

Abstract
Wnt signaling generates patterns in all embryos, from flies to humans, and controls cell fate, proliferation and metabolic homeostasis. Inappropriate Wnt pathway activation results in diseases, including colorectal cancer. The adenomatous polyposis coli (APC) tumor suppressor gene encodes a multifunctional protein that is an essential regulator of Wnt signaling and cytoskeletal organization. Although progress has been made in defining the role of APC in a normal cellular context, there are still significant gaps in our understanding of APC-dependent cellular function and dysfunction. We expanded the APC-associated protein network using a combination of genetics and a proteomic technique called two-dimensional difference gel electrophoresis (2D-DIGE). We show that loss of Drosophila Apc2 causes protein isoform changes reflecting misregulation of post-translational modifications (PTMs), which are not dependent on β-catenin transcriptional activity. Mass spectrometry revealed that proteins involved in metabolic and biosynthetic pathways, protein synthesis and degradation, and cell signaling are affected by Apc2 loss. We demonstrate that changes in phosphorylation partially account for the altered PTMs in APC mutants, suggesting that APC mutants affect other types of PTM. Finally, through this approach Aminopeptidase P was identified as a new regulator of β-catenin abundance in Drosophila embryos. This study provides new perspectives on the cellular effects of APC that might lead to a deeper understanding of its role in development.
KEY WORDS: APC, ApepP, 2D-DIGE, Post-translational modification, Wnt signaling
Summary: The analysis of Drosophila embryos lacking APC reveals a diverse set of post-translational modifications that are dependent on APC, one of which regulates β-catenin abundance.
INTRODUCTION
The canonical Wnt signaling pathway is well conserved in metazoan development, and normal Wnt signaling regulates embryonic patterning by controlling cell fate determination and stem cell behavior (Polakis, 2012; Kozinski and Dobrzyn, 2013). The role of Wnt signaling in metabolic homeostasis has recently become appreciated (Ring et al., 2014). The clinical importance of this pathway has been demonstrated primarily by its inappropriate activation, such as in colorectal cancer, where mutations in the negative regulator and tumor suppressor adenomatous polyposis coli (APC) initiate tumorigenesis in the majority of both sporadic and inherited cases (Logan and Nusse, 2004; Schneikert et al., 2007). In the absence of a Wnt signal, APC proteins promote the assembly, stability and function of the destructosome, a macromolecular assembly that includes the core components APC (also known as APC1 in Drosophila), APC2, Axin and two kinases, namely Glycogen synthase kinase β (GSK3β) and Casein kinase 1 (CK1) (Kunttas-Tatli et al., 2012, 2014; Stamos and Weis, 2013). The active destructosome recruits and phosphorylates the Wnt effector β-catenin (β-cat), targeting it for ubiquitylation and degradation by the proteasome (Fig. 1A). This effectively keeps cytoplasmic β-cat levels low, which in turn prevents ligand-independent activation of Wnt target genes.
Fig. 1.

Summary of the Wnt signaling pathway and 2D-DIGE workflow. (A) The Wnt signaling pathway. When no Wnt ligand is present (left) the destructosome promotes cytoplasmic β-cat degradation. With a Wnt ligand (right), the destructosome is deactivated and cytoplasmic β-cat accumulates, enters the nucleus and activates Wnt target genes. (B) Principal steps in the 2D-DIGE workflow: (1) covalent labeling of protein lysates with either propyl-Cy3 or methyl-Cy5; (2) combine labeled protein lysates; (3) co-electrophorese on a 2DE gel; (4) fluorescent gel imaging of Cy3 (green) and Cy5 (red) (common spots are yellow, whereas difference-proteins appear as green or red); (5) selected spots shown in the inset are quantified using SourceExtractor; (6) concurrently genetically validate the difference-proteins (6a) and identify the proteins by LC-MS/MS (6b).
Ligand-independent activation of Wnt signaling due to loss of APC clearly contributes to colon cancer initiation; however, less is known about how disruption of other cellular functions of APC impacts normal development and cancer. In addition to their role in Wnt signaling, APC proteins also function in actin assembly, cell-to-cell adhesion, and microtubule network formation through interactions with cytoskeletal proteins (Nathke, 2006). Disruption of these functions has negative consequences, including decreased cell migration and differentiation, chromosomal instability, and increased proliferation that may contribute to cancer initiation and progression (Nathke, 2004, 2006). Furthermore, new APC protein domains are still being identified, which in turn are uncovering new protein and pathway interactions that might also contribute (Zhang et al., 2010; Kunttas-Tatli et al., 2014; Preitner et al., 2014).
To better understand the cellular consequences of APC loss that may contribute to normal development, we used Drosophila melanogaster embryogenesis as a model. APC proteins are well conserved between mammals and fruit flies, making Drosophila a tractable system for studying APC-dependent protein networks. As in humans, the Drosophila genome contains two APC genes (Logan and Nusse, 2004): Apc (Apc1) and Apc2. Both APC proteins in Drosophila and mammals are similarly structured and contain many of the same conserved domains. Furthermore, we and others have demonstrated that Drosophila and vertebrate APC proteins are functionally conserved; like vertebrate APC, both fly APC proteins negatively regulate Wnt signaling (Ahmed et al., 1998; McCartney et al., 1999), collaborate with the formin Diaphanous (Webb et al., 2009; Jaiswal et al., 2013) and can localize to microtubules (McCartney et al., 2001; Akong et al., 2002). In addition, Drosophila APC1 binds actin monomers, nucleates F-actin assembly (Jaiswal et al., 2013) and binds the microtubule plus-tip protein EB1 (Webb et al., 2009), like vertebrate APC. Previous studies have examined the global cellular consequence of APC disruption using RNA-seq and two-dimensional difference gel electrophoresis (2D-DIGE) proteomics (Chafey et al., 2009; Wu et al., 2012). However, the results of these studies reflect chronic changes due to the primary and downstream effects of ligand-independent Wnt target gene activation. Here, we use the early Drosophila embryo [0-2 h after egg laying (AEL)] as our model system. At this time in development, zygotic transcription is largely silenced (Qin et al., 2007; Tadros and Lipshitz, 2009), providing a unique opportunity to observe proteome changes that are independent of β-cat transcription.
Proteome analysis provides valuable information about a cell or system that is unseen at the transcriptome level (Vogel and Marcotte, 2012), including changes in post-translational modification (PTM) and protein stability. Moreover, there is very little correlation between mRNA and protein abundance (Anderson and Seilhamer, 1997; Gygi et al., 1999). 2D-DIGE is a powerful technique that can reveal proteomic changes (the ‘difference-proteins') between two or three protein samples run simultaneously on the same gel (Fig. 1B) (Minden, 2012). This technique combined with a high dynamic range fluorescence imaging system can detect as little as 0.2 fmol of protein over a 100,000-fold concentration range (Minden, 2012; Van et al., 2014a). Difference-proteins can then be identified using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). 2D-DIGE is useful in assessing proteome changes in a wide variety of models, including whole organisms such as Drosophila and zebrafish, cell culture and isolated subcellular structures (May et al., 2012; Miller, 2012; Minden, 2012).
Here, we report the initial identification of 16 difference-protein isoform pairs between wild-type and Apc2 mutant Drosophila embryos. All of these difference-proteins were the result of isoform shifts indicative of changes in PTMs. LC-MS/MS of the difference-protein isoform pairs identified protein components of metabolic and biosynthetic pathways, protein synthesis and degradation, cell signaling and the cellular stress response. After rigorous genetic validation of the difference-protein isoform pairs, one protein, Aminopeptidase P (ApepP), was further shown to be a new regulator of β-cat abundance. These results demonstrate that loss of APC influences an array of protein targets post-translationally, leading to a deeper understanding of the global cellular consequences of APC mutations.
RESULTS
Comparison of the Apc2 null and wild-type proteomes
To understand the proteome changes that occur in embryos lacking APC protein, we compared lysates of Apc2 null and wild-type Drosophila embryos (Fig. 1B). Early embryos (0-2 h AEL) were chosen to characterize the earliest detectable proteome changes associated with APC loss. At this stage, the embryos are syncytial and transcription is strongly inhibited, with the exception of some patterning and housekeeping genes (Qin et al., 2007; Tadros and Lipshitz, 2009). Thus, we predicted that the differences between the Apc2 and wild-type proteomes would mostly appear as post-translational changes, rather than protein abundance changes resulting from transcriptional activation of Wnt targets. Lysates from mutant and wild-type embryos were separately labeled with either Cy3 or Cy5 DIGE dyes, which are shown as green and red, respectively. The separately labeled lysates were combined and run on the same 2DE gel. Protein spots that are common to both lysates are yellow, whereas proteins that are enriched in one lysate will appear redder or greener. PTM differences appear as closely spaced reciprocally changing spots: one protein spot increases while its neighbor decreases (Fig. 1B, step 4, white box).
