

Summary
Emerging resistance to first-line antimalarial combination therapies threatens malaria treatment and the global elimination campaign. Improved therapeutic strategies are required to protect existing drugs and enhance treatment efficacy. We report that the piperazine-containing compound ACT-451840 exhibits single-digit nanomolar inhibition of the Plasmodium falciparum asexual blood stages and transmissible gametocyte forms. Genome sequence analyses of in vitro-derived ACT-451840-resistant parasites revealed single nucleotide polymorphisms in pfmdr1, which encodes a digestive vacuole membrane-bound ATP-binding cassette transporter known to alter P. falciparum susceptibility to multiple first-line antimalarials. CRISPR-Cas9 based gene editing confirmed that PfMDR1 point mutations mediated ACT-451840 resistance. Resistant parasites demonstrated increased susceptibility to the clinical drugs lumefantrine, mefloquine, quinine and amodiaquine. Stage V gametocytes harboring Cas9-introduced pfmdr1 mutations also acquired ACT-451840 resistance. These findings reveal that PfMDR1 mutations can impart resistance to compounds active against asexual blood stages and mature gametocytes. Exploiting PfMDR1 resistance mechanisms provides new opportunities for developing disease-relieving and transmission-blocking antimalarials.
Introduction
Malaria exacts a massive toll across tropical and subtropical regions, primarily affecting sub-Saharan Africa. This infectious disease has a reported incidence of 214 million worldwide, and claims an estimated 438,000 lives per year, mostly children under the age of five (WHO, 2015). Artemisinin-based combination therapies are currently recommended as first-line treatment for uncomplicated Plasmodium falciparum malaria infections. Alarmingly, emerging resistance to artemisinin derivatives, which manifests clinically as a delayed rate of parasite clearance in treated patients, is now widespread in Western Cambodia and across the Greater Mekong Subregion (Dondorp et al., 2009; Ashley et al., 2014; Tun et al., 2015). Artemisinin resistance is driven primarily by mutations in the K13-propeller domain (Ariey et al., 2014; Ashley et al., 2014; Straimer et al., 2015). One consequence has been the higher parasite burdens encountered by the combination therapy partner drugs, resulting in increased selective pressure. Very recently, resistance to piperaquine has emerged and spread across western Cambodia, leading to clinical treatment failures with the first-line therapy dihydroartemisinin-piperaquine (Saunders et al., 2014; Leang et al., 2015; Amaratunga et al., 2016). These reports underscore the importance of identifying new chemical entities that can effectively treat multidrug-resistant P. falciparum.
ACT-451840 is a potent piperazine-containing compound that targets all three stages (rings, trophozoites, and schizonts) of P. falciparum asexual blood stage development. Phase I clinical trials with this compound, developed by Actelion Pharmaceuticals with support from the Medicines for Malaria Venture, documented a good safety and tolerability profile with no serious adverse events (Bruderer et al., 2015). Heterologous expression studies found that ACT-213615, a previous analog in this chemical series, bound to the P. falciparum transporter PfMDR1 (multidrug resistance-1), which is a parasite ortholog of the mammalian dug efflux transporter P-glycoprotein 1. That report also showed that parasites with increased pfmdr1 copy number were less susceptible to ACT-213615 (Brunner et al., 2013). PfMDR1 is a ~160 kDa multimembrane-spanning member of the ATP-binding cassette (ABC) transporter superfamily that resides on the membrane of the parasite’s digestive vacuole (DV) (Cowman et al., 1991). Each half of this transporter contains six transmembrane domains (TMD) and one nucleotide-binding domain. PfMDR1 is predicted to facilitate solute movement into the DV (Rohrbach et al., 2006). While its native solute has not been defined, changes in its amino acid sequence and/or expression level (resulting from pfmdr1 copy number variations) can modulate parasite susceptibility to a variety of antimalarials, including lumefantrine (LMF), mefloquine (MFQ), artesunate (AS), quinine (QN), and chloroquine (CQ) (Reed et al., 2000; Sidhu et al., 2006; Sanchez et al., 2008; Veiga et al., 2011; Conrad et al., 2014; Venkatesan et al., 2014; Veiga et al., 2016).
Using resistance selections, whole-genome sequence analysis, and CRISPR-Cas9 based genome editing, we identify and confirm the role of PfMDR1 mutations in ACT-451840 resistance, and observe inverse patterns of parasite susceptibility between this agent and several drugs used in first-line antimalarial therapies. Our results evoke new strategies to combine PfMDR1-interacting drugs to neutralize resistance, and highlight these piperazine-containing compounds as potent inhibitors of both asexual blood stages and the transmissible gametocyte stages.
Results
In vitro resistance selection of P. falciparum to ACT-451840
ACT-451840 is a potent inhibitor of both multidrug-resistant and sensitive P. falciparum asexual blood stage parasites, with in vitro IC50 values in the sub-nanomolar range (7G8: 0.3 nM; Dd2: 0.7 nM; NF54: 0.6 nM). Single-step selection studies to determine the minimum inoculum for resistance (MIR) in vitro were performed by subjecting 2 × 104 to 2 × 109 parasites to ACT-451840 pressure; this experiment was done in triplicate. The MIR for the 7G8 strain subjected to 1.7 nM ACT-451840 was revealed to be 2 × 107, whereas the MIR for the Dd2 and NF54 strains subjected to 2.7 nM was 2 × 106 and 2 × 107 respectively (Supporting Information Table S1). These selective pressures correspond to 4–6× the parental IC50 values. Cultured parasites were generally observed microscopically 17–22 days after initiating drug exposure, although with some selections parasites emerged as late as day 61. Of 44 independent selections, 20 yielded recrudescent parasites (Supporting Information Table S1). In parallel, we also performed step-wise selection experiments on NF54 parasites, beginning with 0.8 nM and gradually increasing to 12.8 nM over a period of 4 months.
In vitro drug-pressured parasites harbor PfMDR1 polymorphisms associated with varying degrees of ACT-451840 resistance
To identify causal mutations, we hybridized genomic DNA (gDNA) from a subset of ACT-451840-resistant lines, selected from the 7G8, Dd2, and NF54 strains, against a P. falciparum microarray containing 4.8 million tiled 25-mer probes (Dharia et al., 2009). Control arrays were hybridized with gDNA from the parental strains. With the single-step mutants, this analysis revealed multiple single nucleotide polymorphisms (SNPs) in the pfmdr1 gene (PF3D7_0523000). Each resistant line harbored a single pfmdr1 nucleotide substitution, as confirmed by Sanger sequencing. When applied to the ACT-451840-resistant parasites obtained with step-wise selection, this analysis identified a double mutation in PfMDR1, namely M841I and M924I. It is possible that due to the prolonged selection process, these mutations evolved sequentially. PfMDR1 mutations and haplotypes for all drug-pressured lines are shown in Fig. 1 and Supporting Information Table S2.
