

Abstract
Human induced pluripotent stem cell (iPSC) models of epilepsy are becoming a revolutionary platform for mechanistic studies and drug discovery. The skyrocketing pace of epilepsy gene discovery is vastly outstripping the development of in vivo animal models. Currently, antiepileptic drug prescribing to patients with specific genetic epilepsies is based on small-scale clinical trials and empiricism; however, rapid production of patient-derived iPSC models will allow for precision therapy. We review iPSC-based studies that have already afforded novel discoveries in diseases with epileptic phenotypes, as well as challenges to using iPSC-based neurological disease models. We also discuss iPSC-derived cardiomyocyte studies of arrhythmia-inducing ion channelopathies that exemplify novel drug discovery and use of multielectrode array technology that can be translated to epilepsy research. Beyond initial studies of Rett, Timothy, Phelan-McDermid and Dravet syndromes, the stage is set for groundbreaking iPSC-based mechanistic and therapeutic discoveries in genetic epilepsies with the potential to impact patient treatment and quality of life.
Keywords: Human Induced Pluripotent Stem Cells, Epilepsy, Patient-Specific Modeling, Channelopathies, Drug Discovery
Introduction to iPSC technology
Modeling genetic epilepsies has long been accomplished through the use of animal models (mainly rodents) and heterologous expression systems. The focus has largely been on genetically modified mice over the past 15 years. Despite many groundbreaking discoveries, these systems present many challenges for fully recapitulating human disease mechanisms and allowing the possibility of mutation-specific drug discovery. The development of human induced pluripotent stem cell (iPSC) technology by Yamanaka and colleagues in 2007 opened the door to the rapid production of patient-specific neuronal models of disease and promises regenerative therapies in the near future [1]. Disease modeling with iPSCs circumvents the issues of murine genetic background and lack of neuronal environment for mouse models and heterologous expression systems, respectively. While whole animal models remain critical for studying acquired epilepsies and examining network-based aspects of all epilepsies, iPSCs are fast becoming a critical platform for mechanistic studies at the cellular and molecular levels, and for drug development (see [2] and Figure 1 therein).
Figure 1. Antiepileptic drug screening of genetic epilepsy patient iPSC-derived neurons by multielectrode array (MEA).
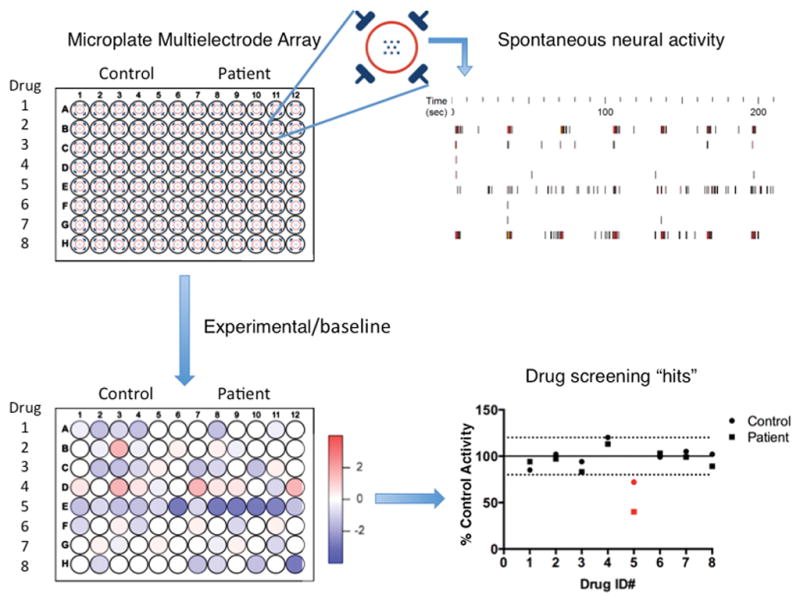
Upper left: Depiction of a 96 well-plate MEA used on the Maestro platform (Axion Biosystems). Each well incorporates 8 recording electrodes with 4 ground electrodes (enlargement, upper middle). Upper right: Representative raster plots generated from 8 recording electrodes on the Maestro platform. Lower left: Drug dependent activity (spike rate) heat map generated by dividing the experimental MEA activity by a baseline read. Lower right: % of control activity is plotted for each drug, and “hits” in red are defined by a predetermined significance threshold. These hits will then be validated by concentration response curves via MEA (spontaneous activity) and patch-clamp recordings (ionic currents).
