

Abstract
The vertebrate small intestine requires an enormous surface area to effectively absorb nutrients from food. Morphological adaptations required to establish this extensive surface include generation of an extremely long tube and convolution of the absorptive surface of the tube into villi and microvilli. In this Review, we discuss recent findings regarding the morphogenetic and molecular processes required for intestinal tube elongation and surface convolution, examine shared and unique aspects of these processes in different species, relate these processes to known human maladies that compromise absorptive function and highlight important questions for future research.
KEY WORDS: Absorptive surface, Intestinal lengthening, Mesenchymal clusters, Microvillus inclusion disease, Short bowel syndrome, Villus morphogenesis
Summary: This Review highlights the latest insights into the mechanisms underlying intestinal morphogenesis - from gut tube lengthening to villus and microvillus formation.
Introduction: development of the absorptive surface
The absorptive surface area of the adult intestine is estimated to be 30 m2 (323 square feet) in humans (Helander and Fandriks, 2014). Significant loss of this surface area can lead to malabsorption or intestinal failure, which is defined as insufficient nutritional uptake to sustain homeostasis (in adults) or growth (in children) (Goulet et al., 2004). Treatment of this condition generally requires prolonged intravenous nutritional supplementation (parenteral nutrition). In severe cases, transplantation is required. Appropriate development and homeostasis of the intestinal surface is therefore crucial for human health.
In vertebrates, generation of intestinal absorptive surface occurs in a stepwise fashion, beginning shortly after gastrulation, with the formation and polarization of the endodermal layer. This endodermal sheet then becomes molded into an epithelial tube that is surrounded by mesoderm. The complex events involved in the formation and subsequent regional patterning of the gut tube have been reviewed in depth elsewhere (Le Guen et al., 2015; Spence et al., 2011a; Zorn and Wells, 2009). Following gut tube patterning, three major morphogenetic processes contribute to the establishment of sufficient absorptive surface area for intestinal function: elongation of the tube, generation of finger-like projections called villi and formation of microvilli (the brush border) on the apical surface of each epithelial cell (Fig. 1).
Fig. 1.

Growth of mouse small intestinal apical surface. (A-C) Small intestinal elongation from E12.5 (A) to E14.5 (B) and E15.5 (C). Schematic illustrations of transverse slices (A′-C′) from each stage, shown in more detail in A″-C″, reveal changes in epithelial cell shape and apical surface expansion over time (epithelium in yellow; apical surface in dark green). The apical surface is flat before villus formation (A″,B″). This early pseudostratified epithelium is highly proliferative and its epithelial cells grow in height, becoming up to 50 µm tall just prior to villus formation (B″). At E15.5, villus emergence expands the apical surface (C′,C″). As villi form epithelial cells at the villus tip transition to a simple columnar epithelium, while the intervillus regions remain pseudostratified (C″). On emerged villi, brush border microvilli further amplify the apical surface area. C‴ represents higher magnification of a portion of two cells within the boxed region in C″; the gray box depicts a tight junction between two neighboring cells.
In this Review, we discuss the molecular and cellular events underlying each of these morphogenetic processes. We highlight how defects in these developmental events lead to disease in human and animal models and summarize how these phenotypes have furthered our understanding of the underlying developmental machinery. Finally, we provide insight into the major open questions in the field.
Gut tube lengthening
The remarkable length of the small intestine contributes greatly to its large absorptive surface area. A consensus estimate from several studies suggests that the average adult human small intestine measures just over 6 m (20 feet) (Gondolesi et al., 2012; Helander and Fandriks, 2014; Hounnou et al., 2002; Weaver et al., 1991). Much of this length is attained prenatally: at birth, the human intestine measures approximately 2.75 m – a remarkable 40% of its adult length (Struijs et al., 2009; Weaver et al., 1991) – and it already exhibits villi and microvilli that further amplify its surface area nearly 100-fold (Helander and Fandriks, 2014). Table 1 summarizes the milestones of intestinal development in humans and mice.
Table 1.
Key milestones of prenatal intestinal development in humans and mice, compiled from Baxter (1953), Grand et al. (1976), Matsumoto et al. (2002) and Moxey and Trier (1978)

Cellular basis of prenatal small intestinal lengthening
Although intestinal elongation continues after birth, events prior to birth are crucial to establish proper intestinal length. Importantly, prenatal gut elongation is a continuous process that takes place in several distinct morphological settings. Substantial tube lengthening takes place before villi form, but elongation continues during elaboration of the villi, as well as after formation of the crypts (flask-like structures located between villi that contain the stem cell population; crypts form early in fetal life in humans but postnatally in mice). During progressive gut lengthening in each setting, epithelial cells have different shapes, the location and size of the epithelial proliferative zones differ, and the surrounding mesenchymal compartment undergoes progressive morphogenesis. Although most of the data discussed here relate to lengthening prior to villus formation, it is important to consider that different factors may drive elongation in each of these three phases.
In all the three morphological settings, cell proliferation plays an important role in driving gut elongation. An important characteristic of the early intestinal epithelium is that it consists of a single layer of pseudostratified cells (each cell contacts both the apical surface and basement membrane) and nuclei are staggered along the apical-basal axis (Grosse et al., 2011) (Box 1). Pseudostratification allows efficient packing of a large number of tall, thin, rapidly dividing cells in a relatively restricted space (Lee and Norden, 2013). As villi begin to emerge, cells at the tips of the outgrowing villi shorten and widen (Walton et al., 2016), expanding the apical surface area of the intestinal tube substantially. Such shape changes might also drive lengthening of the gut tube (Fig. 1C). The morphological and signaling events that control the process of villus emergence are discussed in detail below.
Box 1. Pseudostratification and interkinetic nuclear migration (IKNM).

Prior to villus emergence, the intestinal epithelium is one cell layer thick, as shown in this cross-section of an E14.5 mouse small intestine, in which the tall thin cells are labeled in light green (A,B). Each pseudostratified cell is polarized (individual cells are shown in light green), with an apical (dark green) and basal (gray) surface, as schematically depicted in C. In contrast, in stratified epithelia (D), multiple cell layers are present and only the top layer of cells is apically polarized. Cells of pseudostratified epithelia undergo IKMN, a process in which nuclei move between the basal and apical surfaces in accordance with the cell cycle, as illustrated in E. DNA synthesis occurs basally whereas mitosis occurs apically (Gelbart et al., 2012; Grosse et al., 2011). This constant movement accounts for the staggered appearance of the nuclei in the epithelium and led early researchers to mistakenly characterize the epithelium as stratified (Mathan et al., 1976).
Cell division may also be accompanied by cues that ensure the two daughter cells are vectorially distributed down the long axis of the tube, resulting in lengthening rather than widening of the tube. Several elongating tubes or tissues, such as the mouse kidney tubule (Karner et al., 2009; Saburi et al., 2008), the chick neural tube (Sausedo et al., 1997), the developing vertebrate limb bud (Gros et al., 2010) and the outgrowing forestomach (Matsuyama et al., 2009), require oriented cell division for directional growth. It seems likely that oriented cell division may also play a role in intestinal tube lengthening, although this has yet to be formally demonstrated (Matsuyama et al., 2009).
In some developmental systems, radial intercalation – a type of convergent extension movement – has been proposed to be involved in gut lengthening (Keller et al., 2000). In a three-dimensional tube, radial intercalation involves the convergence of multi-layered (stratified) cells into one layer, leading to simultaneous tube narrowing and elongation (Fig. 2). Such movements are a major driving force for elongation of the early gut tube of the frog (Chalmers and Slack, 2000; Reed et al., 2009). However, intercalation of cell layers does not play a role in the mouse, since the early intestinal epithelium is already a single layer (Grosse et al., 2011).
Fig. 2.

Radial intercalation elongates the Xenopus primitive gut tube. (A,B) Tadpoles at stage 31 (A) and 41 (B); yellow highlights the gut (adapted from Nieuwkoop and Faber, 1994). (C,D) The primitive gut is initially a solid rod (C). Intercalation movements (arrows in C) narrow and elongate the gut and simultaneously generate a lumen (green tube in D). (E,F) During radial intercalation, endoderm cells slide between anterior and posterior neighbors (arrows), elongating the gut along its anterior-posterior axis. Colored cells, initially unpolarized and multi-layered (E), transition to a single layer with an apical (green) surface (F).
Molecular signaling pathways in prenatal intestinal lengthening
Epithelial-mesenchymal signaling crosstalk is crucial for intestinal lengthening and several signaling pathways have been implicated, including Wnt5a, Fgf, Hedgehog and Notch. Of these, Wnt5a, which can act in the non-canonical Jun N-terminal kinase-mediated planar cell polarity (PCP) pathway (Qian et al., 2007), is perhaps the best-studied with respect to its role in intestinal elongation. Wnt5a is expressed by the intestinal mesenchyme during gut elongation and a gut length deficit is seen in its absence (Cervantes et al., 2009). Interestingly, overexpression of Wnt5a ligand (Bakker et al., 2012) also interrupts elongation. Therefore, either a gradient of Wnt5a activity is needed for lengthening, or a very precise concentration of Wnt5a is required. Interestingly, defects in elongation are only seen if Wnt5a is overexpressed prior to embryonic day (E)13.5 (Bakker et al., 2012), indicating that Wnt5a controls proper lengthening of the pseudostratified intestine prior to villus formation, but not after villi emerge.
Three mechanisms were proposed to explain the short intestines seen in Wnt5a−/− mice. First, proliferation was reduced at E11.5, a factor that could substantially affect organ size given the robust proliferation index of the early gut tube (Cervantes et al., 2009). Second, BrdU pulse-chase studies revealed that post-mitotic cells did not effectively integrate into the epithelium, which was characterized as 3-4 cell layers thick (Cervantes et al., 2009). This feature was interpreted as a likely failure of convergence-extension-like movements that would normally reduce the stratified epithelium to a single layer. However, since the wild-type intestinal epithelium was recently shown to be a single layer of cells (Grosse et al., 2011), loss of Wnt5a probably instead causes ectopic stratification; the pile-up of cells could contribute to the elongation defect. Third, the bifurcated gut tube seen in the Wnt5a−/− animal could also contribute to gut shortening by usurping cells normally destined to contribute to a single gut tube.