Fig. 1.
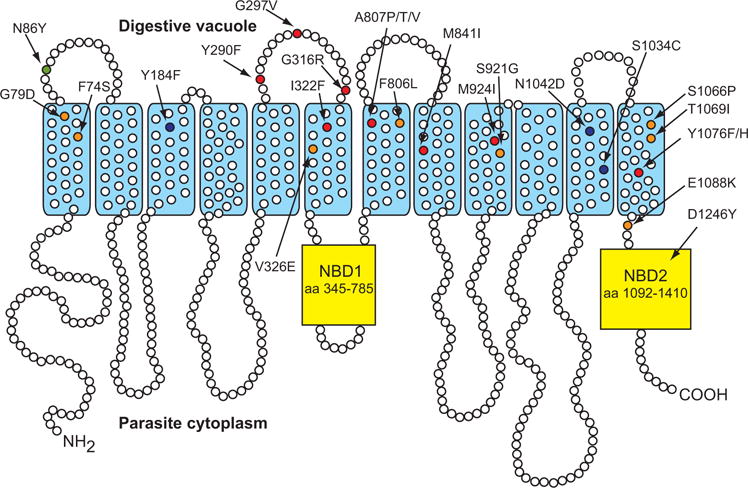
PfMDR1 mutations identified in ACT-451840-selected lines. Common polymorphisms are shown in green (Dd2: N86Y) and blue (7G8: Y184F, S1034C, N1042D, D1246Y).
Polymorphisms selected by ACT-451840 and confirmed phenotypically by IC50 determination are indicated in red (Y290F, G297V, G316R, I322F, A807P, A807T, A807V, M841I, M924I, Y1076F, Y1076H); polymorphisms selected by ACT-451840 but not confirmed phenotypically are indicated in orange (F74S, G79D, V326E, F806L, S921G, S1066P, T1069I, E1088K). NBD, nucleotide binding domain.
Illumina-based paired-end whole-genome sequencing of four resistant clones (recovered from four separate flasks) generated in the 7G8 background confirmed these pfmdr1 polymorphisms and found no other SNPs common to another gene that could account for the gain of ACT-451840 resistance (Supporting Information Table S3). Whole-genome sequencing and tiling array analyses found no evidence of copy number variants that associated with the gain of resistance (our Dd2 1pa clone has three copies of pfmdr1 (Eastman et al., 2011) and was used as an internal control). Nonetheless, it remains possible that ACT-451840 selection also resulted in other genetic changes that might have differed between lines and that contributed to resistance.
Dose-response studies with parasites labeled with SYBR green and subjected to flow cytometry demonstrated a gain of resistance in our drug-pressured clones. 7G8 parasites harboring the PfMDR1 mutations G316R, A807T or M841I showed similar levels of resistance (~30-fold increases in mean IC50 values compared to the parental strain). We observed similar IC50 values in Dd2 mutants expressing the PfMDR1 A807V, M841I or Y1076F mutations (showing ~15-fold increases in mean IC50 values compared to the parent). ACT-451840-resistant lines derived from NF54 parasites showed a wide range in their degrees of resistance to this compound (11- to 100-fold IC50 increases compared to the parent) (Fig. 2A and Supporting Information Table S4). Of our 20 recrudescent lines (Supporting Information Table S1), we tested for shifts in IC50 values for a subset that represented 11 of the 19 novel pfmdr1 mutations (Fig. 1).
Fig. 2.
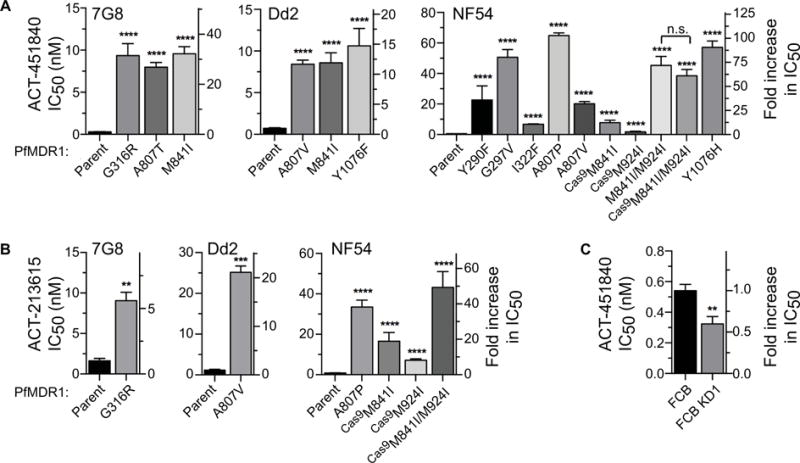
P. falciparum resistance to ACT-451840 and ACT-213615.
A. ACT-451840 potency against ACT-451840-resistant lines selected in 7G8, Dd2, or NF54 strains.
B. Cross-resistance to ACT-213615 in ACT-451840-resistant lines.
C. ACT-451840 sensitivity of FCB and FCB KD1 lines (the latter is a recombinant line that expresses only one of the two pfmdr1 copies present in FCB). Bar graphs display mean IC50 values ± S.E.M. Student’s t-test was performed to compare resistant to parental lines, unless otherwise indicated. **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s. indicates no statistically significant difference (P > 0.05).
pfmdr1 copy number modulates ACT-451840 susceptibility
Prior studies with related piperazine-containing compounds suggest that this class of agents might directly bind to PfMDR1 and thus inhibit its function (Brunner et al., 2013). Our results demonstrated that resistance to ACT-451840 imparted cross-resistance to the analog ACT-213615, implying that both compounds act in a similar manner (Fig. 2B; structures shown in Supporting Information Fig. S1). We also examined ACT-451840 potency against FCB parasites, which have two copies of pfmdr1, and the isogenic line FCB KD1, which was genetically modified to disrupt one of these two copies (Sidhu et al., 2006). FCB KD1 was ~two-fold more susceptible to ACT-451840 (Fig. 2C), implicating a role for pfmdr1 copy number in modulating the potency of compounds in this chemical series.
