

Abstract
Background
Ischemia-reperfusion injury is a major source of morbidity and mortality after lung transplantation. We previously demonstrated a proinflammatory role of adenosine A2B receptor (A2BR) in lung ischemia-reperfusion injury. The current study tests the hypothesis that A2BR antagonism is protective to ischemic lungs after in vivo reperfusion or ex vivo lung perfusion (EVLP).
Methods
Mice underwent lung ischemia-reperfusion with/without administration of ATL802, a selective A2BR antagonist. A murine model of EVLP was also utilized to evaluate rehabilitation of donation after circulatory death (DCD) lungs. DCD lungs underwent ischemia, cold preservation and EVLP with Steen solution with/without ATL802. A549 human type 2 alveolar epithelial cells were exposed to hypoxia-reoxygenation (3hr/1hr) with/without ATL802 treatment. Cytokines were measured in bronchoalveolar lavage fluid and culture media by ELISA.
Results
After ischemia-reperfusion, ATL802 treatment significantly improved lung function (increased pulmonary compliance; reduced airway resistance and pulmonary artery pressure) and significantly attenuated proinflammatory cytokine production, neutrophil infiltration, vascular permeability and edema. ATL802 also significantly improved the function of DCD lungs after EVLP (increased compliance and reduced pulmonary artery pressure). After hypoxia-reoxygenation, A549 cells exhibited robust production of IL-8, a potent neutrophil chemokine, which was significantly attenuated by ATL802.
Conclusions
These results demonstrate that A2BR antagonism attenuates lung ischemia-reperfusion injury and augments reconditioning of DCD lungs by EVLP. The protective effects of ATL802 may involve targeting of A2BRs on alveolar epithelial cells to prevent IL-8 production. A2BR may be a novel therapeutic target for mitigating ischemia-reperfusion injury to increase the success of lung transplantation.
Keywords: Lung ischemia/reperfusion injury, lung transplantation, inflammatory mediators, inflammatory cells, cell signaling
Ischemia-reperfusion injury (IRI) after lung transplantation remains a significant source of morbidity and mortality [1]. Compared to other solid organs, outcomes for lung transplantation are the worst with a five year mortality of 55% [2]. Severe IRI leads to primary graft dysfunction (PGD), which results in acute graft failure and is a significant risk factor for the development of bronchiolitis obliterans syndrome and chronic graft failure; the major cause of mortality beyond one year of transplantation [3–5]. Advancements in understanding the mechanisms involved in lung IRI have revealed a complex inflammatory process involving activation and infiltration of innate immune cells, endothelial and epithelial cell injury, oxidative stress, and cytokine responses [6–9]. To date no therapeutic agent is available to prevent IRI after lung transplantation.
In addition to the existing challenges of IRI after transplantation, the number of patients needing transplantation continues to far exceed donor supply. New advances in lung preservation and utilization of marginal lungs, such as those from donation after circulatory death (DCD), have the potential to reduce this disparity [10]. The development of normothermic ex vivo lung perfusion (EVLP) has provided a unique platform for the evaluation and rehabilitation of marginal (e.g. DCD) donor lungs [11], which could potentially improve donor supply and transplant outcomes [12].
Adenosine is released during cellular stress and inflammation, such as IRI, and is generally considered to mediate anti-inflammatory responses. However, it is now known that pro- or anti-inflammatory actions of adenosine vary according to the tissue and cells involved, the adenosine receptors expressed, and the injury type [13]. Adenosine mediates its effects via four G-protein-coupled receptors (A1R, A2AR, A2BR and A3R), each of which exhibits a distinct pattern of tissue distribution, intracellular signaling and pharmacokinetics. All four adenosine receptors are expressed in lungs of mice [14] and humans [15], and we have demonstrated potent protective effects of A1R, A2AR and A3R agonists in models of lung IRI [16–19].
The role of A2BR is of particular interest given it has the lowest affinity for adenosine and has elevated expression after physiologic stress [20]. A2BR is highly expressed on alveolar type 2 epithelial cells [21], and whether A2BR plays pro- or anti-inflammatory roles in lung injury may depend on several factors such as specific injury conditions and relative contributions of tissue- versus bone marrow-derived cells. A2BRs on bone marrow-derived cells have been shown to evoke anti-inflammatory actions [22, 23], and A2BR activation has been shown to dampen inflammation and enhance barrier function in some models of acute lung injury [24, 25]. However, other studies have demonstrated a proinflammatory role for A2BR [26–28].
