

Abstract
At the Mid-Blastula Transition (MBT), externally developing embryos refocus from increasing cell number to elaboration of the body plan. Studies in Drosophila reveal a sequence of changes in regulators of Cyclin:Cdk1 that increasingly restricts the activity of this cell cycle kinase to slow cell cycles during early embryogenesis. By reviewing these events, we provide an outline of the mechanisms slowing the cell cycle at and around the time of MBT. The perspectives developed should provide a guiding paradigm for the study of other MBT changes as the embryo transits from maternal control to a regulatory program centered on the expression of zygotic genes.
Keywords: Cyclin:Cdk1, Cdc25, S phase checkpoint, DNA replication, Mid-Blastula Transition (MBT)
Unusual biology at the normal beginning of animal life
We are the weird ones. As mammals, we are part of a small, late-evolved clade of organisms that have adopted a derived program of early embryogenesis. Unlike us, the vast majority of existing animals and our ancestors are oviparous: that is, they lay eggs and embryogenesis takes place externally. Importantly, this external development takes place without available nutrients other than those provided in the egg. This restriction necessitates the “big egg” program in which the egg is loaded with everything needed to fund the development of a feeding-competent organism [1]. Thus, eggs are huge cells, but unlike other huge cells, the egg is limited to a post-fertilization diploid content of DNA, which does not have a transcriptional capacity sufficient to make rapid adjustments in the RNAs contained in the massive cytoplasm of the egg [2, 3]. This limitation cripples the impact of transcriptional regulation, a dominant player in the control circuits of normally proportioned cells. Consequently, the egg relies on alternative ways to regulate early events. The mother not only loads the embryo with nutrients needed for development, it also loads the embryo with the components of a pre-arranged regulatory program that uses post-transcriptional mechanisms to regulate the early events of embryogenesis. It is this distortion of usual biology that makes the early embryo so unusual, and it is the reversal of the unusual program of early development that is at the foundation of the mid-blastula transition, or MBT.
Changing the underpinnings of biology
Before transiting to a more normal program of development, the egg needs to normalize the coding capacity of its genome with the cytoplasm under its jurisdiction. It does so by mitotic cycles that amplify the number of nuclei without the doubling of the cytoplasm that usually accompanies growth of other proliferating cells. In a wide range of organisms, these embryonic cell cycles are exceptional: they run without transcriptional inputs, they are exceedingly fast, and they lack the gap phases that usually separate DNA synthesis and mitosis. These features are departures from a canonical cell cycle program that is widely conserved among eukaryotes (Figure 1, Key Figure). This review will focus on the reintroduction of cell cycle controls at the MBT in Drosophila. While not discussed here, mammalian embryogenesis in its protected and nutritious uterine environment is derived from the ancestral programs discussed here [1, 4].
Figure 1. Key Figure. The coordination of early embryonic cell cycle changes with development.
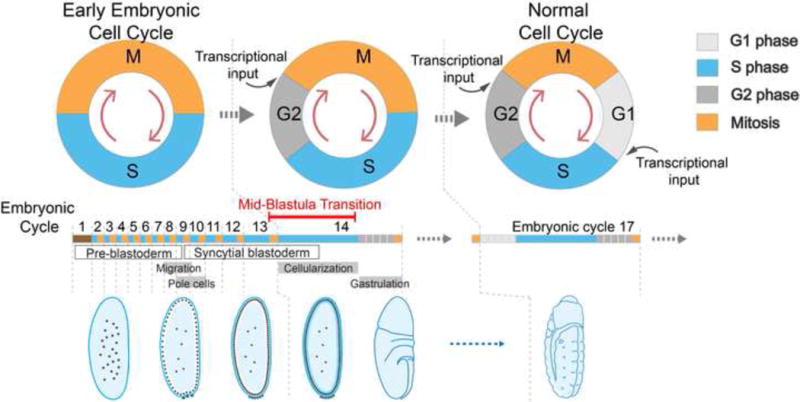
As illustrated schematically, the cell cycle acquires gap phases in two distinct steps as embryogenesis progresses. Separate introduction of the gap phases apparently follows from the fact that the programs controlling G1 and G2 are different, and each requires distinct regulatory modifications. The central bar is a linear representation of the progression of cell cycle phases through the successive cycles of early development. The associated labels and images illustrate the coordination of the cell cycle timeline with developmental events as described in detail in box 1 and summarized briefly in the text.
Embryogenesis in Drosophila melanogaster is fast, and tightly orchestrated. Abandoned in rotting fruit, the 10 μg ovoid fly egg generates an independent larva with complex anatomy in a single day. The early rapid mitotic cycles (cycles 2–9) are periodic (~9min each), synchronous and lack cytokinesis so that the nuclei proliferate in a syncytial cytoplasm. These mitotic cycles produce a cortical shell of 500 nuclei called the blastoderm. The blastoderm nuclei continue to divide rapidly and in near synchrony for four additional cycles (cycles 10–13) that slow incrementally and progressively, to a cycle time of ~21 min (interphase plus mitosis) by cycle 13 (see Figure 1 and Box 1 for details) [5]. These blastoderm cycles present a marvelous experimental system because the single cortical layer of nuclei can be imaged beautifully in real time, and the entirety of the syncytial embryo is accessible to injected markers such as fluorescent proteins, or experimental reagents including RNAi.
Box 1. Early embryonic cell cycle events.
After a specialized first-cycle in which the male and female pronuclei are combined (brown in the timeline of Fig. 1), the nuclei undergo extremely rapid mitotic cycles. These synchronous 9 min cycles are unusual in their regulation and physical circumstances. Nuclei alternate between mitosis (~5.4 min) and S phase (~3.4 min) without intervening gap phases. These mitotic cycles occur without cytokinesis, amplifying nuclei within the large egg cytoplasm as a syncytium. Despite the lack of cellular separation, the organization of the nuclei is stereotyped and dynamic, with a precise temporal program. After 6 divisions deep within the cytoplasm toward the anterior of the embryo where the polar bodies first met, the nuclei spread toward the posterior (bottom left diagram of Fig.1). Toward the completion of each of the subsequent mitoses, the now well-spaced nuclei move outward, reaching the surface at cycle 9. Some specialization of nuclei is already evident. About 100 nuclei don’t make the journey to surface. These nuclei, called yolk nuclei, soon lose their centrosomes and fail at subsequent mitoses, but continue to replicate, becoming polyploid in the process. These nuclei, which have no established contribution to embryogenesis, remain with the central yolky cytoplasm and eventually are lost. Among the nuclei reaching the surface, a few enter a specialized region of cortical cytoplasm at the posterior pole of the egg, which programs them to become the future germline. These nuclei are partially surrounded in membrane in especially prominent protruding cytoplasmic buds [6]. These buds are pinched off as separate cells in conjunction with their tenth mitosis. The majority of nuclei form a well-spaced array near the surface of the embryo called the blastoderm. Here, they continue rapid mitotic cycles, now in “metasynchrony” so that nuclei toward the poles of the embryo are slightly more advanced and a “wave” of mitotic entry can be seen to spread from the poles to the center of the embryo. For four cycles (10–13) these blastoderm nuclei slow incrementally and progressively so that cycle 13 is 21 min long. Although there are preparatory events leading to the MBT and an ensuing progression of “MBT” events, the completion of mitosis 13 and entry into cycle 14 is generally taken as the time marking the MBT (see BOX 2). All of the events through the completion of mitosis 13 can occur in the absence of transcription. Transcription levels are low during these early cell cycles, nonetheless a cascade of expression of important regulatory genes sets the stage for sex determination and morphogenesis [2, 7, 8].