APC2 is the most abundant of the two APC proteins during early embryogenesis, and functions in Wnt signaling and cytoskeletal regulation (McCartney et al., 1999, 2001; Webb et al., 2009; Zhou et al., 2011; Kunttas-Tatli et al., 2012, 2014). 2D-DIGE comparison of Apc2g10 (Apc2 null) (McCartney et al., 2006) and wild-type embryos revealed 16 prominent protein changes observed in 100% of the trials (n=30), all of which appeared to be PTMs as evidenced by two or more protein spots horizontally arranged and changing reciprocally (Fig. 2, white boxes). Such horizontal shifts indicate a change in isoelectric point (pI) or net protein charge, but not protein mass. This set of protein changes was observed in 30 high-quality biological replicates and is composed of moderately to highly abundant proteins. Other, lower abundance protein changes were observed inconsistently, which was likely to be due to gel-to-gel variation.
Fig. 2.
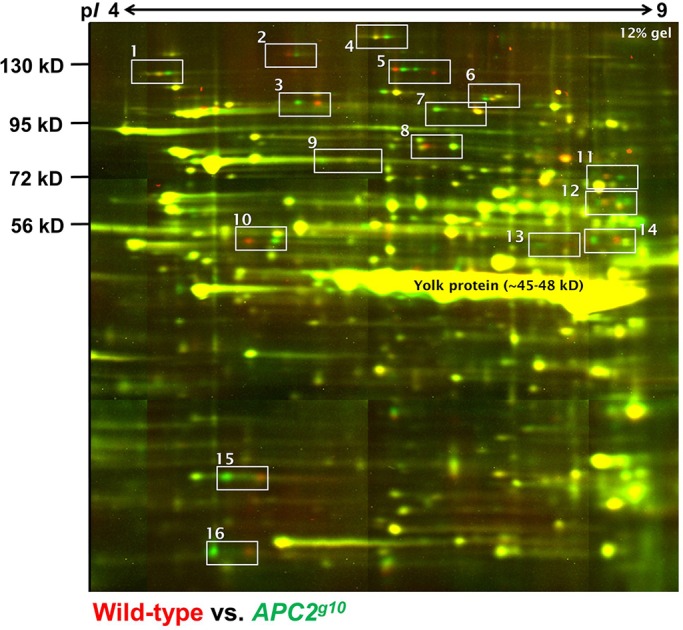
2D-DIGE of Apc2 null versus wild-type Drosophila embryos. A tiled array of a whole gel comparing Apc2g10 (green) and wild-type (red) embryos. The 16 Apc2-dependent difference-protein pairs are demarcated by white boxes. Contrast and brightness were manipulated on the whole-gel images using ImageJ (full details of image analysis are provided in the supplementary Materials and Methods).
The 16 difference-protein pairs in the Apc2g10 embryos were quantified using an open-source astronomy software package, SourceExtractor, previously described (Bertin and Arnouts, 1996; Gong et al., 2004) (Table S1). The typical variation in the measured abundance of common protein spots is less than 4% for 2D-DIGE; thus, the cutoff for a significant total abundance change was set to at least ±10%, which is 2.5 standard deviations from the typical variance (Gong et al., 2004). Just over half of the difference-protein pairs (9 of 16) were close to being perfectly reciprocal changes (<10% total protein change) (Fig. 3A). The remaining seven exhibited a total abundance reduction of >10% (Fig. 3B), suggesting increased protein degradation in Apc2g10 embryos. Thus, the loss of Apc2 in early embryos, when there is no Wnt pathway activation and very little transcription, leads to significant protein changes affecting PTMs that might decrease the stability of a subset of these proteins.
Fig. 3.

Quantification of Apc2-dependent difference-proteins. (A) The fold change for each difference-protein pair as calculated using the Cy3 and Cy5 raw fluorescence intensities. Cy3/Cy5 ratios <1 were converted to negative fold change. (B) Total protein abundance, defined as the sum of the fluorescence intensities of putative difference-protein isoforms, was calculated for each protein region in wild-type and Apc2g10 embryos. The percentage difference in total protein isoform fluorescence relative to wild type was then calculated. Difference-proteins that display a >10% change in total abundance are marked with an asterisk.
Identification of difference-protein isoform pairs in Apc2 null and wild-type proteomes
The difference-protein pairs were analyzed by MS to identify the proteins and confirm that each pair of protein spots represented isoforms. This analysis revealed that some difference-protein pairs contained more than one protein species. A list of protein identifications for the 16 difference-protein isoform pairs was generated from at least two biological replicates (Table 1; Table S2). Protein spots from gels containing reciprocally labeled samples were also analyzed to provide technical replicates, further validating the protein identifications. All identified proteins had a predicted mass and pI relatively well-matched to the observed difference-proteins on the 2DE gel. The identified proteins span a range of cellular functions, from metabolic proteins as the most prominent difference-proteins (43%) to RNA-binding proteins (22%), DNA-binding proteins (13%), chaperones and proteases (9% each) and signaling proteins (4%).
Table 1.
MS identification of Apc2-dependent difference-proteins
We used immunoblot analysis to confirm the identity of one of the difference-proteins for which a Drosophila-specific antibody was available, Calcium-binding protein 1 (CaBP1) (Fig. 4). The immunoblot showed that the acidic isoform is in greater abundance in wild-type embryos, whereas the basic isoform is more abundant in Apc2g10 mutant embryos, confirming our MS identification of CaBP1, and confirming that its isoform distribution is altered in Apc2 null mutants.
Fig. 4.
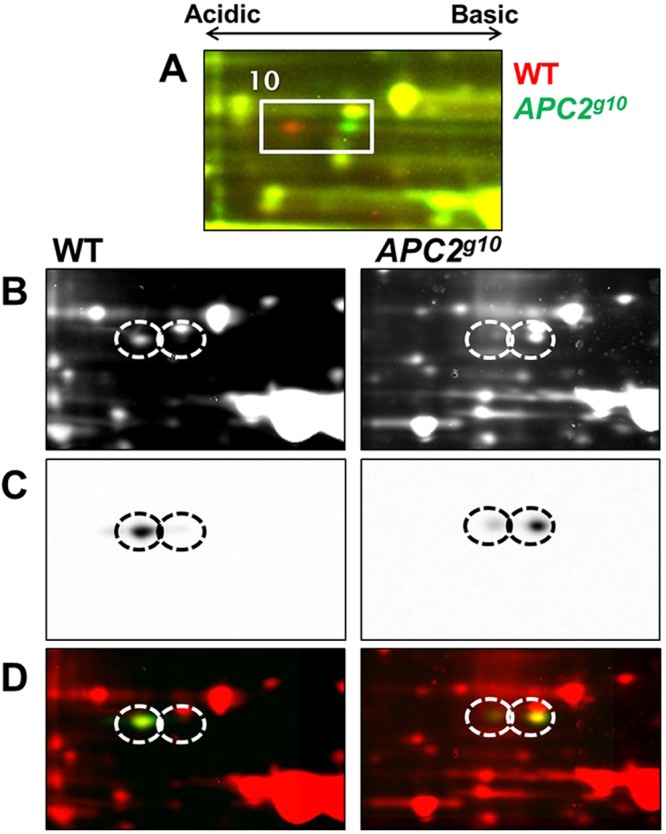
Immunoblot confirmation that CaBP1 is an APC2-dependent difference-protein. (A) 2D-DIGE image of the region containing CaBP1 isoforms. (B) Fluorescent images of wild-type and Apc2g10 embryo lysates that were separately labeled with Cy3 DIGE dye, resolved on different 2DE gels and transferred to nitrocellulose. (C) Immunoblot images of proteins stained with anti-CaBP1 antibody. (D) Superimposed images of total Cy3-labeled protein (red) and the CaBP1 2D immunoblot (green) confirm protein identification by LC-MS/MS. The predominant isoform found in wild-type lysate is the left isoform, whereas the right isoform is predominant in Apc2 null embryos.