To produce human iPSC lines, somatic cells - typically dermal fibroblasts from a skin biopsy or more recently blood-derived hematopoietic cells [3] are forced to express several transcription factors necessary for reprogramming to the pluripotent state (e.g., combinations of OCT3/4, SOX2, KLF4, C-MYC or L-MYC, LIN28, NANOG). After 3–5 weeks in culture, iPSC colonies form at a rate of typically 0.01–0.1% of cells depending upon the technique used to express the transcription factors. These cells are an attractive system for modeling genetic diseases because iPSCs have an identical genome to the donor patient and, using recent protocols that utilize episomal vectors or Sendai virus, contain no exogenous DNA after the reprogramming process [4, 5]. Unlike primary cultures, iPSCs are naturally immortalized due to a high expression of telomerase resulting in a theoretically infinite supply of cells for study [6]. Numerous techniques for differentiating iPSCs into particular neuronal subtypes and glia have been published [7], allowing for modeling disease in many relevant cell types, at a scale previously unattainable from patient tissues. The rare and heterogeneous nature of genetic epilepsies lends itself well to such a model since the production of transgenic animals for all such mutations is logistically untenable.
Rapid progress in genome editing, particularly the utilization of the TALEN (transcription activator-like effector nuclease) or, more recently, the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein-9 nuclease) systems allows for rapid production of “virtual” patient iPSC lines [8]. These lines are generated by editing a wild-type epileptic gene in a control iPSC line to the mutated form found in an epileptic patient. This procedure circumvents difficulties in obtaining patient tissues and allows for comparisons with an isogenic control. Generating additional virtual patients on the same genetic background also allows for controlled comparisons between specific patient mutations. The one downside to virtual patient generation is the loss of patient-specific genetic modifiers. An alternative approach is to generate a patient-specific isogenic line by correcting the patient mutation using CRISPR gene editing [9]. Isogenic controls are vital to definitively determine causality between genotype and phenotype. Currently scarless gene editing of specific mutations (i.e., in the absence of other genome modifications) in iPSCs, which does not allow for use of selectable markers, is inefficient and can be painstakingly difficult for isolating a pure corrected line [10]. However, as gene-editing technology becomes better adapted for iPSCs, isogenic controls will almost certainly become a standard throughout the field.
Genetic epilepsies
Epilepsy is the fourth most common neurological disorder behind Parkinson’s disease, Alzheimer’s disease, and stroke with a prevalence of 7.1 cases per 1000 [11]. Of these four diseases, only epilepsy has a large portion of incidence in early life [12]. This early onset, along with increased mortality in epilepsy, gives it the second highest neurological disease burden behind stroke in terms of years of potential life lost [13]. With 30–40% of cases not adequately controlled with medication, epilepsy poses a huge burden to individuals and families.