The major receptor for Wnt5a, Ror2, is expressed by both intestinal epithelial and mesenchymal cells (Yamada et al., 2010). Many of the phenotypes of the Wnt5a−/− model are recapitulated in Ror2−/− mice, including short intestines. However, loss of Ror2 causes a milder length defect at E13.5 (37% shorter than controls) than loss of Wnt5a (75% shorter), suggesting that additional receptors might be involved. In this regard, it is interesting that mice lacking Ryk, a member of the receptor tyrosine kinase family, have outgrowth defects in multiple organs, including limbs, mandible and snout (Halford et al., 2000), but intestinal length has not been examined in this model. Ryk can also bind Wnt5a and has been implicated in PCP signaling (Andre et al., 2012; Macheda et al., 2012), making this receptor an interesting candidate that could contribute to Wnt5a signaling in the gut.
In addition to Wnt5a, other signaling pathways can affect gut elongation, some of which affect mesenchymal populations. The mesenchyme underlying the early intestinal epithelium is primarily derived from lateral plate mesoderm and is a poorly characterized heterogeneous population of cells that includes fibroblasts, myofibroblasts and ingrowing blood elements and nerves (Fig. 3). Several signaling pathways, including Hedgehog (Hh), Notch and Fgf affect intestinal lengthening in the mouse by altering these mesenchymal cell populations. Hh ligands (Shh and Ihh), which are expressed in the epithelium from early developmental stages, signal in a paracrine fashion to the mesenchyme (Kolterud et al., 2009; Ramalho-Santos et al., 2000). Loss of both Hh ligands or deletion of the major Hh signal transducer, smoothened (Smo), severely reduces mesenchymal proliferation and results in a digestive tract that is 90% shorter than controls – even shorter than the Wnt5a−/− gut (de Santa Barbara et al., 2002; Mao et al., 2010). Similarly, inactivating Notch signaling by deletion of the Notch effector Rbpj only in the mesenchyme diminishes the number of subepithelial fibroblasts and results in intestines that are 20-30% shorter than controls (Kim and Shivdasani, 2011). Finally, removal of epithelial Fgf9 increases Tgf-β signaling, which drives differentiation of mesenchymal cells to myofibroblasts, impairing small intestinal elongation (Geske et al., 2008). Embryos lacking the mesenchymal receptors Fgfr1 and Fgfr2 also exhibit shortened intestines (Geske et al., 2008). While all of these studies demonstrate that the mesenchymal cell populations can influence intestinal lengthening, the underlying mechanisms are unknown.
Fig. 3.

Villus formation and patterning in the mouse. (A) The intestinal epithelium (yellow) is pseudostratified (not shown); both apical (green) and basal (black) epithelial surfaces are flat at E13.5. The epithelium secretes Hh ligands that signal in a paracrine manner to underlying mesenchymal cells (pink). The intestine is encircled by the inner circular muscle (red hatching). (B) At E14.5, Hh signals trigger mesenchymal cells to cluster, beginning in the anterior small intestine. A self-organizing Turing-type field drives cluster patterning via mesenchymal Bmp signaling. Clusters send unknown signals (indicated by ‘?’) back to overlying epithelial cells, causing them to shorten. (C) Progressive shape changes in epithelial cells cause villus emergence at each cluster by E15.5. New clusters form beneath the still-proliferative intervillus epithelium, directing another round of villus emergence. Vasculature (red) and ectodermally derived nerves (blue) connect to the clusters. The inner circular muscle (red hatching) may confine the epithelium, promoting rapid villus emergence. Throughout this time, Pdgfa is expressed by the epithelium and Pdgfra is expressed by mesenchymal clusters. Disruption of this signaling pathway reduces subsequent rounds of villus formation, but does not perturb emergence of the first villi.
To summarize, myriad cell signaling cascades participate in the epithelial-mesenchymal crosstalk that drives intestinal elongation and interruption of these critical signals has major effects on tissue outgrowth. Although intestine-specific mutations in mouse models are beginning to decipher mechanisms underlying intestinal elongation, none of the pathways so far studied are clearly linked to short bowel syndromes in humans, likely because the global disruption of such pathways are often embryonically lethal. Direct identification of genes that cause short bowel phenotypes in humans will allow investigators to develop animal models that allow further dissection of the molecular mechanisms involved in gut lengthening.
Short bowel syndrome
Extreme shortening of the small intestine results in a clinical diagnosis of short bowel syndrome (SBS). SBS may be caused by conditions requiring major intestinal resection, such as necrotizing enterocolitis and intestinal atresia (Davenport and Bianchi, 1990) or by genetic mutations that result in short intestines at birth (e.g. 50 cm compared with 250 cm). The latter condition is termed congenital SBS (CSBS) (Hasosah et al., 2008; van der Werf et al., 2015, 2012). In spite of the advances made in treatment options for SBS, the majority of CSBS patients die within the first year (Sabharwal et al., 2004; van der Werf et al., 2015). Thus, a better understanding of the mechanisms underlying the defects in intestinal lengthening in this condition is essential.
Mutations in two genes have been discovered to cause CSBS in humans (Table 2): CLMP and FLNA (van der Werf et al., 2015, 2012). CLMP encodes the Coxsackie- and adenovirus receptor-like membrane protein, a transmembrane protein that interacts with tight junctions. The disease is inherited in an autosomal recessive manner and patients with CLMP mutations usually lack extra-intestinal symptoms (Van Der Werf et al., 2012). Overexpression of wild-type or mutant CLMP in human T84 cells (which do not normally express the protein) does not affect cell proliferation, migration, viability, or adhesion (van der Werf et al., 2013) and no mouse model has yet been generated for the Clmp mutation. Therefore, further in vivo work will be essential to understand the connection between CLMP and CSBS.
Table 2.
Mutations in patients and mouse models with congenital short bowel syndrome and microvillus inclusion disease
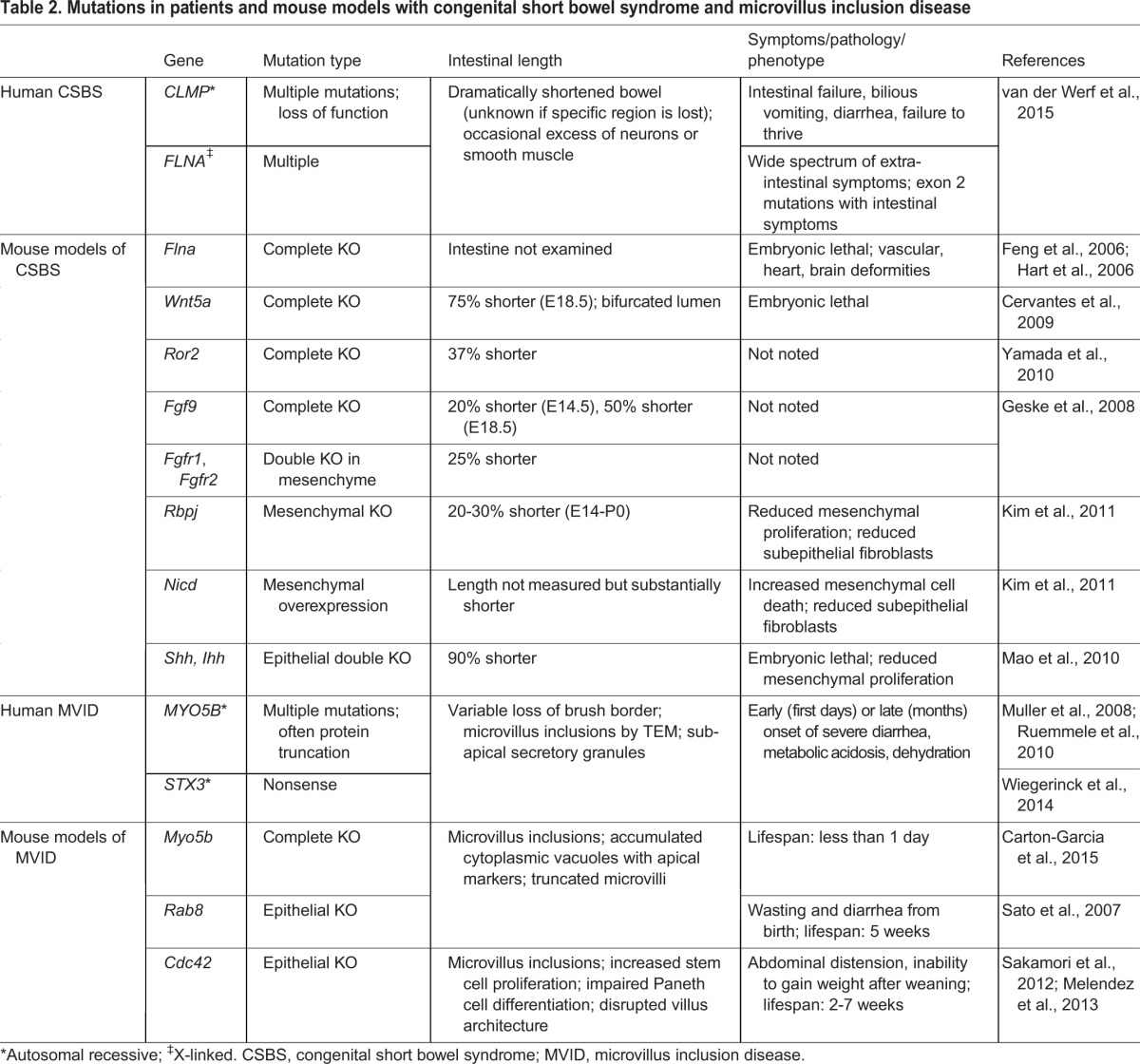
The second gene known to cause human CSBS is FLNA, which encodes Filamin A, a cytoskeletal protein that binds to actin as well as 70 other proteins (Kim and McCulloch, 2011) FLNA is located on the X chromosome and is widely expressed. Male patients with FLNA mutations exhibit phenotypes in many organs; severe neurological and cardiac symptoms are most apparent (van der Werf et al., 2015). Recently, a novel mutation was discovered in one of two known FLNA isoforms in patients with a short bowel, but no extra-intestinal symptoms (van der Werf et al., 2015); this suggests a specific requirement for this longer FLNA isoform in the developing intestine. Although most FLNA mutations seen in patients have not been analyzed in vivo, two mouse models with complete loss of Flna exhibit embryonic lethality in males by E14.5 or E15.5. These models recapitulate the cardiac, brain, skeletal and vascular defects associated with human FLNA mutations, but intestinal length was not examined (Feng et al., 2006; Hart et al., 2006).
