 molecules as coplanar dimers in the crystal structure. Black, C atoms; white, H atoms; green, F atoms; blue, N atoms; red, O atoms.
FIG 3.
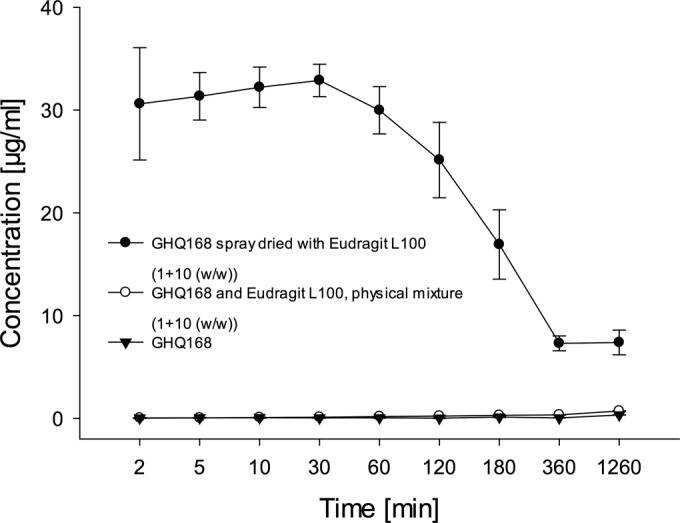
Solubility profile of the GHQ168 spray-dried formulation in comparison to that of the physical mixture of crystalline GHQ168 with Eudragit L100 and that of the crystalline raw substance.
The chemical strategy targeted solubility improvement by structural modification of GHQ168 (Fig. 4) as follows. (i) The introduction of basic moieties (tertiary amines) allowed salt formation, and the resulting GHQ243 and GHQ250 were presented as oxalate salts with a stoichiometry (active pharmaceutical ingredient [API] to oxalate) of 1:1 and 1:1.5, respectively. (ii) The introduction of a polar substituent, i.e., the replacement of a phenyl moiety with a pyridine ring, resulted in GHQ242 and GHQ237, again presented as oxalate salts with a stoichiometry (API to oxalate) of 1:1.5 and 1:0.5, respectively. (iii) The formation of the amidine of GHQ168 resulted in GHQ232, presented as a hydrochloride. (iv) The introduction of an additional carbonyl group in position 2 resulted in GHQ215, an acidic moiety.
FIG 4.

Structures of salt compounds newly synthesized for solubility enhancement.
Details of the synthesis pathways (including the experimental details) are described in the supplemental material. The introduction of a basic functionality in position 7, which is essential for the activity of the topoisomerase inhibitors in clinical treatment (24), always resulted in a substantially decreased activity against T. b. brucei and a lower selectivity index (SI; calculated as the 50% cytotoxic concentration for Vero cells divided by the 50% inhibitory concentration [IC50] of the compound for T. cruzi cells) (9). Thus, this position was not considered for the introduction of a basic substituent.
With the exception of GHQ215, the solubility of the compounds was enhanced by structure variations, particularly for the oxalate salts GHQ242, GHQ243, and GHQ250, as preliminarily assessed by light microscopy (Table 1). However, all new compounds were less active against T. b. brucei and, therefore, displayed a decreased selectivity index (Table 1), which was already reported previously (9). The potentiometric determination of the pKa of GHQ168 did not result in a measurable value between pH 2 and pH 12. After introduction of basic moieties, the pKa values of GHQ242 and GHQ243, the candidates chosen for use in in vivo efficacy studies, were 5.25 ± 0.05 and 6.86 ± 0.06, respectively (data not shown). We detailed the preliminary, microscopic assessment of solubility (Table 1) nephelometrically, with GHQ242 and GHQ243 having aqueous kinetic solubilities of 274 ± 18 μg/ml and 152 ± 1 μg/ml, respectively. Both had a statistically significant and improved kinetic solubility compared to that of GHQ168 (15 ± 1 μg/ml).
TABLE 1.