Only 25 years ago the first genetic cause of epilepsy was discovered [14]. Now, 500 loci are listed as potentially causative when mutated [15], and the pace of epilepsy gene discovery continues to skyrocket. Although epilepsy can be genetic or acquired, childhood epileptic encephalopathies (CEEs) such as Dravet syndrome, infantile spasms, and Lennox-Gastaut syndrome (LGS) are increasingly linked to specific mutations. These disorders also comprise some of the most severe and pharmaco-resistant classes of epilepsy [16]. Recent developments in whole exome and genome sequencing have allowed for the identification of de novo mutations that contribute to the development of CEEs [17–19]. Often after an epilepsy gene is discovered, many more patients are often identified with mutations throughout the gene. In over 80% of Dravet syndrome cases, for example, mutations in the SCN1A gene are causative, and over 1,250 distinct mutations (including missense, frameshift, truncation, deletion, and splice sites) are currently identified in patients with SCN1A-related epilepsies including Dravet syndrome, genetic epilepsy with febrile seizures +, intractable childhood epilepsy with generalized tonic-clonic seizures, and infantile spasms [20, 21]. The impracticality of sufficient animal modeling of many genetic epilepsies is underscored by the massive mutation diversity. Unlike mouse models, the development of an iPSC model can take a month with a distinctly lower cost while preserving the exact genetics of the patient. Additionally, perhaps 20% of CNS genes show distinct cortical expression pattern differences between mouse and human, further highlighting the need for human models [22]. Moreover, heterologous expression systems may fail to determine precise disease mechanisms without the full complement of interacting proteins, splicing patterns, and other factors unique to neurons.
iPS disease models with epileptic phenotypes
Since the advent of iPSC technology, CNS disorders have comprised a large portion of published disease models. This is not surprising considering the limited access and growth potential of primary patient-derived brain samples. For the purposes of this review, we will focus on genetic epilepsies or genetic brain diseases with epileptic symptoms in a subset of patients (summarized in Table 1). Patient-derived iPSC models for many of these diseases have demonstrated altered neuronal morphology including soma size, neurite outgrowth, synapse formation, and dendritic spine length. Additionally, altered spontaneous activity and ion current density have been seen in some of these disease models. Rett syndrome is perhaps the epileptic disorder most studied via iPSCs. Rett syndrome is caused by mutations in the methyl CpG binding protein 2 gene (MeCP2) on the X chromosomes, and 50–90% of Rett syndrome patients have seizures [23]. Rett syndrome iPSC models from several groups show decreases in neuronal soma size, neurite outgrowth, synapse formation, and spontaneous activity compared to isogenic controls [24, 25]. Marchetto et al also demonstrated rescue of the MeCP2 mutation associated phenotypes via insulin-like growth factor-1 (IGF1) treatment [24]. It should be noted that wild-type neurons had similar abnormal morphological phenotypes when co-cultured with iPSC-derived astrocytes from Rett syndrome patients, underscoring the need to consider astrocytic contributions to epileptogenesis in addition to the neuronal contribution [26]. The Hanefeld variant of Rett syndrome is caused by mutations in CDKL5 and typically presents with intractable seizures starting before 6 months of age. Neurons differentiated from a CDKL5 mutant Rett syndrome patient iPSC line and primary neurons from a CDKL5 mutant mouse had reduced synapse formation with increased dendritic spine length [27]. More recent work tied together these two forms of Rett syndrome by showing that the only expression change common to the MeCP2 and CDKL5 variants in iPSCs was an upregulation in GRID1, which encodes for the glutamate D1 receptor. This protein acts as a synaptic adhesion molecule, and MeCP2 binds to the promoter region [28].
Table 1.