Intriguingly, it has been speculated that FLNA might interact with CLMP in apical junctional complexes (van der Werf et al., 2015). Additionally, FLNA associates with the WNT5a receptor, ROR2 and this is required for WNT5a-mediated JNK activation in vitro (Nomachi et al., 2008). This interesting potential connection between human CSBS and Wnt5a signaling warrants further investigation.
Villus morphogenesis
The development of villi – finger-like projections that emerge from the initially flat lumenal surface – further increases the total adult absorptive surface area 10-fold (Fig. 3). Histologically, villi are composed of an epithelial layer and a mesenchymal core. Villus development is primarily a fetal event; animal models of resected intestines suggest that the formation of new villi is relatively inefficient after birth (Clarke, 1972; Forrester, 1972). Here, we review the key morphogenetic events involved in the initiation of villus development and discuss some of the molecular signals that drive this process.
Expansion of apical surface does not occur through formation and fusion of secondary lumens
Early studies of villus formation in the rat led to the idea that the pre-villus epithelium is stratified and that the de novo formation and fusion of secondary lumens ‘carves out’ the initial villi, simultaneously expanding intestinal surface area (Mathan et al., 1976). This process was thought to be defective in mice lacking the apical ezrin/radixin/moesin (ERM) protein ezrin, leading to the formation of isolated lumens and fused villi (Saotome et al., 2004). Loss of Crumbs3, another apical protein, causes a similar fused-villus phenotype (Whiteman et al., 2014). However, more recent analyses, utilizing three-dimensional reconstruction to resolve luminal contiguity, have established that all luminal surfaces are connected during villus morphogenesis (Grosse et al., 2011). Instead of secondary lumina expanding the apical surface, it appears that just prior to villus emergence, numerous membrane invaginations initially extend the apical surface of the pseudostratified tube. The exact morphological events that accompany these membrane invaginations and the signaling events required for their formation remain elusive.
Given that the secondary lumen model appears not to hold, there is renewed interest in understanding the mechanisms underlying villus morphogenesis and patterning. Investigations in two of the best-studied animal models – mouse and chick – reveal that while common signaling pathways appear to be involved in the elaboration of villi in both species, the mechanisms that drive villus patterning differ.
Villus formation and patterning in the mouse
At the initiation of villus morphogenesis in the mouse, the intestinal epithelium is a single pseudostratified layer of tall (50 µm) thin cells, with a flat apical surface, defining a thin lumen (Fig. 3A). As villi emerge, the apical surface becomes highly convoluted and cell height is reduced to about 15 µm. The first step in this process is the aggregation of mesenchymal cells adjacent to the basal surface of the epithelium, forming densely packed mesenchymal ‘clusters’ (Fig. 3B), which express a variety of signaling molecules that act to coordinate villus development (Karlsson et al., 2000; Walton et al., 2012, 2016). Clusters appear first in the duodenum at E14.5 and spread in a wave-like fashion over the entire intestine during the next 36 h (Walton et al., 2012). As clusters form directly beneath the epithelium, the overlying epithelial cells begin to shorten, resulting in gentle basal deformations (Fig. 3B) (Walton et al., 2012). Further shortening and eventual widening of epithelial cells over the clusters may contribute to initial emergence as well as further outgrowth of the villi (Fig. 3C). As villi lengthen and the intestine continues to grow in girth and length, new clusters form under the proliferative and expanding intervillus epithelium, initiating the emergence of new villi between existing villi. This occurs in several rounds, with each successive round approximately doubling the number of villi (Walton et al., 2012).
The aggregation of mesenchymal cells into clusters is dependent upon Hh ligands secreted from the pseudostratified epithelium (Walton et al., 2012). Cells within the clusters are Hh responsive, expressing Gli1, Ptc1 and other components of the Hh signal transduction machinery (Kolterud et al., 2009). Compromised villus formation is noted in vivo when Hh signaling is low (Madison et al., 2005) or absent (Mao et al., 2010). Additionally, in explant cultures of fetal intestine, blocking Hh signaling inhibits cluster formation and villus development. Conversely, application of a Hh agonist increases the size of clusters and emerging villi (Walton et al., 2012). Although Hh signaling is required for agglutination of the clusters, the ‘adhesives’ responsible for adherence of the cluster cells to one another and for adherence of clusters to the epithelium remain unknown.
When mesenchymal cluster size or pattern is altered, villus shape and spacing is disrupted, suggesting that the mesenchymal clusters drive villus morphogenesis (Karlsson et al., 2000; Walton et al., 2012, 2016). For example, mesenchymal cluster cells express Pdgfra and respond to Pdgfa ligands secreted from the epithelium (Karlsson et al., 2000). Loss of Pdgfa or Pdgfra in the mouse results in decreased proliferation of mesenchymal cells, reducing the number of clusters; although the first round of villi form normally, subsequent villi are oddly shaped and irregularly spaced (Karlsson et al., 2000).
Bmp ligands and signaling pathway components are expressed in mesenchymal clusters and must be tightly regulated for proper patterning to occur (Walton et al., 2016). Genetically deleting the Bmp receptor Bmpr1a in the mesenchymal cluster cells results in larger clusters and wider villi (Walton et al., 2016). In vitro, addition of exogenous Bmp ligand prevents clusters (and villi) from forming, while global inhibition of Bmp signaling using the small molecule inhibitor dorsomorphin changes cluster pattern from discrete spots to stripes. This patterning change resembles features seen in a self-organizing Turing field (Walton et al., 2016). Alan Turing first demonstrated mathematically how interactions of an activator and inhibitor, both emanating from the same source, can control the formation of a repeating pattern (Turing, 1952). During cluster patterning, evidence suggests that Bmp ligands act as the Turing inhibitor while modifiers of Bmp signaling, such as twisted gastrulation (Twsg1), may comprise the Turing activator (Walton et al., 2016). Thus, intestinal mesenchymal cluster patterning can be added to a growing list of biological systems that behave in accord with the Turing model, including: patterning of digits (Raspopovic et al., 2014), chick feather buds (Baker et al., 2009), hair follicles (Maini et al., 2006; Sick et al., 2006), zebrafish mesodermal pigment (Eom et al., 2012) and tongue papillae (Zhou et al., 2006).
In summary, murine villus development occurs through rapid conversion of a thick pseudostratified epithelial tube into a regular field of patterned villus domes, a process initiated by epithelially secreted Hh ligands. These paracrine Hh signals cause underlying mesenchymal cells to aggregate into clusters, which spread in wave-like fashion down the gut tube, in a self-organizing manner that is driven by a Bmp-based Turing patterning system. Then, this established cluster pattern is transferred back to the overlying epithelial cells via unknown signals that induce cell shape changes (epithelial cell shortening and widening) leading to villus emergence. This signaling-based patterning system contrasts sharply with the mechanically driven process of villus patterning that characterizes the chick intestine, discussed below.
Villus formation and patterning in the chick
In the chick, tensile forces from the developing visceral muscle layers progressively deform the epithelium into a regular pattern of ridges and then zigzags (Fig. 4). At E7-E8, formation of the inner circular muscle radially confines the rapidly proliferating epithelium, bending the flat surface into longitudinal ridges (Coulombre and Coulombre, 1958; Grey, 1972; Shyer et al., 2013). At E12-E13, the outer longitudinal muscle develops and begins to exert a longitudinal compressive force, causing epithelial ridges to fold into zigzags (Coulombre and Coulombre, 1958; Grey, 1972; Shyer et al., 2013). Finally, at E16, a third inner longitudinal muscle layer forms beneath the epithelium. Coordinating with the development of this last muscle layer, bulges appear along the zigzags that develop into definitive villi by E17 (Coulombre and Coulombre, 1958; Grey, 1972; Shyer et al., 2013). Treatment of the intestinal tube with inhibitors of smooth muscle differentiation prevents these progressive epithelial deformations, whereas confining it in a rigid silk tube in the presence of these inhibitors rescues proper epithelial folding (Shyer, et al., 2013). Thus, in the chick, tensile forces from the developing muscles are necessary and sufficient to deform the epithelium into patterned ridges, zigzags and finally, villi.
Fig. 4.

Villus formation and patterning in the chick. (A-D) Schematic representations of intestinal cross-sections (top) and partial longitudinal views (bottom) illustrate sequential development of patterned epithelial folds that precede villus patterning. (A) At E6, prior to visceral muscle development, a loose mesenchyme (pink) surrounds the epithelium (yellow); the epithelial apical surface (green) is flat. (B) By E8, the outer circular muscle forms, confining the proliferative epithelium and leading to development of longitudinal ridges. (C) Formation of the outer longitudinal muscle at E15 bends the ridges into zigzags. (D) Formation of the inner longitudinal muscle is concomitant with villus emergence. Red arrows indicate the direction of compression conveyed by the addition of each muscle layer; black arrows show forces imposed by previously formed muscles. The progressive muscle-induced epithelial bending creates localized pockets of concentrated Hh signals that drives patterned mesenchymal cluster formation and villus emergence.
During formation of ridges and zigzags, the epithelium continuously secretes Hh ligands. Progressive bending of the epithelial surface creates localized pockets of high Hh that upregulate expression of genes identified as mesenchymal cluster markers in the mouse, such as Bmp4 and Pdgfra (Shyer et al., 2015). Importantly, clusters form only when and where the deformation of the epithelium is substantial enough to create these Hh maxima. Indeed, artificial deformation of the epithelium by placing a wire grid on the open epithelial surface is sufficient to cause precocious formation of clusters and emergence of villi in the chick (Shyer et al., 2015).