We have previously shown that A2BR deficient mice are protected from lung IRI, which appears to be mediated through A2BRs on resident pulmonary cells rather than bone marrow-derived cells [29]. These results suggest that A2BR is proinflammatory in the setting of lung IRI. Thus the current study tests the hypothesis that an A2BR antagonist will attenuate lung IRI and will augment EVLP-mediated rehabilitation of DCD lungs.
Material and Methods
Animals and study design
C57BL/6 wild-type mice (Jackson Laboratory, Bar Harbor, ME) of 8–12 weeks of age were utilized. Three groups of mice underwent sham surgery, left lung IR or left lung IR (n=4-9/group). ATL802 (Lewis and Clark Pharmaceuticals, Charlottesville, VA) was injected intraperitoneally (1mg/kg) five minutes prior to surgery. Using competition binding assays to recombinant mouse A1R, A2AR, A2BR and A3R, ATL802 was previously shown to be a highly potent (Ki = 8.6nM) and selective (>900 fold over other adenosine receptors) antagonist of murine A2BR [21]. A separate study involved three groups of murine DCD lungs that underwent warm ischemia, cold preservation and EVLP with or without 100 nM ATL802 (n=9/group). Doses of ATL802 utilized were based on prior study [21] and preliminary work. This study conformed to the Guide for the Care and Use of Laboratory Animals as published by the National Institutes of Health and was approved by the University of Virginia Institutional Animal Care and Use Committee.
Murine lung IRI model
An in vivo murine model of IRI was used as previously described [8]. Mice were anesthetized with inhalational isoflurane, and orotracheal intubation was performed for mechanical ventilation (Harvard Apparatus Co, South Natick, MA) at 120 strokes/min with room air. Stroke volume was set at 0.5ml, and peak inspiratory pressure limited to <20 cmH2O. Heparin (20 U/kg) was administered via the right external jugular vein, and the left pulmonary hilum was exposed with an anterolateral thoracotomy at the third intercostal space. A 6–0 Prolene suture was passed around the hilum, and suture ends were threaded through 5mm PE-60 tubing to permit hilar occlusion via tightening of the suture; securing it with a surgical clip. Animals were extubated and returned to their cage after receiving analgesia (0.2 mg/kg buprenorphine). After 1 hour of left lung ischemia, mice were reanesthetized, intubated, and the hilar occlusion was released to allow reperfusion. Mice were extubated and returned to their cage for a 2-hour reperfusion period after which pulmonary function was assessed. Sham surgery entailed all of the above steps except hilar occlusion.
Pulmonary function
Pulmonary function was assessed using an isolated, buffer-perfused lung system (Hugo Sachs Elektronik, March-Huggstetten, Germany) as previously described [9]. Mice were anesthetized with ketamine and xylazine. Animals were ventilated via tracheostomy with room air, 100 strokes/min and tidal volume of 7μl/g with a positive end-expiratory pressure of 2 cmH2O. Animals were exsanguinated with transection of the inferior vena cava. The pulmonary artery was cannulated through the right ventricle, and the left ventricle was vented with a small ventriculotomy at the apex of the heart. Lungs were perfused at a constant flow of 60 μl/g/min with KH buffer (335–340 mOsm/kg H2O) at 37°C. The isolated lungs were maintained on the system for a 10-minute equilibration period, after which hemodynamic and pulmonary parameters were recorded by the PULMODYN data acquisition system (Hugo Sachs Elektronik).