Mitosis 13 and transition to cycle 14 is usually taken as the time of the MBT [2, 9, 10]. This is somewhat of an oversimplification since the transition is a progressive process with aspects starting before cycle 14 and others continuing beyond cycle 14 [11, 12]. Nonetheless, cell cycle 14 is marked by major changes (see BOX 2). Importantly, the cell cycle is greatly prolonged, a G2 phase is introduced, and cell membrane ingresses to surround each cortical nucleus. The resulting cellular monolayer begins the morphogenetic movements that shape the embryo during gastrulation (Figure 1). Transcription is greatly increased and cycle 14 is also a time at which many but certainly not all maternally provided gene products are eliminated [13, 14].
BOX 2. Key transformations at the MBT.
At the end of cycle 13 the embryo has entered the MBT. The approximately 8000 nuclei exit mitosis 13 and enter S phase 14, which lasts at least 50 minutes, roughly 40 min longer than the previous S phase. This abrupt extension of S phase occurs as a result of the delayed replication time of heterochromatic regions of the genome. This extension of S phase plays roles in a second change in the cell cycle, the introduction of a G2. Downregulation of the mitotic activator Cdc25 occurs during this S phase, and S phase, apparently acting via checkpoint pathways, restrains the cell cycle until this downregulation has taken effect. If S phase is experimentally bypassed or shortened, the embryo undergoes another rapid and synchronous division that lacks a G2 [23, 24]. If the S phase checkpoint is compromised by mutation, the embryo similarly enters a premature synchronous mitosis [25, 26]. Downregulation of Cdc25 activity involves elimination of the maternal RNAs from two genes, string and twine, as well as abrupt destruction of Twine protein in cycle 14. The elimination of Twine protein plays a critical role during the onset of cell cycle slowing specifically in cycle 14 [24].
The MBT represents a gene expression revolution for the embryo. During cycle 13/14, many zygotic genes become transcribed for the first time, and many maternal products are selectively destroyed [13, 14]. Many other major developmental processes are initiated in cycle 14, the first relatively long cycle. The plasma membrane invaginates to surround individual nuclei in a process termed cellularization that takes about an hour to complete. Cellularization fully encloses the nuclei shortly after the cells enter G2, at which point, dramatic cell movements and epithelial folding reshape the embryo as gastrulation begins. Only then do the newly formed cycle 14 cells enter mitosis, and they do so in a patterned manner that depends on the transcription of the zygotic gene string. This string dependent mitosis is the first division in the embryo requiring a zygotic gene product [27]. Mitoses 14, 15 and 16 are each induced by pulses of string gene expression [12]. The still missing G1 phase of the cell cycle is not introduced until cycle 17.
Cell cycles, particularly mitosis and cytokinesis, can be very disruptive. Indeed, induction of premature mitoses interferes with cellular events during gastrulation and leads to abortion of ongoing nascent transcripts [28], a setback that has a negative effect on the expression of long transcripts. Thus, the addition of more time to interphase in cycle 14 is crucial to numerous MBT and gastrulation processes, including increased gene expression, cellularization, cell shape changes, cell movements, and tissue folding. For example, experimental manipulations or mutations that cause a premature mitosis 14 disrupt ongoing cellularization and ventral furrow formation [24].
Timing Early Development
Descriptions of the MBT in frog from 34 years ago established a framework that still influences ideas about its timing during early embryogenesis [15, 16]. The MBT was initially argued to be a single transition triggered at a unique time. This highlighted the question—what is the timer? Early cycles increase the number of nuclei without increasing the cytoplasm, hence exponentially increasing the nuclear to cytoplasmic ratio (N/C), a potential temporal signal. Manipulations of N/C showed that this parameter has a major impact on MBT timing [10, 11, 17–19]. Ideas about embryonic timing of the MBT have been dominated by a suggestion that the increasing DNA might titrate an unknown maternally provided component whose depletion triggers the transition.
It has, however, long been clear that modeling early embryonic timing as a single transition is an inaccurate presentation of early embryogenesis. Embryogenesis is accurately stereotyped in time and space. Though dramatic morphogenetic events are deferred until after the MBT, the early stages still feature numerous developmental events and these follow a precise schedule (see Box 1 for description). In addition to the cell biological events, many molecular changes mark a temporally complex and precisely coordinated progression of early embryonic development [8, 13, 14, 20–22]. These observations show that time is incrementally differentiated throughout early stages, and that an alarm mechanism that times a single transition would not provide a sufficient explanation for the temporal control of early embryogenesis. Apparently, there is a more sophisticated clock or the progression itself encrypts temporal information.
Justifying a Focus on Cell Cycle Slowing at the MBT
The early descriptions of the MBT suggested that it was a single transition at which multiple things change—the cell cycle slows, transcription is activated and the embryo transits from maternal to zygotic control. Beyond the fact that all these events occur over a short period of development, the justification for aggregating these events as a single transition was based on the idea that they are similarly regulated by the rising N/C. However, as we look with more temporal resolution, the signature events of the MBT are not coincident but progressive and influenced by a succession of inputs. For example, while there is an increase of transcription at the MBT, a cascade of transcriptional regulation occurs prior to the MBT [14]. Thus, transcription per se does not await the MBT [11, 29, 30]. Furthermore, the influence of N/C ratio on the timing of expression of specific genes is very heterogeneous: some show marked changes in response to alteration of nuclear DNA content while others do not, and those that are sensitive to N/C ratio do not turn on expression as a coherent group at one time, but activate according to individual schedules during the progressive changes that mark the MBT [10, 31, 32]. Thus, while changes such as increased phosphorylation of the C-terminal domain of RNA polymerase [33] suggest changes in transcriptional machinery at the MBT, because regulation is complex with individual genes differing in timing of activation and response to change in N/C, activation of zygotic gene expression should not be viewed as a coherent switch.
In Drosophila, the development of haploid embryos revealed the consequence of halving the amount of DNA per nucleus [11]. The outcome is often summarized saying that the haploid embryos undergo fifteen cell cycles before cellularization instead of the normal fourteen—a statement suggesting clear support that N/C controls the MBT. However, the cited paper describes development carefully and reported that haploid embryos begin to cellularize in cycle 14, as do normal diploid embryos. This promptly initiated cellularization is interrupted by an extra syncytial mitosis in the haploid embryos so that it is only completed in the next cell cycle. Thus, the lower N/C of haploid embryos did not delay onset of cellularization but resulted in an additional rapid cell cycle. This seminal paper also pointed out that transcription was not delayed in parallel with deferral of cell cycle slowing. From this work, we suggest that N/C does regulate cell cycle slowing, but its impact on other MBT events might be secondary or at least complex. Indeed, although the simple interpretation tends to be retained, recent data from genome wide studies of the onset of transcription support the suggested complexity of transcriptional activation [10, 14, 17, 31].