In summary, the successive maturation of muscle groups in the chick mechanically deforms the epithelium into zigzags that then pattern the formation of mesenchymal clusters, driving villus outgrowth. In sharp contrast, the murine smooth muscle layers do not develop coordinately with the events of villus formation (Walton et al., 2016). Furthermore, artificial deformation of the epithelium with wire grids is not sufficient to pattern villi in the mouse (Walton et al., 2016). Thus, although Hh signals are absolutely required for cluster formation in both the mouse and the chick, cluster pattern is established by mesenchymal cell signaling in the mouse and by the field of muscular forces in the chick.
These different patterning mechanisms could potentially reflect the distinct morphologies of the epithelium prior to villus emergence in the two species (Box 2). In the mouse, clusters form beneath a thick pseudostratified epithelium; signals from a patterned field of clusters are needed to quickly direct epithelial shape changes to drive villus buckling. In contrast, the chick epithelium is already thin and columnar even as the first ridges are forming, a week before villi emerge. This thinner epithelial layer is flexible enough to allow regular deformation by the developing muscles to pattern the location of forming clusters.
Box 2. Cell shape in chick and mouse pre-villus epithelium.
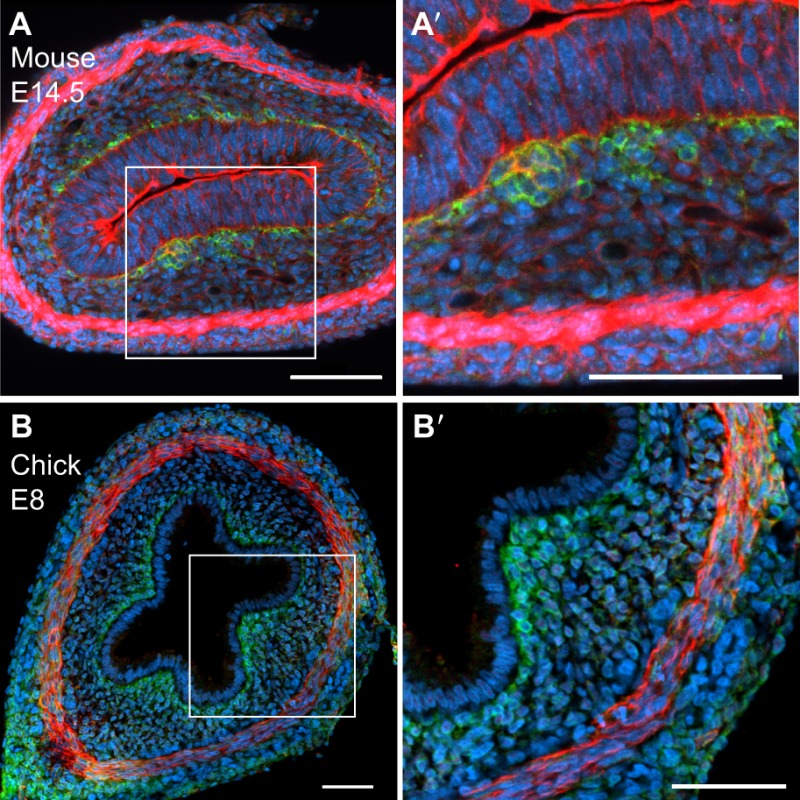
In the mouse, the intestinal epithelium is thick and pseudostratified when the earliest mesenchymal clusters (here stained green with anti-Pdgfra) begin to form at E14.5 (A,A′). Phalloidin (red), which stains F-actin, marks the well-formed inner circular muscle that surrounds the epithelium (at bottom of panel) as well as the epithelial cells, where the apical surface is actin rich. Within the next few hours, these forming mesenchymal clusters will signal to overlying epithelial cells, causing shape changes that convert the rigid pseudostratified epithelium to a more flexible simple columnar epithelium that can bend as villi emerge. In contrast, the chick intestinal epithelium is already a thin, simple columnar epithelium at E8 (ridge stage), even though mesenchymal cluster formation and villus emergence are still a week away (B,B′). The subepithelial mesenchymal cells that will later form clusters are marked in green (anti-Pdgfra), while the inner circular smooth muscle is stained red (anti-aSMA). The thin epithelial cell shape may facilitate the progressive and dramatic epithelial bending that is required for cluster formation and villus emergence by E16.
Additional molecules implicated in villus formation and patterning
Work in mouse models has revealed several mesenchymally expressed transcription factors that are also required for proper villus morphogenesis, including Nkx2.3 (Wang et al., 2000), Foxf1, Foxf2 (Ormestad et al., 2006) and Foxl1 (Kaestner et al., 1997). Since many Nkx and Fox genes are targets of Hh signaling (Xu et al., 2006), it is possible that these mesenchymal factors play key roles in murine villus formation by affecting mesenchymal cluster formation downstream of the Hh pathway. This might also be true in the chick, given the clear role for Hh signaling in villus development in both species.
Epithelially expressed Kruppel-like factor 5 (Klf5) plays a particularly important role in the transition of a proliferative pseudostratified epithelium to differentiated columnar cells during murine villus formation (Bell et al., 2013). In mice lacking Klf5, the epithelium remains proliferative and villus generation is retarded. Pathway analysis suggests that Klf5 normally represses epithelial Foxa1 at the onset of villus formation. Foxa1 is a known repressor of Elf3 (E74-like factor 3), an ETS-family transcription factor that is expressed in epithelial cells beginning at E14.5 (Ng et al., 2002; Tymms et al., 1997). Deletion of Elf3 results in 30% embryonic lethality by E11.5. In surviving fetuses, epithelial cells lacking Elf3 fail to change shape from pseudostratified to columnar; the few villi that do form are decreased in number and abnormal in shape (Ng et al., 2002).
Interestingly, the Klf5-Foxa1-Elf3 axis is independent of epithelial to mesenchymal signaling through the Hh or Pdgf pathways, both of which are intact in the absence of Klf5 (Bell et al., 2013). However, Tgfbr2 is severely decreased in the epithelium of Elf3 mutants and genetic overexpression of Tgfbr2 in the epithelium rescues villus formation in the Elf3−/− model (Flentjar et al., 2007). These findings implicate Tgf-β ligands as potential signals from mesenchymal clusters that could promote cell shape changes in the overlying epithelial cells during villus outgrowth in the mouse.
Restriction of the proliferative compartment during villus emergence in mouse and chick
The process of villus emergence divides the uniformly proliferative epithelium into non-proliferative and proliferative compartments, located on the emerging villi and between villi, respectively. Although this is true in both chick and mouse, the underlying mechanisms once again may differ. In chick, localized environments of high Hh signaling created by the zigzags cause upregulation of Bmp4 expression that suppresses proliferation in the overlying villus tip epithelium, thereby restricting the proliferative compartment to the base of the villi (Shyer et al., 2015). In chick intestinal explants, addition of Bmp4 reduces the epithelial proliferative compartment, while addition of the Bmp inhibitor Noggin expands it (Shyer et al., 2015). In contrast, in the mouse, interrupting the reception of Bmp signals by the epithelium either in vivo or in vitro does not suppress proliferation of epithelial cells, although fused clusters and larger villi are observed (Walton et al., 2016). Therefore, Bmp signaling does not appear to be the signal that suppresses epithelial proliferation during villus outgrowth in the mouse; different signals, likely emanating from the mesenchymal clusters, may be responsible for restriction of this compartment in the murine intestine; the identity of these signals has yet to be uncovered.
In conclusion, it is fascinating that while common signaling pathways (Hh, Bmp, Pdgf) generate villi in both mouse and chick, the mechanisms that determine villus pattern and compartmentalize the epithelial proliferative compartment differ in the two species. Tissue mechanics dominate the chick model: smooth muscle-driven deformation of the epithelium generates the pattern of mucosal folds that trap localized peaks of epithelially secreted Hh, inducing cluster gene expression. In this species, conversion of the epithelium from pseudostratified to columnar precedes mesenchymal cluster formation. Bmp signals from clusters located at the villus tips restrict epithelial proliferation to the intervillus regions. A different mechanism portends in the mouse, where mesenchymal clusters form early in the process and are patterned by a self-organizing field that is controlled by Bmp signaling. In this species, clusters act as signaling hubs to effectively transfer this regular mesenchymal pattern to the epithelium. Signals from clusters (not Bmp, but currently unknown in nature) cause overlying epithelial cells to change shape, becoming columnar, and withdraw from the cell cycle, thereby restricting the proliferating compartment to the intervillus regions. Notably, it has been suggested that villi evolved convergently in the chick and the mouse (Shyer et al., 2015) – a notion that is consistent with the distinct morphologies (Box 2) and the very different mechanisms used by these two species to pattern the villi (Figs 3 and 4).
Formation of the microvillar brush border
The apical surface area of the intestine is further expanded by the formation of microvilli (Fig. 5), which are convoluted apical membrane extensions with cores of crosslinked actin bundles. Microvilli are also found in other polarized epithelia and excellent reviews on microvillar biology are available (Crawley et al., 2014; Sauvanet et al., 2015). Here, we focus on the intestinal phenotypes of relevant mouse models and human conditions in which microvillus structure is perturbed.
Fig. 5.

Microvilli further increase the absorptive surface area of the small intestine. (A) Field of microvilli on the surface of emerging villi in an E15.5 mouse intestine, as seen by scanning electron microscopy; scale bar: 2 µm. (B) Transmission electron microscopy reveals ordered microvilli with dense actin cores; scale bar: 500 nm. (C) Schematic illustration of microvilli with actin cores that are bundled by fimbrin, villin and espin, and crosslinked to the apical membrane (green) by ezrin.
Microvillus formation begins when actin filaments nucleate at electron-dense plaques in the apical actin terminal web (Crawley et al., 2014; Sauvanet et al., 2015). The actin-bundling protein villin is first expressed at E10 and becomes apically concentrated by E13.5 (Ezzell et al., 1989) where it crosslinks and stabilizes actin fibers. Mice lacking villin form relatively normal microvilli, although their actin cores are not tightly bundled (Pinson et al., 1998). Functionally, the mutant microvilli are also more susceptible to in vitro calcium-mediated injury (Ferrary et al., 1999).
Another actin crosslinker, fimbrin, increases microvillus stiffness and order (Crawley et al., 2014; Ezzell et al., 1989). Villin and fimbrin bind to different domains of F-actin and may work cooperatively (Hampton et al., 2008; Sauvanet et al., 2015). In the absence of fimbrin, microvilli are shorter with less well-ordered cores. However, these mice are still viable and fertile, with no detectable gross intestinal defects (Grimm-Gunter et al., 2009).