The difference-protein isoform pairs are due to multiple types of PTMs
Because the APC-containing destructosome includes two kinases, GSK3β and CK1α, we tested the hypothesis that many of the observed isoform differences were due to phosphorylation changes by treating wild-type and Apc2g10 embryo lysates with a broad-spectrum phosphatase, Lambda protein phosphatase (λPP). 2D-DIGE was used to compare λPP-treated wild-type and Apc2 mutant embryo lysates with untreated lysates. Dephosphorylated proteins shift to the right (more basic) relative to untreated lysates, whereas non-phosphorylated (or λPP refractory) protein will be unaffected. The efficacy of phosphatase treatment was confirmed by observing the entire yolk protein population shifting to the right (data not shown). Unfortunately, the λPP reaction conditions interfered with isoelectric focusing in the basic region of the 2DE gels, preventing analysis of seven of the 16 difference-proteins isoform pairs. The acidic half of the gels was well resolved, and we were able to assess the remaining nine difference-proteins isoform pairs. Surprisingly, phosphorylation was found to play a minor role in generating the isoform changes. Below are two examples of what we observed.
Dp1 difference-protein isoforms displayed phosphatase sensitivity (Fig. 5A). Dp1 has two isoforms on 2DE gels: the acidic L isoform, which is elevated in wild-type embryos (Fig. 5A′, left panel), and the basic R isoform, which is elevated in Apc2 mutant embryos (Fig. 5A″, left panel). The L isoform shifted to the right upon λPP treatment, indicating that this isoform is phosphorylated (Fig. 5A′, right panel). The R isoform did not shift upon λPP treatment, indicating that it is not phosphorylated (Fig. 5A″, right panel). Interestingly, in both wild-type and Apc2 mutant embryos conversion of the L isoform to the R is incomplete, suggesting that Dp1 might be partially refractory to λPP or that some fraction of the L protein spot bears a different PTM.
Fig. 5.

APC2-dependent isoform changes are due to phosphorylation and other PTMs. (A) Model for the PTM responsible for Dp1 isoforms. (A′,A″) Dp1 isoforms are affected by phosphorylation, as the left isoform collapses to the right isoform after λPP treatment for both wild-type and Apc2g10 embryo lysates. (B) Model for the PTM(s) responsible for ApepP isoforms. (B′,B″) ApepP isoforms were not affected by λPP treatment, as there were no isoform shifts observed after phosphatase treatment. Difference-protein isoforms (L, left; M, middle; R, right) are circled.
ApepP appears to be insensitive to phosphatase treatment (Fig. 5B). ApepP has three isoforms on 2DE gels. In wild-type embryos the more basic R isoform is elevated (Fig. 5B′, left panel), whereas in Apc2 mutant embryos the acidic L isoform is elevated (Fig. 5B″, left panel). The middle isoform does not significantly change in Apc2 mutant embryos compared with wild type (Fig. 5B, ApepP). Surprisingly, none of the ApepP isoforms shifted upon λPP treatment, suggesting that none is phosphorylated or that ApepP is completely resistant to λPP. This was true for the majority of the resolved difference-proteins isoforms (eight of nine), suggesting that phosphorylation is not the major PTM altered in Apc2 null embryos.
Controlling for genetic background effects
Controlling for genetic variability is necessary to interpret proteomic comparisons between two conditions. We asked if any of the 16 difference-protein isoforms were variably expressed across multiple wild-type strains by comparing the proteome of our w1118 wild-type lab strain (wMc) with two closely related, yet independently segregated, w1118 lines: wD and wP (Fig. 6A; Table S3A). The wP wild-type strain is originally from the Peifer lab (University of North Carolina at Chapel Hill), from which the McCartney w1118 line was derived and used for outcrossing of the Apc2g10 stock. The wD strain was obtained from the Duke University Model Systems Genomics Facility, where our Apc2-FL transgenic line was generated in this genetic background (McCartney et al., 2006), and subsequently crossed into the Apc2g10 background for genetic rescue experiments (see below). Thus, these two independent w1118 strains were the most relevant wild-type genotypes to determine if the protein changes found in our original wMc versus Apc2g10 comparison are variable. From this analysis, we found that wD shares three difference-proteins with Apc2g10 and that wP shares an additional six difference-proteins with Apc2g10 (Table S3A). Because of the possibility that the changes to these nine difference-proteins are not specific to Apc2g10 and instead result from common background effects, we chose to continue our analysis with the remaining wild type-independent (WTI) difference-proteins (1, 4, 7, 9, 11, 12 and 16) that did not appear as changes in any of our three wild-type w1118 strains.
Fig. 6.

Validating the APC-dependent difference-proteins. (A) A decision tree showing the genetic approach taken to validate the APC-dependent difference-proteins. Six of the 16 difference-proteins were concluded to be APC dependent and to be independent of wild-type variation. (B) 2D-DIGE comparisons of FL-GFP-Apc2;Apc2g10 versus Apc2g10 or wild-type embryo lysates were performed to test whether ectopic APC2 could reverse the observed Apc2g10 versus wild-type proteome changes. Shown here are sub-images cropped from tiled array images of whole 2D-DIGE gels. ApepP and Irp-1B exemplify reversal and failure to compensate, respectively.
Validating the APC-dependent difference-protein isoform pairs
We next asked whether the exogenous expression of full-length APC2 protein is able to rescue the remaining seven WTI difference-proteins in Apc2 null embryos (Table S3B) to further confirm their Apc2 specificity. The proteome of Apc2g10 embryos was compared with that of Apc2g10 embryos carrying two copies of a full-length Apc2 transgene (FL-GFP-Apc2;Apc2g10). This Apc2 transgene, which is driven by the native Apc2 promoter, is expressed at endogenous levels (McCartney et al., 2006) and it rescues both the cytoskeletal defects seen in the early embryo and Wnt signaling defects observed later in embryogenesis (Webb et al., 2009; Kunttas-Tatli et al., 2012). Five of the seven WTI difference-proteins, namely CG2918, Dp1, GlyP, ApepP and Cbs/Tcp-1ζ, were reverted by ectopic expression of APC2 (e.g. Fig. 6B, ApepP, left versus middle panel). Further evidence for the effect of ectopic APC2 expression in FL-GFP-Apc2 embryos was provided by the presence of these five difference-protein changes when compared with Apc2 null embryo lysates (Fig. 6B, ApepP, left versus right panel). The remaining two WTI difference-protein pairs, mRpS30 and Jafrac1, were not reversed by ectopic APC2 expression (e.g. Fig. 6B, Irp-1B).
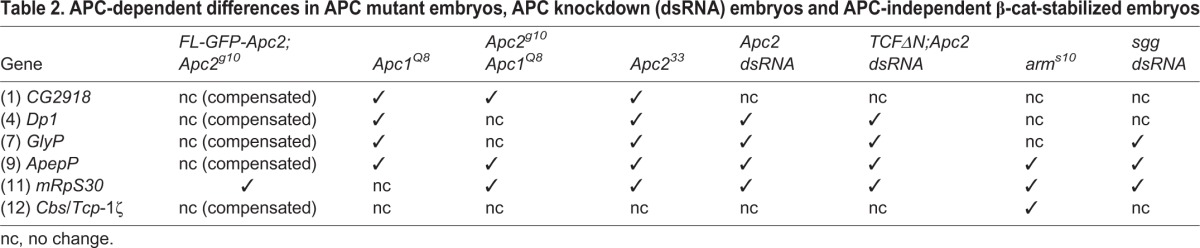
There are two possible explanations why FL-GFP-Apc2 failed to rescue two of the WTI difference-protein pairs in Apc2g10: (1) these protein differences are due to genetic background differences in Apc2g10 that were not present in the other wild-type w1118 lines; or (2) the FL-GFP-APC2 protein has reduced function compared with wild-type APC2, and that the proteomic changes are highly sensitive to APC2 activity. To distinguish between these possibilities, an independent Apc2 null allele in a distinct genetic background was analyzed. Apc233 was created by imprecise excision of a P-element within Apc2, deleting a large portion of the 5′ end of the gene, spanning from the promoter to the fifth Armadillo repeat (Takacs et al., 2008). All WTI difference-proteins, except Jafrac1, were observed in the Apc233 versus wild type comparison, suggesting that they are bona fide Apc2-dependent protein changes. Thus, the failure of FL-GFP-Apc2 to compensate for some of the WTI difference-proteins is likely to be due to insufficient activity (Table S3C). To further support that these changes are APC dependent, a variety of these changes can be reproduced in various APC knockdown situations (Table 2; Table S3). Isoform differences in Dp1, GlyP, ApepP and mRpS30 were recapitulated by knocking down Apc2 with a dsRNA driven by a strong maternal driver (MTD) as compared with wMc. CG2918, Dp1, GlyP and ApepP displayed isoform differences when comparing Apc1Q8 null germ line clone embryos with wMc embryos. Isoform changes in CG2918, ApepP and mRpS30 were observed when comparing Apc2g10Apc1Q8 null germ line clone embryos with wMc embryos. Together, these data strongly support the conclusion that six of the seven WTI difference-protein isoform pairs indeed reflect APC-dependent changes and are not due to genetic background.
