

ABSTRACT
Mutation of the LMNA gene, encoding nuclear lamin A and lamin C (hereafter lamin A/C), is a common cause of familial dilated cardiomyopathy (DCM). Among Finnish DCM patients, the founder mutation c.427T>C (p.S143P) is the most frequently reported genetic variant. Here, we show that p.S143P lamin A/C is more nucleoplasmic and soluble than wild-type lamin A/C and accumulates into large intranuclear aggregates in a fraction of cultured patient fibroblasts as well as in cells ectopically expressing either FLAG- or GFP-tagged p.S143P lamin A. In fluorescence loss in photobleaching (FLIP) experiments, non-aggregated EGFP-tagged p.S143P lamin A was significantly more dynamic. In in vitro association studies, p.S143P lamin A failed to form appropriate filament structures but instead assembled into disorganized aggregates similar to those observed in patient cell nuclei. A whole-genome expression analysis revealed an elevated unfolded protein response (UPR) in cells expressing p.S143P lamin A/C. Additional endoplasmic reticulum (ER) stress induced by tunicamycin reduced the viability of cells expressing mutant lamin further. In summary, p.S143P lamin A/C affects normal lamina structure and influences the cellular stress response, homeostasis and viability.
KEY WORDS: Lamin, Dilated cardiomyopathy, Laminopathy, ER stress, UPR
Highlighted Article: Mutant p.S143P lamin A/C causes familial dilated cardiomyopathy (DCM). It is shown that this protein affects normal lamina structure and influences the cellular stress response, homeostasis and viability.
INTRODUCTION
The nuclear lamina is a protein meshwork located between the inner nuclear membrane and chromatin. The main components of the lamina are the type V intermediate filament proteins called lamins (Goldman et al., 1986). The lamins provide structural shape to the nucleus and are involved in various biological processes, such as chromatin organization, the DNA damage response, gene transcription, cell cycle progression and cell migration (Dechat et al., 2010). In humans, the two major A-type lamins, lamin A and lamin C (hereafter lamin A/C), as well as two minor isoforms, lamin AΔ10 and lamin C2 are produced from the LMNA gene through alternative splicing (Burke and Stewart, 2013). The major B-type lamins, lamin B1 and lamin B2, are encoded by the LMNB1 and LMNB2 genes, respectively. The latter gene can also produce the germ-cell-specific isoform lamin B3 (Furukawa and Hotta, 1993). Structurally, the lamin monomer consists of an α-helical rod domain, flanked by a short unstructured N-terminal domain (‘head’) and a long non-α-helical C-terminal domain (‘tail’) harboring an immunoglobulin-like fold segment (Dechat et al., 2010). The rod domain drives the self-assembly of lamin monomers into dimers in vitro and it is of principal importance for the regular lateral assembly of lamins (Ben-Harush et al., 2009; Heitlinger et al., 1992). Multiple dimers form head-to-tail chains, which can assemble laterally to form filaments and further large paracrystalline arrays (Stuurman et al., 1998). The formation of the lamina in vivo, however, is less well understood (Herrmann and Foisner, 2003). A-type and B-type lamins form separate networks that overlap in the nuclear periphery and interact with various lamina-associated proteins (Shimi et al., 2008). Importantly, a fraction of lamins can also be found throughout the nucleoplasm where they interact with nucleoplasmic binding partners such as lamina-associated polypeptide 2 (LAP2α, also known as TMPO), nuclear actin, chromatin and transcription regulatory proteins (Dorner et al., 2007). Notably, ∼10% of A-type lamins can be extracted using low concentrations of non-ionic detergents (Kolb et al., 2011).
Over 450 mutations in the LMNA gene lead to a group of diseases collectively called laminopathies (http://www.umd.be/LMNA/). These include dilated cardiomyopathy (DCM), skeletal myopathies, familial partial lipodystrophy, peripheral neuropathy and the premature aging disease Hutchinson–Gilford progeria syndrome (Worman, 2012). Mutations in the LMNA gene are, after those in titin (TTN), the second most common cause of familial DCM (Hershberger et al., 2013). Moreover, DCM patients with LMNA mutations frequently suffer from atrioventricular conduction defects and have a significantly increased risk of sudden death due to ventricular arrhythmias (Hershberger et al., 2013). The molecular pathobiology behind LMNA-associated DCM is still poorly understood and remains to be explored. The current knowledge is mostly based on research utilizing a few mouse models such as LMNAH222P/H222P knock-in mice, a model for Emery–Dreifuss muscular dystrophy (EDMD) and DCM (Arimura et al., 2005). Abnormal activation of mitogen-activated protein kinase (MAPK) pathways (ERK1/2, JNK), as well as of Akt and mammalian target of rapamycin (mTOR) has been observed in cardiac muscle from LMNAH222P/H222P mice (Choi and Worman, 2013; Muchir et al., 2009a). Interestingly, drugs inhibiting the MAPK pathway (MEK1 and MEK2) improve cardiac function and prolong survival of LMNAH222P/H222P mice (Wu et al., 2011). In support for this, MAPK inhibitors protect induced pluripotent stem cell (iPSC)-derived cultured LMNAR225X/WT cardiomyocytes from apoptosis after electrical stimulation (Siu et al., 2012). Furthermore, LMNAN195K/N195K mice develop DCM and show alterations in cytoskeletal protein structure and focal adhesions as well as impairment of nuclear translocation and signaling downstream of the mechanosensitive transcription factor megakaryoblastic leukaemia 1 (MKL1) (Ho et al., 2013).
In the Finnish population, the founder mutation p.S143P in LMNA accounts for ∼7% of all the familial DCM cases (Kärkkäinen et al., 2004). The patients carrying the heterozygous p.S143P LMNA mutation show a progressive atrioventricular conduction defect, left ventricular systolic dysfunction and dilatation leading to pacemaker implantation, end-stage heart failure or sudden death. Whether cells expressing p.S143P lamin A/C share any pathognomonic features with other DCM-associated LMNA mutants is unclear, and understanding of the underlying molecular mechanisms is crucial in finding new therapies to treat this devastating disease. Here, we show that the p.S143P lamin A/C is more soluble, more mobile and less incorporated into the lamina than the wild-type lamin A/C. These features are due to inability of mutant lamin to form proper filaments in vitro, which eventually leads to formation of dense lamin aggregates in vitro and in vivo. At the cellular level, expression of p.S143P lamin A/C activates the unfolded protein response (UPR), which has an effect on cell viability. These pathognomonic features might explain how p.S143P lamin A/C eventually causes accumulated cellular stress and cell death in cardiac tissue among DCM patients.
RESULTS
p.S143P mutant lamin A/C localizes preferentially to the nucleoplasm and forms intranuclear aggregates in cells from patients
Confocal microscopy analysis showed that the great majority of cultured p.S143P patient fibroblasts had normal symmetrically shaped, round to oval nuclei (Fig. 1A). However, there was more nucleoplasmic lamin A/C and reduced signal intensity at the lamina region in p.S143P cells when compared to controls (Fig. 1A). In addition, ∼7% of cells from patients contained lamin-A/C-positive aggregates in the nucleoplasm at early passage cultures (Fig. 1B, n=300). These cells typically showed slight condensation of chromatin, suggesting that aggregation of mutant lamin A/C might interfere with chromatin organization. Given that one of the patients had gone through cardiac transplantation surgery, we also stained cardiac tissue from the explanted heart for lamin A. Lamin-A-positive intranuclear aggregates were noticed in 3% (n=300) of cardiomyocytes from the patient with p.S143P mutation, but not in the control tissue obtained from a DCM patient without LMNA mutation (Fig. 1C). This suggests that mutant lamin A/C also forms aggregates in vivo.
Fig. 1.