CRISPR-Cas9-mediated genetic engineering confirms the role of pfmdr1 mutations
To test whether mutations in pfmdr1 suffice to confer ACT-451840 resistance, we genetically engineered the double mutation M841I/M924I into wild-type NF54 parasites. These studies utilized the CRISPR-Cas9 system, which has recently been developed for genome editing in Plasmodium (Ghorbal et al., 2014; Wagner et al., 2014; Zhang et al., 2014). Eight different guide RNAs (gRNAs) were chosen by visually scanning the pfmdr1 gene for GN19GG sequences. gRNA target sequences were 15–708 nucleotides away from the desired mutations and were unique in the P. falciparum genome (Supporting Information Fig. S2). gRNAs were expressed from the T7 promoter on a plasmid that also expressed the Cas9 nuclease. A separate plasmid expressed the T7 RNA polymerase and the 1.5 kb donor sequence, with silent mutations at Cas9 binding sites to prevent Cas9 cleavage of the plasmid or the edited gene (Fig. 3).
Fig. 3.

Gene editing via CRISPR-Cas9.
A. The Cas9 enzyme is encoded on a plasmid that also expresses the gRNA and a hdhfr selectable marker. The donor plasmid contains a template with silent mutations at the gRNA, the PfMDR1 M841I/M924I double mutation, and the bsd selectable marker.
B. Electropherograms showing successful editing of PfMDR1 M841I, M924I, and silent binding site mutations. Gray boxes indicate edited nucleotide substitutions.
In the initial set of transfections, each of the eight pairs of Cas9 and donor plasmids was electroporated into NF54 parasites on two separate occasions. Parasites were then cultured in the presence of 2.7 nM ACT-451840, a concentration that kills sensitive but not resistant parasites, to select for modified resistant parasites. We microscopically observed parasites in 11 of the 16 transfections, of which three incorporated the desired synonymous and non-synonymous mutations. The other eight positive transfections had spontaneous mutations in pfmdr1 without Cas9-introduced silent binding site mutations, resulting in the amino acid substitutions G79D, Y290F (2 transfectants), A807V, A807P, S921G, or S1066P (2 transfectants). Of the three successful editing events, the one with gRNA2 captured only the M841I mutation. The other two employed gRNA8 and captured both M841I and M924I (Supporting Information Table S5). Full-length pfmdr1 sequencing ruled out any additional mutations in this gene.
Because gRNA8 appeared the most effective, we selected this guide for subsequent transfections. To examine whether pfmdr1 editing required direct selection with ACT-451840, we electroporated parasites on two separate days and maintained these with WR99210 and blasticidin for six days, followed by culturing in drug-free media. These agents select for the human dhfr and blasticidin S-deaminase (bsd) markers present on the Cas9 and donor plasmids respectively (Fig. 3A). One of these two transfections successfully captured both the binding site mutations and the M924I mutation (Supporting Information Table S5). This demonstrates that selection for the plasmids alone can suffice to incorporate the desired mutations. Control transfections with solely the donor plasmid (without Cas9) did not result in pfmdr1 gene editing.
Parasites engineered to express the double mutation (NF54 MDR1 Cas9M841I/M924I) showed similar levels of ACT-451840 resistance as compared with mutant parasites resulting from single-step selections (NF54 MDR1 M841I/M924I) (Fig. 2A). These data provide compelling evidence that the PfMDR1 mutations observed in ACT-451840-selected lines were responsible for the gain of resistance, and argue against a major role for any other genetic changes that might have arisen elsewhere in the genome during drug selection. Using our Cas9-edited parasites, we compared the resistance contribution of the individual M841I and M924I mutations. Parasites expressing MDR1 Cas9M841I or MDR1 Cas9M924I both displayed low-level resistance (13-fold and 3-fold increase in resistance compared to parental lines, respectively). The combination of these mutations in the edited MDR1 Cas9M841I/M924I line was synergistic in terms of augmenting ACT-451840 resistance (64-fold increase in resistance compared to parental line) (Fig. 2A and Supporting Information Table S4). Comparable levels of cross-resistance were observed with the analog ACT-213615 (Fig. 2B and Supporting Information Table S6).
ACT-451840 resistance potentiates the action of multiple clinical antimalarial drugs
Given the known involvement of PfMDR1 in multidrug resistance traits (Venkatesan et al., 2014), we profiled our ACT-451840 resistant mutant lines against a panel of clinical antimalarial drugs. These included LMF, MFQ, QN, CQ, its metabolite monodesethyl-chloroquine (md-CQ), monodesethyl-amodiaquine (md-ADQ), halofantrine (HF) and piperaquine (PPQ). We note that PPQ, like ACT-451840, is a piperazine-containing compound (Supporting Information Fig. S1). We also examined AS, an artemisinin derivative that is the core component of several first-line artemisinin-based combination therapies. AS and artemether, employed clinically, are both converted to the active metabolite dihydroartemisinin (Eastman and Fidock, 2009).
When compared to the parental 7G8 strain (that harbors Y184F, S1034C, N1042D and D1246Y), the G316R variant was three times more sensitive to MFQ, and twice as sensitive to LMF and QN (Fig. 4A–C, left panels and Supporting Information Table S6). 7G8 MDR1 G316R showed a trend towards increased sensitivity to HF compared to its parental line, but had no significant effect on parasite susceptibility to CQ, md-CQ, md-ADQ, and PPQ (Supporting Information Fig. S3, left panels). 7G8 MDR1 A807T and 7G8 MDR1 M841I showed trends of mildly increased sensitivity to MFQ (Fig. 4A), but behaved similarly to the 7G8 parent when exposed to LMF, QN, md-CQ, md-ADQ, HF, and PPQ (Fig. 4B and C, and Supporting Information Fig. S3B–E). The differential contributions of assorted PfMDR1 mutations evoke specific alterations in protein-drug interactions, protein conformation or native function that impact drug sensitivities.
Fig. 4.

Sensitivity of ACT-451840-resistant lines to clinical antimalarials. Susceptibility of parental and mutant lines to (A) mefloquine, (B) lumefantrine, and (C) quinine. Bar graphs display mean IC50 values ± S.E.M. Student’s t-test was performed comparing resistant to parental lines. *P < 0.05, **P < 0.005.
The Dd2 strain carries a N86Y mutation, which has been associated with decreased susceptibility to CQ and md-ADQ and increased susceptibility to LMF, dihydroartemisinin, and MFQ (Mungthin et al., 2010; Wurtz et al., 2014; Veiga et al., 2016). Compared to the Dd2 parent, Dd2 MDR1 A807V was two-fold more sensitive to LMF and QN (Fig. 4B and C, middle panels). This mutant showed a non-significant trend towards reduced sensitivity to MFQ (Fig. 4A, middle panel) and HF (Fig. S3D), and no change in susceptibility to CQ, md-CQ, md-ADQ, PPQ, or AS (Supporting Information Fig. S3).