Physicochemical and microbiological data for quinolone amide compounds
| Compound | Mol wt (g/mol) | LogP | Preliminary kinetic solubility (μM) | Melting point (°C) | Albumin binding (%) | IC50 (μM) at: |
CC50a (μM) |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 48 h/72 h for T. b. brucei | 72 h for T. b. rhodesiense | L6 | J774.1A | HEK 293T | HepG2 | ||||||
| GHQ168b | 437.5 | 3.67 | <19 | 171.4 | 80.5 | 0.047 ± 0.00/0.05 ± 0.01 | 0.001 ± 0.0006 | 47.2 ± 9.7 | 57 | 26.30 ± 5.35 | 47.04 ± 3.04 |
| GHQ237 | 420.5 | 3.05 | 100 | 178.1 | 67.6 ± 4.53 | 0.55 ± 0.08/1.02 ± 0.33 | 0.012 ± 0.0025 | >95.2 | >100 | >160 | >160 |
| GHQ242 | 438.5 | 1.74 | >5,000 | 149.0 | 99.95 ± 0.02 | 0.29 ± 0.26/0.76 ± 0.77 | 0.002 ± 0.0004 | 24.3 ± 4.2 | >100 | 86.81 ± 6.43 | 153.31 ± 9.47 |
| GHQ243 | 452.5 | 2.67 | 1,000 | 198.2, 223.0 | 45.9 ± 4.19 | 0.54 ± 0.01/0.63 ± 0.01 | 0.018 ± 0.0094 | 30.7 ± 7.4 | 42.3 | 48.03 ± 1.94 | 48.84 ± 0.12 |
| GHQ250 | 524.6 | 3.41 | >5,000 | 172.3 | 79.1 ± 7.42 | 2.89 ± 0.62/3.37 ± 0.29 | NDc | ND | 39.9 | 10.25 ± 1.56 | 13.12 ± 0.03 |
| GHQ232 | 436.5 | 3.47 | 100 | >300 | 85.8 ± 5.27 | 9.08 ± 0.12/15.05 ± 1.79 | ND | ND | 39.1 | 13.98 ± 0.64 | 19.34 ± 2.91 |
| GHQ215 | 453.5 | 5.53 | 10 | >300 | ND | 3.08 ± 0.69/5.73 ± 0.91 | ND | ND | >100 | 26.59 ± 5.29 | >160 |
CC50, 50% cytotoxic concentration.
Most of the data for GHQ168 were already reported in reference 9 (see compound 29).
ND, not determined.
Albumin binding, metabolism, plasma stability, mutagenicity, and transepithelial transport.
Albumin binding was determined by a continuous titration method (11, 27). Whereas GHQ168 and GHQ243 were 81% and 46% bound to albumin, respectively, GHQ242 was completely bound to albumin at a level exceeding 99% (Table 1). Since GHQ168 was found to be the most active compound in vivo (see below), the oxidative metabolism of GHQ168 was studied. The HPLC-MS full scan analysis revealed 4 peaks (at m/z 454, at m/z 412, and twice at m/z 348) not observed in control reactions. The m/z results suggested metabolites resulting from ring hydroxylation, β-oxidation, and N-dearylation, respectively. Although linear NADPH generation and the activity of microsomal enzymes for 90 min were assessed (data not shown), the decrease in the GHQ168 concentration did not differ significantly after 30, 60, and 90 min (4.7% ± 0.9%), indicating enzyme inhibition by GHQ168 or one of its metabolites. Concerning stability in plasma, no degradation was observed throughout 2 h for GHQ168, GHQ242, and GHQ243.
The mutagenicity of GHQ168 in cultured V79 cells was assessed in the HPRT assay. In light of the GHQ168-mediated inhibition of microsomal enzyme activity and the absence of metabolites with a structural alert for mutagenicity (such as catechols), the HPRT assay was performed in the absence of a metabolically activating system. GHQ168 was tested at concentrations ranging from 2 to 47 μM, and it significantly decreased the plating efficiency and cell numbers at 47 μM and 16 to 47 μM, respectively, as determined by previously described protocols (28). Whereas the known mutagen NQO caused a significant increase in the mutant frequency to 186 ± 42 compared to that for control cells (9 ± 3), the mutant frequency was not affected by treatment with any concentration of GHQ168.