Summary of epileptic disease iPSC studies
| Gene | Disease | % of cases with seizures | Publication(s) | Major findings |
|---|---|---|---|---|
| SCN1A | Dravet syndrome | 100% | Liu et al 2013, Jiao et al 2013, Higurashi et al 2013 | Altered sodium current density, spontaneous activity |
| CDKL5 | Hanefeld variant Rett syndrome | 100% | Livide et al 2014 | Increased dendritic protrusions, decreased synaptic formation |
| UBE3A | Angelman syndrome | 90% | Chamberlain et al 2010 | Parental imprinting maintained during reprogramming |
| MeCP2 | Rett syndrome | 50–90% | Marchetto et al 2010; Cheung et al2011; Ananiev et al 2011; Kim et al2011; Hotta et al 2009 | Reduced soma size, neurite outgrowth, VGLUT1 positive puncta density, and spontaneous postsynaptic currents. IGF1 rescued some phenotypes |
| CYFIP2 | 15q11.2 microdeletion | 66% | Yoon et al 2014 | Altered neuroprogenitor organization and polarity |
| SHANK3 | Phelan-McDermid syndrome | 27% | Shcheglovitov et al 2013 | Defects in excitatory synapse formation |
| CACNA1C | Timothy syndrome | 20% | Pasca et al 2011 | Increased action potential duration, impaired voltage-gated calcium channel inactivation, phenotypes blocked by L-type calcium channel blockers |
| FMR1 | Fragile X | 10–20% | Urbach 2010, Sheridan 2011, J. Liu 2012 | Decreased neurite outgrowth |
The developmental disorder Phelan-McDermid syndrome (PMS) is usually caused by loss of a long arm segment of chromosome 22 and approximately 25% of patients have seizures [29]. The lost chromosome segment contains the synaptic scaffolding gene, SHANK3. One study co-cultured control and patient iPSC-derived cortical neurons with green (GFP) and red (mKate2) reporters to distinguish the two lines [30]. This allowed the authors to perform electrophysiological and cytoarchitectural analyses with reduced variability based on culture conditions such as the culture-specific ratio of glutamatergic and GABAergic cells. PMS patient cells had reduced synapse formation and spontaneous activity, which was rescued by SHANK3 overexpression or by addition of IGF1 [30].
Fragile X syndrome (FXS) is the most common genetic cause of intellectual disability and presents with seizures in 10–20% of cases [31]. Patient iPSC-derived neuronal models of FXS have shown reductions in neurite length and synapse formation as well as increased transient calcium current frequency and amplitude [32]. These differences may be due to a down-regulation of RE1-silencing transcription factor (REST) regulated genes responsible for axon guidance due to lower microRNA-382 expression found in another iPSC study [33].
Modeling ion channelopathies
Epilepsies induced by channelopathies are ripe for both mechanistic and drug screening studies in iPSC-derived neurons and astrocytes. While the functions of many epileptic genes discussed in the previous section are still being worked out, the basic functions of ion channels have been extensively studied and defined. The effects of these mutations are also cell autonomous while the synaptic and connectivity deficits identified in Rett syndrome, FXS, and PMS require networks to produce disease-related excitability phenotypes. Heterogeneity in network formation could, therefore, confound drug screening for synaptic disorders. Additionally, the majority of antiepileptic drugs currently approved by the FDA are known to act on ion channels, particularly inhibition of voltage-gated sodium channels as well as the GABA receptor chloride channel. Although drugs have been indicated or contraindicated for particular types of epilepsies based on small clinical studies, the use of an in vitro drug-screening platform for patients with rare channelopathies should at the very least give a clearer rational for prescribing a particular drug to a particular patient. To this end, we describe the currently reported iPSC-based studies of putative epilepsy-causing channelopathies in Timothy and Dravet syndromes.
Timothy syndrome is caused by mutations in the voltage-gated calcium channel gene CACNA1C encoding the protein CaV1.2. It leads to both cardiac (Long QT syndrome [LQTS]) and neurological (autism and epilepsy) abnormalities. Several studies from Ricardo Dolmetsch describe modeling this disease using iPSCs derived from patients with a G406R mutation in CACNA1C. In cardiomyocytes, the mutation was found to increase action potential durations due to impaired CaV1.2 channel inactivation. This was reversed with the calcium channel blocker, roscovitine [34]. In neurons differentiated from the same patient iPSC lines, action potential width and sustained calcium current were increased. These alterations were blocked by the L-type calcium channel blocker, nimodipine [35]. By showing rescue of disease-related electrophysiological phenotypes, this group has identified a potential therapy for this disease and proof-of-principle for the efficacy of drug development using patient iPSC-derived neurons and cardiomyocytes.