Other experimental manipulations uncouple some MBT events from N/C. When Cdk1 was prematurely downregulated in cycles 11, 12 or 13 by injection of dsRNA targeting all three mitotic cyclins, the mitotic cycle arrested. Nonetheless, the embryos went on to cellularize [29]. Thus, when Cdk1 was downregulated, everything needed for this post-MBT event occurred without reaching the N/C ratio of cycle 14 embryo. Reciprocally, embryos lacking two cell cycle checkpoint functions, Chk1/Grapes and Chk2/Loki, fail to slow down their cell cycle in cycle 14, but they nonetheless activate the transcriptional program characteristic of the post-MBT embryo [34]. Given the lack of concordance in the regulation of different processes upon perturbation, they are not obligatorily co-regulated. Apparently, their normal coordination relies on coupling via secondary regulation rather than induction by a common process. In this context, it might prove profitable to first focus on the regulation of one MBT event and then consider how coordination is achieved.
S phase duration as a timer
Usually the cell cycle is timed at transitions from G1 into S phase or G2 into mitosis. However, in the absence of gap phases, the progressive slowing of cell cycles 10–13 must have a different basis. The key factor is S phase duration. While we are only beginning to understand how it is regulated, S phase duration changes about 100 fold during development and about 15 fold between the early rapid cycles and the first post-MBT S phase (S phase 14) [35–38]. The slowing arises from a feature of S phase that is not widely appreciated. In normal cell cycles, different regions of the genome replicate at different times, and this sequential replication takes a long time. The deferred replication of some sequences is called late replication [39], although the schedule is more elaborate than a simple division into early and late. Various heterochromatic sequences typically exhibit different degrees of deferred replication [36, 40]. However, like almost everything else, this aspect of normal biology is not followed in the early cycles, where many features of heterochromatin including late replication are not evident [41]. In early cycles, replication starts synchronously at many closely spaced origins to rapidly duplicate the entire genome [35]. Incremental slowing of the cycle is due to gradual introduction of delays in the initiation of replication of various blocks of satellite sequence, which are usually constitutively heterochromatic [36, 40].
Mutant phenotypes provided the initial evidence for the importance of S phase duration [25, 26]. Ongoing replication activates a checkpoint that prevents progression to mitosis. In normal cycles that have a temporal leeway between S phase and mitosis, the G2 phase, this S phase checkpoint is dispensable unless replication is disrupted [42]. However, lacking a G2, the early embryonic cycles rely on this checkpoint mechanism to coordinate entry into mitosis with the gradually extending S phase. In checkpoint mutants, cells of the blastoderm embryo enter mitosis prematurely, and by cycle 13, an unmanageable catastrophe occurs as cells try to separate incompletely duplicated chromosomes at mitosis. This shows that the checkpoint is used during these cycles to couple overall cell cycle progress to the time it takes to replicate the genome.
In a direct test of its importance, S phase was deleted. Deleting S phase is different than blocking DNA replication. A block to replication induces the checkpoint and delays the cycle [43], while deletion of S phase fails to provoke an alarm [23]. To delete S phase, geminin, an inhibitor of the licensing process that prepares origins of replication for a round of replication, was injected into embryos. In the cycle following geminin injection, origins lack the factors that trigger initiation of replication, and they also fail to signal to the cell that there is anything that is supposed to be replicated. Without sensing a problem, the cell cycle progresses to mitosis as soon as it is ready, and it executes a peculiar mitosis with unreplicated chromosomes. When S phase is deleted in this way, pre-MBT embryos enter mitosis prematurely, while post-MBT embryos enter mitosis with normal developmentally programed timing [23]. This finding shows a change in the mechanism pacing cell cycle progress at the MBT: prior to the MBT, completing S phase is rate limiting, while afterward new types of temporal control limit cell cycle progress.
Following incremental changes leading up to the MBT, a major jump in S phase duration (to about 50 min) in cell cycle 14 is the first step toward normalizing the cell cycle at the MBT. The importance of this extended S phase to this transition was demonstrated by deleting or shortening it—treatments that led to an extra rapid synchronous cycle [23, 24]. Apparently, lengthening of S phase 14 provides a period of time during which cell cycle control is remodeled to introduce a G2 and the first zygotic control of cell cycle progression [2, 44].
Down regulation of the mitotic kinase is a leading event in the MBT
Befitting its dedication to amplification of the number of nuclei, the early embryo has abundant maternal supplies of RNA and protein for the components activating mitotic progression: Cyclins, Cdk1, and two isoforms of the Cdk1 activating phosphatase, Cdc25, encoded by the twine and string genes [45–47]. Indeed, these are so abundant that one continuing puzzle about the early cycles is how they exit mitosis and sustain an interphase long enough to replicate their DNA (Box 3). But they do, and in each successive blastoderm mitotic cycle the activators of Cdk1 are progressively reduced as if in preparation for the MBT—mitotic destruction of Cyclins becomes more thorough [45], Cdc25/String phosphatase virtually disappears [46], and there is evidence for increasing activation of the checkpoint pathway that acts through a kinase cascade to inhibit Cdk1 by inhibitory phosphorylation [48–50]. These changes, though not completely effective in inhibiting Cyclin:Cdk1, appear to set the stage for the MBT (Figure 2 and Box 3).
BOX 3. Bringing Cdk1 under control at the MBT.
The activity of the key cell cycle kinase, Cdk1, is famously regulated by association with its Cyclin subunit whose mitotic destruction triggers exit from mitosis. However, because Cyclins re-accumulate in interphase, additional regulation is needed (Figure 2). In most cell cycles, as new Cyclin:Cdk complexes are formed in interphase, they are inactivated by inhibitory phosphorylation (T14, Y15) by the Wee-type kinases. At the propitious moment, switch-like Cdc25-mediated dephosphorylation activates preformed Cyclin:Cdk complexes to trigger mitosis [53]. However, Cdc25 is abundant in pre-MBT embryos, and little inhibitory phosphate accumulates on Cdk1. Other factors seem to sustain interphase: loss of an Cdk1 activating phosphate (T161) [45], exclusion of Cyclin:Cdk1 from the nucleus [54, 55], and reversal of Cdk1 actions by the PP2A:B55 phosphatase which is selectively active in interphase [56].
As embryos approach the MBT, the high interphase activity of Cdk1 declines in coordination with N/C and in conjunction with changes in Cdk1 regulators. Cyclins A and B levels first exhibit detectable though slight oscillation in cycle 8. Increasing anaphase declines in these Cyclins suggest a progressive increase in APC/C-mediated degradation in successive mitoses [45]. Since it is the Cyclin associated with mitotic spindles that is degraded [55], the increase in nuclear number might drive increasing destruction. Starting at about cycle 11, Chk1/Grapes increasingly delays mitosis as if this checkpoint kinase is increasingly active [48, 50]. Although not directly measured in Drosophila, increasing DNA in a Xenopus mitotic extract increases Chk1 activation, and Chk1 activity emerges as frog embryos approach the MBT [57, 58]. The checkpoint activator, ATR, is itself activated by DNA damage, which might increase with the total DNA being replicated [59]. Finally, Cdc25/String level peaks at cycle 10 and then declines to low levels during cycles 11 to 13, a decrease that sensitizes the embryo to later loss of Cdc25/Twine (BOX4).