In light of future profiling of the compounds, GHQ168, GHQ242, and GHQ243 were tested for transepithelial transport. GHQ168, GHQ242, and GHQ243 had a flux through Caco-2 cell monolayers (as an in vitro model for intestinal absorption) comparable to that for the positive control, propranolol HCl (see Fig. S4 and Table S2 in the supplemental material). On the basis of the previously demonstrated low solubility and the good permeation observed with the Caco-2 cell monolayer assay, in the absence of information from in vivo studies, GHQ168 can only preliminarily be categorized as Biopharmaceutics Classification System (BCS) class II (low solubility, high permeation).
Cell toxicity and in vitro efficacy.
GHQ168 as well as the new derivatives were characterized for their physicochemical properties (logP, kinetic solubility, melting point) as well as albumin binding, their activity against T. b. brucei and T. b. rhodesiense (72 h), and their cytotoxicity (muscle cells, macrophages, kidney cells, and hepatocytes), which are displayed in Table 1. On the basis of the solubility results and demonstrated in vitro efficacy, spray-dried GHQ168 and the oxalate salts of GHQ242 and GHQ243 were selected for use in in vivo efficacy studies.
Pharmacokinetics.
During the efficacy study with the stringent mouse model, blood samples were collected from the surviving mice at three time points and analyzed by LC/MS/MS (Fig. 5; see also Fig. S7 in the supplemental material). Blood concentrations were about 10-fold higher for GHQ168 (delivered from the spray-dried formulation) and GHQ243 than for GHQ242. An unusual time response of the blood concentration of GHQ242 was observed among the mice. Two of the four mice tested within the GHQ242 group had higher blood concentrations at a subsequent time point than a previous one (see Fig. S7 in the supplemental material; data for these 2 mice are highlighted by arrows). The half-lives of GHQ168 and GHQ243 were comparable, ranging from approximately 1.5 to 3.5 h (Table 2). The pharmacokinetic outcome for GHQ242 was more heterogeneous (see above), such that we cannot decide whether the blood concentrations analyzed 16 h after application were due to metabolism of the compound or due to flip-flop kinetics. In fact, the half-life reported for GHQ242 may reflect metabolism or may reflect another step, such as drug release from a precipitate formed during i.p. injection. As the data obtained were insufficient to clarify this question (data obtained after intravenous [i.v.] administration would be required), we did not calculate the apparent clearance or the apparent volume of distribution for GHQ242, as in the absence of a clear assignment of the terminal half-life to elimination for this compound, possible conclusions may very well be prone to misinterpretation. However, the apparent clearance and the apparent volume of distribution were similar for GHQ168 and GHQ243 (Table 2).
FIG 5.
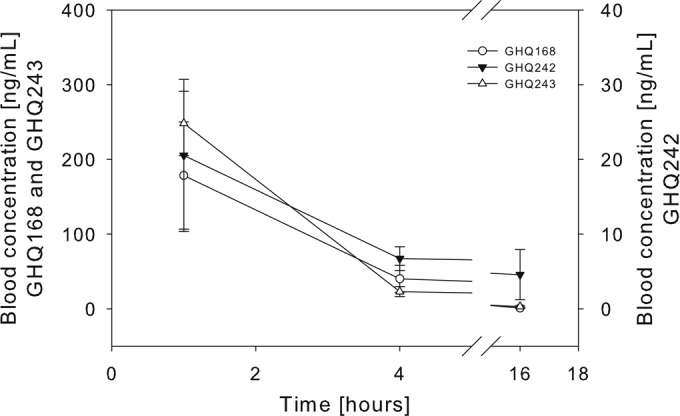
Mean blood concentration-versus-time profile ± SD.
TABLE 2.