Dravet syndrome is an infantile onset epileptic disorder characterized by refractory epilepsy and cognitive dysfunction. As mentioned earlier, approximately 80% of Dravet syndrome cases are caused by mutations in the NaV1.1 voltage-gated sodium channel encoding gene, SCN1A. Several groups have modeled Dravet syndrome using patient-specific iPSCs with various reported phenotypes. Higurashi et al showed impaired action potential generation in primarily GABAergic cells from a patient with a predicted truncation at residue 1645 of the NaV1.1 protein suggesting loss-of-function [36]. In contrast, Jiao et al showed significant increases in spontaneous activity and sodium current density, particularly persistent sodium current, in lines from two patients with missense mutations (F1415I and Q1923R) using iPSC-derived neurons or glutamatergic neurons directly converted from fibroblasts [37]. These reports would suggest loss-of-function mutations lead to decreased GABAergic activity or increased sodium current and hyperexcitability of glutamatergic cells. A third study demonstrated elevated sodium current density and spontaneous activity in both putative GABAergic and glutamatergic cell types with two patient lines with loss-of-function mutations (IVS14+3A>T and truncation Y325X) [38]. This counterintuitive result of sodium channel loss-of-function mutations leading to hyperexcitability may be explained by compensation by the expression of other voltage-gated sodium channels, such as NaV1.6.
While relatively few channelopathies of the CNS have been modeled in iPSCs, many more studies have used iPSC-derived cardiomyocytes to model channelopathies resulting in cardiac dysfunction similar to the Timothy syndrome study discussed earlier [34]. Using iPSC-derived cardiomyocytes from a LQTS patient, a mutation in the KCNH2 potassium channel gene was found to reduce rectifying potassium current needed to repolarize cells leading to prolonged action potential duration [39]. These cells were then used at the single and multicellular level to assay drugs known to alter LQTS. This model faithfully recapitulated the expected drug effects and allowed for characterization of novel therapeutics. Similar studies have also been carried out in patients with SNC5A and KCNQ1 mutations [40, 41].
Challenges to iPSC-based epilepsy modeling
While patient-derived iPSCs are fast becoming an economical and feasible model of human disease and pharmacology, many challenges for their use in modeling genetic epilepsies remain to be overcome. First, iPSC lines can have variable expression profiles and differentiation potential. One study demonstrated decreased efficiency of iPSCs, as compared to hES cells, in their ability to differentiate into PAX6+ neural progenitors, an important first step in differentiating many CNS neuronal subtypes [42]. This variability is due, in part, to epigenetic differences from the donor somatic cell retained from incomplete reprogramming [43]. Gene expression profiling, such as with the Pluritest, is now being used to validate individual iPSC lines by comparing gene expression data to a large set of ES cell lines [44]. These efforts should decrease some differentiation variability; however, heterogeneity is a common theme in differentiation of ESCs and iPSCs into neuronal subtypes [45]. No culture is a pure population unless flow cytometry techniques are used. Unfortunately, few neuronal or astrocytic subtypes have a distinguishing extracellular protein to allow for antibody-dependent flow cytometry, and mature neurons cannot tolerate such sorting. Most sorting procedures rely on either pan-neuronal markers or promoter driven fluorescent proteins.
A critical issue for epilepsy studies is that human iPSC-derived neurons take dramatically more time for electrophysiological properties and synaptic connections to mature in culture compared to mouse iPSCs [46]. This prolongation is presumably due to developmental timing that matches in vivo human development. Many compounds have been used to accelerate the rate of maturation including gamma secretase inhibitors (DAPT and compound E) to block notch signaling. Neurotophic factors including brain-derived neurotrophic factor (BDNF), glial-derived neurotrophic factor (GDNF), nerve growth factor (NGF), and neurotrophin-3 (NT3) all increase neuronal survival as well as maturation. Neuronal maturation is also enhanced when iPSC-derived neural progenitors are co-cultured with either human or rodent astrocytes, as measured by spontaneous firing, and sodium and potassium current amplitude [47]. Techniques are also available to differentiate astrocytes from patient iPSC lines allowing for patient-specific astrocytic cultures. Such disease-specific astrocytes will be particularly important in disorders such as Rett syndrome that have an astrocytic component to epileptogenesis, malformations of cortical development [48] or possibly patients with mutations in astrocytic channels involved in glutamate cycling, disorders of which may contribute to both genetic and acquired epilepsies.