Espin, a third actin-binding protein found in microvilli, attaches to actin monomers. It is recruited after villin and fimbrin (Bartles et al., 1998; Loomis et al., 2006, 2003). Mice lacking espin have normal microvillus structure (Zheng et al., 2000). Interestingly, even if villin, fimbrin and espin are all removed from the intestinal epithelium, only modest changes in microvillus structure result, similar to those seen with fimbrin loss alone. Therefore, compensation by other, unknown molecules is likely (Revenu et al., 2012).
The apical ERM protein ezrin crosslinks actin to the apical membrane. In Ezr−/− mice, villus architecture is perturbed and microvilli are shortened, reflecting its important role in their formation and/or stabilization (Saotome et al., 2004). Ezrin is stabilized in its active conformation through phosphorylation at threonine 567 and binding to phosphatidylinositol 4,5-bisphosphate (PIP2) at the apical surface (Fehon et al., 2010; Jayasundar et al., 2012). Similarly, Crb3 is also required for proper elaboration of the brush border, as microvilli are shortened in Crb3−/− mice, similar to Ezr−/− animals (Whiteman et al., 2014), although the underlying mechanisms are unclear.
Overall, the intestinal brush border is a particularly robustly engineered system that ensures the presence of thousands of well-ordered microvilli on the surface of each cell. How brush border formation is initiated and how uniform microvillar width and length is established is still a mystery. However, recent work has increased our understanding of microvillar packing, revealing protocadherin-like proteins that link the external membrane at the tip of each microvillus to its neighbors to establish the well-ordered, tightly packed lawn of structures (reviewed in Crawley et al., 2014). Failure to establish this tip-targeted complex in patients with Usher Syndrome causes defects in hearing (affecting stereocilia) as well as perturbed gastrointestinal function due to brush border dysmorphology (Bitner-Glindzicz et al., 2000; Hussain et al., 2004). This connection has also been modeled in mice (Crawley et al., 2014). Finally, once formed, the microvillus domain must be maintained; a job that is accomplished by the apical recycling system. Mutations in that system can cause microvillus inclusion disease (MVID) – a condition in which the intestinal brush border is defective.
Microvillus formation and absorptive disease
The essential function of microvilli in amplifying surface area is emphasized by the phenotype of patients with MVID, who present with congenital diarrhea in the first days (early-onset form) to months (late-onset) of life (Ruemmele et al., 2010). The defining histological features of MVID are substantial loss of the brush border, presence of intracellular vacuoles with microvilli (microvillus inclusions) and subapical secretory granules in enterocytes (Muller et al., 2008; Ruemmele et al., 2010; Wiegerinck et al., 2014). Prognosis is poor and treatment is often limited to long-term total parenteral nutrition (Ruemmele et al., 2010).
The most common genetic cause of MVID is an autosomal recessive mutation in Myosin Vb (MYO5B), an unconventional myosin that is essential for organelle transport and apical endosome recycling (Muller et al., 2008; Ruemmele et al., 2010). Knockdown of MYO5B in Caco2 cells causes microvillus inclusions and loss of microvilli (Ruemmele et al., 2010). Recently, a Myo5b−/− mouse model was described (Carton-Garcia et al., 2015). These mice die within 12 h and recapitulate the histological characteristics of human MVID (Carton-Garcia et al., 2015). Further study of this system may allow mechanistic insight into early-onset MVID.
Mutations in Syntaxin 3 (STX3) also cause MVID; the phenotype is indistinguishable from MYO5B-mediated MVID. STX3 is part of the SNARE machinery for vesicular delivery and its loss is thought to arrest trafficking of apically destined vesicles. An Stx3 null mouse model has not been developed, but expression of truncated STX3 in Caco2 cells results in mispolarization and formation of microvillus inclusions via a dominant-negative mechanism (Wiegerinck et al., 2014).
Two other mouse models exhibit phenotypes similar to MVID, but do not correspond with identified human mutations. First, germline loss of Rab8, which is involved in recycling apical endosomes, causes extreme diarrhea and progressive wasting from birth. Cytoplasmic apical protein accumulation and microvillus inclusions are present (Sato et al., 2007). Further mechanistic work will be required to clarify the connection between Rab8, Myo5b and Stx3 in the context of microvillus formation. Second, mice conditionally lacking Cdc42, a small Rho GTPase (generated using the Villin-Cre driver to remove Cdc42 in late gestation), fail to gain weight and die at 2-7 weeks of age (Melendez et al., 2013; Sakamori et al., 2012). As in human MVID, microvillus inclusions form, enterocytes are incorrectly polarized and villus architecture is disrupted (Melendez et al., 2013). Lack of Cdc42 causes incorrect trafficking of Rab8 to the midbody in the stem cells, resulting in a failure of cytokinesis and likely inducing apoptosis as well as epithelial dysmorphogenesis. The molecular connection between loss of Cdc42 expression, MVID and the other observed phenotypes in differentiated cells needs to be further examined.
Conclusions and future considerations
While our understanding of intestinal morphogenesis is improving, there are still significant gaps in our knowledge and a need for more detailed understanding of the mechanisms underlying the generation of intestinal surface area. Since surface expansion occurs progressively, defects that occur early affect the entire template for surface growth and therefore have a devastating impact on intestinal function. This highlights a need to better understand how mutations in genes such as Clmp and Flna can compromise intestinal elongation and to flesh out the pathways upon which these factors impinge. Indeed, the kinetics of tissue expansion and clonal growth in the early epithelium have yet to be carefully investigated.
Both tissue elongation and villus morphogenesis rely on epithelial-mesenchymal crosstalk and the manner in which each layer contributes to the complex signaling events is still unclear. The mesenchymal cluster that drives villus emergence in both mouse and chick is a fascinating signaling hub that plays multiple roles, establishing pattern in the mesenchyme (at least in the mouse) and transferring that pattern to the epithelium (in mouse and chick). More work is needed to understand what glues the cluster cells together, how it becomes so tightly adhered to the basement membrane, and which signals it uses to control overlying epithelial cell shape and proliferation.
Interestingly, there is more than one way to make a villus. In chick, tissue forces play a key patterning role, compared with the mouse model, where signaling is paramount. Of course, both signaling and mechanical forces certainly play into each model. Villus morphogenesis occurs in a confined space and tissue forces must clearly shape the process – even in the mouse, where it does not appear to be the primary driving force as in chick. The power of mechanical models to clarify the effects of these forces has been demonstrated and these models have led to novel insights regarding the signal concentrating effects of mechanically bending an epithelium (Shyer et al., 2015, 2013).
Finally, once mature apical structures are built, there is a need for continuous homeostatic mechanisms to maintain them. This aspect is driven home by the severe phenotypes of patients with MVID in which apical recycling is defective. While we have highlighted the important role of the brush border in the amplification of intestinal surface area, this structure also functions as a barrier to prevent pathogenic bacteria from attaching to the epithelial cells (Crawley et al., 2014). Thus, more research into the dynamic aspects of microvillus turnover and the linkages between the apical recycling machinery, the actin-bundling proteins and the microvillar tip complexes is clearly warranted.
From a translational perspective, an important future goal is to develop strategies for the generation of functional tissue to replace diseased or missing intestines. One promising avenue is the development of ‘organoid’ or ‘tissue-derived epithelial organoid’ (previously called enteroid) systems to grow intestinal tissue in vitro (Box 3). While progress is being made, it is not yet clear whether these systems will indeed be useful for the large-scale replacement of absorptive surface, since they typically grow as spheres with restricted internal apical surfaces. Additionally, many such in vitro systems lack other features that may be important to villus morphogenesis, including smooth muscle and a vascular and nerve supply. Improving these systems, to make clinically viable tools for intestinal surface area replacement will require a more detailed knowledge of the fundamental processes directing intestinal tube generation and elongation, villus formation and microvillus elaboration.
Box 3. Engineering intestines.
Advances in bioengineered human intestinal tissues provide new possibilities for prospective treatment of short gut syndrome and intestinal disease and offer valuable in vitro models for the study of basic intestinal cell biology (Sinagoga and Wells, 2015; Wells and Spence, 2014). Three main formats predominate. Tissue engineered small intestines (TESIs) are generated from a slurry of adult whole intestine and include all cell types (Spurrier and Grikscheit, 2013). Tissue-derived epithelial organoids (also called enteroids) are derived from isolated adult intestinal stem cells already committed to an endodermal fate and are grown without mesenchyme in medium supplemented to favor undifferentiated intestinal stem cells (Ootani et al., 2009; Sato et al., 2009). Organoids are derived from pluripotent cells (e.g. human embryonic or induced pluripotent stem cells), driven towards mesendodermal fates by treatment with cytokines and cell signaling inhibitors, generating both endoderm and mesoderm (Sato et al., 2009; Spence et al., 2011b). Epithelial organoids tend to form simple proliferative cysts, while organoids and TESIs often exhibit crypt-like structures. Although none of these engineered intestinal constructs generate villi in vitro, transplantation into a vascular-rich host leads to villus development; villus core components from the host provide necessary structures and/or signals to drive villus morphogenesis (Finkbeiner et al., 2015, 2012; Watson et al., 2014). Future work will be needed to determine the identity and source of these cues.
Acknowledgements
The authors would like to thank Dr Cliff Tabin and Tyler Huycke for stimulating discussions. Images were collected in the Microscopy and Image Analysis Laboratory at the University of Michigan Medical School.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
Work in our labs receives funding from the National Institutes of Health [R01 DK089933 and F30 DK100125]; American Society for Parenteral and Enteral Nutrition Rhoads Research Foundation; University of Michigan Gastrointestinal Peptide Center Pilot Feasibility Award [funded through an NIDDK/NIH Research Core Grant [P30 DK034933]. Deposited in PMC for release after 12 months.