PK parameters for GHQ168, GHQ242, and GHQ243a
| Compound | Mouse no. | t1/2 (h) | CL (ml/h) | V (liters/kg BW) | AUC0–∞ (ng · h/ml) |
|---|---|---|---|---|---|
| GHQ168 | 1 | 1.6 | 93 | 8 | 945 |
| 2 | 2.3 | 118 | 15 | 742 | |
| 3 | 2.6 | 125 | 19 | 701 | |
| GHQ242 | 1 | * | * | * | * |
| 2 | 3.7 | 4,792 | 1,014 | 120 | |
| 3 | 23 | 4,232 | 5,608 | 135 | |
| 4 | 2.3 | 2,686 | 357 | 213 | |
| GHQ243 | 1 | 2.4 | 402 | 55 | 1,350 |
| 2 | 3.1 | 819 | 148 | 663 | |
| 3 | 2.3 | 693 | 92 | 784 | |
| 4 | 1.8 | 830 | 86 | 6,542 |
t1/2, half-life; CL, clearance; V, volume of distribution; BW, body weight; AUC0–∞, area under the concentration-time curve from time zero to infinity. *, calculation of PK parameters was not possible due to flip-flop kinetics.
In vivo efficacy studies.
The dose and its regimen were simulated through PBPK modeling prior to in vivo studies (see Fig. S5A to C in the supplemental material). Two in vivo models were used and are referred to as the stringent mouse model, in which 8 doses were administered twice daily throughout days 3 to 6 postinfection, and the early-treatment model, in which 8 doses were administered twice daily throughout days 1 to 4. Treatment with all three compounds tested, especially GHQ168, showed in vivo activity, whereas no treatment did not (Fig. 6A). All infected mice treated with compound GHQ168 were free of parasites for 10 days (tail blood examinations; microscopy test) and relapsed on day 14. One mouse treated with GHQ168 died after the 5th treatment without parasitemia. Test animals treated with compounds GHQ242 and GHQ243 did not show parasitemia on day 7 but relapsed on day 10 (GHQ242) and day 14 (GHQ243) postinfection. The mean survival durations (MSDs) of mice in the study with the stringent mouse model and medicated with GHQ168, GHQ242, GHQ243, and the control treatment were 17, 14, 12, and 9 days, respectively (Table 3), meaning that statistically significant differences were observed for GHQ168 versus the control (P < 0.001), whereas insignificant results were obtained for GHQ242 and GHQ243 versus the control. On the basis of this outcome and corroboration of the results with those of the in vitro microbiological assays, GHQ168 was further profiled in the early-treatment model (Fig. 6B). GHQ168 administration prevented parasitemia in all animals (Fig. 6B). However, one mouse died after the 5th dose and two mice died after the 8th dose but showed no external signs of toxicity or parasitemia. The MSDs for the three remaining mice were >32 days (end of study; all surviving mice were free of parasites, as determined by tests of tail blood). This resulted in statistically significant differences in the MSD for GHQ168 versus that for the control (P < 0.001). A similar efficacy of melarsoprol (2 mg/kg) and pentamidine isethionate (5 mg/kg) was reported in previous studies for surviving mice (28). However, in this study, the mice were infected with T. b. brucei.
FIG 6.

(A) Activities of GHQ168, GHQ242, and GHQ243 in the stringent mouse model (n = 4). Statistically significant differences in MSD between the group treated with GHQ168 (17 days) and the control group (9 days) were observed (P < 0.001). (B) Mice treated with GHQ168 in the easy-to-cure mouse model (n = 6). Again, statistically significant differences in MSD were observed between the two groups (P < 0.001).
TABLE 3.
Trial design and results of the in vivo efficacy studies
| Study model | Compound | Treatment period (days) postinfection | Dose (mg/kg b.i.d.a) | No. of mice cured/no. infected | MSDb | MRDc |
|---|---|---|---|---|---|---|
| Stringent | Control | 0/4 | 9 | |||
| GHQ168 | 3–6 | 3.5 | 0/4d | 17 | 14 | |
| GHQ242 | 3–6 | 22.9 | 0/4 | 14 | 10 | |
| GHQ243 | 3–6 | 21.7 | 0/4 | 12 | 11 | |
| Early treatment | Control | 0/4 | 7 | |||
| GHQ168 | 1–4 | 3.5 | 3/6e | >32 | >32 |
b.i.d., twice a day.
MSD, mean survival duration (days postinfection).
MRD, mean relapse duration (days), where relapse is defined as the presence of parasites in blood.