Despite the current advances in iPSC-derived neuronal culture, many groups report depolarized resting potentials and small percentages of neurons that undergo evoked firing in patch clamp recordings. These issues are not typical of primary neuronal cultures, demonstrating a challenge to fully differentiating iPSC-derived neurons. In comparison to the electrophysiological recordings of human fetal neurons, the properties seen may merely recapitulate the properties of neurons in utero [49]. However, recently, a cell culture medium has been developed that appears to stimulate maturation and support electrophysiological function of iPSC-derived neurons, suggesting that this obstacle with the previously reported culture conditions will soon be overcome [50].
The Future
The future of iPSC-based modeling of genetic epilepsies portends high-throughput screening for new pharmaceuticals for rare diseases leading to precision therapy. These studies are underway in human iPSC-derived cardiomyocytes in patients with genetic arrhythmias via multielectrode array (MEA) recordings. MEAs allow for real-time spontaneous activity measurement from hundreds of neurons simultaneously under normal culture conditions. If the issues discussed in the previous section are adequately overcome (i.e., cell type heterogeneity, neuronal maturation), the effects of compound libraries on neuronal activity can be assayed and compared between control and patient cells. Finding patient-specific activity phenotypes would allow for the identification of pharmaceuticals that specifically target the patient disease mechanism with limited effects on normal neuronal activity. The best example to date was performed in SOD1 mutant amyotrophic lateral sclerosis (ALS) patient iPSC-derived neurons [51]. This study identified a hyperexcitable phenotype in the patient cells using a MEA platform. Patch clamp recordings identified reduced potassium current density as the most likely cause, and a potassium channel opener, retigabine, blocked the hyperexcitablity. In the same way, we anticipate that iPSC epilepsy models will produce activity dependent phenotypes on MEAs that can subsequently be leveraged in drug-screening assays (as depicted in Figure 1). Before large screenings can be performed, the sensitivity of the assay to detect changes in activity must be determined. Neurotoxicological screening in rat primary cortical cultures showed that a MEA identified 87% of known positive compounds and 100% of negative compounds using 3–7 wells with 64 electrodes per well [52]. These data suggest that a small number of samples can determine a “hit” on a large drug screen; however, similar studies must be performed in human iPSC-derived neurons to determine their screening potential compared to rodent primary neurons.
Uses for human iPSCs in modeling epilepsies go beyond routine cell culture and MEA recordings. Recently a group has reported the generation of cerebral organoids by three-dimensional culture [53]. Remarkably, the cultures preserved the dorsal forebrain-like progenitor zone and primitive cortical laminar patterning, as well as regional marker (albeit not structural) specification of the hippocampus and ventral forebrain. This model system will be important for genetic neurodevelopmental disorders that often incorporate both epilepsy and cortical developmental abnormalities. Cerebral organoids are amenable to calcium imaging [53] and acute in vitro slice recordings to assess cortical excitability. Alternatively, patient iPSC-derived neurons can also be transplanted in utero into rodent models allowing for in vivo analysis of developmental integration of neurons and their electrophysiological properties as has been previously performed [54, 55]. One must keep in mind that animals injected with human cells should not be bred to avoid potential germline transmission, and the number of cells injected should be limited to avoid drastically altering brain morphology and function.
Conclusion
iPSC technology provides an unprecedented ability to model genetic disease. A growing number of epileptic disease models using patient-derived neurons and astrocytes have already provided novel insights into disease mechanisms. However, the vast number of de novo epileptic gene discoveries being made in recent years suggests that the field has only scratched the surface of iPSC-based model potential in the genetic epilepsies. Despite the list of challenges and caveats to using iPSC models, this burgeoning field has already demonstrated rapid solutions to early challenges and promises continued progress. We contend that the current state of iPSC modeling will soon allow for drug discovery that will improve therapy in patients with severe genetic epilepsies.