NF54 carries the canonical wild-type haplotype and is a drug-sensitive line. Compared to the NF54 parent, the Cas9-modified lines MDR1 Cas9M841I and NF54 MDR1 Cas9M924I were each two-fold more sensitive to md-ADQ. This increased sensitivity, however, was not observed when both mutations were co-expressed in NF54 MDR1 Cas9M841I/M924I. These polymorphisms did not significantly affect NF54 susceptibility to CQ, md-CQ, HF, PPQ or AS (for PPQ we also saw no effect with A807P; Supporting Information Fig. S3, right panels).
In summary, multiple ACT-451840-conferring resistance mutations were observed to increase parasite susceptibility to a range of antimalarial drugs including MFQ, LMF, md-ADQ and/or QN. None of these mutations mediated reduced susceptibility to the antimalarials tested herein, including the piperazine-containing drug PPQ.
ACT-451840-resistant mutations in PfMDR1 result in reduced parasite fitness in vitro
To examine whether ACT-451840-resistance conferring mutations are associated with a fitness cost in vitro, we performed direct co-competition assays between isogenic mutants and their parental strains. Changes over time in the prevalence of distinct pfmdr1 alleles were assessed by pyrosequencing, a technique that can distinguish the proportion of dissimilar nucleotides at a specified position (Zhou et al., 2006). Results showed increased proportions of the parental allele over a 30-generation period in all three genetic backgrounds (Supporting Information Fig. S4A). When compared to their respective parental strains, 7G8 MDR1 M841I, Dd2 MDR1 A807V, and NF54 MDR1 Cas9M841I/M924I experienced 5.3%, 4.2%, and 1.5% mean relative fitness costs per generation (Supporting Information Fig. S4B and C; calculations defined in Experimental Procedures).
PfMDR1 mutations protect mature gametocytes from the potent transmission-blocking activity of ACT-451840
Antimalarials that act on targets in the DV such as CQ, ADQ and QN are active against asexual blood stage parasites and early gametocytes, but lose potency against the transmissible stage V gametocytes (Butcher, 1997; Bousema and Drakeley, 2011). This reduced potency coincides with the cessation of hemoglobin degradation in these mature stages (Hanssen et al., 2012). In light of our evidence that the DV-resident transporter PfMDR1 modulates parasite susceptibility to ACT-451840, we hypothesized that this compound might be active on early but not mature gametocytes. We tested this by exposing early and late gametocytes from both Dd2 and NF54 to a range of ACT-451840 concentrations. Results, surprisingly, showed low nanomolar IC50 values against both immature stages and mature stage V gametocytes (Fig. 5 and Supporting Information Table S7). The potent activity of ACT-451840 on stage V gametocytes is in marked contrast to previous reports that mature gametocytes are generally refractory to antimalarials (Adjalley et al., 2011). Notably, PfMDR1 mutations protected P. falciparum early and late gametocytes from ACT-451840 in both the Dd2 and NF54 backgrounds (Fig. 5 and Supporting Information Table S7). These data evoke the possibility that PfMDR1 might play an important role in stage V gametocytes.
Fig. 5.
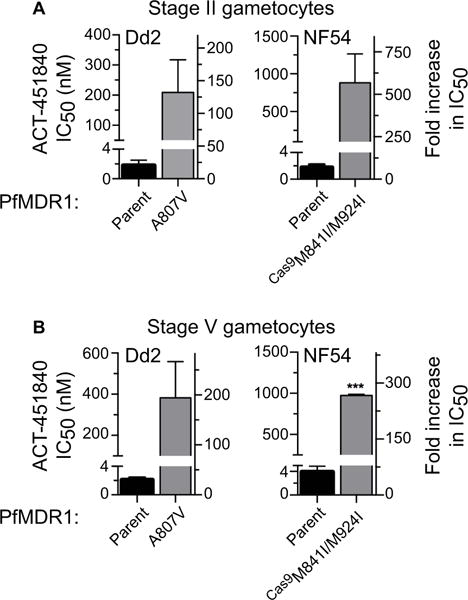
ACT-451480 activity in gametocytes. In vitro drug resistance studies examining susceptibility to ACT-451840 in (A) stage II and (B) stage V gametocytes. Bar graphs display mean IC50 values ± S.E.M. Student’s t-test was performed comparing mutant to parental lines. ***P < 0.0005.
To address this point, we examined the DV in ultrastructural sections from synchronous stage IV and stage V gametocytes. Results confirmed that immature (stage IV) gametocytes harbored one or more DVs, characterized by the presence of hemozoin crystals (Fig. 6A). The detection of cytostomes in stage IV gametocytes (harvested at days 6–7 of development) provided evidence of ongoing endocytosis of host cytoplasm (see indent in Fig. 6A). DVs were also observed in mature (stage V) gametocytes (Fig. 6B). The presence of intact membrane surrounding these DVs suggested the structural integrity of these subcellular organelles.
Fig. 6.
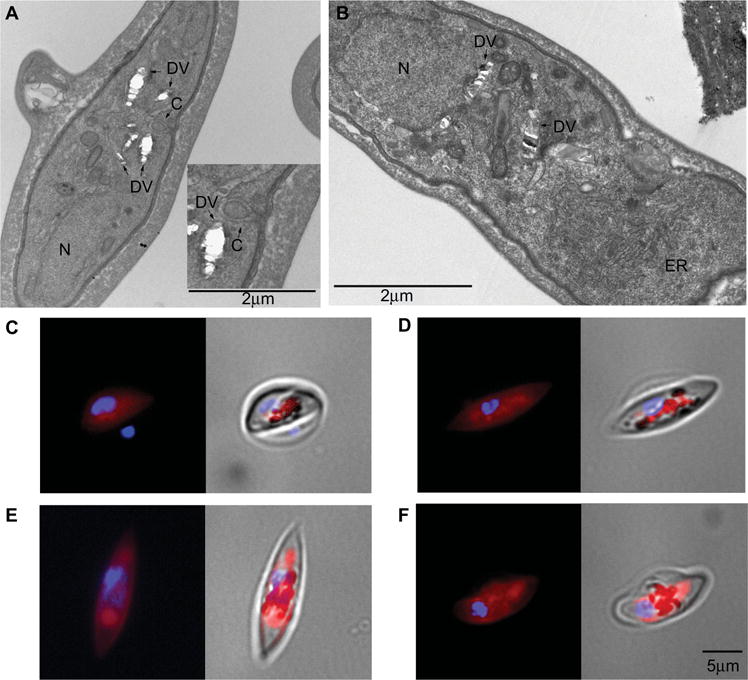
Presence of the DV in developing gametocytes.