Genetic manipulations of maternally contributed Cdk1 regulators show their importance to MBT timing. A reduced dose of maternal Cdc25 resulted in premature MBT, while an increased dose of Cdc25/Twine delayed cellularization to cycle 15 in some embryos [46]. Similarly, some embryos with reduced Cyclin cellularize a cell cycle early [45]. Finally, embryos lacking Chk1 function fail to properly slow blastoderm cell cycles or execute the MBT [25, 26]. Thus, all three of these Cyclin:Cdk1 regulators influence MBT timing. If they act through Cdk1, manipulations in one regulator might compensate for defects in another. Indeed, reduction in Cyclin suppresses checkpoint mutants [60, 61].
The blastoderm reductions in Cdk1 activity are gradual, giving the appearance of progressive titration. This is followed by a more abrupt change in early cycle 14 when Cdc25/Twine is destabilized [47, 51]. If this decline of Cdc25/Twine is overridden by injection of mRNA, S phase is shortened and G2 eliminated. Thus, MBT requires decline of Cdc25/Twine [24]. Other events in cycle 14 appear to synergize with Twine reduction to suppress Cdk1, including the zygotic expression of the Cdk inhibitor, Fruhstart [46, 52].
Figure 2. Regulatory processes impinging on Cdk1 kinase activity.
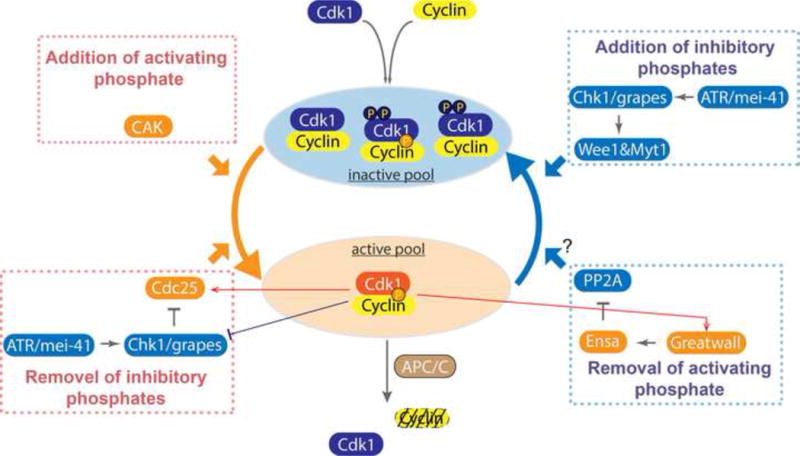
The active form of Cdk1 (in amber oval) has a Cyclin partner, the activating phosphate on the T161 of its T-loop, and is not modified at the inhibitory sites T14 and Y15 whose phosphorylation occludes ATP from the catalytic center. Production of this active form is promoted by the production of Cyclin, the addition of the activating phosphate by Cyclin:Cdk Activating Kinase (CAK), and the Cdc25 dual specificity phosphatase that removes the inhibitory phosphates. Its activity is suppressed by the Wee1 and Myt1 kinases that add inhibitory phosphates, and by destruction of its Cyclin partner in an APC/C stimulated reaction at mitotic exit. Additionally, other important regulators govern the activity of these primary regulators. Protein Phosphatase 2A:B55 (PP2A) counters the action of Cdk1 by removing phosphates from its substrates, and we hypothesis that this phosphatase also removes the activating phosphate from Cdk1 to downregulate its activity. Additionally, the checkpoint kinase Chk1 modulates the activity of the Wee1 kinase and Cdc25 phosphatase to indirectly inhibit activation of Cyclin:Cdk1.
Manipulations of the gene dose of Cdk1 activators suggest that levels of Cdc25 and Cyclins influence the timing of the MBT [24, 45, 46]. The dose of maternal Twine has a particularly strong effect (Box 3). Twine protein is abruptly destabilized a few minutes into cycle 14 and the Twine RNA is degraded shortly later [46, 47, 51]. If downregulation of Twine is experimentally overridden, embryos undergo a short S phase 14 followed by an additional syncytial mitosis. Reciprocally, experimental downregulation of Cdk1 in early cycles prolongs S phase, suggesting that this activity is responsible for the unusual speed of DNA replication in early S phases. Thus, the so-called ‘mitotic kinase’ has an S phase accelerating function, and downregulation of its activator at the MBT is required for the transition to a longer S phase [24, 47].
There appears to be different phases in the downregulation of Cdk1—gradual diminishing Cdk1 activity sets the stage for an abrupt suppression of Cdk1 upon Twine destruction, an event that is reinforced by zygotic expression of a Cdk1 inhibitor, Fruhstart [52]. A variety of genetic and experimental manipulations support the conclusion that downregulation of this central cell cycle kinase underlies the slowing of the cell cycle at the MBT (Box 3).
Cell cycle slowing following the MBT
Upon elimination of the maternally provided activator of mitosis, Cdc25 phosphatase, two inhibitory kinases, Wee1 and Myt1, stifle the activity of Cyclin:Cdk1 and stall cell cycle progression after S phase 14, creating the G2 [44, 45, 49, 62]. Cells only re-enter mitosis when triggered by a transcriptional input giving new expression of Cdc25/String [27]. This expression occurs in temporally-regulated pulses whose timing defines the spatially programmed schedule of mitoses 14 [12, 27, 63]. This is the first transcriptional requirement for cell cycle progression during development (Figure 1), and it provides a paradigm for the maternal to zygotic transition (MZT), when control of a process switches from maternal to zygotic regulation.
While the MZT is often discussed as an embryo wide event, the schedule and steps leading to loss of maternal gene function vary from gene to gene. Hence different processes and different cell cycle events undergo an MZT at different times. The complexity of the MZT is well illustrated by the loss of the maternal contributions of different cell cycle regulators (BOX 4).
BOX 4. How the cell cycle transits from maternal to zygotic control.
Many maternally supplied mRNAs are eliminated near the time of the MBT but with timing that varies from gene to gene [13]. The consequence of mRNA loss awaits decay of the protein, and even then will have no immediate relevance if the gene product is not needed at the time of its disappearance. Finally, replacement by zygotic expression can mask loss of maternal function [10]. Several maternal RNAs encoding cell cycle regulators disappear at the MBT with different consequences exemplifying these various factors.
Decay of Cdc25 activity
Early mitotic cycles proceed independently of zygotic gene expression. Cycle 14 is the first cycle to depend on gene expression, and the only zygotic factor needed is the Cdc25 phosphatase. The disappearance of maternal Cdc25 (both String and Twine) creates this zygotic dependence and underlies the switch to zygotic dependence (MZT) for control of the cell cycle. The string RNA disappears within the first few minutes of cell cycle 14. The association of this disappearance with the time of the MBT attracted early attention [68].