References
- Andre P., Wang Q., Wang N., Gao B., Schilit A., Halford M. M., Stacker S. A., Zhang X. and Yang Y. (2012). The Wnt coreceptor Ryk regulates Wnt/planar cell polarity by modulating the degradation of the core planar cell polarity component Vangl2. J. Biol. Chem. 287, 44518-44525. 10.1074/jbc.M112.414441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker R. E., Schnell S. and Maini P. K. (2009). Waves and patterning in developmental biology: vertebrate segmentation and feather bud formation as case studies. Int. J. Dev. Biol. 53, 783-794. 10.1387/ijdb.072493rb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker E. R. M., Raghoebir L., Franken P. F., Helvensteijn W., van Gurp L., Meijlink F., van der Valk M. A., Rottier R. J., Kuipers E. J., van Veelen W. et al. (2012). Induced Wnt5a expression perturbs embryonic outgrowth and intestinal elongation, but is well-tolerated in adult mice. Dev. Biol. 369, 91-100. 10.1016/j.ydbio.2012.06.007 [DOI] [PubMed] [Google Scholar]
- Bartles J. R., Zheng L., Li A., Wierda A. and Chen B. (1998). Small espin: a third actin-bundling protein and potential forked protein ortholog in brush border microvilli. J. Cell Biol. 143, 107-119. 10.1083/jcb.143.1.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter J. S. (1953). Frazer's Manual of Embryology The Development of the Human Body, 3rd edn. London: Bailliere, TIndall and Cox. [Google Scholar]
- Bell S. M., Zhang L., Xu Y., Besnard V., Wert S. E., Shroyer N. and Whitsett J. A. (2013). Kruppel-like factor 5 controls villus formation and initiation of cytodifferentiation in the embryonic intestinal epithelium. Dev. Biol. 375, 128-139. 10.1016/j.ydbio.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner-Glindzicz M., Lindley K. J., Rutland P., Blaydon D., Smith V. V., Milla P. J., Hussain K., Furth-Lavi J., Cosgrove K. E., Shepherd R. M. et al. (2000). A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat. Genet. 26, 56-60. 10.1038/79178 [DOI] [PubMed] [Google Scholar]
- Cartón-García F., Overeem A. W., Nieto R., Bazzocco S., Dopeso H., Macaya I., Bilic J., Landolfi S., Hernandez-Losa J., Schwartz S. Jr. et al. (2015). Myo5b knockout mice as a model of microvillus inclusion disease. Sci. Rep. 5, 12312 10.1038/srep12312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes S., Yamaguchi T. P. and Hebrok M. (2009). Wnt5a is essential for intestinal elongation in mice. Dev. Biol. 326, 285-294. 10.1016/j.ydbio.2008.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers A. D. and Slack J. M. (2000). The Xenopus tadpole gut: fate maps and morphogenetic movements. Development 127, 381-392. [DOI] [PubMed] [Google Scholar]
- Clarke R. M. (1972). The effect of growth and of fasting on the number of villi and crypts in the small intestine of the albino rat. J. Anat. 112, 27-33. [PMC free article] [PubMed] [Google Scholar]
- Coulombre A. J. and Coulombre J. L. (1958). Intestinal development. I. Morphogenesis of the villi and musculature. J. Embryol. Exp. Morphol. 6, 403-411. [PubMed] [Google Scholar]
- Crawley S. W., Mooseker M. S. and Tyska M. J. (2014). Shaping the intestinal brush border. J. Cell Biol. 207, 441-451. 10.1083/jcb.201407015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport M. and Bianchi A. (1990). Congenital intestinal atresia. Br. J. Hosp. Med. 44, 174, 176, 178-180. [PubMed] [Google Scholar]
- de Santa Barbara P., van den Brink G. R. and Roberts D. J. (2002). Molecular etiology of gut malformations and diseases. Am. J. Med. Genet 115, 221-230. 10.1002/ajmg.10978 [DOI] [PubMed] [Google Scholar]
- Eom D. S., Inoue S., Patterson L. B., Gordon T. N., Slingwine R., Kondo S., Watanabe M. and Parichy D. M. (2012). Melanophore migration and survival during zebrafish adult pigment stripe development require the immunoglobulin superfamily adhesion molecule Igsf11. PLoS Genet. 8, e1002899 10.1371/journal.pgen.1002899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzell R. M., Chafel M. M. and Matsudaira P. T. (1989). Differential localization of villin and fimbrin during development of the mouse visceral endoderm and intestinal epithelium. Development 106, 407-419. [DOI] [PubMed] [Google Scholar]
- Fehon R. G., McClatchey A. I. and Bretscher A. (2010). Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276-287. 10.1038/nrm2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Chen M. H., Moskowitz I. P., Mendonza A. M., Vidali L., Nakamura F., Kwiatkowski D. J. and Walsh C. A. (2006). Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc. Natl. Acad. Sci. USA 103, 19836-19841. 10.1073/pnas.0609628104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrary E., Cohen-Tannoudji M., Pehau-Arnaudet G., Lapillonne A., Athman R., Ruiz T., Boulouha L., El Marjou F., Doye A., Fontaine J.-J. et al. (1999). In vivo, villin is required for Ca(2+)-dependent F-actin disruption in intestinal brush borders. J. Cell Biol. 146, 819-830. 10.1083/jcb.146.4.819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S. R., Zeng X.-L., Utama B., Atmar R. L., Shroyer N. F. and Estes M. K. (2012). Stem cell-derived human intestinal organoids as an infection model for rotaviruses. MBio 3, e00159-12 10.1128/mBio.00159-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S. R., Hill D. R., Altheim C. H., Dedhia P. H., Taylor M. J., Tsai Y.-H., Chin A. M., Mahe M. M., Watson C. L., Freeman J. J. et al. (2015). Transcriptome-wide analysis reveals hallmarks of human intestine development and maturation in vitro and in vivo. Stem Cell Rep. 4, 1140-1155. 10.1016/j.stemcr.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flentjar N., Chu P.-Y., Ng A. Y.-N., Johnstone C. N., Heath J. K., Ernst M., Hertzog P. J. and Pritchard M. A. (2007). TGF-betaRII rescues development of small intestinal epithelial cells in Elf3-deficient mice. Gastroenterology 132, 1410-1419. 10.1053/j.gastro.2007.02.054 [DOI] [PubMed] [Google Scholar]
- Forrester J. M. (1972). The number of villi in rat's jejunum and ileum: effect of normal growth, partial enterectomy, and tube feeding. J. Anat. 111, 283-291. [PMC free article] [PubMed] [Google Scholar]
- Gelbart M. A., He B., Martin A. C., Thiberge S. Y., Wieschaus E. F. and Kaschube M. (2012). Volume conservation principle involved in cell lengthening and nucleus movement during tissue morphogenesis. Proc. Natl. Acad. Sci. USA 109, 19298-19303. 10.1073/pnas.1205258109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske M. J., Zhang X., Patel K. K., Ornitz D. M. and Stappenbeck T. S. (2008). Fgf9 signaling regulates small intestinal elongation and mesenchymal development. Development 135, 2959-2968. 10.1242/dev.020453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondolesi G., Ramisch D., Padin J., Almau H., Sandi M., Schelotto P. B., Fernandez A., Rumbo C. and Solar H. (2012). What is the normal small bowel length in humans? first donor-based cohort analysis. Am. J. Transplant. 12 Suppl. 4, S49-S54. 10.1111/j.1600-6143.2012.04148.x [DOI] [PubMed] [Google Scholar]
- Goulet O., Ruemmele F., Lacaille F. and Colomb V. (2004). Irreversible intestinal failure. J. Pediatr. Gastroenterol. Nutr. 38, 250-269. 10.1097/00005176-200403000-00006 [DOI] [PubMed] [Google Scholar]
- Grand R. J., Watkins J. B. and Torti F. M. (1976). Development of the human gastrointestinal tract. A review. Gastroenterology 70, 790-810. [PubMed] [Google Scholar]
- Grey R. D. (1972). Morphogenesis of intestinal villi. I. Scanning electron microscopy of the duodenal epithelium of the developing chick embryo. J. Morphol. 137, 193-213. 10.1002/jmor.1051370206 [DOI] [PubMed] [Google Scholar]
- Grimm-Gunter E. M. S., Revenu C., Ramos S., Hurbain I., Smyth N., Ferrary E., Louvard D., Robine S. and Rivero F. (2009). Plastin 1 binds to keratin and is required for terminal web assembly in the intestinal epithelium. Mol. Biol. Cell 20, 2549-2562. 10.1091/mbc.E08-10-1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros J., Hu J. K.-H, Vinegoni C., Feruglio P. F., Weissleder R. and Tabin C. J. (2010). WNT5A/JNK and FGF/MAPK pathways regulate the cellular events shaping the vertebrate limb bud. Curr. Biol. 20, 1993-2002. 10.1016/j.cub.2010.09.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse A. S., Pressprich M. F., Curley L. B., Hamilton K. L., Margolis B., Hildebrand J. D. and Gumucio D. L. (2011). Cell dynamics in fetal intestinal epithelium: implications for intestinal growth and morphogenesis. Development 138, 4423-4432. 10.1242/dev.065789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford M. M., Armes J., Buchert M., Meskenaite V., Grail D., Hibbs M. L., Wilks A. F., Farlie P. G., Newgreen D. F., Hovens C. M. et al. (2000). Ryk-deficient mice exhibit craniofacial defects associated with perturbed Eph receptor crosstalk. Nat. Genet. 25, 414-418. 10.1038/78099 [DOI] [PubMed] [Google Scholar]
- Hampton C. M., Liu J., Taylor D. W., DeRosier D. J. and Taylor K. A. (2008). The 3D structure of villin as an unusual F-Actin crosslinker. Structure 16, 1882-1891. 10.1016/j.str.2008.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart A. W., Morgan J. E., Schneider J., West K., McKie L., Bhattacharya S., Jackson I. J. and Cross S. H. (2006). Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum. Mol. Genet. 15, 2457-2467. 10.1093/hmg/ddl168 [DOI] [PubMed] [Google Scholar]
- Hasosah M., Lemberg D. A., Skarsgard E. and Schreiber R. (2008). Congenital short bowel syndrome: a case report and review of the literature. Can. J. Gastroenterol. 22, 71-74. 10.1155/2008/590143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helander H. F. and Fändriks L. (2014). Surface area of the digestive tract - revisited. Scand. J. Gastroenterol. 49, 681-689. 10.3109/00365521.2014.898326 [DOI] [PubMed] [Google Scholar]