(A, B) Ultrastructural analyses of stage IV and stage V gametocytes (3D7 strain).
A. Active endocytosis of infected erythrocyte cytoplasm in stage IV gametocytes through a cytostome (indent of panel A) and the accumulation of hemozoin in membrane-surrounded vesicles constituting the fragmented DV typical of gametocytes are observed.
B. Hemozoin-containing vesicles are enclosed in a membrane in stage V gametocytes. C: cytostome, ER: endoplasmic reticulum, DV: digestive vacuole, and N: nucleus. Magnification bar: 2 μm.
C–F. LysoTracker staining of gametocytes at stages (C) II, (D) III, (E) IV, and (F) V of maturation. A tight colocalization of LysoTracker fluorescence and dark hemozoin granules is evident in stage II and III gametocytes, whereas in stages IV and V areas of the acidic DV compartment are devoid of pigment granules. Magnification bar: 5 μm.
To further examine whether these DV structures are physiologically intact, we stained live gametocytes with LysoTracker, a lysosomotropic dye that has been used to mark this highly acidic compartment in P. falciparum asexual blood stages (Bohorquez et al., 2012). Lyso-Tracker fluorescence was observed in a defined area that co-localized with hemozoin granules in all stages of gametocyte development (Fig. 6C–F). Intriguingly, some LysoTracker-positive regions were devoid of hemozoin pigment in mature (stage V) gametocytes (Fig. 6E and F), suggesting reduced levels of hemoglobin degradation that did not yield observable hemozoin crystals. Alternatively, these regions might represent distinct acidic compartments that do not mediate hemoglobin degradation. These results provide evidence that stage V gametocytes harbor intact DVs that retain their characteristic acidic property. These findings support the hypothesis that PfMDR1 is functional and mediates resistance to ACT-451840 in asexual blood stages and throughout gametocyte development.
Discussion
Our study shows that the piperazine-containing compound ACT-451840 is a potent antimalarial active against both asexual blood stage parasites (Fig. 2 and Supporting Information Table S4) and sexual stage gametocytes (Fig. 5 and Supporting Information Table S7). ACT-451840 was derived from ACT-213615, an earlier member of this chemical class that was shown to physically interact with PfMDR1 (Brunner et al., 2013). Consistent with these prior data, resistance to ACT-451840 was obtained in lines observed to have mutations in PfMDR1, whose role was confirmed using CRISPR-Cas9 based gene editing (Fig. 2). Of note, resistance was quite moderate, with ACT-451840-pressured lines showing mean IC50 values below 11 nM in the 7G8 and Dd2 backgrounds, and in the range of 7–65 nM in NF54 parasites (Fig. 2, Supporting Information Table S4). Of particular interest were the mutations at residues 807 and 841, which appeared in all three strains and which were associated with varying degrees of parasite resistance to ACT-451840. These data suggest specific contributions of these residues as well as strain-dependent differences (we note that these strains differ in their baseline PfMDR1 haplotype; Supporting Information Table S2). Mutations in PfMDR1 have previously been shown to modulate P. falciparum susceptibilities to a range of first-line antimalarials (Reed et al., 2000; Sidhu et al., 2005; Veiga et al., 2011; Conrad et al., 2014; Veiga et al., 2016), to similar or lesser degrees. Our data reveal that PfMDR1 point mutations causal for ACT-451840 resistance can augment the potency of several clinically deployed antimalarials, including LMF, MFQ, and QN (Fig. 4). These patterns of inverse susceptibility depended on both the PfMDR1 haplotype and the genetic background of the parasite strain.
While our selection experiments did not identify pfmdr1 amplification as a causal mechanism of resistance, our data show that increased pfmdr1 copy number can modestly reduce ACT-451840 potency (Fig. 2C). This compound might therefore prove to be slightly less active against multicopy pfmdr1 parasites, which are commonly found in parts of Asia subject to LMF or MFQ pressure (Veiga et al., 2016). Importantly, pfmdr1 gene amplification is very rare in Africa (Venkatesan et al., 2014), where the enhanced potency of LMF against ACT-451840-resistant parasites evokes an interesting scenario of opposing selective pressures that might be therapeutically beneficial. This concept is already being applied in ongoing clinical trials that are testing the benefit of adding ADQ to artemether plus LMF (ClinicalTrials.gov, identifier no. NCT02453308), based in part on the earlier observation that ADQ and LMF exert opposing selections on both PfMDR1 and PfCRT haplotypes (Venkatesan et al., 2014).
We note that our ACT-451840 resistance selections were performed at concentrations that corresponded to 4–6 times the IC50 values for the parental lines (7G8, Dd2 and NF54). Our data with isogenic FCB parasites that differ in pfmdr1 copy number showed that gene duplication resulted in only a two-fold increase in IC50 value (Fig. 2C). That result suggests that small copy number amplifications would have not survived our higher selective pressures, which yielded only pfmdr1 point mutations that were associated with greater levels of resistance.
ACT-451840 potently inhibited both early and mature gametocytes. This gametocytocidal activity was also observed in a recent clinical trial with P. falciparum-infected humans, which showed that a single dose of ACT-451840 that was sub-curative of asexual P. falciparum parasitemia eliminated all detectable gametocytes. The absence of these sexual stages was determined by RT-PCR based detection of the mature gametocyte marker pfs25 (Krause et al., 2016). Furthermore, standard membrane feeding assays have recently shown that ACT-451840 can inhibit P. falciparum stage V gametocyte transmission to Anopheles mosquitoes at concentrations of 100 nM or lower (A. Le Bihan and S. Wittlin, pers comm.), thereby demonstrating potent transmission-blocking potential.