p.S143P mutant lamin A/C is more nucleoplasmic than WT lamin A/C and forms aggregates in cultured fibroblasts from patients. (A) Confocal sections of cultured fibroblasts from a healthy control and a patient carrying the p.S143P mutation stained for lamin A/C. DNA is visualized with Hoechst 33342. Scale bars: 5 µm. (B) Confocal projection showing a p.S143P patient fibroblast stained for lamin A/C, lamin B1 and DNA. Lamin-A/C-positive aggregates are detectable in the lamina region. Scale bar: 5 µm. (C) Immunohistochemical lamin A staining from the left ventricle tissue of a DCM patient tested negative for LMNA mutations (WT) and from a patient with the p.S143P mutation. Lamin-A-positive nucleoplasmic aggregates (arrowheads) are visible at high magnification. Scale bars: 50 µm (low magnification), 10 µm (high magnification). (D) Cultured p.S143P patient and control fibroblasts were treated with 0.5% Triton X-100 before fixation and stained for lamin A/C. The fluorescence intensities at the lamina and nucleoplasm were determined and the values were normalized to those of untreated control cells. Examples of the chosen areas for measurement are shown in red. Scale bar: 10 µm. (E) Mean±s.e.m. intensity values in the nucleoplasm and at the lamina region were determined from untreated and Triton-X-100-treated control cells and cells from patients (n=20 for both cell lines). *P<0.05; **P<0.01; ***P<0.001; ns, not significant (Student's t-test). (F) Cultured control and patient fibroblasts were harvested and extracted with either 0.1% Triton X-100 or 0.1% Triton X-100+500 mM NaCl (soluble fractions). The fractions were separated by centrifugation; 2% of the original sample, 15% of each soluble fraction and 2% of the pellet (insoluble fraction) were separated by SDS-PAGE and analyzed by immunoblotting.
During interphase, the nuclear lamina is a highly stable structure and resistant to extraction with non-ionic detergents and high concentrations of salt (Lehner et al., 1986). The localization and solubility of lamin A/C in the patient and control cells was further studied by measuring lamin A/C staining intensities in the nucleoplasm and at the lamina region using confocal sections of individual cells fixed either before or after treatment with 0.5% Triton X-100 (Fig. 1D,E). In unextracted cells from patients, the lamin A/C signal was significantly higher in the nucleoplasm (P<0.001) and reduced at the lamina region (P=0.012) when compared to unextracted control cells. This shows that p.S143P lamin A/C to a significant extent mislocalizes to the nucleoplasm. In Triton-X-100-extracted control cells, the lamin A/C signal was reduced by 24% in the nucleoplasm (P=0.068) and 1% at the lamina region (P=0.855). In comparison, the lamin A/C signal in Triton-X-100-extracted cells from patients was reduced by 71% in the nucleoplasm (P<0.001) and 23% at the lamina region (P=0.001) suggesting that mutant lamin A/C is less tightly bound to intranuclear elements and to the lamina.
These results were supported by a biochemical solubility assay where cultured cells were harvested and sequentially extracted first with a low Triton X-100 concentration (0.1%) and then with Triton X-100 in high-salt solution (Fig. 1F). The soluble fractions obtained from each step after centrifugation and the remaining pellet fractions were further analyzed by immunoblotting. In control cells, lamin A/C was essentially recovered in the insoluble pellet, whereas lamin A/C was found to some extent in the detergent- and high-salt-soluble supernatant fraction in cells from patients, demonstrating a higher solubility of lamin A/C in the cells from patients relative to controls. These results suggest that the p.S143P mutation might interfere with the assembly and incorporation of lamin A/C into the lamina.
p.S143P mutation alters the localization and mobility of lamin A/C in transfected cells
To confirm the impact of the p.S143P mutation on lamin A/C localization, we transfected control fibroblasts with pFLRU lentiviral vectors encoding either FLAG-tagged wild-type lamin A (FLAG–WT-LA) or p.S143P lamin A (FLAG–p.S143P-LA). FLAG–WT-LA localized mainly to the lamina and to a lesser extent to the nucleoplasm (Fig. 2A). In contrast, FLAG–p.S143P-LA was evenly distributed throughout the nucleus and less prominent at the lamina region. In ∼10% of FLAG–p.S143P-expressing cells, large nucleoplasmic aggregates were observed whereas such nuclear structures were not detected in FLAG–WT-LA-expressing cells (Fig. 2A; P<0.0004, n=300). Similarly, in HeLa cells transiently transfected with either GFP-tagged wild-type (GFP–WT-LA) or mutant lamin A (GFP–p.S143P-LA), the latter was more prominent in the nucleoplasm and formed nucleoplasmic aggregates in ∼10% of the cells (Fig. 2B). Solid electron-dense aggregates at the lamina region were also detected by transmission electron microscopy (TEM) in HeLa cells expressing either FLAG- or GFP-tagged p.S143P lamin A (Fig. 2C,D). Given that antibodies might not penetrate the interior of such dense aggregates, this is likely to explain the ‘ring-like’ staining pattern frequently seen with anti-lamin A/C and -FLAG antibodies on large aggregates (Figs 1B and 2A). These results confirm that the p.S143P mutation interferes with intranuclear localization of lamin A/C and induces lamin aggregation.
Fig. 2.

FLAG-tagged and GFP-tagged p.S143P lamin A form intranuclear aggregates. (A) Fibroblasts from a healthy donor were transduced with lentiviral vectors encoding either FLAG-tagged WT lamin A or p.S143P lamin A and stained with anti-FLAG and anti-lamin A (LA) antibodies, and DAPI. Single mid-plane confocal sections and maximum projections are shown. Scale bar: 10 μm. (B) HeLa cells were transfected with either GFP–WT-LA or GFP–p.S143P-LA and stained with anti-lamin B1 (LB1) antibodies and DAPI. Single mid-plane confocal sections and maximum projections are shown. Scale bar: 10 μm. (C,D) TEM analysis of HeLa cells transfected with either FLAG-tagged WT lamin A or p.S143P lamin A (C) or GFP-tagged WT lamin A or p.S143P lamin A (D) showing electron-dense nuclear aggregates at the lamina region in mutant-transfected cells. Scale bars: 5 µm (upper left panel in C), 2 µm (other panels in C,D).
To investigate the impact of the p.S143P mutation on the dynamics and mobility of lamin A in living cells, we performed fluorescence loss in photobleaching (FLIP) analysis on HeLa cells transiently transfected with either GFP-tagged WT lamin A or p.S143P lamin A. One half of the nucleus was repeatedly photobleached for 8-s time intervals (Fig. 3A) and the fluorescence signals were determined in the nucleoplasmic and lamina regions from non-bleached areas before and after each photobleaching (Fig. 3B,C). In both the nucleoplasm and lamina, the fluorescence loss of non-aggregated GFP–p.S143P-LA was more significant than that of GFP–WT-LA (∼92% versus ∼65%, and ∼75% versus ∼31%, respectively). These results reveal that the p.S143P lamin A exhibits higher mobility and is less firmly anchored at the nuclear envelope in interphase nuclei than its WT counterpart.
Fig. 3.

GFP–p.S143P-LA is more mobile than GFP–WT-LA. (A) HeLa cells transiently expressing either GFP-tagged WT or p.S143P lamin A were used for fluorescence loss in photobleaching (FLIP) experiments. GFP-tagged lamin A was photobleached in the boxed areas every 8 s, immediately followed by image acquisition. Fluorescence signals were measured in a non-bleached area of the lamina and the nucleoplasm. (B) The fluorescence intensities of GFP–WT-LA in nucleoplasm (open circle) and the lamina (open circles) and GFP–p.S143P-LA in nucleoplasm (filled circle) and the lamina (filled circles) were plotted as a function of time. Mean values from seven different cells are shown in the graphs. (C) The fluorescence loss of GFP–WT-LA and GFP–p.S143P-LA in the nucleoplasm and lamina were calculated by curve-fitting and are plotted in the histograms (mean±s.d.). The difference in fluorescence loss between GFP–WT-LA and GFP–p.S143P-LA was statistically significant (**P<0.005, Student's t-test).
p.S143P alters assembly properties of lamin A/C in vitro
Owing to increased solubility and mobility found in living cells, we speculated that the assembly of p.S143P-lamin A into higher-order structures, such as dimeric protofilaments, single filaments and their highly organized, laterally associated filament arrays (referred to as paracrystals) might be affected. To test this hypothesis in detail, bacterially expressed recombinant WT lamin A and p.S143P lamin A were assembled in vitro and the assembly products were analyzed with TEM. After 10 min under lamin-A-specific filament assembly conditions, the WT lamin A formed typical extended filaments with a diameter of ∼10 nm (Fig. 4A), very similar to those originally found with lamin A from rat liver nuclei (Aebi et al., 1986). In contrast, the p.S143P lamin A did not form any extended filaments but only unordered, partially fibrillary associations during the same period of time, indicating a disruption of the regular assembly pathway of lamin dimers under these conditions (Fig. 4D). Obviously, the precise lateral contacts needed to structurally stabilize assembly intermediates for regular elongation failed. When the assembly of complex paracrystalline fiber arrays was investigated, which involves dialysis of reconstituted dimers from high-salt conditions (300 mM NaCl) to more physiological buffer conditions (50 mM NaCl), the WT lamin A assembled into long laterally associated fiber arrays with the typical 24.5 nm axial-repeat pattern (Fig. 4B,C). The distinct organization of dimeric and tetrameric strands in these structures has recently been elucidated by cryo-electron tomography (Ben-Harush et al., 2009). In stark contrast p.S143P lamin A formed disorganized, often roundish electron-dense aggregates of heterogeneous size and shape (Fig. 4E). These aggregates were on occasion connected by long fibrillar cables lacking the typical axial-repeat texture (Fig. 4F). Hence, longitudinal assembly by the head-to-tail association of dimers does occur, but the precise lateral arrangement of dimeric or tetrameric protofilaments is corrupted.