Ex vivo lung perfusion (EVLP) model
A murine model of ex vivo lung perfusion (EVLP) was utilized as previously described [30]. Mice (n=9/group) were anesthetized with inhalational isoflurane and euthanized by cervical dislocation followed by a 15-minute period of warm ischemia. The mediastinum was incised, and the left atrium was vented with atriotomy. Lungs were perfused in situ with 3ml cold Perfadex solution (XVIVO Perfusion Inc., Englewood CO) supplemented with THAM solution (XVIVO Perfusion Inc., Englewood CO). The chest was covered with ice, and lungs underwent cold static preservation for 60 minutes. Lungs were then placed on an isolated lung perfusion system (Hugo Sachs Elektronik, March-Huggstetten, Germany) and perfused with either Krebs-Henseleit (KH) buffer or Steen solution (XVIVO Perfusion Inc., Englewood, CO) at a constant rate of 60μl/g body weight/minute. Steen solution was supplemented with 10,000 IU heparin, 500mg cefazolin, and 500mg methylprednisolone per 1500ml to model clinical EVLP protocols. Perfusate was warmed in the circuit from 4°C to 37°C over approximately a 10 minute period. EVLP continued for 60 minutes. Lung function was measured during EVLP as described above, and values obtained after 60 minutes of EVLP are reported.
Bronchoalveolar lavage (BAL)
After pulmonary function measurements, the right lung was occluded and the left lung lavaged with 0.4ml normal saline. BAL fluid was centrifuged, and supernatant was stored at −80°C until further analysis.
Myeloperoxidase and cytokine measurements
Myeloperoxidase levels in BAL fluid were measured using a mouse ELISA kit (Hycult Biotech, Uden, Netherlands). Cytokine concentrations in BAL fluid were measured using a mouse cytokine multiplex ELISA panel (Bio-Rad Laboratories, Hercules, CA) as previously described [31].
Pulmonary vascular permeability
Using separate groups of animals (n=5/group), vascular permeability was estimated using the Evans blue dye extravasation technique as previously described [8]. Evans blue (20mg/kg, Sigma, St. Louis, MO) was injected retro-orbitally, and after 30 minutes pulmonary vasculature was perfused for 10 minutes with PBS to remove intravascular dye. Lungs were homogenized in PBS to extract the Evans blue and centrifuged. Absorption of Evans blue was measured in supernatant at 620nm and corrected for the presence of heme pigments: A620 (corrected) = A620 − (1.426 × A740 + 0.030). The concentration of Evans blue was determined according to a standard curve.
Immunohistochemistry
Using separate groups of animals, lungs were fixed in 10% formalin for 24 hours, placed in 70% ethanol and paraffin embedded. Immunostaining of lung sections was performed using rat anti-mouse neutrophil antibody (Ly-6B.2, AbD Serotec, Raleigh, NC) as described previously [7]. Alkaline phosphatase-conjugated anti-rat IgG (Sigma) was used as the secondary antibody. Signals were detected using Fast-Red (Sigma), and sections were counterstained with hematoxylin. A blinded investigator obtained counts of neutrophils per high-powered field. Five semi-standardized fields per lung were counted at 20X magnification, and the mean of these values established the final value per animal.
Cell culture and hypoxia-reoxygenation (HR) model
A549 cells (human alveolar type 2 epithelial cell line; ATCC, Manassas, VA) were grown in Dulbecco's Modified Eagle's Medium with 4.5g/L glucose, 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen Corporation, Carlsbad, CA) in a humidified incubator (37°C, 5% CO2). The A549 cell line, derived from a human alveolar cell carcinoma, has many properties of ATII cells [32] and was utilized because they are the most widely used in vitro model for type 2 pulmonary alveolar epithelial cells. For exposure to HR, cultures were placed in a hypoxic chamber (Billups-Rothenberg, Del Mar, CA) and purged with 95% N2 and 5% CO2 for 25 min to establish hypoxia as described previously [33]. The chamber was placed in a 37°C incubator for 3 hours. Reoxygenation was achieved by removing the cultures from the hypoxic chamber and placing them in a normoxic, humidified incubator (37°C, 5% CO2) for 1 hour. The partial percentage of O2 in the culture media immediately after 3 hours hypoxia was consistently found to be 5% versus 21% in normoxic cultures.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 6.0, and data are presented as the mean ± SEM. Analyses were performed using one-way ANOVA with post-hoc Tukey's multiple comparisons test. A p<0.05 was considered significant.