One mouse died during the treatment but did not have parasites in its blood. Data for mice that died during the treatment but without parasites were excluded from the calculation of MSD and MRD, as the cause of death was unrelated to parasitemia.
Three mice died during and after the treatment but did not have parasites in their blood. Data for mice that died during the treatment but without parasites were excluded from the calculation of MSD and MRD, as the cause of death was unrelated to parasitemia.
DISCUSSION
In a previous study, a morpholino-substituted quinolone amide (Fig. 1) was identified to be highly active against T. b. brucei and T. b. rhodesiense (9). However, this substance was practically insoluble in aqueous media (Table 1), thereby making in vivo studies challenging. Thus, the aim of the study reported here was to overcome the solubility limitations and to characterize the compound class concerning physicochemical and biological properties as well as profiling these in in vivo pharmacokinetic and efficacy experiments for the first time.
The solubility could be enhanced by using an appropriate formulation (Fig. 3) as well as by making structural variations to the hit/lead compound (Fig. 4). The introduction of amino functions as well as the replacement of phenyl rings with pyridine rings especially resulted in compounds with lower lipophilicities and, hence, increased solubilities, and the oxalate salts of the active bases particularly increased the solubility. However, the solubility enhancement by these structural modifications was achieved at the expense of activity, i.e., a decrease of the IC50s of at least 10-fold toward T. b. brucei (Table 1). Since the cytotoxicity, determined in L6 cells, macrophages, kidney cells, and hepatocytes (Table 1), was found to be almost equal to or even higher than that of the lead compound, the selectivity deteriorated. Nevertheless, compounds GHQ242 and GHQ243 were considered for use in in vivo studies because both substances showed (i) rather high levels of activity against T. b. rhodesiense (IC50s, 2 nM and 18 nM for GHQ242 and GHQ243, respectively, after 72 h), (ii) low cytotoxicity, and (iii) sufficient solubility, as determined by PKPD modeling. Since GHQ168 still ranked among the most active compounds with an IC50 value (72 h) of 1 nM against T. b. rhodesiense, a formulation improving the kinetic solubility by a factor of 6,000 was developed (Fig. 3), thereby opening this promising compound to efficacy studies. The spray-drying technology has been applied industrially for decades, and in the meantime, spray drying has become a well-understood process that is easily scaled up from developmental batch sizes. The spray drying of GHQ168 was a robust and efficient process, facilitating the amorphization of the drug substance while maintaining its stability in Eudragit L100. In order to avoid the use of blood concentration-time levels beyond the therapeutic window, the corresponding doses for the in vivo studies for intraperitoneal administration as well as the study design were predicted by means of PBPK simulations.
Since pharmacokinetic properties govern the activities of drugs, the most important properties were evaluated. Whereas GHQ168 and GHQ243 exhibited high and moderate levels of human serum albumin binding (81% and 46%), an almost exhaustive albumin binding was found for GHQ242. Therefore, the fraction unbound was very low for GHQ242, and provided that the dissociation kinetics (which were not determined) are slow, this pattern may potentially challenge the use of this compound in the future, at least for parenteral use. Interestingly, the pharmacokinetic profile of GHQ242 reflected observations analogous to those found for albumin binding. In spite of the fact that the same dose (in moles) of GHQ242 as GHQ168 and GHQ243 was applied, PK concentrations were roughly 10-fold lower upon i.p. administration. Possible explanations for this low PK concentration profile include (i) local precipitation upon i.p. injection (suggested by the fluctuating PK profile [see below]) or (ii) extensive binding to the extracellular matrix and other surfaces at the site of injection and/or at other sites (suggested by the albumin binding [see above]), or (iii) both. One may speculate that the initially paradox observation that following GHQ242 administration some blood concentrations exceeded those in previous samples reflects slow drug release from precipitated drug following i.p. injection. Further studies, including analysis of pharmacokinetic profiles following i.v. administration, are required to detail these findings. However, in light of the possibility of GHQ242 precipitation at the injection site, the half-life calculated from the pharmacokinetic profile (Fig. 5; see also Fig. S7 in the supplemental material) might not reflect metabolism (i.e., the elimination half-life) but might reflect the slow release of molecules from the GHQ242 precipitate at the injection site (the half-life of molecules being released from the precipitate). With that in mind, we did not calculate the apparent clearance and apparent volume of distribution for GHQ242 because, since data from i.v. profiles were missing, we were unable to clearly assign the terminal half-life to metabolism or drug release kinetics from a possible precipitate formed upon administration. These pharmacokinetic data sets indicated that the successful development of GHQ242 as a parenteral form is more risky than the development of GHQ168 or GHQ243 as a parenteral form. However, GHQ242 may be successfully presented as an oral formulation, thereby overcoming the possibility of local precipitation upon injection. The possible successful use of GHQ242 as an oral formulation was also suggested by the permeation results from the Caco-2 cell monolayer studies (see Fig. S4 in the supplemental material).