Table 2.
APC-dependent differences in APC mutant embryos, APC knockdown (dsRNA) embryos and APC-independent β-cat-stabilized embryos
The APC-dependent difference-protein isoform pairs do not require β-cat transcription
Previous proteomics of APC mutant tissue identified a number of proteins with elevated levels, presumably resulting from the transcriptional activation of Wnt target genes via β-cat (Chafey et al., 2009; Hammoudi et al., 2013). In our analysis, only two of the APC-dependent difference-proteins displayed a total abundance change in the Apc2 mutant consistent with reduced protein stability (mRpS30 and Cbs/Tcp-1ζ, Fig. 3B). This, coupled with the suppression of zygotic transcription in 0-2 h embryos, suggested that the APC-dependent protein isoform differences were not a result of β-cat-dependent transcriptional activation. To test this hypothesis directly, we examined whether the APC-dependent isoform differences required the activation of Wnt target genes by expressing a dominant-negative form of TCF (TCFΔN). TCF is essential for β-cat-mediated transcription in embryos (Fig. 1A). The complexity of the genetics prevented us from performing this experiment in the Apc2 null background, and instead we reduced APC2 activity using dsRNA. Embryos derived from MTD-GAL4>UAS-Apc2dsRNA females exhibited the same protein isoform differences for Dp1, GlyP, ApepP and mRpS30 as Apc2 null embryos (Table 2). It is likely that CG2918 and Cbs/Tcp-1ζ are unchanged due to insufficient knockdown of Apc2 with the dsRNA.
Next, we generated embryos derived from MTD-GAL4>UAS-TCFΔN;UAS-Apc2dsRNA females to simultaneously knock down Apc2 and block Wnt-dependent transcription from the start of oogenesis. When we compared the proteomes of TCFΔN;Apc2dsRNA embryos and Apc2dsRNA embryos, all four Apc2-dependent protein changes that we observed in both Apc2 null and Apc2dsRNA embryos were present (Table 2). This suggests that these four proteomic changes are unaffected by the blockade of β-cat/TCF-dependent transcription. Importantly, if we crossed the MTD-GAL4>UAS-TCFΔN;UAS-Apc2dsRNA females to males carrying tubulin-GAL4 to drive ubiquitous expression of both TCFΔN and Apc2dsRNA zygotically past 2 h of development, we find that a portion of these embryos die with a cuticle phenotype consistent with the TCFΔN block of Wnt-dependent transcription (a lawn of denticles, data not shown; Parker et al., 2002). Although we cannot rule out the possibility that CG2918 and Cbs/Tcp-1ζ behave differently, these results strongly support the hypothesis that the APC-dependent difference-protein isoform pairs are not a consequence of transcriptional activation through β-cat and TCF. Interestingly, APC-dependent protein isoform changes of GlyP, ApepP, mRpS30 and Cbs/Tcp-1ζ are also observed in embryos knocked down for GSK3β (Sgg in Drosophila) or expressing a stabilized form of β-cat [Armadillo (Arm) in Drosophila, arms10], as two APC-independent ways to accumulate stabilized β-cat (Table 2) (Pai et al., 1997). This suggests that the accumulation of stabilized β-cat, but not β-cat-dependent transcriptional activation, is required for the APC-dependent difference-protein isoforms.
ApepP is a novel regulator of β-cat levels during embryogenesis
To investigate the functional relationship between APC2 and our APC-dependent difference-proteins, we asked if mutants in any of these genes could affect APC-dependent processes, such as regulating the stability of β-cat. Because ApepP was affected in all backgrounds where components of the Wnt pathway were manipulated (Table 2), we predicted that ApepP might be responding to changes in the destructosome and/or to the accumulation of stabilized Arm. Therefore, ApepP itself might play a role in Wnt pathway regulation. To test this, we examined the effect of two P-element insertions into ApepP, namely ApepPEY02585 (ApepPEY) and ApepPMI01970 (ApepPMI), on Wnt-dependent patterning and morphogenesis in the embryo.
ApepPEY is a maternal-effect embryonic semi-lethal allele (Fig. S1F; 54% of progeny die as embryos) bearing a P-element insertion in the 5′-UTR that reduces ApepP protein abundance and alters its isoform distribution (Fig. 7G). The cuticles of these maternally and zygotically ApepPEY dead embryos resembled those of embryos with mild overactivation of the Wnt pathway (McCartney et al., 2006; Swarup and Verheyen, 2012). These phenotypes include a reduction in body length, defects in head morphogenesis and head holes, and loss of denticles (Fig. 7B). In addition, fusion of the first and second denticle rows was frequently observed (Fig. 7B′, arrowhead), and is known to result from the expression of stabilized Arm in these rows of cells (Pai et al., 1997), although we do not see this in Apc2 mutants (McCartney et al., 2006). ApepPEY cuticles also exhibited large body holes that we do not find in Apc2 mutants (Fig. 7C,C′, circled). All of these defects were also visible in ApepPMI embryos carrying an independent P-element insertion in the 5′-end of the ApepP coding region and in ApepPEY/ApepPMI transheterozygotes, suggesting that these defects are due to ApepP disruption (Fig. S1B-D).
Fig. 7.

ApepPEY embryos display Wnt activation phenotypes similar to Apc2 mutant embryos. (A-F) Anterior is to the left and posterior is to the right. (A-C′) Wild-type (A) and ApepPEY mutant (B,C) cuticles. ApepPEY cuticles exhibit head defects, fusion of the first and second denticle rows (compare A′ with B′, arrowhead) and denticle loss (B′, circled). ApepPEY cuticles also have body holes (C,C′, circled). (D) Dose reduction of Apc2 in ApepPEY embryos modifies the ApepPEY phenotype. This example has head defects and denticle loss. (E,F) Wild-type (E) Arm protein localization and accumulation reflect patterned Wnt pathway activation, whereas ApepPEY mutant embryos (F) exhibit a more uniform accumulation of Arm protein, as exemplified in the traces beneath. (G) 2D-DIGE comparison of wild-type and ApepPEY embryos showing that total ApepP protein is decreased in mutant embryos and that there is a shift in isoform distribution toward the left isoform in ApepPEY embryos. (H-J) Anti-Arm immunoblots of embryo lysates at 0-2 h (I) and 4-6 h (J) AEL. At 0-2 h AEL Arm significantly accumulates in both mutants, consistent with reduced Arm degradation, whereas at 4-6 h AEL Arm levels are decreased in both mutants (H; n=3 replicates).
The ApepP gene lies within an intron of the gene tweek, which plays crucial roles in synaptic vesicle recycling (Verstreken et al., 2009). The EY02585 insertion fails to complement point mutants specifically affecting tweek (tweek 1 and tweek 2) (Verstreken et al., 2009), suggesting that EY02585 disrupts both ApepP and tweek. The primary lethal phase of tweek mutants is larval through pharate adult (Verstreken et al., 2009). Because rescuing transgenes do not exist to separate ApepP and tweek function (Verstreken et al., 2009), we asked whether the embryonic lethal phenotypes that we observed in EY02585 embryos could be the result of tweek disruption by assessing embryonic lethality in the progeny of tweek 1/+ or tweek 2/+ parents (Fig. S2A). Although there was some embryonic lethality (tweek 1=4% and tweek 2=16%), the cuticle phenotypes do not resemble those of EY02585 embryos (Fig. S2C,D). Therefore, our data strongly suggest that the embryonic lethality and cuticle phenotypes associated with EY02585 are the result of disrupting ApepP, and not tweek, during embryogenesis.