Results
Lung function after IR is improved by ATL802
Mice displayed significant pulmonary dysfunction after IR as indicated by increased airway resistance and pulmonary artery pressure as well as decreased pulmonary compliance (Figure 1). Pulmonary compliance was significantly decreased after IR vs. Sham (3.41±0.10 vs. 9.57±0.83 μl/cm H2O, p<0.0001), which was significantly increased by ATL802 (5.39±0.57 μl/cm H2O, p=0.022). Airway resistance was significantly increased after IR vs. Sham (2.04±0.17 vs. 1.23±0.08 cm H2O/μl/sec, p=0.009), which was significantly reduced by ATL802 (1.43±0.12 cm H2O/μl/sec, p=0.015). Pulmonary artery pressure was significantly increased after IR vs. Sham (11.24±0.65 vs. 5.55±0.15 cm H2O, p<0.0001), which was significantly reduced by ATL802 (7.01±0.54 cm H2O, p=0.0001).
Figure 1.

Lung function after IR is improved by ATL802. Mice exhibited significant lung dysfunction after IR, which was significantly attenuated by ATL802 treatment. Compared to untreated mice after IR, ATL802 treatment significantly increased pulmonary compliance and significantly decreased airway resistance and pulmonary artery pressure. *p<0.01 vs. Sham, #p<0.03 vs. IR, n=4-9/group.
ATL802 attenuated expression of proinflammatory cytokines after IR
Expression of KC (CXCL1), IL-6, IL-1β, MCP-1 (CCL2), MIP-1α (CCL3), and RANTES (CCL5) in BAL fluid were significantly induced after IR, and all were significantly attenuated by ATL802 treatment (Figure 2). KC expression increased after IR vs. Sham (6347±1628 vs. 1442±518 pg/ml, p=0.013) and was reduced by ATL802 (1579±395, p=0.011). IL-6 production increased after IR vs. Sham (3062±655 vs. 356±70 pg/ml, p=0.003) and was reduced by ATL802 (694±260, p=0.007). IL-1β expression increased after IR vs. Sham (141.8±11.8 vs. 71.0±18.4 pg/ml, p=0.0002) and was reduced by ATL802 (80.8±6.5, p=0.0003). MCP-1 increased after IR vs. Sham (608±246 vs. 32.4±8.3 pg/ml, p=0.045) and was reduced by ATL802 (40.7±13.3, p=0.048). MIP-1α increased after IR vs. Sham (358±39.4 vs. 155±42.0 pg/ml, p=0.013) and was reduced by ATL802 (152±36.8, p=0.012). RANTES production increased after IR vs. Sham (88.3±23.3 vs. 22.0±5.9 pg/ml, p=0.0002) and was reduced by ATL802 (27.0±5.6, p=0.0003).
Figure 2.

Cytokine production after IR is significantly attenuated by ATL802. IR resulted in significantly elevated levels of proinflammatory cytokines (KC, IL-6, IL-1β, MCP-1, MIP-1α and RANTES) in bronchoalveolar lavage fluid, which were all significantly attenuated by ATL802 treatment. *p<0.05 vs. Sham, #p<0.05 vs. IR, n=4–5/group.
Neutrophil infiltration and activation after IR is attenuated by ATL802
Robust infiltration of neutrophils occurred after IR; accompanied by significantly elevated myeloperoxidase levels (Figure 3). Neutrophils per high-powered field (HPF) increased after IR vs. Sham (190.0±38.1 vs. 64.7±10.1, p=0.001), which was significantly reduced by ATL802 (69.5±5.5, p=0.012). Myeloperoxidase, an enzyme abundantly present in neutrophil azurophilic granules, was measured as an indicator of neutrophil activation. Myeloperoxidase increased after IR vs. Sham (74.8±15.0 vs. 17.8±2.8 pg/ml, p=0.0008) and was significantly reduced by ATL802 (18.5±1.4, p=0.0013) (Figure 3).
Figure 3.

ATL802 attenuates infiltration and activation of neutrophils after IR. Lung sections were immunostained for neutrophils (red color), and representative images are shown on top (20x magnification). Quantification of neutrophils per high-powered field (HPF) demonstrated significant neutrophil infiltration in lungs after IR, which was significantly attenuated by ATL802 treatment (*p=0.001 vs. Sham, #p<0.012 vs. IR, n=4/group). In addition, myeloperoxidase levels in BAL fluid were significantly elevated after IR, which was significantly attenuated by ATL802 treatment (**p=0.0008 vs. Sham, ##p=0.0013 vs. IR, n=4–6/group).