The target site of these compounds is still unknown. Previously performed studies point to the kinetoplast as a possible target, as these quinolone amide derivatives impact kinetoplast segregation (9). However, sensitivity measurements with acriflavine-induced dyskinetoplastic T. b. brucei revealed a decrease of the IC50, but the IC50 was still in the low micromolar range (data not shown), indicating that the compounds are still highly active and may address another lethal target. This remains to be elucidated.
GHQ168 and its related molecules differ from the available quinolones marketed today, in that the free carboxyl group of the marketed quinolones is derivatized and therefore does not address the topoisomerase target in trypanosomes. Due to the absence of the carboxyl group in GHQ168, this compound might be associated with fewer or no side effect issues due to the complexation of Mg2+ and Ca2+; this will need to be experimentally confirmed in future studies.
The small number of animals exposed to these novel quinolones is a limitation of this study. Therefore, positive trends may be assumed from the data sets, but larger studies need to be performed to corroborate the findings and to obtain a final conclusion. As pointed out above, no final conclusion regarding the pharmacokinetic profile for GHQ242 can be drawn, as flip-flop kinetics may have occurred due to the possible precipitation of the i.p. administered molecule. Lastly, the solubility challenges for GHQ168 observed during the serum albumin binding studies render the outcome of this experiment for this compound questionable.
The in vivo studies suggested a statistically significant impact of GHQ168 on the mean survival time compared to that for the untreated controls in both efficacy studies (Fig. 6A and B). However, statistical calculations based on the small data set are critical and require careful interpretation. Future studies need to confirm the evidence provided here. Following i.p. administration of GHQ168, this test substance kept the mice free of parasites over a period of 14 days, on average (stringent mouse model), and over more than 32 days (easy-to-cure model). However, some mice without parasitemia died during the treatment period. Although no external signs of toxicity were observed in the efficacy studies (and in the PK studies), future studies need to detail the cause of these events. GHQ242 and GHQ243 had a positive trend on the MSD, but the outcome was not significantly different from that for the control (Fig. 6A). This observation suggests that GHQ242's 10 times higher in vitro activity against T. b. rhodesiense than that of GHQ243 (Table 1) compensated for its 10 times lower exposure than that of GHQ243 (Table 2, area under the concentration-time curve).
In conclusion, the quinolone amide skeleton was found to be a promising starting point for the development of highly active antitrypanosomal compounds. This study identified a lead compound with promising pharmacokinetic properties and efficacy, in that curing of T. b. rhodesiense-infected mice was achieved. The development of even more active compounds and compounds with greater solubility by variation of the skeleton is in progress, as is the search for the detailed mode of action.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elena Katzowitsch and Tobias Ölschläger (Institute for Molecular Infection Biology, University of Würzburg) for the cytotoxicity screening, Jennifer Rath and Antje Fuss (Medical Mission Institute, Würzburg, Germany) for testing the compounds against T. b. brucei, Alena Zikova (University of South Bohemia, Czech Budejovice, Czech Republic) for the dyskinetoplast experiments, Annette Albrecht (Institute of Pharmacy and Food Chemistry, University of Würzburg) for testing GHQ168 in the HPRT assay, Marius Gareis (Institute of Pharmacy and Food Chemistry, University of Würzburg) for performing the studies on the oxidative metabolism of GH168, and Lena Lauber for support with the Caco-2 cell assays. V79 cells were kindly provided by H. Glatt, German Institute of Human Nutrition, Potsdam, Germany.