Mature gametocytes are refractory to heme-targeting antimalarials (Butcher, 1997; Bousema and Drakeley, 2011), and the observation that hemoglobin digestion no longer takes place beyond stage IV of gametocyte development has led to the suggestion that the DV is no longer functional in these stages (Hanssen et al., 2012). Indeed, pfmdr1 transcripts were found to be down regulated in stage V gametocytes, as detected by strand-specific RNA-seq Illumina-based sequencing (Lopez-Barragan et al., 2011). Nevertheless, a proteomic analysis of synchronized parasites observed comparable levels of PfMDR1 peptide in trophozoites, early (stage I) and mature (stage V) gametocytes (Silvestrini et al., 2010). Measurable hemoglobin digestion and morphological evidence of cytostomes engulfing infected red blood cell cytoplasm in stage II and III gametocytes (Lanfrancotti et al., 2007; Hanssen et al., 2012), and stage IV gametocytes (Fig. 6A), argue for the presence of an active DV in all immature gametocyte stages. The observation that ACT-451840 is a potent inhibitor of early gametocytes and that mutant PfMDR1 confers resistance in these stages was therefore not surprising. In contrast, in stage V gametocytes, the observed activity of ACT-451840 and the increased mean IC50 values conferred by the PfMDR1 mutations suggest that this transporter might remain functionally important in late gametocytes. Our electron microscopy and LysoTracker staining studies provide evidence of intact and acidic DVs in stage V gametocytes (Fig. 6A–F). Collectively, these data suggest a DV function involving PfMDR1 in mature gametocytes that is separate from hemoglobin metabolism. These findings open new perspectives for identifying combinations of drugs that interfere with DV physiology, exert opposing selective forces on the multidrug resistance transporter PfMDR1, and augment cure of symptomatic asexual blood stage infections with transmission-blocking properties.
Experimental procedures
Generation of ACT-451840-resistant parasites
Dd2, 7G8 and NF54 intra-erythrocytic parasites were cultured in vitro as described (Fidock et al., 1998). Recently cloned parasites, starting at 2% parasitemia, were exposed to constant selective pressure (7G8: 1.7 nM ACT-451840 (6×IC50); Dd2: 2.7 nM ACT-451840 (4×IC50); NF54: 2.7 nM ACT-451840 (5×IC50)). Initial parasite inocula ranged from 2 × 104 to 2 × 109, and were tested in triplicates. Media was changed every 24 h for the first 6 days and parasitemia closely monitored to prevent parasite overgrowth. Media was then changed every 48 h and cultures were monitored for evidence of regrowth. Experiments were carried out to 60 days, or until parasites recrudesced. NF54 was also subjected to step-wise selection, in which parasites were initially exposed to 1×IC90 (0.8 nM). When parasites recrudesced and expanded to ≥3% parasitemia at this concentration, drug pressure was successively increased two-fold, until a final concentration of 16×IC90 (12.8 nM). Parasites that recrudesced at 16×IC90 were selected for further analyses.
Whole-genome sequence analyses and pfmdr1 genotyping
Parasite gDNA was hybridized to Affymetrix P. falciparum microarrays as described (Dharia et al., 2009). The PfGe-nominator software was used to extract meaning from raw data. SNP detection for Dd2 samples was set with a cut off P-value of 10−25. SNP detection for 7G8 and NF54 samples were decreased to a cut off P-value of 10−28. Whole genome paired-end Illumina-based multiplex sequencing was performed on purified parasite gDNA prepared using the Illumina Nextera XT kit. To genotype pfmdr1, gDNA was prepared using DNeasy Blood & Tissue Kits (Qiagen) and the PCR amplified locus was sequenced using primers detailed in Supporting Information Table S8.
In vitro drug resistance studies
Asexual parasites were subjected to in vitro SYBR-green-based drug assays as described in (Ekland et al., 2011), except that parasites were assayed at 72 h post-drug exposure. Gametocytes were generated by nutrient deprivation (Fivelman et al., 2007). Stage II gametocytes were purified from uninfected erythrocytes over a 60% Percoll density gradient (Kariuki et al., 1998). Stage V gametocytes were obtained by purifying stage II gametocytes by MACS Separation Columns CS (Miltenyi Biotec) and allowed to mature for an additional 8 days. Gametocytes were exposed to drugs and cell viability was measured using a parasite lactate dehydrogenase (pLDH) assay as described (D’Alessandro et al., 2013), except that drug was washed out after 48 h, and cell viability was measured after an additional 72 h. IC50 values were obtained by non-linear regression analyses (GraphPad Prism 6.0). The total number of replicates indicated in the appropriate tables represents biological replicates, each consisting of technical duplicates.
Fitness assays
ACT-451840-resistant lines and their parental counterparts were mixed in a 1:1 ratio and cultured for 30 generations. gDNA was harvested regularly every two to three days, using DNeasy Blood & Tissue Kits (Qiagen). The proportion of parental to mutant allele was determined by pyrosequencing polymorphic pfmdr1 sites (Zhou et al., 2006) using PyroMark Q96 ID reagents (Qiagen). SNPs within pfmdr1 codons 807, 841, and 924 were examined in Dd2, 7G8, and NF54 pairs, respectively, using primers detailed in Supporting Information Table S8. Pyrosequencing was performed on a Qiagen Pyrosequencing PSQ 96. The natural logarithm (ln) of the proportion of mutant parasites (pt) with respect to parental parasites (qt), ln (pt/qt), was plotted against generation (t). The slope (m) of the line of best fit was determined using Graphpad Prism 6.0. The relative fitness (ω) of each mutant allele is equivalent to em (Hartl et al., 1985). The fitness cost per generation (s) was calculated as s = ω−1 (Gabryszewski et al., 2016).
CRISPR-Cas9-mediated genetic engineering
Eight unique (https://chopchop.rc.fas.harvard.edu) potential CRISPR sites were identified in pfmdr1, detailed in Supporting Information Fig. S2. gRNA constructs that help Cas9 target CRISPR sites were synthesized with overhanging BbsI sites, and inserted behind a T7 promoter in the pCas9-hdhfr plasmid that co-expressed the Cas9 enzyme and the human dhfr selectable marker. A 1.5 kb fragment of pfmdr1 containing the G2523T/G2772A ACT-451840 resistance-conferring mutations (M841I/M924I) was amplified from the step-wise selection of resistant NF54 parasites using 5′-GAGCTCGTACTATAAGATATGCCAATACA (SacI restriction site underlined) and 5′-GACGTCCATTTGGTCTTGAAATATAACGG (AatII restriction site underlined). From this fragment, we generated eight different constructs, each containing one of the eight sequences with silent binding site mutations (Supporting Information Fig. S2). Mutations were introduced using the Q5 Site-Directed Mutagenesis Kit (NEB). A total of 7–10 bp changes resulting in silent mutations were engineered to protect the site from further Cas9 cleavage. The resulting pfmdr1 donor fragment containing G2523T, G2772A, and one of the eight protected CRISPR sites was inserted using SacI and AatII restriction sites into the pT7Pol-bsd plasmid, which expresses the blasticidin S-deaminase selectable marker. NF54 parasites co-electroporated with the pCas9-gRNA-hdhfr and pT7Pol-donor-bsd plasmids were cultured in 2.7 nM ACT-451840-supplemented media for 60 days, or selected with 2.5 nM WR99210 and 2 μg ml−21 blasticidin for 6 days, then cultured in drug-free media.