Pulmonary vascular permeability after IR is reduced by ATL802
Vascular permeability as assessed by Evans blue dye extravasation significantly increased after IR vs. Sham (27.63±2.67 vs. 0.082±0.46 μg/gm tissue, p<0.0001), which was significantly attenuated by ATL802 (6.83±1.42, p<0.0001) (Figure 4).
Figure 4.
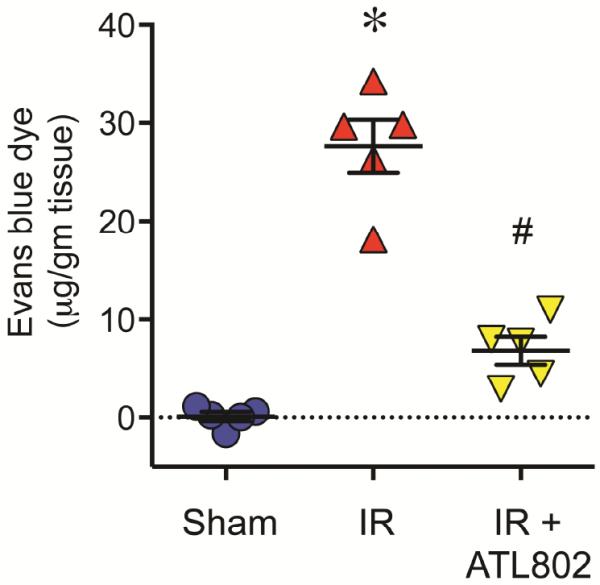
Pulmonary vascular permeability after IR is attenuated by ATL802. Vascular permeability was assessed using Evans blue dye extravasation technique. IR resulted in significant vascular permeability, which was significantly reduced by ATL802 treatment. *p<0.0001 vs. Sham, #p<0.0001 vs. IR, n=5/group.
ATL802 augmented lung function after EVLP
Using a murine model of EVLP, DCD lungs underwent 1 hour of EVLP with either standard KH buffer or Steen solution. EVLP with Steen solution significantly improved lung function versus KH buffer as shown by significantly increased pulmonary compliance (3.79±0.05 vs. 2.29±0.14 μl/cm H2O, p<0.0001) and decreased pulmonary artery pressure (9.74±0.13 vs. 13.90±0.12 cm H2O, p<0.0001) (Figure 5). Pulmonary compliance (4.53±0.08 μl/cm H2O, p=0.0001) and pulmonary artery pressure (8.03±0.09 cm H2O, p<0.0001) were further and significantly improved by EVLP with Steen solution supplemented with 100 nM ATL802 versus EVLP alone (Figure 5).
Figure 5.

ATL802 improves lung function after EVLP of warm ischemic lungs. Lungs underwent 15 minutes of warm ischemia, 1 hour of cold preservation and 1 hour of EVLP using either standard Krebs-Henseleit buffer (KH) buffer or Steen solution. EVLP with Steen solution significantly improved lung function (elevated pulmonary compliance and reduced pulmonary artery pressure) versus KH buffer, which was further and significantly improved by supplementing the Steen solution with 100 nM ATL802. *p<0.0001 vs. EVLP KH, #p<0.0001 vs. EVLP Steen, n=9/group.
Activation of A549 cells by HR is attenuated by ATL802
Using an in vitro surrogate model of lung IR in which cells are exposed to 3 hrs hypoxia and 1 hour reoxygenation (HR), exposure of A549 cells to HR induced significant IL-8 expression versus normoxic controls (105.6±6.1 vs. 7.5±2.8 pg/ml) (Figure 6). The activation of A549 cells (IL-8 production) by HR was significantly attenuated by treatment with 100nM (47.6±6.5 pg/ml) and 200nM (49.8±2.8 pg/ml) ATL802.
Figure 6.