Fig. 4.

p.S143P lamin A/C is unable to spontaneously assemble into normal filament polymers in vitro. Electron microscope images of negatively stained WT lamin A (LA) (A–C,G), p.S143P lamin A (D–F,H) and a 1:1 mixture of WT lamin A and p.S143P lamin A (I) assemblies formed in vitro. (A,D) Filament-forming conditions. (B,C,E–I) Paracrystal assembly. Scale bars: 0.5 μm (A,B,D,E), 0.2 μm (C,F), 0.25 μm (G–I).
In order to mimic the condition when both proteins are synthesized in similar amounts, we analyzed how WT lamin A and p.S143P lamin A behave in a coassembly situation. Both proteins were mixed in their soluble state in 1:1 ratio and dialyzed stepwise into physiological conditions (100 mM NaCl). Here, the WT lamin A formed the typical paracrystalline fiber arrays (Fig. 4G), whereas the p.S143P lamin A never yielded this high degree of organization exhibiting fibrillar strands loosely associating laterally (Fig. 4H). Most notably, the mixture of both proteins also never attained the highly organized lateral association of elongated fibers indicating that the mutant lamin dominantly impacts on the lateral association (Fig. 4I). At 50 mM NaCl, when the paracrystal formation of WT lamin A is further enhanced (Fig. 4B), only huge irregular aggregates were found (data not shown). Moreover, forcing the formation of heterodimers consisting of WT and mutant protein, by mixing them in 8 M urea and reconstituting them together, generated practically identical assembly pattern to that shown in Fig. 4I (data not shown). In summary, these results demonstrate that the p.S143P mutation impairs the ability of lamin A to form well organized, laterally ordered filament arrays, the endpoint of which in vitro is paracrystalline filament arrays. Thus, this change in the intrinsic properties of the mutant protein (i.e. the breaking of the α-helical structure in coil 1B) provides a molecular explanation for the lamin A/C aggregates observed in mutant-expressing cells.
The impact of the p.S143P mutation on gene expression
Several studies have shown that mutations in lamin A/C cause altered gene expression (Muchir et al., 2007; Puckelwartz et al., 2011). To study whether p.S143P lamin A/C affects general transcription we carried out a whole-genome expression analysis on patient and control cell lines using an Illumina Human HT-12 v.4 expression beadchip. Validation of the data showed high correlation (r=0.9667) using Pearson metrics (Fig. 5A) and a heatmap showed two different clusters representing controls and patients (Fig. 5B). The full ranked gene list, ordered in decreased order of significance, was analyzed for functional trends using the GSEA software (www.broadinstitute.org/gsea/). The analysis was focused on a curated set with Reactome pathway and false discovery rate (FDR) with q-value of <0.05 (Croft et al., 2014; Milacic et al., 2012). This generated 27 different gene sets that are shown in Table S1. The leading-edge genes for all gene sets were further valuated and the most significant genes with a fold change >1.4 and P-value <0.05 are listed in Fig. 5C. The list includes genes involved in the UPR (XBP1, EXOSC7 and ASNS) and genes affected by the UPR such as CDK5RAP2 and APP, which are involved in ER-stress-mediated apoptosis, and CHST3, which is upregulated by ER stress (Kang et al., 2012; Okuda et al., 2014; Takahashi et al., 2009). The other genes on the list are involved in lysosomal degradation of heparan sulfate and dermatan sulfate (IDS), glycosylation (MAN1A1), ATP release (PANX1) and RNA polymerase II stimulation (TCEA1). We decided to focus more carefully on the UPR-related genes and the enrichment score from the GSEA analysis of the UPR is shown in Fig. 5D. This analysis runs down the ranked lists and the leading-edge proteins are the hits before the enrichment score for the gene set.
Fig. 5.

Whole-genome gene expression analysis of patient and control fibroblasts identifies ER-stress-related pathways. (A) Pearson metric analysis of seven p.S143P and four control cell lines show high overall correlation between the samples. (B) Heatmap of microarray (47323 transcripts) clustering cell lines into two categories. (C) Table of leading-edge genes with fold change (FC) >1.4 and P<0.05. (D) GSEA enrichment scores (ES) for UPR. The green line shows the enrichment score and the black lines represent hits as the analysis runs down the ranked list. (E) Schematic image of XBP1 mRNA splicing and structure of XBP1u and XBP1s proteins. Both proteins contain a basic leucine zipper (bZIP) domain. In addition, XBP1u contains a hydrophobic region (HR) that attaches it to the ER membrane whereas XBP1s contains a transcriptional activation domain (AD). Primers designed for total and spliced XBP1 mRNA are shown below mRNA (black bars). (F) qRT-PCR analysis of genes involved in the UPR. The mean±s.d. of fold change values from the different analyses is shown. (G) Western blot analysis of ER-stress-related proteins phosphorylated (p)eIF2α and XBP1 from two patient cell lines and from age-matched controls. eIF2α is phosphorylated and XBP1 increasingly spliced (XBP1s) in cells from patients. Actin was used as a loading control.
ER stress is typically caused by an accumulation of unfolded or misfolded proteins in the ER that further leads to activation of UPR. The most fundamental ER stress sensor is inositol-requiring enzyme 1 (IRE1α, also known as ERN1) that, upon ER stress, mediates a cytosolic splicing of transcription factor X-box-binding protein 1 (XBP1) mRNA (Calfon et al., 2002; Lee et al., 2002). In unstressed cells the mRNA resides in the cytosol as a precursor called unspliced XBP1 (XBP1u) mRNA. When IRE1α senses ER stress, a 26 nucleotide intron (XBP1 c.530-c.557) is cleaved off, generating the activated form, spliced XBP1 (XBP1s) mRNA (Fig. 5E) (Yoshida et al., 2001). Both proteins are translated although the unconventional translational splicing creates a frame-shift downstream in the coding sequence thus generating a longer sequence for XBP1s (Fig. 5E).
The change in expression of genes involved in the UPR were further chosen for confirmation using quantitative real-time PCR (qRT-PCR) (Fig. 5F). The exosome component 7 (EXOSC7) and phenol sulfotransferase 1A, member 4 (SULT1A4) were found to be upregulated in the qRT-PCR analysis, whereas aspargine synthase (ASNS) and signal sequence receptor (SSR1) were downregulated in cells from patients relative to controls (Fig. 5F). XBP1 expression was downregulated, confirming the gene expression results. This prompted us to design primers specific for the spliced XBP1s mRNA (Fig. 5E). There was a 4.1-fold increase in expression of XBP1s mRNA and also significantly more XBP1s protein in cells from patients than from controls, suggesting an activation of the UPR (Fig. 5F,G). Accordingly, eukaryotic translation initiation factor 2A (eIF2α), which represses protein translation during ER stress, was hyperphosphorylated in cells from patients indicating an elevated UPR (Fig. 5G).
The expression of p.S143P lamin A/C sensitizes cells to additional stress
In order to confirm that UPR-related signaling is due to mutant lamin A/C, we further transduced control fibroblasts with the FLRU double vectors encoding either FLAG–WT-LA or FLAG–p.S143P-LA and short hairpin RNA (shRNA) targeted to 3′UTR of LMNA to silence endogenous lamin A/C. The cells (p4) were followed for 2 days and further analyzed by western blotting. Similar to untreated cells from patients, we noticed upregulation of phosphorylated (p)eIF2α and XBP1s in mutant-expressing cells (Fig. 6A). In addition, immunofluorescence showed an increased level of nuclear XBP1 in FLAG–p.S143P-LA-expressing cells when compared to controls (Fig. 6B). This nuclear translocation of XBP1 was most evident in FLAG–p.S143P-LA-expressing cells showing nuclear lamin aggregates (Fig. 6B). These results show that ectopic expression of p.S143P lamin A leads to an UPR in cultured normal cells.
Fig. 6.