We gratefully acknowledge the Deutsche Forschungsgemeinschaft (SFB 630) and the Bayerische Forschungsstiftung (grant Springs and Parachutes) for financial support, as well as ACC GmbH (Analytical Clinical Concepts GmbH, Leidersbach, Germany) for financial support, support with statistics (Volker Guth), and support with analytical work (Martin Barkworth).
We declare no conflict of interest.
Funding Statement
Financial support (consumables) and equipment were received from the funding sources.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01757-15.
REFERENCES
- 1.Franco JR, Simarro PP, Diarra A, Jannin JG. 2014. Epidemiology of human African trypanosomiasis. Clin Epidemiol 6:257–275. doi: 10.2147/CLEP.S39728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stich A, Ponte-Sucre A, Holzgrabe U. 2013. Do we need new drugs against human African trypanosomiasis? Lancet Infect Dis 13:733–734. doi: 10.1016/S1473-3099(13)70191-9. [DOI] [PubMed] [Google Scholar]
- 3.Pena I, Pilar Manzano M, Cantizani J, Kessler A, Alonso-Padilla J, Bardera AI, Alvarez E, Colmenarejo G, Cotillo I, Roquero I, de Dios-Anton F, Barroso V, Rodriguez A, Gray DW, Navarro M, Kumar V, Sherstnev A, Drewry DH, Brown JR, Fiandor JM, Julio Martin J. 2015. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource. Sci Rep 5:8771. doi: 10.1038/srep08771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maser P, Wittlin S, Rottmann M, Wenzler T, Kaiser M, Brun R. 2012. Antiparasitic agents: new drugs on the horizon. Curr Opin Pharmacol 12:562–566. doi: 10.1016/j.coph.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Bringmann G, Hoerr V, Holzgrabe U, Stich A. 2003. Antitrypanosomal naphthylisoquinoline alkaloids and related compounds. Pharmazie 58:343–346. [PubMed] [Google Scholar]
- 6.Nenortas E, Kulikowicz T, Burri C, Shapiro TA. 2003. Antitrypanosomal activities of fluoroquinolones with pyrrolidinyl substitutions. Antimicrob Agents Chemother 47:3015–3017. doi: 10.1128/AAC.47.9.3015-3017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keiser J, Burri C. 2001. Evaluation of quinolone derivatives for antitrypanosomal activity. Trop Med Int Health 6:369–389. doi: 10.1046/j.1365-3156.2001.00713.x. [DOI] [PubMed] [Google Scholar]
- 8.Ma X, Zhou W, Brun R. 2009. Synthesis, in vitro antitrypanosomal and antibacterial activity of phenoxy, phenylthio or benzyloxy substituted quinolones. Bioorg Med Chem Lett 19:986–989. doi: 10.1016/j.bmcl.2008.11.078. [DOI] [PubMed] [Google Scholar]
- 9.Hiltensperger G, Jones NG, Niedermeier S, Stich A, Kaiser M, Jung J, Puhl S, Damme A, Braunschweig H, Meinel L, Engstler M, Holzgrabe U. 2012. Synthesis and structure-activity relationships of new quinolone-type molecules against Trypanosoma brucei. J Med Chem 55:2538–2548. doi: 10.1021/jm101439s. [DOI] [PubMed] [Google Scholar]
- 10.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 11.Heinze A, Holzgrabe U. 2006. Determination of the extent of protein binding of antibiotics by means of an automated continuous ultrafiltration method. Int J Pharm 311:108–112. doi: 10.1016/j.ijpharm.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 12.Hubatsch I, Ragnarsson EG, Artursson P. 2007. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc 2:2111–2119. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- 13.Raz B, Iten M, Grether-Buhler Y, Kaminsky R, Brun R. 1997. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T. b. rhodesiense and T. b. gambiense) in vitro. Acta Trop 68:139–147. doi: 10.1016/S0001-706X(97)00079-X. [DOI] [PubMed] [Google Scholar]