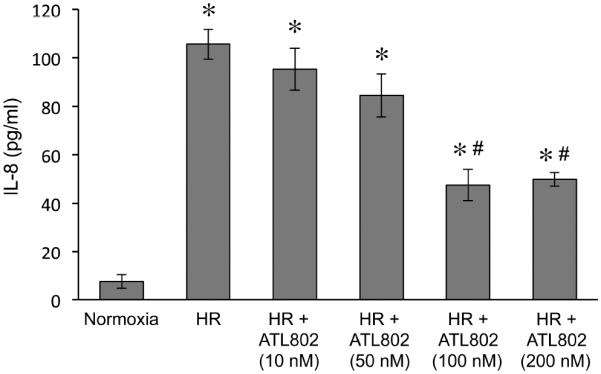
Secretion of IL-8 by alveolar type 2 epithelial cells after hypoxia-reoxygenation (HR) is attenuated by ATL802. A549 cells exhibited significant production of IL-8 by HR versus normoxia, which was significantly attenuated by ATL802 (100 and 200 nM). *p<0.0001 vs. Normoxia, #p<0.0001 vs. HR, n=8 replicates/group.
Comment
These results support our prior study that demonstrated a proinflammatory role for A2BRs on resident, non-immune cells in lung IRI [29]. The current study demonstrated that selective antagonism of A2BR with ATL802 significantly attenuates lung dysfunction, inflammation and vascular permeability after IR. In addition, ATL802 augmented EVLP-mediated reconditioning of DCD lungs. Finally, ATL802 was shown to block IL-8 production by A549 cells after HR, suggesting that a mechanism of protection from IRI by ATL802 may involve preventing the release of potent neutrophil chemokines such as IL-8 from alveolar epithelial cells. This is supported by the observation that ATL802 blocked KC production and neutrophil infiltration after lung IR. These results suggest that A2BR antagonists may be an effective therapeutic strategy for rehabilitating DCD lungs by EVLP and mitigating lung IRI to increase the success of lung transplantation.
The A2BR has been implicated in both anti- and pro-inflammatory responses, highlighting the complexity of adenosine receptor signaling in disease and injury. An anti-inflammatory role for alveolar epithelial cell A2BR signaling was described by Hoegl and colleagues in a two-hit model of acute lung injury [34], and work from Zhou and colleagues support an anti-inflammatory role for A2BR in a bleomycin model of acute lung injury but a pro-fibrotic role in chronic lung disease [35]. However, many studies suggest that A2BRs modulate adenosine-dependent regulation of proinflammatory paracrine factors. For example, activation of A2BRs was shown to evoke the production of proinflammatory cytokines by a variety of cells: IL-1β and IL-8 by mast cells [36], MCP-1 and IL-6 by airway smooth muscle cells [37], IL-19 by bronchial epithelial cells [28], IL-6 by lung fibroblasts [27] and IL-8 by endothelial cells [38]. In addition, Kolachala et al. demonstrated that the A2BR antagonist ATL801 significantly reduced the extent and severity of murine colitis and attenuated IL-6 and KC levels in colonic organ cultures [26]. Similarly, we observed that ATL802 attenuated the production of KC (murine homolog of IL-8), IL-1β, IL-6, MIP-1α, RANTES and MCP-1 in murine lungs after IR.
Work by Wang et al. suggests that A2BR may exist as a multiprotein complex in intestinal epithelia [39], which may result in varying downstream effects depending on interactions with various stimuli. Inflammation typically entails activation of immune cells and induction of cytokines, which has been reflected in the role of A2BR in myeloid cells [22, 40]. Pulmonary A2BRs appear to modulate a localized inflammatory response involving enhanced secretion of chemokines and cytokines, which is consistent with our results in the lung IRI model in which A2BR antagonism attenuated the induction of pro-inflammatory cytokines after IR. Thus adenosine and A2BR responses to stress are complex and may be dependent on relative extent of acute, systemic stress involving myeloid cells, or localized responses within the tissue.