Expression of FLAG–p.S143P-LA causes upregulation and increased nuclear localization of ER-stress-related markers. (A) Western blot analysis of fibroblasts expressing either FLAG–WT-LA or FLAG–p.S143P-LA. The ER-stress-related proteins phosphorylated (p)eIF2α and the spliced form of XBP1 are detected in FLAG–p.S143P-LA-expressing cells. GAPDH was used as loading control. (B) Confocal microscopy image of FLAG–WT-LA-and FLAG–p.S143P-LA-expressing cells stained for FLAG and XBP1. The cells with intranuclear aggregates show increased amounts of nuclear XBP1. Scale bar: 10 µm. (C) The number of cells with lamin A/C intranuclear aggregates was determined before and after treatment with tunicamycin (Tm) for 4, 6 and 24 h (n=300). (D) The number of viable adherent cells was determined in FLAG–WT-LA- and FLAG–p.S143P-LA-expressing cell cultures and in primary patient and control fibroblasts before and after treatment with tunicamycin for 24 h. (E) The number of cells with aggregates was determined in FLAG–p.S143P-LA-expressing fibroblasts treated with either proteasome inhibitor (MG132), autophagy inhibitor wortmannin (WM) or autophagy enhancer rapamycin (RM) for 24 h. (F) Untreated or tunicamycin-treated FLAG–p.S143P-LA-expressing cells show reduced number of cells with aggregates after incubation with 4-PBA for 4 h. The mean±s.d. from three separate experiments are shown for C–F. *P<0.05, **P<0.01 (Student's t-test).
Expression of FLAG–p.S143P-LA in normal control fibroblasts generated intranuclear lamin A/C aggregates in 13% of cells in early passage cultures 11 days after transduction whereas similar structures were not detected in FLAG–WT-LA-expressing cells (P<0.0004, n=300). We speculated that additional induction of ER stress could further increase the number of cells with lamin A/C aggregates. To test this, the cells expressing either FLAG–WT-LA or FLAG–p.S143P-LA were treated with the ER stress inducer tunicamycin, which inhibits protein N-glycosylation (Dorner et al., 1990). After 4 h of tunicamycin treatment the number of cells with FLAG-positive aggregates increased to 17.3% in mutant-expressing cells but decreased significantly thereafter (5.3% at 24 h, Fig. 6C). Because this decrease could be due to increased cell death, we further analyzed the number of viable adherent cells prior to and after treatment with tunicamycin. There was a significant reduction in the cell number in both FLAG–WT-LA- and FLAG–p.S143P-LA-expressing cell cultures after 24 h treatment with tunicamycin (Fig. 6D). However, the number of mutant-expressing cells was reduced to a higher degree (47.5% versus 57.7%, P<0.03). Similarly, there was a significant difference on cell viability between patient and control fibroblasts after tunicamycin treatment (40.8% versus 64.7%, P<0.01, Fig. 6D). In summary, the cells expressing p.S143P lamin A are more sensitive to additional ER stress induced with tunicamycin and this might reduce the cell viability.
Upon ER stress, misfolded proteins are targeted to ER-associated degradation (ERAD), which degrades proteins through the proteasome–ubiquitin pathway (Sommer and Jentsch, 1993; Werner et al., 1996). To confirm this, we treated the fibroblasts with a proteasome inhibitor (MG132) for 24 h, which significantly increased the number of cells with aggregates (from 5% to 8.3%, P<0.01) in FLAG–p.S143P-LA-expressing cells (Fig. 6E). However, neither treatment with the autophagy inhibitor wortmannin nor the autophagy enhancer rapamycin for 24 h had a significant effect on the numbers of cells with aggregates (Fig. 6E). These results suggest that the mutant lamin A is degraded through the proteasome–ubiquitin machinery. To further test how attenuation of ER stress affects aggregate formation, we treated the FLAG–p.S143P-LA-expressing cells with the small-molecule chaperone 4-phenylbutyric acid (4-PBA), which inhibits the UPR. 4-PBA treatment for 4 h significantly decreased the number of cells with lamin A/C aggregates both in tunicamycin-treated (10% vs 2.8%, P<0.01, Fig. 6F) and untreated cells (6.17% to 1.41%, P<0.01, Fig. 6F). In summary, these results indicate that ER stress promotes aggregation of p.S143P lamin A/C.
DISCUSSION
In this study, we have analyzed in detail the impact of the c.427T>C LMNA mutation (p.S143P) on the molecular properties of lamin A/C, nuclear structure and gene expression. The motivation of the study was that p.S143P is the most prevalent DCM-associated mutation in the Finnish population (Kärkkäinen et al., 2004). Our imaging data demonstrates that the p.S143P lamin A/C is enriched in the nucleoplasm and appears less abundant at the nuclear lamina when compared to WT lamin A/C. Extraction of cells with non-ionic detergent showed a dramatic reduction in the nucleoplasmic pool of p.S143P lamin A/C and to a lesser extent at the lamina, indicating that the mutant lamin A/C is more soluble and less tightly incorporated into the lamina. Concordantly, in transfected cells the nucleoplasmic non-aggregated pool of GFP–p.S143P-LA appeared highly mobile in live-cell imaging suggesting that it is not as strongly bound to stable intranuclear elements (e.g. chromatin) but rather floats in the nucleoplasm and thus exhibits a different molecular organization. Interestingly, p.S143P-lamin A/C also formed nucleoplasmic aggregates in a fraction of the patient and transduced cells in early passage cultures. Moreover, the outcome of the in vitro assembly experiments points to substantial alterations in the assembly properties of the mutated protein and hence provides a starting point for the explanation of the features observed in cells (Fig. 4). p.S143P lamin A failed to accomplish proper interactions from the earliest stage of assembly, suggesting that the ‘nucleoplasmicity’ is due to impaired recognition by the assembly machinery. Instead of forming long fibers containing laterally interacting head-to-tail lamin polymers, the p.S143P-lamin A eventually formed roundish aggregates of up to 1 µm in diameter similar to the aggregates observed in patient and FLAG–p.S143P-LA-expressing cells.
Serine 143 is in a c-position of the heptad repeat pattern in the coil 1B of the α-helical rod, and the introduction of a proline in this position would be expected to disturb the α-helical structure in this segment of the protein. Single amino acid changes do not usually impact coiled-coil formation as demonstrated for other intermediate filament proteins such as desmin (Bär et al., 2006). However, as the geometry of the coiled-coil dimer is altered locally by the proline residue, it seems plausible that the mutation terminates normal lateral association reactions of lamin A/C and leads to disorganized assembly during filament elongation, although the head-to-tail overlap reaction is not directly affected. Regarding the possible phosphorylation of serine 143, recent studies have found p.S143 not to be a phosphorylation site (Kochin et al., 2014).
Out of various LMNA mutations causing DCM, ∼64% are found in the rod domain (Scharner et al., 2010). The closest DCM mutations to p.S143P are p.E161K (Sebillon et al., 2003), p.N195K (Östlund et al., 2001), p.R190Q (Cowan et al., 2010) and p.R189W (Botto et al., 2010). In the close proximity, p.E145K mutation causes progeria with early onset of cardiovascular defects, and cultured cells from a patient with this mutation showed highly lobulated nuclei and clustered chromosome organization (Taimen et al., 2009). Different from p.S143P, the p.E145K lamin A is capable of forming filament structures but the lateral assembly of protofilaments is disorganized in vitro (Taimen et al., 2009). Interestingly, another mutation in residue 143, of serine to phenylalanine, results in skeletal myopathy with mild progeroid features (Kandert et al., 2007). Surprisingly, the p.S143F lamin A/C incorporates into the lamina but leads to large nuclear herniations or ‘blebs’ that were not detected in cells expressing p.S143P lamin A (Bercht Pfleghaar et al., 2015). These examples show that the localization, as well as the molecular properties, of the substituted amino acid are highly crucial and might have profound effects on the assembly. The fact that so many DCM-associated LMNA mutations cause impaired assembly indicates that incompletely assembled and mechanically fragile lamina might reduce cellular stress tolerance, in particular of cells that are under permanent mechanical stress such as muscle cells, which would be the precursor to myopathy. In support for this, heterozygous LMNA+/− mice, which express ∼50% less lamin A/C, have impaired activation of mechanosensitive genes (Cupesi et al., 2010), reduced contractility of cardiomyocytes and eventually develop atrioventricular block and DCM (Nikolova et al., 2004; Wolf et al., 2008). Therefore, the current hypothesis on the pathogenesis of DCM holds that LMNA mutation cause haploinsufficiency or/and dominant-negative structural effects leading to a mechanically unstable lamina and/or alterations in gene expression (Cattin et al., 2013). Both these features have also been observed in cells expressing p.S143P lamin A/C.