Transmission electron microscopy
Stage IV and V gametocytes were Percoll-purified and processed as described (Perry and Gilbert, 1979). Briefly, cells were fixed with 2.5% glutaraldehyde, 2% paraformaldehyde and 2 mM CaCl2 in 0.1 M sodium cacodylate buffer (pH 7.4) overnight at 4°C. Parasites were washed in cacodylate buffer and postfixed with 1% OsO4 in 0.1 M sodium cacodylate buffer for 1 h at room temperature, treated with 1% tannic acid in 0.05 M cacodylate buffer for 30 min and rinsed in 1% sodium sulphate in 0.05 cacodylate for 10 min. Fixed specimens were washed, dehydrated through a graded series of ethanol solutions (30–100% ethanol) and embedded in Agar 100 (Agar Scientific, U.K.). Ultrathin sections were prepared using a MT-2B Ultramicrotome (UC6 - Leica), stained with uranyl acetate and lead citrate and examined using an EM 208 Philips electron microscope.
LysoTracker staining
1 × 105 gametocytes at stages II, III, IV and V were loaded with 100 nM of the acidotropic fluorescent dye LysoTracker Red DND-99 in complete medium and incubated for 2 h at 37°C. During the last 15 min 10 μg ml−1 of Hoechst 33258 was added to stain nuclei, followed by two washes in 1×PBS. A Leica DMRB microscope was used to visualize live samples. Fluorescence images were acquired using a Leica DFC340 FX camera through a Leica PL FLUOTAR 100× objective. Filters used to detect LysoTracker Red DND-99 were: EX: 515–560, EM: 590 long-pass filter. Filters used to detect Hoechst 33258 were: EX: 340–380, EM 425 long-pass filter.
Statistical analysis
Data were expressed as mean ± S.E.M. and analyzed using a Student’s t-test using GraphPad Prism 6.0.
Supplementary Material
Acknowledgments
We thank Drs. Paul Roepe (Georgetown University), Didier Leroy, Benjamin Blasco and Xavier Ding (Medicines for Malaria Venture, MMV) for many helpful discussions. We also thank Dr. Liyong Deng at Columbia University for providing access to pyrosequencing. Support for this work was provided by the NIH (R01 AI50234 to D.A.F. and R01 AI103058 to E.A.W. and D.A.F.) as well as by MMV (to D.A.F.). We also gratefully acknowledge support from the Bill & Melinda Gates Foundation (OPP1040398 and OPP1040394 to D.A.F. to P.A. respectively).
Footnotes
The authors declare no competing financial interests.
Author contributions
C.L.N., G.S., M.C.S.L., E.A.W., P.A., and D.A.F. conceived and designed the experiments. C.L.N., G.S., M.J.A., S.E.B., L.B., S.W., R.G.K., and V.C.C. acquired the data. C.L.N., G.S., M.C.S.L., S.E.B., V.C.C., A.L.B., M.C., E.A.W., P.A., and D.A.F. analyzed and interpreted the data. C.L.N., G.S., P.A., and D.A.F. wrote the paper, with input from other authors. All authors approved the final manuscript.
Data and materials availability
Resistant P. falciparum lines will be deposited to the Malaria Research and Reference Reagent Resource Center (MR4) and will be made available upon request to D.A.F.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Adjalley SH, Johnston GL, Li T, Eastman RT, Ekland EH, Eappen AG, et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc Natl Acad Sci USA. 2011;108:E1214–1223. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaratunga C, Lim P, Suon S, Sreng S, Mao S, Sopha C, et al. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis. 2016;16:357–365. doi: 10.1016/S1473-3099(15)00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohorquez EB, Chua M, Meshnick SR. Quinine localizes to a non-acidic compartment within the food vacuole of the malaria parasite Plasmodium falciparum. Malar J. 2012;11:350. doi: 10.1186/1475-2875-11-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousema T, Drakeley C. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin Microbiol Rev. 2011;24:377–410. doi: 10.1128/CMR.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer S, Hurst N, de Kanter R, Miraval T, Pfeifer T, Donazzolo Y, Dingemanse J. First-in-humans study of the safety, tolerability, and pharmacokinetics of ACT-451840, a new chemical entity with antimalarial activity. Antimicrob Agents Chemother. 2015;59:935–942. doi: 10.1128/AAC.04125-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner R, Ng CL, Aissaoui H, Akabas MH, Boss C, Brun R, et al. UV-triggered affinity capture identifies interactions between the Plasmodium falciparum multidrug resistance protein 1 (PfMDR1) and antimalarial agents in live parasitized cells. J Biol Chem. 2013;288:22576–22583. doi: 10.1074/jbc.M113.453159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher GA. Antimalarial drugs and the mosquito transmission of Plasmodium. Int J Parasitol. 1997;27:975–987. doi: 10.1016/s0020-7519(97)00079-9. [DOI] [PubMed] [Google Scholar]
- Conrad MD, LeClair N, Arinaitwe E, Wanzira H, Kakuru A, Bigira V, et al. Comparative impacts over 5 years of artemisinin-based combination therapies on Plasmodium falciparum polymorphisms that modulate drug sensitivity in Ugandan children. J Infect Dis. 2014;210:344–353. doi: 10.1093/infdis/jiu141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman AF, Karcz S, Galatis D, Culvenor JG. A P-glycoprotein homologue of Plasmodium falciparum is localized on the digestive vacuole. J Cell Biol. 1991;113:1033–1042. doi: 10.1083/jcb.113.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro S, Silvestrini F, Dechering K, Corbett Y, Parapini S, Timmerman M, et al. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J Antimicrob Chemother. 2013;68:2048–2058. doi: 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- Dharia NV, Sidhu AB, Cassera MB, Westenberger SJ, Bopp SE, Eastman RT, et al. Use of high-density tiling microarrays to identify mutations globally and elucidate mechanisms of drug resistance in Plasmodium falciparum. Genome Biol. 2009;10:R21. doi: 10.1186/gb-2009-10-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol. 2009;7:864–874. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman RT, Dharia NV, Winzeler EA, Fidock DA. Piperaquine resistance is associated with a copy number variation on chromosome 5 in drug-pressured Plasmodium falciparum parasites. Antimicrob Agents Chemother. 2011;55:3908–3916. doi: 10.1128/AAC.01793-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekland EH, Schneider J, Fidock DA. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 2011;25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Wellems TE. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol Pharmacol. 1998;54:1140–1147. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- Fivelman QL, McRobert L, Sharp S, Taylor CJ, Saeed M, Swales CA, et al. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol Biochem Parasitol. 2007;154:119–123. doi: 10.1016/j.molbiopara.