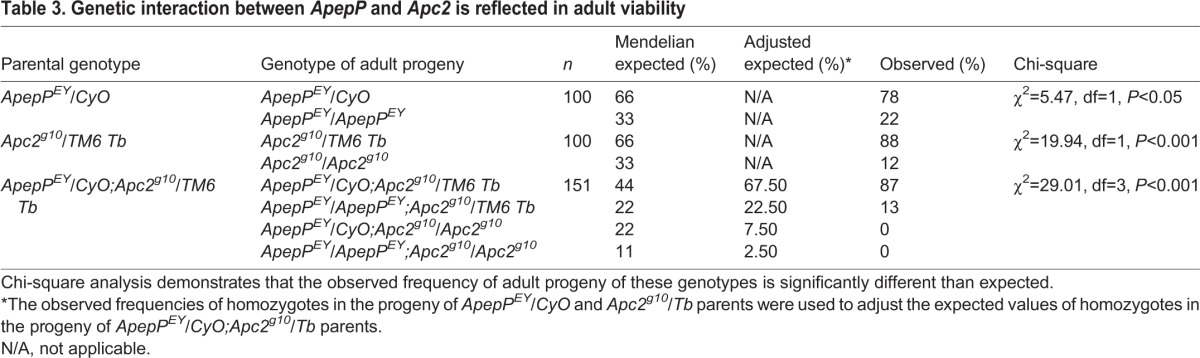
Because defects in ApepP mutants resembled the effects of mild inappropriate Wnt pathway activation, we predicted that reduction of Apc2 in ApepPEY homozygous embryos might enhance the ApepPEY phenotypes. To test this, we first assessed the cuticle phenotypes of the progeny of ApepPEY/ApepPEY;Apc2g10/+ parents. Because the progeny of Apc2g10/+ parents are 97% embryonic viable with no visible cuticle defects in the few embryos that fail to hatch (data not shown), any phenotypes that we see in embryos from ApepPEY/ApepPEY;Apc2g10/+ parents will be due to the ApepPEY/ApepPEY genotype or its genetic interaction with Apc2g10. Dose reduction of Apc2 did not enhance the embryonic lethality of ApepPEY embryos (Fig. S1F; 48% of progeny die), but the ApepPEY cuticle defects were modified. The large ApepPEY body holes were almost completely absent and denticle row fusion was strongly reduced, but defects in head morphogenesis, like those in Apc2 mutants, were more prevalent (Fig. 7D). Although the resulting phenotype is complex, and does not precisely resemble either the ApepP or the Apc2 mutant phenotype, it is consistent with a genetic interaction as Apc2g10 appears to modify the effects of ApepPEY. We found a clearer and more striking example of genetic interaction between ApepP and Apc2 in adults; whereas both ApepPEY and Apc2g10 homozygotes are separately adult viable (although not at the expected Mendelian ratios; Table 3) (this work; McCartney et al., 2006; Kunttas-Tatli et al., 2012), neither ApepPEY/ApepPEY;Apc2g10 nor ApepPEY/CyO;Apc2g10 survived to pupal stages (n=151; Table 3). This suggests that any disruption of ApepP drives Apc2 null homozygotes to complete zygotic lethality, and that a simple dose reduction of Apc2 significantly suppresses the adult viability of ApepPEY/ApepPEY (Table 3). Together, these data suggest that ApepP and Apc2 interact genetically and that their functional relationship might be in the negative regulation of Wnt signaling.
Table 3.
Genetic interaction between ApepP and Apc2 is reflected in adult viability
Consistent with this hypothesis, ApepPEY mutants exhibited dramatically elevated levels of Arm similar to Apc2 mutants (Fig. 7I). At 0-2 h, ApepP mutants exhibited a ∼6-fold increase in Arm protein relative to wild type (Fig. 7H). Apc2 null mutants exhibited a ∼15-fold increase in Arm (Fig. 7H). Wild-type embryos at 4-6 h accumulated Arm in ectodermal stripes due to Wnt signaling as a result of destructosome deactivation (Fig. 7E). In strong Apc2 hypomorphs, this pattern of Arm accumulation is lost and all cells appear to accumulate uniform levels of Arm (McCartney et al., 2006). Similarly, 92% of ApepPEY/ApepPEY embryos (n=51) show a visible disruption in the pattern of Arm accumulation, with a decrease in stripe (peak)/inter-stripe (valley) ratio (Fig. 7E versus F plot profiles) consistent with a uniform pattern of Arm accumulation. Surprisingly, both ApepP mutants and Apc2 null mutants show a decrease in overall Arm protein levels at this time compared with wild type (∼2-fold and ∼4-fold, respectively; Fig. 7H,J). Overall, these data strongly support the hypothesis that, like APC2, ApepP plays an important role in regulating β-cat protein levels to prevent ligand-independent activation of Wnt signaling during embryogenesis.
DISCUSSION
APC proteins exhibit remarkable functional breadth, playing key roles in such diverse areas of cell biology as Wnt signaling, microtubule stability and actin assembly (McCartney and Nathke, 2008; Cadigan and Peifer, 2009; Ring et al., 2014). Surprisingly, new roles for APC proteins are still being discovered (Preitner et al., 2014). This functional complexity makes it challenging to develop a comprehensive understanding of the cellular consequences of APC mutations, and how these changes affect cancer initiation and progression. To address these gaps, we conducted a comparative proteomic analysis using 2D-DIGE in Drosophila embryos null for APC proteins and discovered a set of APC-dependent protein isoform changes due to PTMs. 2D-DIGE comparisons of livers isolated from wild-type and Apc knockout mice 7 days post-knockout revealed significant protein abundance changes (Chafey et al., 2009). Many of these changes are likely to result from transcriptional activation by β-cat.
In 0-2 h Drosophila embryos transcription is largely silent, and more acute difference-protein isoforms were revealed as a result of changes in PTMs. We genetically validated that a subset of these difference-protein isoforms were due to the loss of APC activity. Furthermore, we also demonstrated that the validated APC-dependent difference-protein isoform pairs are very unlikely to be the direct or indirect result of β-cat transcriptional activation. First, none of the difference-protein isoform pairs had elevated total protein abundance. Second, we reproduced four out of six APC-dependent difference-protein isoform changes (GlyP, ApepP, mRpS30 and Cbs/Tcp-1ζ) when also expressing a dominant-negative form of the TCF protein in an Apc2 reduced background (the remaining two could not be assessed; Table 2). This suggested that the APC-dependent protein isoform changes were likely to be the result of disrupting APC non-destructosome functions. However, when we examined proteomic changes in embryos reduced for GSK3β activity, or expressing a stabilized form of Arm, we were surprised to find that GlyP, ApepP, mRpS30 and Cbs/Tcp-1ζ exhibited an APC mutant isoform profile. These data suggest that the modifications of these four difference-proteins might be a result of merely elevating β-cat in the cytoplasm. If this is the case, then it suggests that during normal development in cells in which the Wnt pathway is activated, the destructosome is deactivated and Arm is stabilized, GlyP, ApepP, mRpS30 and Cbs/Tcp-1ζ will undergo the same protein isoform changes that we observe during ligand-independent stabilization of β-cat. This raises the intriguing possibility that these protein isoform changes play a role in the cellular response to Wnt pathway activation via a non-transcriptional mechanism. The remaining two APC-dependent difference-protein isoforms, CG2918 and Dp1, did not respond to either β-cat manipulation, suggesting that these might result from disruption of other activities of APC, such as cytoskeletal functions.
Loss of APC proteins impacts phosphorylation and other PTMs
Phosphorylation is the predominant PTM in eukaryotic cells (Khoury et al., 2011). Because APC proteins are complexed with kinases GSK3β and CK1 in the destructosome, and APC enhances GSK3β activity in vitro (Zumbrunn et al., 2001; Ha et al., 2004; Rao et al., 2008; Valvezan et al., 2012), we predicted that the difference-protein isoforms were primarily due to changes in phosphorylation. Thus, we were surprised to find that this does not appear to be the case for the majority of isoform changes: of the nine difference-proteins that we could assess under the conditions of λPP treatment, only one isoform change (Dp1) appears to be due to phosphorylation differences, whereas the remaining difference-proteins are thus predicted to be modified by alternative PTMs. As the second most prominent reversible PTM, acetylation might contribute to some of the difference-protein isoform changes. Interestingly, acetylation plays a role in β-cat signaling at the level of the TCF complex (Levy et al., 2004), and appears to play a role in Wnt-mediated breast cancer proliferation (Wang et al., 2014). Finally, ApepP-dependent cleavage of short N-terminal peptides may alter the pI of a protein but not its apparent molecular weight on 2DE gels, which would appear as a horizontal shift, reminiscent of common PTMs. A subset of the APC-dependent protein isoform changes also appear in ApepP mutants (data not shown), suggesting that these changes could result from changes to ApepP activity. Identifying the mechanisms by which APC regulates these PTMs is vital to understanding the non-transcriptional effects of APC loss in normal development, as well as in the onset of cancer.