- Hounnou G., Destrieux C., Desme J., Bertrand P. and Velut S. (2002). Anatomical study of the length of the human intestine. Surg. Radiol. Anat. 24, 290-294. 10.1007/s00276-002-0057-y [DOI] [PubMed] [Google Scholar]
- Hussain K., Bitner-Glindzicz M., Blaydon D., Lindley K. J., Thompson D. A., Kriss T., Rajput K., Ramadan D. G., Al-Mazidi Z., Cosgrove K. E. et al. (2004). Infantile hyperinsulinism associated with enteropathy, deafness and renal tubulopathy: clinical manifestations of a syndrome caused by a contiguous gene deletion located on chromosome 11p. J. Pediatr. Endocrinol. Metab. 17, 1613-1621. [DOI] [PubMed] [Google Scholar]
- Jayasundar J. J., Ju J. H., He L., Liu D., Meilleur F., Zhao J., Callaway D. J. E. and Bu Z. (2012). Open conformation of ezrin bound to phosphatidylinositol 4,5-bisphosphate and to F-actin revealed by neutron scattering. J. Biol. Chem. 287, 37119-37133. 10.1074/jbc.M112.380972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner K. H., Silberg D. G., Traber P. G. and Schütz G. (1997). The mesenchymal winged helix transcription factor Fkh6 is required for the control of gastrointestinal proliferation and differentiation. Genes Dev. 11, 1583-1595. 10.1101/gad.11.12.1583 [DOI] [PubMed] [Google Scholar]
- Karlsson L., Lindahl P., Heath J. K. and Betsholtz C. (2000). Abnormal gastrointestinal development in PDGF-A and PDGFR-(alpha) deficient mice implicates a novel mesenchymal structure with putative instructive properties in villus morphogenesis. Development 127, 3457-3466. [DOI] [PubMed] [Google Scholar]
- Karner C. M., Chirumamilla R., Aoki S., Igarashi P., Wallingford J. B. and Carroll T. J. (2009). Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat. Genet. 41, 793-799. 10.1038/ng.400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R., Davidson L., Edlund A., Elul T., Ezin M., Shook D. and Skoglund P. (2000). Mechanisms of convergence and extension by cell intercalation. Philos. Trans. R Soc. Lond. B Biol. Sci. 355, 897-922. 10.1098/rstb.2000.0626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. and McCulloch C. A. (2011). Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 585, 18-22. 10.1016/j.febslet.2010.11.033 [DOI] [PubMed] [Google Scholar]
- Kim T.-H. and Shivdasani R. A. (2011). Genetic evidence that intestinal Notch functions vary regionally and operate through a common mechanism of Math1 repression. J. Biol. Chem. 286, 11427-11433. 10.1074/jbc.M110.188797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.-H., Kim B.-M., Mao J., Rowan S. and Shivdasani R. A. (2011). Endodermal Hedgehog signals modulate Notch pathway activity in the developing digestive tract mesenchyme. Development 138, 3225-3233. 10.1242/dev.066233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolterud A., Grosse A. S., Zacharias W. J., Walton K. D., Kretovich K. E., Madison B. B., Waghray M., Ferris J. E., Hu C., Merchant J. L. et al. (2009). Paracrine Hedgehog signaling in stomach and intestine: new roles for hedgehog in gastrointestinal patterning. Gastroenterology 137, 618-628. 10.1053/j.gastro.2009.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guen L., Marchal S., Faure S. and de Santa Barbara P. (2015). Mesenchymal-epithelial interactions during digestive tract development and epithelial stem cell regeneration. Cell. Mol. Life Sci. 72, 3883-3896. 10.1007/s00018-015-1975-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. O. and Norden C. (2013). Mechanisms controlling arrangements and movements of nuclei in pseudostratified epithelia. Trends Cell Biol. 23, 141-150. 10.1016/j.tcb.2012.11.001 [DOI] [PubMed] [Google Scholar]
- Loomis P. A., Zheng L., Sekerková G., Changyaleket B., Mugnaini E. and Bartles J. R. (2003). Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J. Cell Biol. 163, 1045-1055. 10.1083/jcb.200309093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis P. A., Kelly A. E., Zheng L., Changyaleket B., Sekerková G., Mugnaini E., Ferreira A., Mullins R. D. and Bartles J. R. (2006). Targeted wild-type and jerker espins reveal a novel, WH2-domain-dependent way to make actin bundles in cells. J. Cell Sci. 119, 1655-1665. 10.1242/jcs.02869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheda M. L., Sun W. W., Kugathasan K., Hogan B. M., Bower N. I., Halford M. M., Zhang Y. F., Jacques B. E., Lieschke G. J., Dabdoub A. et al. (2012). The Wnt receptor Ryk plays a role in mammalian planar cell polarity signaling. J. Biol. Chem. 287, 29312-29323. 10.1074/jbc.M112.362681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison B. B., Braunstein K., Kuizon E., Portman K., Qiao X. T. and Gumucio D. L. (2005). Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development 132, 279-289. 10.1242/dev.01576 [DOI] [PubMed] [Google Scholar]
- Maini P. K., Baker R. E. and Chuong C. M. (2006). Developmental biology. The Turing model comes of molecular age. Science 314, 1397-1398. 10.1126/science.1136396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J., Kim B. M., Rajurkar M., Shivdasani R. A. and McMahon A. P. (2010). Hedgehog signaling controls mesenchymal growth in the developing mammalian digestive tract. Development 137, 1721-1729. 10.1242/dev.044586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathan M., Moxey P. C. and Trier J. S. (1976). Morphogenesis of fetal rat duodenal villi. Am. J. Anat. 146, 73-92. 10.1002/aja.1001460104 [DOI] [PubMed] [Google Scholar]
- Matsumoto A., Hashimoto K., Yoshioka T. and Otani H. (2002). Occlusion and subsequent re-canalization in early duodenal development of human embryos: integrated organogenesis and histogenesis through a possible epithelial-mesenchymal interaction. Anat. Embryol. 205, 53-65. 10.1007/s00429-001-0226-5 [DOI] [PubMed] [Google Scholar]
- Matsuyama M., Aizawa S. and Shimono A. (2009). Sfrp controls apicobasal polarity and oriented cell division in developing gut epithelium. PLoS Genet. 5, e1000427 10.1371/journal.pgen.1000427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez J., Liu M., Sampson L., Akunuru S., Han X., Vallance J., Witte D., Shroyer N. and Zheng Y. (2013). Cdc42 coordinates proliferation, polarity, migration, and differentiation of small intestinal epithelial cells in mice. Gastroenterology 145, 808-819. 10.1053/j.gastro.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxey P. C. and Trier J. S. (1978). Specialized cell types in the human fetal small intestine. Anat. Rec. 191, 269-285. 10.1002/ar.1091910302 [DOI] [PubMed] [Google Scholar]
- Muller T., Hess M. W., Schiefermeier N., Pfaller K., Ebner H. L., Heinz-Erian P., Ponstingl H., Partsch J., Röllinghoff B., Köhler H. et al. (2008). MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat. Genet. 40, 1163-1165. 10.1038/ng.225 [DOI] [PubMed] [Google Scholar]
- Ng A.-Y., Waring P., Ristevski S., Wang C., Wilson T., Pritchard M., Hertzog P. and Kola I. (2002). Inactivation of the transcription factor Elf3 in mice results in dysmorphogenesis and altered differentiation of intestinal epithelium. Gastroenterology 122, 1455-1466. 10.1053/gast.2002.32990 [DOI] [PubMed] [Google Scholar]
- Nieuwkoop P. D. and Faber J. (1994). Normal Table of Xenopus Laevis (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis. New York: Garland Pub. [Google Scholar]
- Nomachi A., Nishita M., Inaba D., Enomoto M., Hamasaki M. and Minami Y. (2008). Receptor tyrosine kinase Ror2 mediates Wnt5a-induced polarized cell migration by activating c-Jun N-terminal kinase via actin-binding protein filamin A. J. Biol. Chem. 283, 27973-27981. 10.1074/jbc.M802325200 [DOI] [PubMed] [Google Scholar]
- Ootani A., Li X., Sangiorgi E., Ho Q. T., Ueno H., Toda S., Sugihara H., Fujimoto K., Weissman I. L., Capecchi M. R. et al. (2009). Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 15, 701-706. 10.1038/nm.1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormestad M., Astorga J., Landgren H., Wang T., Johansson B. R., Miura N. and Carlsson P. (2006). Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development 133, 833-843. 10.1242/dev.02252 [DOI] [PubMed] [Google Scholar]
- Pinson K. I., Dunbar L., Samuelson L. and Gumucio D. L. (1998). Targeted disruption of the mouse villin gene does not impair the morphogenesis of microvilli. Dev. Dyn. 211, 109-121. [DOI] [PubMed] [Google Scholar]
- Qian D., Jones C., Rzadzinska A., Mark S., Zhang X., Steel K. P., Dai X. and Chen P. (2007). Wnt5a functions in planar cell polarity regulation in mice. Dev. Biol. 306, 121-133. 10.1016/j.ydbio.2007.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho-Santos M., Melton D. A. and McMahon A. P. (2000). Hedgehog signals regulate multiple aspects of gastrointestinal development. Development 127, 2763-2772. [DOI] [PubMed] [Google Scholar]
- Raspopovic J., Marcon L., Russo L. and Sharpe J. (2014). Modeling digits. Digit patterning is controlled by a Bmp-Sox9-Wnt Turing network modulated by morphogen gradients. Science 345, 566-570. 10.1126/science.1252960 [DOI] [PubMed] [Google Scholar]
- Reed R. A., Womble M. A., Dush M. K., Tull R. R., Bloom S. K., Morckel A. R., Devlin E. W. and Nascone-Yoder N. M. (2009). Morphogenesis of the primitive gut tube is generated by Rho/ROCK/myosin II-mediated endoderm rearrangements. Dev. Dyn. 238, 3111-3125. 10.1002/dvdy.22157 [DOI] [PubMed] [Google Scholar]