The gene expression analysis demonstrated elevated UPR in p.S143P patient fibroblasts compared to the controls (Fig. 5). The p.S143P cells showed an increase in spliced XBP1 mRNA, a unique UPR-linked transcript whose splicing is dependent on the phosphorylation of eIF2α (Majumder et al., 2012). Accordingly, immunoblotting revealed upregulation of spliced XBP1 and hyperphosphorylation of eIF2α in mutant-expressing cells relative to controls. The activation of the UPR in fibroblasts overexpressing FLAG–p.S143P-LA suggests that the mutant lamin truly elicits ER stress. Similarly, the number of cells with intranuclear lamin A aggregates increased when cells were treated with a chemical inducer of ER stress, indicating that overload or saturation of ER stress correlates with the formation of lamin A/C aggregates in cells. As we demonstrated that the p.S143P lamin A/C is unable to incorporate properly into the lamina, the mislocalized mutant lamin A/C might directly or indirectly lead to ER stress. Furthermore, the viability of cells expressing p.S143P lamin A was decreased in the presence of tunicamycin relative to controls. ER-stressed cells expressing a misfolded protein typically experience cycles of activation and recovery, pending between maintaining the protein level and degrading unfolded protein. This physiological challenge might threaten the long-term survival of the cells and also sensitize them to additional stress. The UPR is associated with the pathophysiology of the heart and is involved in several processes such as ischemia–reperfusion, cardiac hypertrophy, heart failure, atherosclerosis and cardiomyopathy (Glembotski, 2007). In a study of explanted hearts from patients suffering from DCM, ER stress markers (GRP78, XBP1 and ATF6) were upregulated (Ortega et al., 2014). Furthermore, the expression of a mutant Lys-Asp-Glu-Leu (KDEL) receptor in mice activated ER stress and the mice developed DCM (Hamada et al., 2004). The mutant KDEL receptor disturbed retrograde transport between Golgi and ER and misfolded proteins accumulated in the ER. Our study is focused on an in vitro model and distinct cell types. However, the examples mentioned above show that observed alterations in ER-stress-related signaling might also be relevant for cardiac homeostasis. In further support for this, overactivation of MAPK signaling due to LMNA p.H222P mutation has been phenocopied in other cell types, such as HeLa cells in vitro, showing that the signaling defects caused by mutant lamin A/C are not restricted to cardiac tissue (Muchir et al., 2009b).
In summary, the p.S143P mutant protein is incapable of incorporating into the lamina, which might cause a mechanically fragile lamina and a reduction in cellular stress tolerance. These changes ultimately lead to the hallmark symptoms that reveal themselves in late life in DCM patients, such as decreased cardiac contractile force and irreversible cardiac tissue injury. In vitro assembly shows that the mutant lamin A/C aggregates also into large accumulations as readily observed in cells from patients as well as in transfected cells. The aggregation and mislocalization of mutant lamin also has an impact on cellular homeostasis, which we demonstrate by showing an increased activity of the UPR. It is possible that the UPR is unable to dispose of the misfolded protein through proteasomal degradation, intensifying the pathogenic state further and possibly being one reason behind the cardiac tissue injury. Understanding how aggregation of mutant lamin A/C leads to ER stress would potentially provide a target for therapy to inhibit the ER-stress-mediated cell injury in the future.
MATERIALS AND METHODS
Patients and isolation of primary cell lines
This study was approved by the Ethics Committees of the Hospital District of Helsinki and Uusimaa (HUS 387/13/03/2009) and Kuopio University Hospital. All procedures were undertaken with informed consent and according to the principles expressed in the Declaration of Helsinki. Seven patients carrying the p.S143P LMNA mutation were enrolled in the study. The age, gender and clinical symptoms of the patients are provided in Table S2. Skin biopsies from the patients and healthy donors were taken under local anesthesia (1% lidocaine), washed twice with PBS, cut into small pieces and attached to the bottom of the culture dish. MEM supplemented with 20% fetal bovine serum (FBS), 2 mM glutamine, vitamin solution, non-essential amino acids, and penicillin-streptomycin (all Invitrogen) was added and the fibroblasts were isolated by trypsination 2–3 weeks later after reaching confluent culture. Paraffin-embedded cardiac tissues were obtained from the archives of the Department of Pathology, Helsinki University Hospital.
Cell culture
Fibroblasts were cultured in MEM as described above. Lentiviral particles were produced in 293-T cells and used 1:1 for transduction of normal fibroblasts (Alastalo et al., 2015). HeLa cells were cultured in DMEM (supplemented with 10% FBS and penicillin-streptomycin) and transfected with TransIt HeLa Monster kit (Mirus, Madison, USA) according to the manufacturer's protocol. The medium was changed 16 h later and the cells were fixed or used for live-cell imaging at 48 h after transfection. ER stress was induced with 2 µg ml−1 tunicamycin (Sigma-Aldrich) for different time points. Cell counts were determined with a Bürker chamber. MG132 (10 µM, Sigma-Aldrich), wortmannin (10 µM, Enzo Life Sciences) and rapamycin (80 ng ml−1, Sigma-Aldrich) were all used for 24 h. For inhibition of ER stress, 10 µM 4-phenylbutyric acid (Sigma-Aldrich) was used for 4 h.
Plasmids
The CMV2-FLAG–LA and pEGFP-myc-LA constructs were cloned as described elsewhere (Moir et al., 2000; Taimen et al., 2009). The p.S143P mutation was introduced using Quick-Change II XL Site-Directed Mutagenesis kit (Agilent Technologies, CA) and the primers 5′-GGCTCTGCTGAACCCCAAGGAGGCCGC-3′ and 5′-GCGGCCTCCTTGGGGTTCAGCAGAGCC-3′. Inserts of wild-type (WT) lamin A (NM_005572.3) and p.S143P lamin A were subcloned into the lentiviral vector pFLAG-FLRU (kindly provided by Yunfeng Feng, Washington University, St Louis, MO) (Feng et al., 2010). The plasmids were used for lentiviral transduction as such or in combination with a shRNA-lamin A/C-knockdown insert. The shRNA for lamin A/C knockdown was: 5′-AGCAGTCTCTGTCCTTCGA-3′ with a loop of 5′-GCTTCCTGTCAC-3′ (Shimi et al., 2008). For the in vitro filament assembly analysis, the coding sequence (CDS) of wild-type lamin A with the last 18 codons removed was subcloned into the pET 24a plasmid.
Immunofluorescence and immunohistochemistry
Cells were grown on glass coverslips and fixed in 10% formalin followed by permeabilization with 0.1% Triton X-100. The primary antibodies used were mouse monoclonal anti-lamin A/C (1:100, clone 4C11, Cell Signaling), rabbit polyclonal anti-lamin A (1:1000, clone 26300, Abcam), goat polyclonal anti-LB1 (1:100, clone M20, Santa Cruz Biotechnology), mouse monoclonal anti-FLAG (1:250, clone SIG-25, Sigma-Aldrich), mouse monoclonal anti-FLAG (1:1600, clone 9A3, Cell Signaling) and polyclonal rabbit anti-X-box binding protein 1 (XBP1) (1:50, clone M-186, Santa Cruz Biotechnology). The secondary antibodies included rabbit and goat anti-mouse-IgG conjugated to Alexa Fluor 488, rabbit anti-goat-IgG conjugated to Alexa Fluor 555 and goat anti-rabbit-IgG conjugated to Alexa Fluor A555 (Molecular Probes). Hoechst 33342 (1 µg ml−1) or DAPI was used to visualize DNA. The cell images were collected with Zeiss LSM510 and LSM780 confocal microscopes as well as a 3i and Zeiss spinning disk confocal microscope, and were analyzed with the Zeiss Zen 2012 software and ImageJ. Paraffin-embedded cardiac tissue was stained with a mouse monoclonal anti-lamin A/C (1:1000, clone 626, Cell Signaling) antibody using a Ventana automated staining machine. The primary antibody was detected with Vectastain horseradish peroxidase (HRP)-conjugated anti-mouse-IgG secondary antibodies. The slides were counterstained with hematoxylin and observed with a Leica DM3000 microscope equipped with a Leica DFC 450 digital camera and Leica Application Suite version 4.0 (Leica Microsystems, Wetzlar, Germany).