Our study has several limitations. There is a potential for ventilator-induced injury in this model, however intubation time was minimized to reduce this risk. In addition, the lung function measurements reflect function of both right and left lungs although only the left lung underwent IR. However, both lungs are equally injured in the DCD model of EVLP where ATL802 was protective. Despite these limitations we were able to clearly demonstrate impaired lung function after IR that was ameliorated by ATL802 treatment. Finally, future studies will be required to determine the optimal methods for in vivo ATL802 treatment. The dose of ATL802 utilized was initially based on prior study [21], and follow-up preliminary studies using glucose bioassay data demonstrated a functional response to ATL802 that was optimal at the 1mg/kg dose (data not shown). The stability of ATL802 in serum has not been determined yet, but it is likely to have a half-life of 2–4 hours in blood based upon data with other compounds of similar nature such as ATL801 [26]. Repeat dosing may not be required as the purpose of treatment is to preempt and mitigate the damaging effects of the initial reperfusion insult, but future studies will need to confirm this. Our data suggests that, if possible, treatment of the donor would be protective. More likely, ATL802 would be administered (e.g. as an infusion) for a relatively short duration to the recipient immediately following lung transplant. It is unlikely that there would be significant side effects because ATL802 is an antagonist and thus there is less concern for off-target effects, which would be of more concern for an agonist.
In conclusion, the current study demonstrates a proinflammatory role of A2BR in lung IRI as exhibited by pulmonary dysfunction, cytokine expression, neutrophil infiltration, and vascular permeability, which were all attenuated by A2BR antagonism. These proinflammatory effects may be attributable, at least in part, to activation of A2BRs on alveolar epithelial cells and subsequent induction of IL-8 production. Our results suggest that A2BR may be a novel therapeutic target to augment EVLP-mediated reconditioning of DCD lungs and to mitigate lung IRI, leading to improved survival of lung transplant patients by preventing primary graft dysfunction.
Acknowledgments
This work was funded by the National Institutes of Health, grant numbers R01HL119218 (V.E.L. and I.L.K.) and T32 HL007849 (I.L.K.) as well as a University of Illinois Pillsbury grant (M.E.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Article Type = Original Article/STSA 2015 Oral Presentation Discussion included in text file.
Presented at the Basic Science Forum of the Sixty-second Annual Meeting of the Southern Thoracic Surgical Association, Orlando, FL, Nov 4–7, 2015.
References
- 1.Ailawadi G, Lau CL, Smith PW, et al. Does reperfusion injury still cause significant mortality after lung transplantation? J Thorac Cardiovasc Surg. 2009;137:688–94. doi: 10.1016/j.jtcvs.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Christie JD, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: twenty-seventh official adult lung and heart-lung transplant report--2010. J Heart Lung Transplant. 2010;29:1104–18. doi: 10.1016/j.healun.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Christie JD, Sager JS, Kimmel SE, et al. Impact of primary graft failure on outcomes following lung transplantation. Chest. 2005;127:161–5. doi: 10.1378/chest.127.1.161. [DOI] [PubMed] [Google Scholar]
- 4.Fiser SM, Tribble CG, Long SM, et al. Ischemia-reperfusion injury after lung transplantation increases risk of late bronchiolitis obliterans syndrome. Ann Thorac Surg. 2002;73:1041–7. doi: 10.1016/s0003-4975(01)03606-2. [DOI] [PubMed] [Google Scholar]
- 5.Kreisel D, Krupnick AS, Puri V, et al. Short- and long-term outcomes of 1000 adult lung transplant recipients at a single center. J Thorac Cardiovasc Surg. 2011;141:215–22. doi: 10.1016/j.jtcvs.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Ross SD, Tribble CG, Gaughen JR, Jr., Shockey KS, Parrino PE, Kron IL. Reduced neutrophil infiltration protects against lung reperfusion injury after transplantation. Ann Thorac Surg. 1999;67:1428–33. doi: 10.1016/s0003-4975(99)00248-9. [DOI] [PubMed] [Google Scholar]