However, the String protein declines earlier and is extremely low at the time of the MBT [45]. Furthermore, early knockdown of string RNA by RNAi does not influence cell cycle progression [47]. Thus, inactivation of maternally provided String is required, but the abrupt destruction of its RNA early in cycle 14 is less crucial than once thought.
The early decline in Cdc25/String has little immediate consequence because Cdc25/Twine remains [47]. Twine mRNA is also degraded in cell cycle 14, but after the MBT, too late to be the key factor in the prolongation of S phase 14 [46]. Furthermore, simultaneous RNAi knockdown of string and twine RNAs prior to MBT neither substantially reduced Twine protein, nor advanced cell cycle slowing [47], suggesting that Twine protein is relatively stable during early divisions, and that the abrupt destabilization of Twine in early cycle 14 is the event coordinated with the prolongation of the cell cycle [47, 51].
Decay of mitotic Cyclins
Abundant maternal supplies of RNA sustain a dynamic pool of the three mitotic Cyclins, Cyclin A, Cyclin B, and Cyclin B3. Early knockdown of these RNAs rapidly arrests syncytial cycles in an interphase state [29]. Maternal Cyclin RNAs disappear in cell cycle 14, but zygotic expression appears to immediately replace these RNAs in all cells of the embryo, and this expression does not provide spatial and temporal programming information [69, 70].
Installation of S phase Cyclin Oscillations
Cyclin E triggers G1 cells to progress into S phase. It is ubiquitously present until just before the appearance of the G1 phase following mitosis 16. While maternal Cyclin E mRNA is lost during cell cycle 14, it is replaced by zygotic expression [71]. However, there appears to be no immediate consequence of its disappearance as embryos lacking a zygotic copy of cyclin E progress to G1 of cell cycle 17 before exhibiting a defect. While persistence of the protein might explain the delayed phenotype, antibody staining suggests that Cyclin E declines rapidly [72], and a zygotic cyclin E phenotype was observed in cycle 14 upon inhibition of other Cyclins [73]. Thus, it seems likely that Cyclin E is dispensable during the early cell cycles of Drosophila (Sprenger et al., 1997), and that appearance of a phenotype in cycle 17 is the result of other changes in cell cycle regulators that introduce a G1 (see text) and simultaneously create a context in which Cyclin E is indispensable.
After three G2 regulated cell cycles (14, 15 and 16), cells enter the first G1 phase after mitosis 16 (Figure 1). Preparations for the onset of G1 begin in the earlier cycles. In cycle 15, a DNA-replication coupled destruction mechanism downregulates E2F1, a transcriptional factor that promotes S phase [64]. In cycle 16, a key S phase promoting Cyclin—Cyclin E—is downregulated, and two important inhibitors of S phase, the Cyclin E/Cdk2 inhibitor Dacapo and the APC/C activator Cdh1/Fizzy-related, accumulate in cell cycle 16 [65–67]. Consequently, following mitosis 16, cells enter interphase 17 without the wherewithal to immediately begin S phase and arrest in G1. Thus, like the earlier introduction of the G2 phase, introduction of G1 involves several changes that retool operation of central cell cycle mechanisms. Together, multiple regulatory changes contribute to slowing of the early embryonic cycles and these occur in a stereotyped developmental progression over time.
Cell cycle slowing at the MBT and the N/C
As described above, cell cycle slowing at the MBT is coupled with downregulation of Cyclin:Cdk1 and a resulting extension of S phase due to onset of late replication. As outlined above and detailed in Box 3, a progression of changes in Cdk1 regulators impinge on this downregulation. Since the destruction of Twine is delayed until cycle 15 in a haploid embryo, one or more of the steps leading to this destruction is apparently coupled to N/C and arguments have been advanced for more than one possible influence of nuclear density.
The progressive increase in mitotic Cyclin destruction, which occurs in association with spindle structures, suggests that increasing nuclei promotes increasing Cyclin destruction. A small fraction of the embryos from mothers that are heterozygous for both cyclin A and cyclin B mutations undergo MBT a cycle early [45]. This dose dependence shows that MBT timing is sensitive to Cyclin levels, suggesting that the increasing decline of Cyclins during the blastoderm cycles impacts the timing of MBT.
DNA dependent activation of the S phase checkpoint also appears to have timing inputs. The checkpoint activating kinase, ATR/Mei-41, is activated by DNA—particularly DNA with interruptions coated by single stranded DNA binding protein [59]. In Xenopus Chk1 kinase activity increases in the final cycles before the MBT, and addition of increasing DNA levels to a mitotic extract system promoted checkpoint activation [58]. Recent work in Drosophila described binding of RNA polymerase to DNA in cycle 13, and suggested a model in which these transcription complexes interfere with replication forks and activate the checkpoint [74]. Although more complex than simply assessing the N/C ratio, the proposed model accounts for several subtle features of DNA dose effects on cell cycle slowing.
The various observations suggest that a sequence of regulatory changes synergizes to reduce Cdk1 kinase activity and slow the cell cycle at the MBT (Figure 2). Cyclin destruction and S phase checkpoint activation might couple the process to N/C, and destruction of Twine protein in cycle 14 seems to provide the coup de grace that allows accumulation of inhibitory phosphorylation on Cdk1 to fully inactivate it in cycle 14. Finally, destruction of Twine RNA and expression of the Cdk1 inhibitor Fruhstart backup and reinforce the transition.
Alternative proposal for triggering the MBT
It was long ago proposed that increasing DNA would titrate required cell cycle regulators, particularly DNA replication factors, to slow the cell cycle at the MBT. Numerous candidates for titration have been advanced. These include a pool of histone proteins that is largely consumed about the time of the MBT [75, 76], and a pool of deoxynucleotides that is similarly consumed as the embryo approaches cycle 13 (our unpublished observation). While titration of such components might make a contribution, for example by increasing replicative stress and promoting activation of the S phase checkpoint, the number of rapid early cell cycles in Drosophila does not appear to be strictly limited. Treatments such as inhibition of transcription, and experimental supplementation with Twine activity result in an additional rapid cell cycle [24, 36]. If titration is involved, we suggest that it acts indirectly to promote the decline of Cdk1, as might occur if DNA dependent processes activate the S phase checkpoint.
An interesting recent paper reported that supplementing a Xenopus embryo with four replication proteins is sufficient to drive additional rapid cycles [77]. While this report advocated these replication proteins as the components titrated to trigger the MBT, we suggest an alternative interpretation more congruent with findings in Drosophila. These four proteins are activators of the pre-replication complex (pre-RC) that assembles at origin sequences. Cdk1 and DDK kinases trigger this activation step. Overexpression of these replication proteins promotes this activation step, and could bypass its normal regulation [78, 79]. We suggest that the reported finding represents such a bypass and that it identifies the regulated step—a step that is promoted by Cyclin:Cdk1 activity as suggested by work in Drosophila.
The cell cycle focus we have adopted in this review does not delve into the events that control the major increase in transcription that occurs at the MBT or the events governing the elimination of maternal functions [13, 14, 17, 80, 81]. We do however, expect that multiple levels of interactions will interconnect and coordinate these different events as the embryo undergoes its massive transformation and refocuses its efforts from producing lots of cells to directing the development fates of those cells.