ApepP regulates β-catenin abundance
ApepP is a metalloaminopeptidase that removes amino acids adjacent to the N-termini of peptides with a prolyl residue (Yaron, 1987; Yaron and Naider, 1993). ApepP is ubiquitous and conserved from bacteria to vertebrates (Kulkarni and Deobagkar, 2002; Ersahin et al., 2003, 2005; Zheng et al., 2005). Mammalian ApepP exists in two forms, cytosolic and membrane bound, and functions in the maturation of active peptides, including hormones (Bradykinin), neuropeptides (Substance P) and neurotransmitters (Medeiros and Turner, 1994; Yoshimoto et al., 1994; Kim et al., 2000). The cytosolic form of Drosophila ApepP is functionally conserved with human APEPP (XPNPEP1) and can hydrolyze both Bradykinin and Substance P (Kulkarni and Deobagkar, 2002). Our results suggest that ApepP is necessary for β-cat regulation, as ApepP mutants display an array of Wnt activation phenotypes, accumulate Arm in the early embryo like Apc2 mutants, and genetically interact with Apc2g10 (Fig. 7, Table 2). Of the three ApepP isoforms seen in our DIGE analysis comparing wild-type embryos with Apc2 mutant embryos, the right and left isoforms change whereas the middle isoform does not (Fig. 6B). These isoform differences in ApepP were also observed with Sgg knockdown and the expression of stabilized Arm (Table 2). From these observations, we propose a model whereby ApepP protein isoforms, presumably a reflection of ApepP enzymatic activity, are regulated in part by the stabilization of β-cat. However, the fact that disruption of ApepP activity in the P-element insertion mutants appears to stabilize Arm and activate Wnt signaling suggests that this is a complicated, possibly feedback-driven, pathway. Thus, the proper regulation of ApepP activity is required to prevent the inappropriate stabilization of β-cat and downstream Wnt pathway activation. Interestingly, there is a set of proline residues located at the N-terminus of Arm (residues 20, 21, 24 and 25), suggesting that Arm could be a direct target of ApepP. Because nothing is currently known about how ApepP activity is regulated, to begin to test this model we must first identify the APC-dependent PTM modification and assess the enzymatic activity of these different isoforms of ApepP in vitro.
Wild-type variable, APC-like difference-protein isoforms
We were surprised to find that over half of the difference-protein isoforms identified in Apc2 mutant embryos were also variable in different w1118 strains (Table S3A). Because all of the original 16 difference-proteins were observed in some form of APC knockdown in multiple genetic backgrounds (Table S3C,D), we further characterized a subset of variable difference-proteins [Aats-gly (GlyRS), CaBP1 and UCH] by comparing isoform ratios and total abundance within the different w1118 strains and between Apc2 null embryos (data not shown). Interestingly, the wild-type variable difference-protein isoforms displayed different total abundance and isoform changes than Apc2 null embryos. This distinction suggests that these variable difference-proteins might be particularly sensitive to developmental perturbations. Further investigation of wild type-to-wild type proteomic variability is required to elucidate the meaning of this intriguing observation.
Conclusions
Taken together, our results suggest that loss of APC has diverse post-translational consequences for the proteome over a relatively short period of development that are independent of β-cat-driven transcriptional activation. The discovery of these APC-dependent PTM changes was made possible by 2D-DIGE, which maintains proteins in their intact state throughout the separation and detection processes. It is very unlikely that these PTM-dependent changes would have been detected by bottom-up, MS-based proteomics because proteins are fragmented prior to analysis. A single PTM that alters the charge of a protein will be readily detectable by 2D-DIGE, whereas this PTM change translates into one modified peptide out of all the peptides generated by trypsin digestion of a whole proteome and thus the likelihood of detecting a peptide carrying an unknown PTM change is exceptionally low. Therefore, combining 2D-DIGE and MS is the best route to identifying such PTM changes. This powerful technique might prove useful for dissecting the global cellular consequences of genetic mutations in other oncogenes, tumor suppressors and disease genes with implications for understanding the etiology of normal development and disease.
MATERIALS AND METHODS
Fly genetics and stocks
Fly stocks were sourced or generated as described in the supplementary Materials and Methods.
2D-DIGE
For 2D-DIGE, 200 embryos were collected at 1.5 h intervals, following a 1 h pre-collection period. All samples were visually inspected to remove old and damaged embryos. Embryos were dechorionated and washed three times with 0.01% CHAPS and 5 mM HEPES pH 8.0 buffer. Lysis buffer (7 M urea, 2 M thiourea, 4% CHAPS, 10 mM DTT and 10 mM Na-Hepes pH 8.0) was then added at 0.5 µl per embryo. On ice, the embryos were homogenized manually with a fitted pestle. Embryo lysates were adjusted in protein concentration to 2 mg/ml. Protein lysate solutions containing a total of 100 µg protein were labeled with 2 µl of 1 mM propyl-Cy3-NHS or 0.83 mM methyl-Cy5-NHS (CyDye DIGE Fluors; GE Healthcare) as described previously (Unlu et al., 1997). Two-dimensional electrophoresis (2DE) was performed according to Van et al. (2014b). For further details of 2D-DIGE procedures, including image analysis and subsequent difference-protein identification, see the supplementary Materials and Methods.
Analysis of ApepP mutant fly embryos
12 h collections of wild-type and homozygous mutant ApepP and tweek embryos were used for cuticle preparations as described (Wieschaus and Nusslein-Volhard, 1998). For immunofluorescence, stage 6-8 (4-6 h at 29°C) wild-type and homozygous mutant ApepP embryos were dechorionated and fixed in a 1:1 heptane:37% formaldehyde solution (McCartney et al., 1999). Mouse anti-Arm (N27A1, 1:250) antibodies were obtained from the Developmental Studies Hybridoma Bank (DSHB). Secondary antibodies were conjugated with various Alexa dyes (Invitrogen, 1:1000). Embryos were imaged with a spinning disc confocal microscope (Solamere Technology Group) with a QICAM-IR camera (Qimaging) on a Zeiss Axiovert 200M, using QED InVivo software.
For ApepP immunoblot analysis, lysates from 40 wild-type, ApepP mutant or Apc2 mutant embryos were prepared in 2× Laemmli sample buffer treated with 0.1% Protease Inhibitor Cocktail (Sigma) and separated by SDS-PAGE. Membranes were immunoblotted using anti-Arm (1:250), with an anti-tubulin (1:100, DSHB) loading control. Chemiluminescence was detected using an LAS-300 Fujifilm luminescent image analyzer (Formally Fuji, now GE Healthcare). For further details, see the supplementary Materials and Methods.
Phosphatase treatment
Wild-type and Apc2g10 embryo lysates were treated with λPP and analyzed by 2D-DIGE as described in the supplementary Materials and Methods.
CaBP1 analysis
Wild-type and Apc2 embryo lysates were labeled with Cy3, run on 2DE gels, immunoblotted for CaBP1 and luminescence detected as described above. For details, see the supplementary Materials and Methods.
Acknowledgements
We thank the reviewers for comments on the manuscript; the J.S.M. and B.M.M. lab members for useful discussions and criticisms during the preparation of the manuscript; Olivia Molinar, Vinitha Ganesan, Anna Grace Pyzel, Machika Kaku and Dr Phu Van for help with 2D-DIGE; and the Woolford, Ettensohn, Lopez, Hinman, Linstedt, Puthenveedu and Lee labs for reagents and equipment use.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
E.K.-T. made the initial observation. M.A.B., B.M.M. and J.S.M. developed the experimental approach. M.A.B., D.R.S., A.P., S.L.S., H.M.K. and D.A.V. performed experiments and data analysis. M.A.B., B.M.M. and J.S.M. prepared and edited the manuscript.