- Revenu C., Ubelmann F., Hurbain I., El-Marjou F., Dingli F., Loew D., Delacour D., Gilet J., Brot-Laroche E., Rivero F. et al. (2012). A new role for the architecture of microvillar actin bundles in apical retention of membrane proteins. Mol. Biol. Cell 23, 324-336. 10.1091/mbc.E11-09-0765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruemmele F. M., Müller T., Schiefermeier N., Ebner H. L., Lechner S., Pfaller K., Thöni C. E., Goulet O., Lacaille F., Schmitz J. et al. (2010). Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum. Mutat. 31, 544-551. 10.1002/humu.21224 [DOI] [PubMed] [Google Scholar]
- Sabharwal G., Strouse P. J., Islam S. and Zoubi N. (2004). Congenital short-gut syndrome. Pediatr. Radiol. 34, 424-427. 10.1007/s00247-003-1087-2 [DOI] [PubMed] [Google Scholar]
- Saburi S., Hester I., Fischer E., Pontoglio M., Eremina V., Gessler M., Quaggin S. E., Harrison R., Mount R. and McNeill H. (2008). Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat. Genet. 40, 1010-1015. 10.1038/ng.179 [DOI] [PubMed] [Google Scholar]
- Sakamori R., Das S., Yu S., Feng S., Stypulkowski E., Guan Y., Douard V., Tang W., Ferraris R. P., Harada A. et al. (2012). Cdc42 and Rab8a are critical for intestinal stem cell division, survival, and differentiation in mice. J. Clin. Invest. 122, 1052-1065. 10.1172/JCI60282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome I., Curto M. and McClatchey A. I. (2004). Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev. Cell 6, 855-864. 10.1016/j.devcel.2004.05.007 [DOI] [PubMed] [Google Scholar]
- Sato T., Mushiake S., Kato Y., Sato K., Sato M., Takeda N., Ozono K., Miki K., Kubo Y., Tsuji A. et al. (2007). The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature 448, 366-369. 10.1038/nature05929 [DOI] [PubMed] [Google Scholar]
- Sato T., Vries R. G., Snippert H. J., van de Wetering M., Barker N., Stange D. E., van Es J. H., Abo A., Kujala P., Peters P. J. et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262-265. 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- Sausedo R. A., Smith J. L. and Schoenwolf G. C. (1997). Role of nonrandomly oriented cell division in shaping and bending of the neural plate. J. Comp. Neurol. 381, 473-488. [DOI] [PubMed] [Google Scholar]
- Sauvanet C., Wayt J., Pelaseyed T. and Bretscher A. (2015). Structure, regulation, and functional diversity of microvilli on the apical domain of epithelial cells. Annu. Rev. Cell Dev. Biol. 31, 593-621. 10.1146/annurev-cellbio-100814-125234 [DOI] [PubMed] [Google Scholar]
- Shyer A. E., Tallinen T., Nerurkar N. L., Wei Z., Gil E. S., Kaplan D. L., Tabin C. J. and Mahadevan L. (2013). Villification: how the gut gets its villi. Science 342, 212-218. 10.1126/science.1238842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer A. E., Huycke T. R., Lee C., Mahadevan L. and Tabin C. J. (2015). Bending gradients: how the intestinal stem cell gets its home. Cell 161, 569-580. 10.1016/j.cell.2015.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sick S., Reinker S., Timmer J. and Schlake T. (2006). WNT and DKK determine hair follicle spacing through a reaction-diffusion mechanism. Science 314, 1447-1450. 10.1126/science.1130088 [DOI] [PubMed] [Google Scholar]
- Sinagoga K. L. and Wells J. M. (2015). Generating human intestinal tissues from pluripotent stem cells to study development and disease. EMBO J. 34, 1149-1163. 10.15252/embj.201490686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J. R., Lauf R. and Shroyer N. F. (2011a). Vertebrate intestinal endoderm development. Dev. Dyn. 240, 501-520. 10.1002/dvdy.22540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J. R., Mayhew C. N., Rankin S. A., Kuhar M. F., Vallance J. E., Tolle K., Hoskins E. E., Kalinichenko V. V., Wells S. I., Zorn A. M. et al. (2011b). Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105-109. 10.1038/nature09691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurrier R. G. and Grikscheit T. C. (2013). Tissue engineering the small intestine. Clin. Gastroenterol. Hepatol. 11, 354-358. 10.1016/j.cgh.2013.01.028 [DOI] [PubMed] [Google Scholar]
- Struijs M.-C., Diamond I. R., de Silva N. and Wales P. W. (2009). Establishing norms for intestinal length in children. J. Pediatr. Surg. 44, 933-938. 10.1016/j.jpedsurg.2009.01.031 [DOI] [PubMed] [Google Scholar]
- Turing A. M. (1952). The chemical basis of morphogenesis. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 237, 37-72. 10.1098/rstb.1952.0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymms M. J., Ng A. Y. N., Thomas R. S., Schutte B. C., Zhou J., Eyre H. J., Sutherland G. R., Seth A., Rosenberg M., Papas T. et al. (1997). A novel epithelial-expressed ETS gene, ELF3: human and murine cDNA sequences, murine genomic organization, human mapping to 1q32.2 and expression in tissues and cancer. Oncogene 15, 2449-2462. 10.1038/sj.onc.1201427 [DOI] [PubMed] [Google Scholar]
- Van Der Werf C. S., Wabbersen T. D., Hsiao N.-H., Paredes J., Etchevers H. C., Kroisel P. M., Tibboel D., Babarit C., Schreiber R. A., Hoffenberg E. J. et al. (2012). CLMP is required for intestinal development, and loss-of-function mutations cause congenital short-bowel syndrome. Gastroenterology 142, 453-462.e3. 10.1053/j.gastro.2011.11.038 [DOI] [PubMed] [Google Scholar]
- Van Der Werf C. S., Hsiao N.-H., Conroy S., Paredes J., Ribeiro A. S., Sribudiani Y., Seruca R., Hofstra R. M. W., Westers H. and van Ijzendoorn S. C. D. (2013). CLMP is essential for intestinal development, but does not play a key role in cellular processes involved in intestinal epithelial development. PLoS ONE 8, e54649 10.1371/journal.pone.0054649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Werf C. S., Halim D., Verheij J. B. G. M., Alves M. M. and Hofstra R. M. W. (2015). Congenital Short Bowel Syndrome: from clinical and genetic diagnosis to the molecular mechanisms involved in intestinal elongation. Biochim. Biophys. Acta 1852, 2352-2361. 10.1016/j.bbadis.2015.08.007 [DOI] [PubMed] [Google Scholar]
- Walton K. D., Kolterud A., Czerwinski M. J., Bell M. J., Prakash A., Kushwaha J., Grosse A. S., Schnell S. and Gumucio D. L. (2012). Hedgehog-responsive mesenchymal clusters direct patterning and emergence of intestinal villi. Proc. Natl. Acad. Sci. USA 109, 15817-15822. 10.1073/pnas.1205669109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton K. D., Whidden M., Kolterud A., Shoffner S. K., Czerwinski M. J., Kushwaha J., Parmar N., Chandhrasekhar D., Freddo A. M., Schnell S. et al. (2016). Villification in the mouse: bmp signals control intestinal villus patterning. Development. 143, 427-436. 10.1242/dev.130112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.-C., Biben C., Robb L., Nassir F., Barnett L., Davidson N. O., Koentgen F., Tarlinton D. and Harvey R. P. (2000). Homeodomain factor Nkx2–3 controls regional expression of leukocyte homing coreceptor MAdCAM-1 in specialized endothelial cells of the viscera. Dev. Biol. 224, 152-167. 10.1006/dbio.2000.9749 [DOI] [PubMed] [Google Scholar]
- Watson C. L., Mahe M. M., Múnera J., Howell J. C., Sundaram N., Poling H. M., Schweitzer J. I., Vallance J. E., Mayhew C. N., Sun Y. et al. (2014). An in vivo model of human small intestine using pluripotent stem cells. Nat. Med. 20, 1310-1314. 10.1038/nm.3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver L. T., Austin S. and Cole T. J. (1991). Small intestinal length: a factor essential for gut adaptation. Gut 32, 1321-1323. 10.1136/gut.32.11.1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J. M. and Spence J. R. (2014). How to make an intestine. Development 141, 752-760. 10.1242/dev.097386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman E. L., Fan S., Harder J. L., Walton K. D., Liu C.-J., Soofi A., Fogg V. C., Hershenson M. B., Dressler G. R., Deutsch G. H. et al. (2014). Crumbs3 is essential for proper epithelial development and viability. Mol. Cell. Biol. 34, 43-56. 10.1128/MCB.00999-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegerinck C. L., Janecke A. R., Schneeberger K., Vogel G. F., van Haaften-Visser D. Y., Escher J. C., Adam R., Thöni C. E., Pfaller K., Jordan A. J. et al. (2014). Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology 147, 65-68.e10. 10.1053/j.gastro.2014.04.002 [DOI] [PubMed] [Google Scholar]
- Xu J., Srinivas B. P., Tay S. Y., Mak A., Yu X., Lee S. G. P., Yang H., Govindarajan K. R., Leong B., Bourque G. et al. (2006). Genomewide expression profiling in the Zebrafish embryo identifies target genes regulated by Hedgehog signaling during vertebrate development. Genetics 174, 735-752. 10.1534/genetics.106.061523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M., Udagawa J., Matsumoto A., Hashimoto R., Hatta T., Nishita M., Minami Y. and Otani H. (2010). Ror2 is required for midgut elongation during mouse development. Dev. Dyn. 239, 941-953. 10.1002/dvdy.22212 [DOI] [PubMed] [Google Scholar]
- Zheng L., Sekerková G., Vranich K., Tilney L. G., Mugnaini E. and Bartles J. R. (2000). The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102, 377-385. 10.1016/S0092-8674(00)00042-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liu H.-X. and Mistretta C. M. (2006). Bone morphogenetic proteins and noggin: inhibiting and inducing fungiform taste papilla development. Dev. Biol. 297, 198-213. 10.1016/j.ydbio.2006.05.022 [DOI] [PubMed] [Google Scholar]
- Zorn A. M. and Wells J. M. (2009). Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol. 25, 221-251. 10.1146/annurev.cellbio.042308.113344 [DOI] [PMC free article] [PubMed] [Google Scholar]