Detergent extraction
Fibroblasts were grown on glass coverslips in two sets. Before fixation with 4% paraformaldehyde, one set was subjected to 0.5% Triton X-100 at 20°C for 5 min. After fixation both sets were permeabilized with 0.1% Triton X-100 and stained with an antibody against lamin A/C and with DAPI for DNA. Stacked images were captured with a Zeiss LSM780 confocal microscope. The mid-plane section was selected for analysis with ImageJ by measuring the average pixel intensity in a 1000-pixel selection of the lamina and nucleoplasm, respectively, excluding the nucleoli. Values were collected from 20 cells from each set (n=20). The data were normalized to untreated control cells for both the nucleoplasmic pool and the lamina pool, respectively. Statistical analysis of the mean±s.e.m. was carried out with a Student's t-test. Graphpad Prism (GraphPad Software, CA) was used for statistical analysis. For the biochemical solubility assay, cells were washed once with PBS and harvested by trypsinization. Cell pellets were sequentially extracted with 0.1% Triton X-100 and then with 0.1% Triton X-100 plus 500 mM NaCl in lysis buffer [10 mM Tris-HCl pH 7.4, 2 mM MgCl2, 1× protease inhibitor (Roche)] for 5 min on ice. The insoluble pellet and the soluble supernatant were separated with centrifugation and analyzed by immunoblotting.
Immunoblotting
Whole-cell lysates were solubilized in M-PER mammalian protein extraction reagent (Thermo Fisher Scientific, MA, USA) complemented with 1× protease and 1× phosphatase inhibitors. Protein concentration was measured using a NanoDrop spectrophotometer ND-1000. Cell lysates were mixed with 2× SDS-PAGE sample buffer and 20 µg protein was run on a 4–10% gradient gel (BioRad, CA). The primary antibodies were: mouse monoclonal anti-lamin A/C (1:1000, clone 4C11, Cell Signaling), mouse monoclonal anti-lamin A/C (1:150, clone 626, Novocastra, Wetzlar, Germany), mouse monoclonal anti-actin (1:1000, clone AC-40, Sigma-Aldrich), rabbit monoclonal anti-peIF2α (1:1000, clone 119A11, Cell Signaling), rabbit polyclonal anti-eIF2α (1:300, clone FL-315, Santa Cruz Biotechnology), rabbit polyclonal anti-XBP1 (1:200, clone M-186, Santa Cruz Biotechnology), and mouse monoclonal anti-FLAG (1:1000, clone SIG-25, Sigma-Aldrich). Secondary antibodies were HRP-conjugated donkey anti-rabbit-IgG and sheep anti-mouse-IgG (both from GE Healthcare).
TEM
Transfected HeLa cells were grown on Mattek gridded coverslips to facilitate localization of the same cells in fluorescence and TEM. The samples were fixed with 5% glutaraldehyde in 0.16 M s-collidine buffer, pH 7.4, postfixed with 2% OsO4 containing 3% potassium ferrocyanide for 2 h, dehydrated with a series of increasing ethanol concentrations (70%, 96% and twice at 100%) and embedded with a 45359 Fluka Epoxy Embedding Medium kit. Thin sections were cut with an ultramicrotome to a thickness of 70 nm, and stained with 1% uranyl acetate and 0.3% lead citrate. The images were collected with a JEOL JEM-1400 Plus TEM equipped with a OSIS Quemesa 11 Mpix bottom-mounted digital camera operated at 80 kV acceleration voltage.
Live-cell imaging
HeLa cells transiently expressing either GFP–WT-LA or GFP–p.S143P-LA were grown on LAB-TEK (Nalge Nunc International, Rochester, NY) single-well chambered coverslips. During imaging, the cells were maintained in Phenol-Red-free Leibovitz's L-15 Medium (Life Technologies) supplemented with 10% FCS and the temperature was maintained at 37°C with an air stream incubator (Nevtek, Burnsville, VA). The procedure for FLIP experiments was as previously described (Shimi et al., 2004). Briefly, GFP fluorescence in one half of the nucleus was repeatedly photobleached for 8 s with 488 nm of light every 8 s and time-lapse images were acquired after each photobleaching using an LSM 510 META microscope (Zeiss) equipped with an oil immersion objective lens (100×, 1.45 NA, PlanApo, Zeiss). Fluorescence intensity in the unbleached half of the nucleus was measured as a function of time. For data analysis, fluorescence intensity measured at every time point was normalized by the initial fluorescence intensity after extraction of the background signal. Curve-fitting with the following equation for one-phase decay was computationally calculated using GraphPad Prism6 (GraphPad Software).
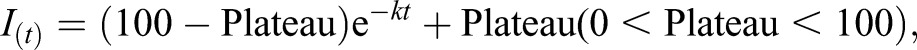 |
where I(t) is normalized fluorescence intensity at a time t, Plateau is the I value when t is infinite, k is the rate constant, which is expressed in reciprocal of time units. Fluorescence loss is calculated as the difference between 100 and Plateau. Statistical values were determined using the Student's t-test.
Protein chemical method
Recombinant proteins were produced according to standard procedures for the pET system (Foeger et al., 2006). For filament assembly, the protein was diluted to 0.2 g/l in storage buffer (10 mM Tris-HCl, pH 7.5, 300 mM NaCl, 8 M urea) and immediately dialyzed for 1 h at 37°C into dimer buffer (25 mM Tris-HCl buffer, pH 8.0, containing 250 mM NaCl, 1 mM DTT). The protein was further dialyzed at 37°C for 50 min into 25 mM Mes-NaOH, 250 mM NaCl, 1 mM DTT with different pH values (6.5, 7.0, 7.5 or 8.0). Samples were fixed for 1 min by addition of an equal volume of 0.2% glutaraldehyde. For paracrystal assembly, protein in storage buffer (0.5 g/l) was dialyzed into Tris buffer (10 mM Tris-HCl, pH 7.4, 2 mM EDTA and 1 mM DTT) containing 300 mM NaCl at 4°C overnight. The protein was diluted to 0.1 g/l and then dialyzed at room temperature into Tris-buffer with a consecutively reduced NaCl concentration (250 mM, 200 mM, 150 mM, 100 mM and 50 mM) for 20 min each. At each stage, samples were removed for electron microscopy. In the case of paracrystal formation, samples were used without fixation.
Microarray analysis
Patient and control fibroblasts were harvested at passage 4–5. Total RNA was extracted using an RNeasy kit (Qiagen, Hilden, Germany) and RNA was quality-tested with a NanoDrop spectrophotometer ND-1000. A whole-genome gene expression analysis using an Ilumina Human HT-12 v.4 Expression beadchip and data analysis was made at the Finnish microarray and Sequencing Centre (http://www.btk.fi/microarray-and-sequencing/). Further processing of the genes was made with the Gene Set Enrichment Analysis (GSEA) software and MSigDB (http://www.broadinstitute.org/gsea/index.jsp). The enrichment scores were calculated with permutation number set at 1000. The data was analyzed with a focus on curated gene sets from the Reactome pathway. The heatmap was created with a two-sided t-test using the GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E/) and the Pearson correlation analysis was performed with MeV software (Saeed et al., 2003).
qRT-PCR
1 µg RNA was converted into cDNA using a Tetro cDNA Synthesis kit (Bioline, London, UK). The amplification step was performed using a DyNAmo HS SYBR green qPCR kit (Thermo Fisher Scientific) following the manufacturer's protocol. The amplification was performed in triplicates; 15 min at 95°C followed by 40 cycles of 15 s at 94°C, 20 s at 60°C, 30 s at 72°C and finally 20 min at 80°C. The melting curve was assessed by 95°C for 15 s, 60°C for 15 s and 95°C for 15 s. Target genes were normalized to actin expression and values calculated using the 2−ΔΔCt method. The primers used were for EXOSC7, 5′-GAAGACCTCCGTGTGGATG-3 and 5′-CCACATCAGTTTCCACTTCG-3′; SULT1A4, 5′-ACGTGGCGGTCTCCTACTAC-3′ and 5′-GACACTTCTCCAGCCATGAA-3′; ASNS, 5′-TGGATACACCAACTGCTGCT-3′ and 5′-CGTGCACCTTCTTATGGTTG-3′; SSR1, 5′-CGGCAATGTATTCCAAGATG-3′ and 5′-AGGAGTTGATGAAGGCCAAC-3′; XBP1t, 5′-GGAGTTAAGACAGCGCTTGG-3′ and 5′-CACTGGCCTCACTTCATTCC-3′; and for XBP1s, 5′-CTGAGTCCGCAGCA GGTGCAG-3′ and 5′-ATCCATGGGGAGATGTTCTGG-3′.