Acknowledgments
Grants: Supported by NS065450 (JMP) and NS087181 (JMP)
Footnotes
Andrew Tidball: Conception and design, manuscript writing
Jack Parent: Conception and design, financial support, manuscript writing
Disclosure of Potential Conflicts of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Marchetto MC, Winner B, Gage FH. Pluripotent stem cells in neurodegenerative and neurodevelopmental diseases. Human Molecular Genetics. 2010;19:R71–R76. doi: 10.1093/hmg/ddq159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okita K, Yamakawa T, Matsumura Y, et al. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31:458–466. doi: 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- 4.Nishimura K, Sano M, Ohtaka M, et al. Development of defective and persistent Sendai virus vector a unique gene delivery/expression system ideal for cell reprogramming. Journal of Biological Chemistry. 2011;286:4760–4771. doi: 10.1074/jbc.M110.183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okita K, Matsumura Y, Sato Y, et al. A more efficient method to generate integration-free human iPS cells. Nature methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 6.Marion RM, Strati K, Li H, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell stem cell. 2009;4:141–154. doi: 10.1016/j.stem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Srikanth P, Young-Pearse TL. Stem Cells on the Brain: Modeling Neurodevelopmental and Neurodegenerative Diseases Using Human Induced Pluripotent Stem Cells. Journal of neurogenetics. 2014;28:5–29. doi: 10.3109/01677063.2014.881358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ran FA, Hsu PD, Lin C-Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwank G, Koo B-K, Sasselli V, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell stem cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Miyaoka Y, Chan AH, Judge LM, et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nature methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirtz D, Thurman D, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326–337. doi: 10.1212/01.wnl.0000252807.38124.a3. [DOI] [PubMed] [Google Scholar]
- 12.Olafsson E, Ludvigsson P, Hesdorffer D, et al. Incidence of unprovoked seizures and epilepsy in Iceland and assessment of the epilepsy syndrome classification: a prospective study. The Lancet Neurology. 2005;4:627–634. doi: 10.1016/S1474-4422(05)70172-1. [DOI] [PubMed] [Google Scholar]
- 13.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–1485. doi: 10.1111/epi.12666. [DOI] [PubMed] [Google Scholar]
- 14.Shoffner JM, Lott MT, Lezza AM, et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA Lys mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 15.Noebels J. Pathway-driven discovery of epilepsy genes. Nature neuroscience. 2015;18:344–350. doi: 10.1038/nn.3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiron C, Dulac O. The pharmacologic treatment of Dravet syndrome. Epilepsia. 2011;52:72–75. doi: 10.1111/j.1528-1167.2011.03007.x. [DOI] [PubMed] [Google Scholar]
- 17.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nature genetics. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1. 6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Frontiers in genetics. 2013:4. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phenome E Consortium EK. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujiwara T, Sugawara T, Mazaki-Miyazaki E, et al. Mutations of sodium channel αsubunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic clonic seizures. Brain. 2003;126:531–546. doi: 10.1093/brain/awg053. [DOI] [PubMed] [Google Scholar]
- 21.Meng H, Xu HQ, Yu L, et al. The SCN1A Mutation Database: Updating Information and Analysis of the Relationships Among Genotype, Functional Alteration, and Phenotype. Human mutation. 2015 doi: 10.1002/humu.22782. [DOI] [PubMed] [Google Scholar]
- 22.Zeng H, Shen EH, Hohmann JG, et al. Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures. Cell. 2012;149:483–496. doi: 10.1016/j.cell.2012.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dolce A, Ben-Zeev B, Naidu S, et al. Rett syndrome and epilepsy: an update for child neurologists. Pediatric neurology. 2013;48:337–345. doi: 10.1016/j.pediatrneurol.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Marchetto MC, Carromeu C, Acab A, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheung AY, Horvath LM, Grafodatskaya D, et al. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Human molecular genetics. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maezawa I, Swanberg S, Harvey D, et al. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. The Journal of Neuroscience. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricciardi S, Ungaro F, Hambrock M, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1 PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nature Cell Biology. 