Concluding remarks
Autonomous development of an egg into a functional organism places extreme demands on the capabilities of a single cell. These demands distort the biology of early embryos, which is devoted to the subdivision of their massive cytoplasm into multiple cells that provide the fodder for morphogenesis. The speed of early development and dramatic changes have led to a notion that an extraordinary retooling of biological regulation is made at a single transition, the MBT, and activation of transcription is often viewed as the focus of this this transition. We advocate a different view. Cell cycle regulation is retooled to achieve transcriptional independence in the early embryo in order to drive the rapid exponential increase in cells. The resulting exponential expansion of the number of nuclei and amount of DNA increases transcriptional capabilities by thousands of fold, ultimately allowing zygotic gene expression to take on its commanding role in the direction of subsequent embryogenesis. The early embryo has an enormous stockpile of maternally contributed cell cycle regulators. Mitotic regulators have a profound impact on normal cells as they drive restructuring of the nucleus, an interruption in transcription, and repurposing of the cytoskeleton to promote mitosis. It should not be surprising that the high levels of these cell cycle regulators might also have a dominating influence on the biology of the early embryo. Since the central event of the MBT is slowing of the cell cycle, we suspect that this is also the central regulatory event in the MBT. Indeed, a progressive program down regulates the dominating activity of cyclin:Cdk1 and the timing of the MBT is influenced by modulating steps that promote this down regulation. Furthermore, recent work has shown that cyclin:Cdk1, which is viewed as the mitotic kinase, also dramatically accelerates S phase by promoting the firing of otherwise late replicating origins: hence, its down regulation can account for the observed changes in all parts of the cell cycle. This cell cycle slowing is integrated with a rapid but progressive program in which post-transcriptional mechanisms of regulation are replaced by regulated zygotic transcription. It appears that the increasing number of nuclei feeds back to promote the down regulation of the drivers of the cell cycle as well as to increase the transcriptional capacity, which, in turn, feeds back to slow the cell cycle by expressing inhibitors of cell cycle activators. The apparent switch-like properties of the MBT are likely to result from these large scale feedback interactions that reinforce the initial changes, which we suggest down regulate maternally contributed cell cycle drivers in processes coupled to the increase in the number of nuclei. While we advocate this cell-cycle-centric position as the “motivator” of the MBT, we suggest that dissection of the process as a sophisticated purposeful developmental progression with many interacting components will be more productive than approaching it as a single abrupt transformation of biological regulation (See Outstanding Questions).
Highlights.
Inhibitors of Cyclin:Cdk1 rise and activators fall to slow embryonic cell cycles at the MBT
Downregulation of Cyclin:Cdk1 first prolongs S phase, then introduces a G2 phase
Following the MBT, further changes in cell cycle regulators introduce a G1 phase
Outstanding questions.
What is the mechanism regulating String degradation in the blastoderm cycles?
What is controlling the abrupt Twine destruction in early cycle 14?
How do other MBT inputs influence the core cell cycle machinery?
How does the gradual normalization of embryonic cell cycles interact with other developmental events, such as cell fate determination and pattern formation?
Trends Box.
Developmental constraints force the early egg to adopt an unusual biology.
Normal biological controls are introduced in a rapid but progressive sequence that we exemplify by changes in key cell cycle regulators that slow and regularize the cell cycle.
This succession of regulatory changes contrasts to the usual depiction of the rapid transformation prior to gastrulation as a monolithic switch called the MBT.
While there are some particularly decisive steps the entire progression is temporally stereotyped.
Thus the MBT, while rapid and transformative, should be considered as a developmental process that engages much of the biology of embryo.
Progressive changes in Cdk1, an influential cell cycle kinase, coordinate much of this developmental program.
Acknowledgments
We thank all the members of the O’Farrell lab for the inspiring discussions. This project has been supported by National Institutes of Health grant GM037193 (to P.H.O).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement: None of the authors have conflicts of interest with regard to this report.
References
- 1.O’Farrell PH. Growing an Embryo from a Single Cell: A Hurdle in Animal Life. Cold Spring Harb Perspect Biol. 2015;7(11) doi: 10.1101/cshperspect.a019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrell JA, O’Farrell PH. From egg to gastrula: how the cell cycle is remodeled during the Drosophila mid-blastula transition. Annu Rev Genet. 2014;48:269–94. doi: 10.1146/annurev-genet-111212-133531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodland H. The translational control phase of early development. Biosci Rep. 1982;2(7):471–91. doi: 10.1007/BF01115246. [DOI] [PubMed] [Google Scholar]
- 4.O’Farrell PH, Stumpff J, Su TT. Embryonic cleavage cycles: how is a mouse like a fly? Curr Biol. 2004;14(1):R35–45. doi: 10.1016/j.cub.2003.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foe VE, O GM, Edgar BA. Mitosis and morphogenesis in the Drosophila embryo: Point and counterpoint. In: Arias MBaAM., editor. The Development of Drosophila melanogaster. Cold Spring Harbor Laboratory Press; 1993. pp. 149–300. [Google Scholar]
- 6.Cinalli RM, Lehmann R. A spindle-independent cleavage pathway controls germ cell formation in Drosophila. Nat Cell Biol. 2013;15(7):839–45. doi: 10.1038/ncb2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott MP, O’Farrell PH. Spatial programming of gene expression in early Drosophila embryogenesis. Annu Rev Cell Biol. 1986;2:49–80. doi: 10.1146/annurev.cb.02.110186.000405. [DOI] [PubMed] [Google Scholar]
- 8.Salz HK, Erickson JW. Sex determination in Drosophila: The view from the top. Fly (Austin) 2010;4(1):60–70. doi: 10.4161/fly.4.1.11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bashirullah A, et al. Joint action of two RNA degradation pathways controls the timing of maternal transcript elimination at the midblastula transition in Drosophila melanogaster. EMBO J. 1999;18(9):2610–20. doi: 10.1093/emboj/18.9.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X, et al. Coupling of zygotic transcription to mitotic control at the Drosophila mid-blastula transition. Development. 2009;136(12):2101–10. doi: 10.1242/dev.034421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edgar BA, Kiehle CP, Schubiger G. Cell cycle control by the nucleo-cytoplasmic ratio in early Drosophila development. Cell. 1986;44(2):365–72. doi: 10.1016/0092-8674(86)90771-3. [DOI] [PubMed] [Google Scholar]