Acknowledgements
We want to thank Sinikka Collanus (University of Turku) for her help with the immunohistohemical stainings and Dr Yunfeng Feng (Washington University, St Louis) for the lentiviral vectors. We acknowledge Markku Saari and Jouko Sandholm at the Cell Imaging Core (CIC) at the Turku Centre for Biotechnology (CBT) for assistance with imaging. We want to thank Markus Peurla and Jenni Laine at the Faculty of Medicine, University of Turku, for assistance with electron microscopy. We also want to thank the Finnish Microarray and Sequencing Centre at the CBT for assistance with microarray data. The collaboration between Taimen and Herrmann groups is part of NANONET COST Action BM1002.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
G.W., J.G., H.H. and P.T. designed and performed research, and wrote the manuscript. L.V., S.P.L., A.K., T.S. and M.M. performed experiments. T.H., M.K., L.O. and J.K. recruited patients and provided material. J.E.E. and R.D.G. provided material and contributed to writing the manuscript. All co-authors approved the final version of the manuscript.
Funding
This study was supported by the Sigrid Jusélius Foundation (Sigrid Juséliuksen Säätiö) (to P.T.); the Finnish Medical Foundation (Suomen Lääketieteen Säätiö) (to P.T.); the Academy of Finland (Suomen Akatemia) [grant number 290160 (to P.T.)]; the Finnish Foundation for Cardiovascular Research (Sydäntutkimussäätiö) (to P.T.); Paavo Nurmi Foundation (to P.T.); Medicinska Understödsföreningen Liv och Hälsa r.f. (to G.W.); Svenska Kulturfonden (to J.G.); the Hospital District of Southwest Finland, Turku University Hospital ERVA funds (to P.T.); the Progeria Research Foundation (to R.D.G); the National Institutes of Health - National Institute of General Medical Sciences (to R.D.G.); and the German Research Foundation (Deutsche Forschungsgemeinschaft) [grant number HE 1853, to H.H.]. Deposited in PMC for release after 12 months.
Data availability
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE82290.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.184150.supplemental
References
- Aebi U., Cohn J., Buhle L. and Gerace L. (1986). The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323, 560-564. 10.1038/323560a0 [DOI] [PubMed] [Google Scholar]
- Alastalo T.-P., West G., Li S.-P., Keinänen A., Helenius M., Tyni T., Lapatto R., Turanlahti M., Heikkilä P., Kääriäinen H. et al. (2015). LMNA mutation c.917T>G (p.L306R) leads to deleterious hyper-assembly of lamin A/C and associates with severe right ventricular cardiomyopathy and premature aging. Hum. Mutat. 36, 694-703. 10.1002/humu.22793 [DOI] [PubMed] [Google Scholar]
- Arimura T., Helbling-Leclerc A., Massart C., Varnous S., Niel F., Lacène E., Fromes Y., Toussaint M., Mura A.-M., Keller D. I. et al. (2005). Mouse model carrying H222P-lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 14, 155-169. 10.1093/hmg/ddi017 [DOI] [PubMed] [Google Scholar]
- Bär H., Mücke N., Ringler P., Müller S. A., Kreplak L., Katus H. A., Aebi U. and Herrmann H. (2006). Impact of disease mutations on the desmin filament assembly process. J. Mol. Biol. 360, 1031-1042. 10.1016/j.jmb.2006.05.068 [DOI] [PubMed] [Google Scholar]
- Ben-Harush K., Wiesel N., Frenkiel-Krispin D., Moeller D., Soreq E., Aebi U., Herrmann H., Gruenbaum Y. and Medalia O. (2009). The supramolecular organization of the C. elegans nuclear lamin filament. J. Mol. Biol. 386, 1392-1402. 10.1016/j.jmb.2008.12.024 [DOI] [PubMed] [Google Scholar]
- Bercht Pfleghaar K., Taimen P., Butin-Israeli V., Shimi T., Langer-Freitag S., Markaki Y., Goldman A. E., Wehnert M. and Goldman R. D. (2015). Gene-rich chromosomal regions are preferentially localized in the lamin B deficient nuclear blebs of atypical progeria cells. Nucleus 6, 66-76. 10.1080/19491034.2015.1004256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto N., Vittorini S., Colombo M. G., Biagini A., Paradossi U., Aquaro G. and Andreassi M. G. (2010). A novel LMNA mutation (R189W) in familial dilated cardiomyopathy: evidence for a ‘hot spot’ region at exon 3: a case report. Cardiovasc. Ultrasound 8, 9 10.1186/1476-7120-8-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke B. and Stewart C. L. (2013). The nuclear lamins: flexibility in function. Nat. Rev. Mol. Cell Biol. 14, 13-24. 10.1038/nrm3488 [DOI] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G. and Ron D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92-96. 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- Cattin M.-E., Muchir A. and Bonne G. (2013). ‘State-of-the-heart’ of cardiac laminopathies. Curr. Opin. Cardiol. 28, 297-304. 10.1097/HCO.0b013e32835f0c79 [DOI] [PubMed] [Google Scholar]
- Choi J. C. and Worman H. J. (2013). Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy 9, 110-111. 10.4161/auto.22403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan J., Li D., Gonzalez-Quintana J., Morales A. and Hershberger R. E. (2010). Morphological analysis of 13 LMNA variants identified in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Circ. Cardiovasc. Genet. 3, 6-14. 10.1161/CIRCGENETICS.109.905422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft D., Mundo A. F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M. R. et al. (2014). The reactome pathway knowledgebase. Nucleic Acids Res. 42, D472-D477. 10.1093/nar/gkt1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupesi M., Yoshioka J., Gannon J., Kudinova A., Stewart C. L. and Lammerding J. (2010). Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J. Mol. Cell. Cardiol. 48, 1290-1297. 10.1016/j.yjmcc.2009.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechat T., Adam S. A., Taimen P., Shimi T. and Goldman R. D. (2010). Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2, a000547 10.1101/cshperspect.a000547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner A. J., Wasley L. C., Raney P., Haugejorden S., Green M. and Kaufman R. J. (1990). The stress response in chinese hamster ovary cells. regulation of ERp72 and protein disulfide isomerase expression and secretion. J. Biol. Chem. 265, 22029-22034. [PubMed] [Google Scholar]
- Dorner D., Gotzmann J. and Foisner R. (2007). Nucleoplasmic lamins and their interaction partners, LAP2alpha, rb, and BAF, in transcriptional regulation. FEBS J. 274, 1362-1373. 10.1111/j.1742-4658.2007.05695.x [DOI] [PubMed] [Google Scholar]
- Edgar R., Domrachev M. and Lash A.E. (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207-210. 10.1093/nar/30.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Nie L., Thakur M. D., Su Q., Chi Z., Zhao Y. and Longmore G. D. (2010). A multifunctional lentiviral-based gene knockdown with concurrent rescue that controls for off-target effects of RNAi. Genomics Proteomics Bioinformatics 8, 238-245. 10.1016/S1672-0229(10)60025-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foeger N., Wiesel N., Lotsch D., Mücke N., Kreplak L., Aebi U., Gruenbaum Y. and Herrmann H. (2006). Solubility properties and specific assembly pathways of the B-type lamin from caenorhabditis elegans. J. Struct. Biol. 155, 340-350. 10.1016/j.jsb.2006.03.026 [DOI] [PubMed] [Google Scholar]
- Furukawa K. and Hotta Y. (1993). cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J. 12, 97-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glembotski C. C. (2007). Endoplasmic reticulum stress in the heart. Circ. Res. 101, 975-984. 10.1161/CIRCRESAHA.107.161273 [DOI] [PubMed] [Google Scholar]
- Goldman A. E., Maul G., Steinert P. M., Yang H. Y. and Goldman R. D. (1986). Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc. Natl. Acad. Sci. USA 83, 3839-3843. 10.1073/pnas.83.11.3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada H., Suzuki M., Yuasa S., Mimura N., Shinozuka N., Takada Y., Suzuki M., Nishino T., Nakaya H., Koseki H. et al. (2004). Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol. Cell. Biol. 24, 8007-8017. 10.1128/MCB.24.18.8007-8017.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitlinger E., Peter M., Lustig A., Villiger W., Nigg E. A. and Aebi U. (1992). The role of the head and tail domain in lamin structure and assembly: analysis of bacterially expressed chicken lamin A and truncated B2 lamins. J. Struct. Biol. 108, 74-91. 10.1016/1047-8477(92)90009-Y [DOI] [PubMed] [Google Scholar]