2012;14:911–923. doi: 10.1038/ncb2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livide G, Patriarchi T, Amenduni M, et al. GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. European Journal of Human Genetics. 2015;23:195–201. doi: 10.1038/ejhg.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhar S, Del Gaudio D, German J, et al. 22q13. 3 deletion syndrome: clinical and molecular analysis using array CGH. American Journal of Medical Genetics Part A. 2010;152:573–581. doi: 10.1002/ajmg.a.33253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shcheglovitov A, Shcheglovitova O, Yazawa M, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berry-Kravis E, Potanos K. Psychopharmacology in fragile X syndrome present and future. Mental Retardation and Developmental Disabilities Research Reviews. 2004;10:42–48. doi: 10.1002/mrdd.20007. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Kościelska KA, Cao Z, et al. Signaling defects in iPSC-derived fragile X premutation neurons. Human molecular genetics. 2012:dds207. doi: 10.1093/hmg/dds207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halevy T, Czech C, Benvenisty N. Molecular Mechanisms Regulating the Defects in Fragile X Syndrome Neurons Derived from Human Pluripotent Stem Cells. Stem cell reports. 2015;4:37–46. doi: 10.1016/j.stemcr.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paşca SP, Portmann T, Voineagu I, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nature medicine. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higurashi N, Uchida T, Lossin C, et al. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol brain. 2013;6:19. doi: 10.1186/1756-6606-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiao J, Yang Y, Shi Y, et al. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Human molecular genetics. 2013;22:4241–4252. doi: 10.1093/hmg/ddt275. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Lopez-Santiago LF, Yuan Y, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Annals of neurology. 2013;74:128–139. doi: 10.1002/ana.23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 40.Davis RP, Casini S, van den Berg CW, et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. 2012;125:3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092. [DOI] [PubMed] [Google Scholar]
- 41.Moretti A, Bellin M, Welling A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. New England Journal of Medicine. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 42.Hu B-Y, Weick JP, Yu J, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proceedings of the National Academy of Sciences. 2010;107:4335–4340. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Müller F-J, Schuldt BM, Williams R, et al. A bioinformatic assay for pluripotency in human cells. Nature methods. 2011;8:315–317. doi: 10.1038/nmeth.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandoe J, Eggan K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nature neuroscience. 2013;16:780–789. doi: 10.1038/nn.3425. [DOI] [PubMed] [Google Scholar]
- 46.Nicholas CR, Chen J, Tang Y, et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang X, Zhou L, Wagner AM, et al. Astroglial cells regulate the developmental timeline of human neurons differentiated from induced pluripotent stem cells. Stem cell research. 2013;11:743–757. doi: 10.1016/j.scr.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sukigara S, Dai H, Nabatame S, et al. Expression of Astrocyte-Related Receptors in Cortical Dysplasia With Intractable Epilepsy. Journal of Neuropathology & Experimental Neurology. 2014;73:798–806. doi: 10.1097/NEN.0000000000000099. [DOI] [PubMed] [Google Scholar]
- 49.Moore AR, Filipovic R, Mo Z, et al. Electrical excitability of early neurons in the human cerebral cortex during the second trimester of gestation. Cerebral Cortex. 2009;19:1795–1805. doi: 10.1093/cercor/bhn206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bardy C, van den Hurk M, Eames T, et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proceedings of the National Academy of Sciences. 2015:201504393. doi: 10.1073/pnas.1504393112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wainger BJ, Kiskinis E, Mellin C, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell reports. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McConnell ER, McClain MA, Ross J, et al. Evaluation of multi-well microelectrode arrays for neurotoxicity screening using a chemical training set. Neurotoxicology. 2012;33:1048–1057. doi: 10.1016/j.neuro.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lancaster MA, Renner M, Martin C-A, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Espuny-Camacho I, Michelsen KA, Gall D, et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013;77:440–456. doi: 10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 55.Cunningham M, Cho J-H, Leung A, et al. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell stem cell. 2014;15:559–573. doi: 10.1016/j.stem.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]