- 14.Papadopoulou MV, Bloomer WD, Lepesheva GI, Rosenzweig HS, Kaiser M, Aguilera-Venegas B, Wilkinson SR, Chatelain E, Ioset JR. 2015. Novel 3-nitrotriazole-based amides and carbinols as bifunctional antichagasic agents. J Med Chem 58:1307–1319. doi: 10.1021/jm5015742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larson EM, Doughman DJ, Gregerson DS, Obritsch WF. 1997. A new, simple, nonradioactive, nontoxic in vitro assay to monitor corneal endothelial cell viability. Invest Ophthalmol Vis Sci 38:1929–1933. [PubMed] [Google Scholar]
- 16.Muth M, Hoerr V, Glaser M, Ponte-Sucre A, Moll H, Stich A, Holzgrabe U. 2007. Antitrypanosomal activity of quaternary naphthalimide derivatives. Bioorg Med Chem Lett 17:1590–1593. doi: 10.1016/j.bmcl.2006.12.088. [DOI] [PubMed] [Google Scholar]
- 17.Bradley MO, Bhuyan B, Francis MC, Langenbach R, Peterson A, Huberman E. 1981. Mutagenesis by chemical agents in V79 Chinese hamster cells: a review and analysis of the literature. A report of the Gene-Tox Program. Mutat Res 87:81–142. [DOI] [PubMed] [Google Scholar]
- 18.Schumacher DM, Metzler M, Lehmann L. 2005. Mutagenicity of the mycotoxin patulin in cultured Chinese hamster V79 cells, and its modulation by intracellular glutathione. Arch Toxicol 79:110–121. doi: 10.1007/s00204-004-0612-x. [DOI] [PubMed] [Google Scholar]
- 19.Di L, Kerns EH, Hong Y, Chen H. 2005. Development and application of high throughput plasma stability assay for drug discovery. Int J Pharm 297:110–119. doi: 10.1016/j.ijpharm.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 20.Jones HM, Gardner IB, Watson KJ. 2009. Modelling and PBPK simulation in drug discovery. AAPS J 11:155–166. doi: 10.1208/s12248-009-9088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poulin P, Theil FP. 2002. Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci 91:1358–1370. doi: 10.1002/jps.10128. [DOI] [PubMed] [Google Scholar]
- 22.Poulin P, Theil FP. 2002. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci 91:129–156. doi: 10.1002/jps.10005. [DOI] [PubMed] [Google Scholar]
- 23.Berezhkovskiy LM. 2004. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci 93:1628–1640. doi: 10.1002/jps.20073. [DOI] [PubMed] [Google Scholar]
- 24.Berezhkovskiy LM. 2004. Determination of volume of distribution at steady state with complete consideration of the kinetics of protein and tissue binding in linear pharmacokinetics. J Pharm Sci 93:364–374. doi: 10.1002/jps.10539. [DOI] [PubMed] [Google Scholar]
- 25.Brun R, Schumacher R, Schmid C, Kunz C, Burri C. 2001. The phenomenon of treatment failures in human African trypanosomiasis. Trop Med Int Health 6:906–914. doi: 10.1046/j.1365-3156.2001.00775.x. [DOI] [PubMed] [Google Scholar]
- 26.Williams RO III, Watts AB, Miller DA. 2012. Formulating poorly water soluble drugs. Springer, New York, NY. [Google Scholar]
- 27.Ponte-Sucre A, Gulder T, Wegehaupt A, Albert C, Rikanovic C, Schaeflein L, Frank A, Schultheis M, Unger M, Holzgrabe U, Bringmann G, Moll H. 2009. Structure-activity relationship and studies on the molecular mechanism of leishmanicidal N,C-coupled arylisoquinolinium salts. J Med Chem 52:626–636. doi: 10.1021/jm801084u. [DOI] [PubMed] [Google Scholar]
- 28.Milliken GA, Johnson DE. 2009. Analysis of messy data: designed experiments. Chapman & Hall/CRC, Boca Raton, FL. [Google Scholar]
- 29.Baltz T, Baltz D, Giroud C, Crockett J. 1985. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J 4:1273–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.