- Herrmann H. and Foisner R. (2003). Intermediate filaments: novel assembly models and exciting new functions for nuclear lamins. Cell. Mol. Life Sci. 60, 1607-1612. 10.1007/s00018-003-3004-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger R. E., Hedges D. J. and Morales A. (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531-547. 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- Ho C. Y., Jaalouk D. E., Vartiainen M. K. and Lammerding J. (2013). Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507-511. 10.1038/nature12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandert S., Luke Y., Kleinhenz T., Neumann S., Lu W., Jaeger V. M., Munck M., Wehnert M., Muller C. R., Zhou Z. et al. (2007). Nesprin-2 giant safeguards nuclear envelope architecture in LMNA S143F progeria cells. Hum. Mol. Genet. 16, 2944-2959. 10.1093/hmg/ddm255 [DOI] [PubMed] [Google Scholar]
- Kang M.-J., Chung J. and Ryoo H. D. (2012). CDK5 and MEKK1 mediate pro-apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat. Cell Biol. 14, 409-415. 10.1038/ncb2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkkäinen S., Heliö T., Miettinen R., Tuomainen P., Peltola P., Rummukainen J., Ylitalo K., Kaartinen M., Kuusisto J., Toivonen L. et al. (2004). A novel mutation, Ser143Pro, in the lamin A/C gene is common in finnish patients with familial dilated cardiomyopathy. Eur. Heart J. 25, 885-893. 10.1016/j.ehj.2004.01.020 [DOI] [PubMed] [Google Scholar]
- Kochin V., Shimi T., Torvaldson E., Adam S. A., Goldman A., Pack C. G., Melo-Cardenas J., Imanishi S. Y., Goldman R. D. and Eriksson J. E. (2014). Interphase phosphorylation of lamin A. J. Cell. Sci. 127, 2683-2696. 10.1242/jcs.141820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb T., Maass K., Hergt M., Aebi U. and Herrmann H. (2011). Lamin A and lamin C form homodimers and coexist in higher complex forms both in the nucleoplasmic fraction and in the lamina of cultured human cells. Nucleus 2, 425-433. 10.4161/nucl.2.5.17765 [DOI] [PubMed] [Google Scholar]
- Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T., Yoshida H., Mori K. and Kaufman R. J. (2002). IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452-466. 10.1101/gad.964702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner C. F., Furstenberger G., Eppenberger H. M. and Nigg E. A. (1986). Biogenesis of the nuclear lamina: in vivo synthesis and processing of nuclear protein precursors. Proc. Natl. Acad. Sci. USA 83, 2096-2099. 10.1073/pnas.83.7.2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder M., Huang C., Snider M. D., Komar A. A., Tanaka J., Kaufman R. J., Krokowski D. and Hatzoglou M. (2012). A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol. Cell. Biol. 32, 992-1003. 10.1128/MCB.06665-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milacic M., Haw R., Rothfels K., Wu G., Croft D., Hermjakob H., D'Eustachio P. and Stein L. (2012). Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers 4, 1180-1211. 10.3390/cancers4041180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir R. D., Yoon M., Khuon S. and Goldman R. D. (2000). Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 151, 1155-1168. 10.1083/jcb.151.6.1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchir A., Pavlidis P., Decostre V., Herron A. J., Arimura T., Bonne G. and Worman H. J. (2007). Activation of MAPK pathways links LMNA mutations to cardiomyopathy in emery-dreifuss muscular dystrophy. J. Clin. Invest. 117, 1282-1293. 10.1172/JCI29042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchir A., Shan J., Bonne G., Lehnart S. E. and Worman H. J. (2009a). Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet. 18, 241-247. 10.1093/hmg/ddn343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchir A., Wu W. and Worman H. J. (2009b). Reduced expression of A-type lamins and emerin activates extracellular signal-regulated kinase in cultured cells. Biochim. Biophys. Acta 1792, 75-81. 10.1016/j.bbadis.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova V., Leimena C., McMahon A. C., Tan J. C., Chandar S., Jogia D., Kesteven S. H., Michalicek J., Otway R., Verheyen F. et al. (2004). Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest. 113, 357-369. 10.1172/JCI200419448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H., Tatsumi K., Horii-Hayashi N., Morita S., Okuda-Yamamoto A., Imaizumi K. and Wanaka A. (2014). OASIS regulates chondroitin 6-O-sulfotransferase 1 gene transcription in the injured adult mouse cerebral cortex. J. Neurochem. 130, 612-625. 10.1111/jnc.12736 [DOI] [PubMed] [Google Scholar]
- Ortega A., Roselló-Lletí E., Tarazón E., Molina-Navarro M. M., Martínez-Dolz L., González-Juanatey J. R., Lago F., Montoro-Mateos J. D., Salvador A., Rivera M. et al. (2014). Endoplasmic reticulum stress induces different molecular structural alterations in human dilated and ischemic cardiomyopathy. PLoS ONE 9, e107635 10.1371/journal.pone.0107635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Östlund C., Bonne G., Schwartz K. and Worman H. J. (2001). Properties of lamin A mutants found in emery-dreifuss muscular dystrophy, cardiomyopathy and dunnigan-type partial lipodystrophy. J. Cell. Sci. 114, 4435-4445. [DOI] [PubMed] [Google Scholar]
- Puckelwartz M. J., Depreux F. F. S. and McNally E. M. (2011). Gene expression, chromosome position and lamin A/C mutations. Nucleus 2, 162-167. 10.4161/nucl.2.3.16003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A. I., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., Thiagarajan M. et al. (2003). TM4: a free, open-source system for microarray data management and analysis. BioTechniques 34, 374-378. [DOI] [PubMed] [Google Scholar]
- Scharner J., Gnocchi V. F., Ellis J. A. and Zammit P. S. (2010). Genotype-phenotype correlations in laminopathies: how does fate translate? Biochem. Soc. Trans. 38, 257-262. 10.1042/BST0380257 [DOI] [PubMed] [Google Scholar]
- Sebillon P., Bouchier C., Bidot L. D., Bonne G., Ahamed K., Charron P., Drouin-Garraud V., Millaire A., Desrumeaux G., Benaiche A. et al. (2003). Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J. Med. Genet. 40, 560-567. 10.1136/jmg.40.8.560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi T., Koujin T., Segura-Totten M., Wilson K. L., Haraguchi T. and Hiraoka Y. (2004). Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J. Struct. Biol. 147, 31-41. 10.1016/j.jsb.2003.11.013 [DOI] [PubMed] [Google Scholar]
- Shimi T., Pfleghaar K., Kojima S.-I., Pack C.-G., Solovei I., Goldman A. E., Adam S. A., Shumaker D. K., Kinjo M., Cremer T. et al. (2008). The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 22, 3409-3421. 10.1101/gad.1735208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu C.-W., Lee Y.-K., Ho J. C.-Y., Lai W.-H., Chan Y.-C., Ng K.-M., Wong L.-Y., Au K.-W., Lau Y.-M., Zhang J. et al. (2012). Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 4, 803-822. 10.18632/aging.100503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer T. and Jentsch S. (1993). A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature 365, 176-179. 10.1038/365176a0 [DOI] [PubMed] [Google Scholar]
- Stuurman N., Heins S. and Aebi U. (1998). Nuclear lamins: their structure, assembly, and interactions. J. Struct. Biol. 122, 42-66. 10.1006/jsbi.1998.3987 [DOI] [PubMed] [Google Scholar]
- Taimen P., Pfleghaar K., Shimi T., Moller D., Ben-Harush K., Erdos M. R., Adam S. A., Herrmann H., Medalia O., Collins F. S. et al. (2009). A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc. Natl. Acad. Sci. USA 106, 20788-20793. 10.1073/pnas.0911895106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Niidome T., Akaike A., Kihara T. and Sugimoto H. (2009). Amyloid precursor protein promotes endoplasmic reticulum stress-induced cell death via C/EBP homologous protein-mediated pathway. J. Neurochem. 109, 1324-1337. 10.1111/j.1471-4159.2009.06067.x [DOI] [PubMed] [Google Scholar]
- Werner E. D., Brodsky J. L. and McCracken A. A. (1996). Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA 93, 13797-13801. 10.1073/pnas.93.24.13797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C. M., Wang L., Alcalai R., Pizard A., Burgon P. G., Ahmad F., Sherwood M., Branco D. M., Wakimoto H., Fishman G. I. et al. (2008). Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell. Cardiol. 44, 293-303. 10.1016/j.yjmcc.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman H. J. (2012). Nuclear lamins and laminopathies. J. Pathol. 226, 316-325. 10.1002/path.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W., Muchir A., Shan J., Bonne G. and Worman H. J. (2011). Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 123, 53-61. 10.1161/CIRCULATIONAHA.110.970673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T. and Mori K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881-891. 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]