- 12.Edgar BA, O’Farrell PH. The three postblastoderm cell cycles of Drosophila embryogenesis are regulated in G2 by string. Cell. 1990;62(3):469–80. doi: 10.1016/0092-8674(90)90012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laver JD, et al. Regulation and Function of Maternal Gene Products During the Maternal-to-Zygotic Transition in Drosophila. Curr Top Dev Biol. 2015;113:43–84. doi: 10.1016/bs.ctdb.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Harrison MM, Eisen MB. Transcriptional Activation of the Zygotic Genome in Drosophila. Curr Top Dev Biol. 2015;113:85–112. doi: 10.1016/bs.ctdb.2015.07.028. [DOI] [PubMed] [Google Scholar]
- 15.Newport J, Kirschner M. A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell. 1982;30(3):675–86. doi: 10.1016/0092-8674(82)90272-0. [DOI] [PubMed] [Google Scholar]
- 16.Newport J, Kirschner M. A major developmental transition in early Xenopus embryos: II. Control of the onset of transcription. Cell. 1982;30(3):687–96. doi: 10.1016/0092-8674(82)90273-2. [DOI] [PubMed] [Google Scholar]
- 17.Blythe SA, Wieschaus EF. Coordinating Cell Cycle Remodeling with Transcriptional Activation at the Drosophila MBT. Curr Top Dev Biol. 2015;113:113–48. doi: 10.1016/bs.ctdb.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Lu X, Drocco J, Wieschaus EF. Cell cycle regulation via inter-nuclear communication during the early embryonic development of Drosophila melanogaster. Cell Cycle. 2010;9(14):2908–10. doi: 10.4161/cc.9.14.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erickson JW, Quintero JJ. Indirect effects of ploidy suggest X chromosome dose, not the X:A ratio, signals sex in Drosophila. PLoS Biol. 2007;5(12):e332. doi: 10.1371/journal.pbio.0050332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welte MA. As the fat flies: The dynamic lipid droplets of Drosophila embryos. Biochim Biophys Acta. 2015;1851(9):1156–85. doi: 10.1016/j.bbalip.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horard B, Loppin B. Histone storage and deposition in the early Drosophila embryo. Chromosoma. 2015;124(2):163–75. doi: 10.1007/s00412-014-0504-7. [DOI] [PubMed] [Google Scholar]
- 22.Johnstone O, Lasko P. Translational regulation and RNA localization in Drosophila oocytes and embryos. Annu Rev Genet. 2001;35:365–406. doi: 10.1146/annurev.genet.35.102401.090756. [DOI] [PubMed] [Google Scholar]
- 23.McCleland ML, Shermoen AW, O’Farrell PH. DNA replication times the cell cycle and contributes to the mid-blastula transition in Drosophila embryos. J Cell Biol. 2009;187(1):7–14. doi: 10.1083/jcb.200906191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farrell JA, et al. Embryonic onset of late replication requires Cdc25 down-regulation. Genes & Development. 2012;26(7):714–725. doi: 10.1101/gad.186429.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sibon OC, V, Stevenson A, Theurkauf WE. DNA-replication checkpoint control at the Drosophila midblastula transition. Nature. 1997;388(6637):93–7. doi: 10.1038/40439. [DOI] [PubMed] [Google Scholar]
- 26.Yu KR, Saint RB, Sullivan W. The Grapes checkpoint coordinates nuclear envelope breakdown and chromosome condensation. Nat Cell Biol. 2000;2(9):609–15. doi: 10.1038/35023555. [DOI] [PubMed] [Google Scholar]
- 27.Edgar BA, O’Farrell PH. Genetic control of cell division patterns in the Drosophila embryo. Cell. 1989;57(1):177–87. doi: 10.1016/0092-8674(89)90183-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shermoen AW, O’Farrell PH. Progression of the cell cycle through mitosis leads to abortion of nascent transcripts. Cell. 1991;67(2):303–10. doi: 10.1016/0092-8674(91)90182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCleland ML, O’Farrell PH. RNAi of mitotic cyclins in Drosophila uncouples the nuclear and centrosome cycle. Curr Biol. 2008;18(4):245–54. doi: 10.1016/j.cub.2008.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimelman D, Kirschner M, Scherson T. The events of the midblastula transition in Xenopus are regulated by changes in the cell cycle. Cell. 1987;48(3):399–407. doi: 10.1016/0092-8674(87)90191-7. [DOI] [PubMed] [Google Scholar]
- 31.Lott SE, et al. Noncanonical compensation of zygotic X transcription in early Drosophila melanogaster development revealed through single-embryo RNA-seq. PLoS Biol. 2011;9(2):e1000590. doi: 10.1371/journal.pbio.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collart C, et al. High-resolution analysis of gene activity during the Xenopus mid-blastula transition. Development. 2014;141(9):1927–39. doi: 10.1242/dev.102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seydoux G, Dunn MA. Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development. 1997;124(11):2191–201. doi: 10.1242/dev.124.11.2191. [DOI] [PubMed] [Google Scholar]
- 34.Takada S, et al. grp (chk1) replication-checkpoint mutations and DNA damage trigger a Chk2-dependent block at the Drosophila midblastula transition. Development. 2007;134(9):1737–44. doi: 10.1242/dev.02831. [DOI] [PubMed] [Google Scholar]
- 35.Blumenthal AB, Kriegstein HJ, Hogness DS. The units of DNA replication in Drosophila melanogaster chromosomes. Cold Spring Harb Symp Quant Biol. 1974;38:205–23. doi: 10.1101/sqb.1974.038.01.024. [DOI] [PubMed] [Google Scholar]
- 36.Shermoen AW, McCleland ML, O’Farrell PH. Developmental control of late replication and S phase length. Curr Biol. 2010;20(23):2067–77. doi: 10.1016/j.cub.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eaton ML, et al. Chromatin signatures of the Drosophila replication program. Genome Res. 2011;21(2):164–74. doi: 10.1101/gr.116038.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lubelsky Y, et al. DNA replication and transcription programs respond to the same chromatin cues. Genome Res. 2014;24(7):1102–14. doi: 10.1101/gr.160010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pope BD, Aparicio OM, Gilbert DM. SnapShot: Replication timing. Cell. 2013;152(6):1390–1390 e1. doi: 10.1016/j.cell.2013.02.038. [DOI] [PubMed] [Google Scholar]
- 40.Yuan K, Shermoen AW, O’Farrell PH. Illuminating DNA replication during Drosophila development using TALE-lights. Curr Biol. 2014;24(4):R144–5. doi: 10.1016/j.cub.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan K, O’Farrell PH. TALE-light imaging reveals maternally guided, H3K9me2/3-independent emergence of functional heterochromatin in Drosophila embryos. Genes Dev. 2016;30(5):579–93. doi: 10.1101/gad.272237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banga SS, Shenkar R, Boyd JB. Hypersensitivity of Drosophila mei-41 mutants to hydroxyurea is associated with reduced mitotic chromosome stability. Mutat Res. 1986;163(2):157–65. doi: 10.1016/0027-5107(86)90044-8. [DOI] [PubMed] [Google Scholar]
- 43.Fasulo B, et al. Chk1 and Wee1 kinases coordinate DNA replication, chromosome condensation, and anaphase entry. Mol Biol Cell. 2012;23(6):1047–57. doi: 10.1091/mbc.E11-10-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouldin CM, Kimelman D. Cdc25 and the importance of G2 control: insights from developmental biology. Cell Cycle. 2014;13(14):2165–71. doi: 10.4161/cc.29537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar BA, et al. Distinct molecular mechanism regulate cell cycle timing at successive stages of Drosophila embryogenesis. Genes Dev. 1994;8(4):440–52. doi: 10.1101/gad.8.4.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edgar BA, Datar SA. Zygotic degradation of two maternal Cdc25 mRNAs terminates Drosophila’s early cell cycle program. Genes Dev. 1996;10(15):1966–77. doi: 10.1101/gad.10.15.1966. [DOI] [PubMed] [Google Scholar]