Funding
This work used the UPCI Cancer Biomarkers Facility, which is supported in part by award P30CA047904 from the National Institutes of Health. D.R.S. and A.P. were supported by a Howard Hughes Medical Institute Undergraduate Education Program Award [grant number 52006917]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.130567.supplemental
References
- Ahmed Y., Hayashi S., Levine A. and Wieschaus E. (1998). Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell 93, 1171-1182. 10.1016/S0092-8674(00)81461-0 [DOI] [PubMed] [Google Scholar]
- Akong K., Grevengoed E. E., Price M. H., McCartney B. M., Hayden M. A., DeNofrio J. C. and Peifer M. (2002). Drosophila APC2 and APC1 play overlapping roles in wingless signaling in the embryo and imaginal discs. Dev. Biol. 250, 91-100. 10.1006/dbio.2002.0776 [DOI] [PubMed] [Google Scholar]
- Anderson L. and Seilhamer J. (1997). A comparison of selected mRNA and protein abundances in human liver. Electrophoresis 18, 533-537. 10.1002/elps.1150180333 [DOI] [PubMed] [Google Scholar]
- Bertin E. and Arnouts S. (1996). SExtractor: software for source extraction. Astronom. Astrophys. Suppl. Ser. 117, 393-404. 10.1051/aas:1996164 [DOI] [Google Scholar]
- Cadigan K. M. and Peifer M. (2009). Wnt signaling from development to disease: insights from model systems. Cold Spring Harb. Perspect. Biol. 1, a002881 10.1101/cshperspect.a002881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chafey P., Finzi L., Boisgard R., Caüzac M., Clary G., Broussard C., Pégorier J.-P., Guillonneau F., Mayeux P., Camoin L. et al. (2009). Proteomic analysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics 9, 3889-3900. 10.1002/pmic.200800609 [DOI] [PubMed] [Google Scholar]
- Chou T. B. and Perrimon N. (1996). The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster. Genetics 144, 1673-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersahin C., Szpaderska A. M. and Simmons W. H. (2003). Rat and mouse membrane aminopeptidase P: structure analysis and tissue distribution. Arch. Biochem. Biophys. 417, 131-140. 10.1016/S0003-9861(03)00348-5 [DOI] [PubMed] [Google Scholar]
- Ersahin C., Szpaderska A. M., Orawski A. T. and Simmons W. H. (2005). Aminopeptidase P isozyme expression in human tissues and peripheral blood mononuclear cell fractions. Arch. Biochem. Biophys. 435, 303-310. 10.1016/j.abb.2004.12.023 [DOI] [PubMed] [Google Scholar]
- Gong L., Puri M., Unlu M., Young M., Robertson K., Viswanathan S., Krishnaswamy A., Dowd S. R. and Minden J. S. (2004). Drosophila ventral furrow morphogenesis: a proteomic analysis. Development 131, 643-656. 10.1242/dev.00955 [DOI] [PubMed] [Google Scholar]
- Gygi S. P., Rochon Y., Franza B. R. and Aebersold R. (1999). Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720-1730. 10.1128/MCB.19.3.1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha N.-C., Tonozuka T., Stamos J. L., Choi H.-J. and Weis W. I. (2004). Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol. Cell 15, 511-521. 10.1016/j.molcel.2004.08.010 [DOI] [PubMed] [Google Scholar]
- Hammoudi A., Song F., Reed K. R., Jenkins R. E., Meniel V. S., Watson A. J. M., Pritchard D. M., Clarke A. R. and Jenkins J. R. (2013). Proteomic profiling of a mouse model of acute intestinal Apc deletion leads to identification of potential novel biomarkers of human colorectal cancer (CRC). Biochem. Biophys. Res. Commun. 440, 364-370. 10.1016/j.bbrc.2013.08.076 [DOI] [PubMed] [Google Scholar]
- Jaiswal R., Stepanik V., Rankova A., Molinar O., Goode B. L. and McCartney B. M. (2013). Drosophila homologues of adenomatous polyposis coli (APC) and the formin diaphanous collaborate by a conserved mechanism to stimulate actin filament assembly. J. Biol. Chem. 288, 13897-13905. 10.1074/jbc.M113.462051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A., Baliban R. C. and Floudas C. A. (2011). Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep. 1, srep00090 10.1038/srep00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. S., Kumar S., Simmons W. H. and Brown N. J. (2000). Inhibition of aminopeptidase P potentiates wheal response to bradykinin in angiotensin-converting enzyme inhibitor-treated humans. J. Pharmacol. Exp. Therap. 292, 295-298. [PubMed] [Google Scholar]
- Kozinski K. and Dobrzyn A. (2013). Wnt signaling pathway--its role in regulation of cell metabolism. Postepy Hig. Med. Dosw. 67, 1098-1108. 10.5604/17322693.1077719 [DOI] [PubMed] [Google Scholar]
- Kulkarni G. V. and Deobagkar D. D. (2002). A cytosolic form of aminopeptidase P from Drosophila melanogaster: molecular cloning and characterization. J. Biochem. 131, 445-452. 10.1093/oxfordjournals.jbchem.a003120 [DOI] [PubMed] [Google Scholar]
- Kunttas-Tatli E., Zhou M.-N., Zimmerman S., Molinar O., Zhouzheng F., Carter K., Kapur M., Cheatle A., Decal R. and McCartney B. M. (2012). Destruction complex function in the Wnt signaling pathway of Drosophila requires multiple interactions between Adenomatous polyposis coli 2 and Armadillo. Genetics 190, 1059-1075. 10.1534/genetics.111.133280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunttas-Tatli E., Roberts D. M. and McCartney B. M. (2014). Self-association of the APC tumor suppressor is required for the assembly, stability, and activity of the Wnt signaling destruction complex. Mol. Biol. Cell 25, 3424-3436. 10.1091/mbc.e14-04-0885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy L., Wei Y., Labalette C., Wu Y., Renard C.-A., Buendia M. A. and Neuveut C. (2004). Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol. Cell. Biol. 24, 3404-3414. 10.1128/MCB.24.8.3404-3414.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan C. Y. and Nusse R. (2004). The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781-810. 10.1146/annurev.cellbio.20.010403.113126 [DOI] [PubMed] [Google Scholar]
- May C., Brosseron F., Chartowski P., Meyer H. E. and Marcus K. (2012). Differential proteome analysis using 2D-DIGE. Methods Mol. Biol. 893, 75-82. 10.1007/978-1-61779-885-6_6 [DOI] [PubMed] [Google Scholar]
- McCartney B. M. and Nathke I. S. (2008). Cell regulation by the Apc protein: Apc as master regulator of epithelia. Curr. Opin. Cell Biol. 20, 186-193. 10.1016/j.ceb.2008.02.001 [DOI] [PubMed] [Google Scholar]
- McCartney B. M., Dierick H. A., Kirkpatrick C., Moline M. M., Baas A., Peifer M. and Bejsovec A. (1999). Drosophila APC2 is a cytoskeletally-associated protein that regulates wingless signaling in the embryonic epidermis. J. Cell Biol. 146, 1303-1318. 10.1083/jcb.146.6.1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney B. M., McEwen D. G., Grevengoed E., Maddox P., Bejsovec A. and Peifer M. (2001). Drosophila APC2 and Armadillo participate in tethering mitotic spindles to cortical actin. Nat. Cell Biol. 3, 933-938. 10.1038/ncb1001-933 [DOI] [PubMed] [Google Scholar]
- McCartney B. M., Price M. H., Webb R. L., Hayden M. A., Holot L. M., Zhou M., Bejsovec A. and Peifer M. (2006). Testing hypotheses for the functions of APC family proteins using null and truncation alleles in Drosophila. Development 133, 2407-2418. 10.1242/dev.02398 [DOI] [PubMed] [Google Scholar]
- Medeiros M. S. and Turner A. J. (1994). Post-secretory processing of regulatory peptides: the pancreatic polypeptide family as a model example. Biochimie 76, 283-287. 10.1016/0300-9084(94)90159-7 [DOI] [PubMed] [Google Scholar]
- Miller I. (2012). Application of 2D DIGE in animal proteomics. Methods Mol. Biol. 854, 373-396. 10.1007/978-1-61779-573-2_26 [DOI] [PubMed] [Google Scholar]
- Minden J. S. (2012). Two-dimensional difference gel electrophoresis. Methods Mol. Biol. 869, 287-304. 10.1007/978-1-61779-821-4_24 [DOI] [PubMed] [Google Scholar]