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Gabryszewski SJ, Modchang C, Musset L, Chookajorn T, Fidock DA. Combinatorial genetic modeling of pfcrt-mediated drug resistance evolution in Plasmodium falciparum. Mol Biol Evol. 2016 doi: 10.1093/molbev/msw037. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol. 2014;32:819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- Hanssen E, Knoechel C, Dearnley M, Dixon MW, Le Gros M, Larabell C, Tilley L. Soft X-ray microscopy analysis of cell volume and hemoglobin content in erythrocytes infected with asexual and sexual stages of Plasmodium falciparum. J Struct Biol. 2012;177:224–232. doi: 10.1016/j.jsb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Dykhuizen DE, Dean AM. Limits of adaptation - the evolution of selective neutrality. Genetics. 1985;111:655–674. doi: 10.1093/genetics/111.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariuki MM, Kiaira JK, Mulaa FK, Mwangi JK, Wasunna MK, Martin SK. Plasmodium falciparum: purification of the various gametocyte developmental stages from in vitro-cultivated parasites. Am J Trop Med Hyg. 1998;59:505–508. doi: 10.4269/ajtmh.1998.59.505. [DOI] [PubMed] [Google Scholar]
- Krause A, Dingemanse J, Mathis A, Marquart L, Mohrle JJ, McCarthy JS. Pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of Actelion-451840 in an induced blood stage malaria study in healthy subjects. Br J Clin Pharmacol. 2016 doi: 10.1111/bcp.12962. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfrancotti A, Bertuccini L, Silvestrini F, Alano P. Plasmodium falciparum: mRNA co-expression and protein co-localisation of two gene products upregulated in early gametocytes. Exp Parasitol. 2007;116:497–503. doi: 10.1016/j.exppara.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Leang R, Taylor WR, Bouth DM, Song L, Tarning J, Char MC, et al. Evidence of Plasmodium falciparum malaria multidrug resistance to artemisinin and piperaquine in western Cambodia: dihydroartemisinin-piperaquine open-label multicenter clinical assessment. Antimicrob Agents Chemother. 2015;59:4719–4726. doi: 10.1128/AAC.00835-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barragan MJ, Lemieux J, Quinones M, Williamson KC, Molina-Cruz A, Cui K, et al. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics. 2011;12:587. doi: 10.1186/1471-2164-12-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungthin M, Khositnithikul R, Sitthichot N, Suwandittakul N, Wattanaveeradej V, Ward SA, Na-Bangchang K. Association between the pfmdr1 gene and in vitro artemether and lumefantrine sensitivity in Thai isolates of Plasmodium falciparum. Am J Trop Med Hyg. 2010;83:1005–1009. doi: 10.4269/ajtmh.2010.10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MM, Gilbert AB. Yolk transport in the ovarian follicle of the hen (Gallus domesticus): lipoprotein-like particles at the periphery of the oocyte in the rapid growth phase. J Cell Sci. 1979;39:257–272. doi: 10.1242/jcs.39.1.257. [DOI] [PubMed] [Google Scholar]
- Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403:906–909. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- Rohrbach P, Sanchez CP, Hayton K, Friedrich O, Patel J, Sidhu AB, et al. Genetic linkage of pfmdr1 with food vacuolar solute import in Plasmodium falciparum. EMBO J. 2006;25:3000–3011. doi: 10.1038/sj.emboj.7601203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez CP, Rotmann A, Stein WD, Lanzer M. Polymorphisms within PfMDR1 alter the substrate specificity for anti-malarial drugs in Plasmodium falciparum. Mol Microbiol. 2008;70:786–798. doi: 10.1111/j.1365-2958.2008.06413.x. [DOI] [PubMed] [Google Scholar]
- Saunders DL, Vanachayangkul P, Lon C. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med. 2014;371:484–485. doi: 10.1056/NEJMc1403007. [DOI] [PubMed] [Google Scholar]
- Sidhu AB, Valderramos SG, Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005;57:913–926. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- Sidhu AB, Uhlemann AC, Valderramos SG, Valderramos JC, Krishna S, Fidock DA. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J Infect Dis. 2006;194:528–535. doi: 10.1086/507115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestrini F, Lasonder E, Olivieri A, Camarda G, van Schaijk B, Sanchez M, et al. Protein export marks the early phase of gametocytogenesis of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics. 2010;9:1437–1448. doi: 10.1074/mcp.M900479-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, et al. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science. 2015;347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tun KM, Imwong M, Lwin KM, Win AA, Hlaing TM, Hlaing T, et al. Spread of artemisinin-resistant Plasmodium falciparum in Myanmar: a cross-sectional survey of the K13 molecular marker. Lancet Infect Dis. 2015;15:415–421. doi: 10.1016/S1473-3099(15)70032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga MI, Ferreira PE, Jornhagen L, Malmberg M, Kone A, Schmidt BA, et al. Novel polymorphisms in Plasmodium falciparum ABC transporter genes are associated with major ACT antimalarial drug resistance. PLoS One. 2011;6:e20212. doi: 10.1371/journal.pone.0020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga MI, Dhingra SK, Henrich PP, Straimer J, Gnadig N, Uhlemann AC, et al. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun. 2016 doi: 10.1038/ncomms11553. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg. 2014;91:833–843. doi: 10.4269/ajtmh.14-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JC, Platt RJ, Goldfless SJ, Zhang F, Niles JC. Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nat Methods. 2014;11:915–918. doi: 10.1038/nmeth.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. World Malaria Report 2015. 2015 [WWW document]. URL http://www.who.int/malaria/publications/world-malaria-report-2015/report/en.
- Wurtz N, Fall B, Pascual A, Fall M, Baret E, Camara C, et al. Role of pfmdr1 in in vitro Plasmodium falciparum susceptibility to chloroquine, quinine, monodesethylamodiaquine, mefloquine, lumefantrine, and dihydroartemisinin. Antimicrob Agents Chemother. 2014;58:7032–7040. doi: 10.1128/AAC.03494-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Xiao B, Jiang Y, Zhao Y, Li Z, Gao H, et al. Efficient editing of malaria parasite genome using the CRISPR/Cas9 system. MBio. 2014;5:e01414–01414. doi: 10.1128/mBio.01414-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Poe AC, Limor J, Grady KK, Goldman I, McCollum AM, et al. Pyrosequencing, a high-throughput method for detecting single nucleotide polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes of Plasmodium falciparum. J Clin Microbiol. 2006;44:3900–3910. doi: 10.1128/JCM.01209-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.