- 47.Farrell JA, O’Farrell PH. Mechanism and regulation of Cdc25/Twine protein destruction in embryonic cell-cycle remodeling. Curr Biol. 2013;23(2):118–26. doi: 10.1016/j.cub.2012.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji JY, Squirrell JM, Schubiger G. Both cyclin B levels and DNA-replication checkpoint control the early embryonic mitoses in Drosophila. Development. 2004;131(2):401–11. doi: 10.1242/dev.00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stumpff J, et al. Drosophila Wee1 kinase regulates Cdk1 and mitotic entry during embryogenesis. Curr Biol. 2004;14(23):2143–8. doi: 10.1016/j.cub.2004.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crest J, et al. Onset of the DNA replication checkpoint in the early Drosophila embryo. Genetics. 2007;175(2):567–84. doi: 10.1534/genetics.106.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Talia S, et al. Posttranslational control of Cdc25 degradation terminates Drosophila’s early cell-cycle program. Curr Biol. 2013;23(2):127–32. doi: 10.1016/j.cub.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grosshans J, Muller HA, Wieschaus E. Control of cleavage cycles in Drosophila embryos by fruhstart. Dev Cell. 2003;5(2):285–94. doi: 10.1016/s1534-5807(03)00208-9. [DOI] [PubMed] [Google Scholar]
- 53.O’Farrell PH. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001;11(12):512–9. doi: 10.1016/s0962-8924(01)02142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Royou A, et al. Grapes(Chk1) prevents nuclear CDK1 activation by delaying cyclin B nuclear accumulation. J Cell Biol. 2008;183(1):63–75. doi: 10.1083/jcb.200801153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang J, Raff JW. The disappearance of cyclin B at the end of mitosis is regulated spatially in Drosophila cells. EMBO J. 1999;18(8):2184–95. doi: 10.1093/emboj/18.8.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glover DM. The overlooked greatwall: a new perspective on mitotic control. Open Biol. 2012;2(3):120023. doi: 10.1098/rsob.120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimuta K, et al. Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition. EMBO J. 2002;21(14):3694–703. doi: 10.1093/emboj/cdf357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peng A, Lewellyn AL, Maller JL. Undamaged DNA transmits and enhances DNA damage checkpoint signals in early embryos. Mol Cell Biol. 2007;27(19):6852–62. doi: 10.1128/MCB.00195-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hekmat-Nejad M, et al. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol. 2000;10(24):1565–73. doi: 10.1016/s0960-9822(00)00855-1. [DOI] [PubMed] [Google Scholar]
- 60.Sibon OC, et al. The Drosophila ATM homologue Mei-41 has an essential checkpoint function at the midblastula transition. Curr Biol. 1999;9(6):302–12. doi: 10.1016/s0960-9822(99)80138-9. [DOI] [PubMed] [Google Scholar]
- 61.Yuan K, Farrell JA, O’Farrell PH. Different cyclin types collaborate to reverse the S-phase checkpoint and permit prompt mitosis. Journal of Cell Biology. 2012;198(6):973–980. doi: 10.1083/jcb.201205007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Campbell SD, et al. Drosophila Wee1 kinase rescues fission yeast from mitotic catastrophe and phosphorylates Drosophila Cdc2 in vitro. Mol Biol Cell. 1995;6(10):1333–47. doi: 10.1091/mbc.6.10.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edgar BA, Lehman DA, O’Farrell PH. Transcriptional regulation of string (cdc25): a link between developmental programming and the cell cycle. Development. 1994;120(11):3131–43. doi: 10.1242/dev.120.11.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shibutani S, Swanhart LM, Duronio RJ. Rbf1-independent termination of E2f1-target gene expression during early Drosophila embryogenesis. Development. 2007;134(3):467–78. doi: 10.1242/dev.02738. [DOI] [PubMed] [Google Scholar]
- 65.de Nooij JC, Letendre MA, Hariharan IK. A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell. 1996;87(7):1237–47. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- 66.Lane ME, et al. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell. 1996;87(7):1225–35. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- 67.Sigrist SJ, Lehner CF. Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell. 1997;90(4):671–81. doi: 10.1016/s0092-8674(00)80528-0. [DOI] [PubMed] [Google Scholar]
- 68.O’Farrell PH, et al. Directing cell division during development. Science. 1989;246(4930):635–40. doi: 10.1126/science.2683080. [DOI] [PubMed] [Google Scholar]
- 69.Lehner CF, O’Farrell PH. Expression and function of Drosophila cyclin A during embryonic cell cycle progression. Cell. 1989;56(6):957–68. doi: 10.1016/0092-8674(89)90629-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lehner CF, O’Farrell PH. The roles of Drosophila cyclins A and B in mitotic control. Cell. 1990;61(3):535–47. doi: 10.1016/0092-8674(90)90535-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Richardson HE, et al. A Drosophila G1-specific cyclin E homolog exhibits different modes of expression during embryogenesis. Development. 1993;119(3):673–90. doi: 10.1242/dev.119.3.673. [DOI] [PubMed] [Google Scholar]
- 72.Knoblich JA, et al. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77(1):107–20. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 73.Vidwans SJ, et al. Sister chromatids fail to separate during an induced endoreplication cycle in Drosophila embryos. Curr Biol. 2002;12(10):829–33. doi: 10.1016/s0960-9822(02)00845-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blythe SA, Wieschaus EF. Zygotic genome activation triggers the DNA replication checkpoint at the midblastula transition. Cell. 2015;160(6):1169–81. doi: 10.1016/j.cell.2015.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gunesdogan U, Jackle H, Herzig A. Histone supply regulates S phase timing and cell cycle progression. Elife. 2014;3:e02443. doi: 10.7554/eLife.02443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Amodeo AA, et al. Histone titration against the genome sets the DNA-to-cytoplasm threshold for the Xenopus midblastula transition. Proc Natl Acad Sci U S A. 2015;112(10):E1086–95. doi: 10.1073/pnas.1413990112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collart C, et al. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science. 2013;341(6148):893–6. doi: 10.1126/science.1241530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanaka S, et al. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature. 2007;445(7125):328–32. doi: 10.1038/nature05465. [DOI] [PubMed] [Google Scholar]
- 79.Zegerman P, Diffley JF. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature. 2007;445(7125):281–5. doi: 10.1038/nature05432. [DOI] [PubMed] [Google Scholar]
- 80.Langley AR, et al. New insights into the maternal to zygotic transition. Development. 2014;141(20):3834–41. doi: 10.1242/dev.102368. [DOI] [PubMed] [Google Scholar]
- 81.Martinho RG, Guilgur LG, Prudencio P. How gene expression in fast-proliferating cells keeps pace. Bioessays. 2015;37(5):514–24. doi: 10.1002/bies.201400195. [DOI] [PubMed] [Google Scholar]