- Nathke I. S. (2004). The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu. Rev. Cell Dev. Biol. 20, 337-366. 10.1146/annurev.cellbio.20.012103.094541 [DOI] [PubMed] [Google Scholar]
- Nathke I. (2006). Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat. Rev. Cancer 6, 967-974. 10.1038/nrc2010 [DOI] [PubMed] [Google Scholar]
- Okada R., Nagaosa K., Kuraishi T., Nakayama H., Yamamoto N., Nakagawa Y., Dohmae N., Shiratsuchi A. and Nakanishi Y. (2012). Apoptosis-dependent externalization and involvement in apoptotic cell clearance of DmCaBP1, an endoplasmic reticulum protein of Drosophila. J. Biol. Chem. 287, 3138-3146. 10.1074/jbc.M111.277921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai L. M., Orsulic S., Bejsovec A. and Peifer M. (1997). Negative regulation of Armadillo, a Wingless effector in Drosophila. Development 124, 2255-2266. [DOI] [PubMed] [Google Scholar]
- Parker D. S., Jemison J. and Cadigan K. M. (2002). Pygopus, a nuclear PHD-finger protein required for Wingless signaling in Drosophila. Development 129, 2565-2576. [DOI] [PubMed] [Google Scholar]
- Polakis P. (2012). Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 4, a008052 10.1101/cshperspect.a008052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preitner N., Quan J., Nowakowski D. W., Hancock M. L., Shi J., Tcherkezian J., Young-Pearse T. L. and Flanagan J. G. (2014). APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 158, 368-382. 10.1016/j.cell.2014.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X., Ahn S., Speed T. P. and Rubin G. M. (2007). Global analyses of mRNA translational control during early Drosophila embryogenesis. Genome Biol. 8, R63 10.1186/gb-2007-8-4-r63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P. R., Makhijani K. and Shashidhara L. S. (2008). Human APC sequesters beta-catenin even in the absence of GSK-3beta in a Drosophila model. Oncogene 27, 2488-2493. 10.1038/sj.onc.1210890 [DOI] [PubMed] [Google Scholar]
- Ring A., Kim Y.-M. and Kahn M. (2014). Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. 10, 512-525. 10.1007/s12015-014-9515-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneikert J., Grohmann A. and Behrens J. (2007). Truncated APC regulates the transcriptional activity of beta-catenin in a cell cycle dependent manner. Hum. Mol. Genet. 16, 199-209. 10.1093/hmg/ddl464 [DOI] [PubMed] [Google Scholar]
- Speicher K. D., Kolbas O., Harper S. and Speicher D. W. (2000). Systematic analysis of -peptide recoveries from in-gel digestions for protein identifications in proteome studies. J. Biomol. Techniq. 11, 74-86. [PMC free article] [PubMed] [Google Scholar]
- Stamos J. L. and Weis W. I. (2013). The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 5, a007898 10.1101/cshperspect.a007898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup S. and Verheyen E. M. (2012). Wnt/Wingless signaling in Drosophila. Cold Spring Harb. Perspect. Biol. 4, a007930 10.1101/cshperspect.a007930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros W. and Lipshitz H. D. (2009). The maternal-to-zygotic transition: a play in two acts. Development 136, 3033-3042. 10.1242/dev.033183 [DOI] [PubMed] [Google Scholar]
- Takacs C. M., Baird J. R., Hughes E. G., Kent S. S., Benchabane H., Paik R. and Ahmed Y. (2008). Dual positive and negative regulation of wingless signaling by adenomatous polyposis coli. Science 319, 333-336. 10.1126/science.1151232 [DOI] [PubMed] [Google Scholar]
- Unlu M., Morgan M. E. and Minden J. S. (1997). Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis 18, 2071-2077. 10.1002/elps.1150181133 [DOI] [PubMed] [Google Scholar]
- Valvezan A. J., Zhang F., Diehl J. A. and Klein P. S. (2012). Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. J. Biol. Chem. 287, 3823-3832. 10.1074/jbc.M111.323337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van P. T., Bass V., Shiwarski D., Lanni F. and Minden J. (2014a). High dynamic range proteome imaging with the structured illumination gel imager. Electrophoresis 35, 2642-2655. 10.1002/elps.201400126 [DOI] [PubMed] [Google Scholar]
- Van P. T., Ganesan V., Bass V., Parthasarathy A., Schlesinger D. and Minden J. S. (2014b). In-gel equilibration for improved protein retention in 2DE-based proteomic workflows. Electrophoresis 35, 3012-3017. 10.1002/elps.201400256 [DOI] [PubMed] [Google Scholar]
- van de Wetering M., Cavallo R., Dooijes D., van Beest M., van Es J., Loureiro J., Ypma A., Hursh D., Jones T., Bejsovec A. et al. (1997). Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 88, 789-799. 10.1016/S0092-8674(00)81925-X [DOI] [PubMed] [Google Scholar]
- Verstreken P., Ohyama T., Haueter C., Habets R. L. P., Lin Y. Q., Swan L. E., Ly C. V., Venken K. J. T., De Camilli P. and Bellen H. J. (2009). Tweek, an evolutionarily conserved protein, is required for synaptic vesicle recycling. Neuron 63, 203-215. 10.1016/j.neuron.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C. and Marcotte E. M. (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227-232. 10.1038/nrg3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. H., Li N., Wei Y., Li Q. R. and Yu Z. P. (2014). beta-catenin deacetylation is essential for WNT-induced proliferation of breast cancer cells. Mol. Med. Rep. 9, 973-978. 10.3892/mmr.2014.1889 [DOI] [PubMed] [Google Scholar]
- Webb R. L., Zhou M.-N. and McCartney B. M. (2009). A novel role for an APC2-Diaphanous complex in regulating actin organization in Drosophila. Development 136, 1283-1293. 10.1242/dev.026963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieschaus E. and Nusslein-Volhard C. (1998). Looking at embryos. In Drosophila: A Practical Approach (ed. D. B. Roberts), pp. 179-214. Oxford: Oxford University Press. [Google Scholar]
- Wu Y., Wang X., Wu F., Huang R., Xue F., Liang G., Tao M., Cai P. and Huang Y. (2012). Transcriptome profiling of the cancer, adjacent non-tumor and distant normal tissues from a colorectal cancer patient by deep sequencing. PLoS ONE 7, e41001 10.1371/journal.pone.0041001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron A. (1987). The role of proline in the proteolytic regulation of biologically active peptides. Biopolymers 26 Suppl., S215-S222. 10.1002/bip.360260019 [DOI] [PubMed] [Google Scholar]
- Yaron A. and Naider F. (1993). Proline-dependent structural and biological properties of peptides and proteins. Crit. Rev. Biochem. Mol. Biol. 28, 31-81. 10.3109/10409239309082572 [DOI] [PubMed] [Google Scholar]
- Yoshimoto T., Orawski A. T. and Simmons W. H. (1994). Substrate specificity of aminopeptidase P from Escherichia coli: comparison with membrane-bound forms from rat and bovine lung. Arch. Biochem. Biophys. 311, 28-34. 10.1006/abbi.1994.1204 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Roe S. M., Diogon M., Kong E., El Alaoui H. and Barford D. (2010). Molecular structure of the N-terminal domain of the APC/C subunit Cdc27 reveals a homo-dimeric tetratricopeptide repeat architecture. J. Mol. Biol. 397, 1316-1328. 10.1016/j.jmb.2010.02.045 [DOI] [PubMed] [Google Scholar]
- Zheng Y., Roberts R. J., Kasif S. and Guan C. (2005). Characterization of two new aminopeptidases in Escherichia coli. J. Bacteriol. 187, 3671-3677. 10.1128/JB.187.11.3671-3677.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.-N., Kunttas-Tatli E., Zimmerman S., Zhouzheng F. and McCartney B. M. (2011). Cortical localization of APC2 plays a role in actin organization but not in Wnt signaling in Drosophila. J. Cell Sci. 124, 1589-1600. 10.1242/jcs.073916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumbrunn J., Kinoshita K., Hyman A. A. and Nathke I. S. (2001). Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr. Biol. 11, 44-49. 10.1016/S0960-9822(01)00002-1 [DOI] [PubMed] [Google Scholar]