- 7.Sharma AK, LaPar DJ, Zhao Y, et al. Natural killer T cell-derived IL-17 mediates lung ischemia-reperfusion injury. Am J Respir Crit Care Med. 2011;183:1539–49. doi: 10.1164/rccm.201007-1173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Sharma AK, Marshall M, Kron IL, Laubach VE. NADPH oxidase in bone marrow-derived cells mediates pulmonary ischemia-reperfusion injury. Am J Respir Cell Mol Biol. 2009;40:375–81. doi: 10.1165/rcmb.2008-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao M, Fernandez LG, Doctor A, et al. Alveolar macrophage activation is a key initiation signal for acute lung ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1018–26. doi: 10.1152/ajplung.00086.2006. [DOI] [PubMed] [Google Scholar]
- 10.Cypel M, Keshavjee S. Strategies for safe donor expansion: donor management, donations after cardiac death, ex-vivo lung perfusion. Curr Opin Organ Transplant. 2013;18:513–7. doi: 10.1097/MOT.0b013e328365191b. [DOI] [PubMed] [Google Scholar]
- 11.Steen S, Sjoberg T, Pierre L, Liao Q, Eriksson L, Algotsson L. Transplantation of lungs from a non-heart-beating donor. Lancet. 2001;357:825–9. doi: 10.1016/S0140-6736(00)04195-7. [DOI] [PubMed] [Google Scholar]
- 12.Cypel M, Rubacha M, Yeung J, et al. Normothermic ex vivo perfusion prevents lung injury compared to extended cold preservation for transplantation. Am J Transplant. 2009;9:2262–9. doi: 10.1111/j.1600-6143.2009.02775.x. [DOI] [PubMed] [Google Scholar]
- 13.Schepp CP, Reutershan J. Bench-to-bedside review: adenosine receptors--promising targets in acute lung injury? Crit Care. 2008;12:226. doi: 10.1186/cc6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan M, Qin W, Mustafa SJ. Characterization of adenosine receptor(s) involved in adenosine-induced bronchoconstriction in an allergic mouse model. Am J Physiol Lung Cell Mol Physiol. 2003;284:L1012–9. doi: 10.1152/ajplung.00353.2002. [DOI] [PubMed] [Google Scholar]
- 15.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci U S A. 1993;90:10365–9. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez LG, Sharma AK, LaPar DJ, Kron IL, Laubach VE. Adenosine A1 receptor activation attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2013;145:1654–9. doi: 10.1016/j.jtcvs.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gazoni LM, Walters DM, Unger EB, Linden J, Kron IL, Laubach VE. Activation of A1, A2A, or A3 adenosine receptors attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2010;140:440–6. doi: 10.1016/j.jtcvs.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaPar DJ, Laubach VE, Emaminia A, et al. Pretreatment strategy with adenosine A2A receptor agonist attenuates reperfusion injury in a preclinical porcine lung transplantation model. J Thorac Cardiovasc Surg. 2011;142:887–94. doi: 10.1016/j.jtcvs.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulloy DP, Sharma AK, Fernandez LG, et al. Adenosine A3 receptor activation attenuates lung ischemia-reperfusion injury. Ann Thorac Surg. 2013;95:1762–7. doi: 10.1016/j.athoracsur.2013.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beukers MW, den Dulk H, van Tilburg EW, Brouwer J, Ijzerman AP. Why are A(2B) receptors low-affinity adenosine receptors? Mutation of Asn273 to Tyr increases affinity of human A(2B) receptor for 2-(1-Hexynyl)adenosine. Mol Pharmacol. 2000;58:1349–56. doi: 10.1124/mol.58.6.1349. [DOI] [PubMed] [Google Scholar]
- 21.Cagnina RE, Ramos SI, Marshall MA, Wang G, Frazier CR, Linden J. Adenosine A2B receptors are highly expressed on murine type II alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L467–74. doi: 10.1152/ajplung.90553.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang D, Zhang Y, Nguyen HG, et al. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–23. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryzhov S, Zaynagetdinov R, Goldstein AE, et al. Effect of A2B adenosine receptor gene ablation on proinflammatory adenosine signaling in mast cells. J Immunol. 2008;180:7212–20. doi: 10.4049/jimmunol.180.11.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–35. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–15. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolachala V, Ruble B, Vijay-Kumar M, et al. Blockade of adenosine A2B receptors ameliorates murine colitis. Br J Pharmacol. 2008;155:127–37. doi: 10.1038/bjp.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol. 2005;32:2–8. doi: 10.1165/rcmb.2004-0103OC. [DOI] [PubMed] [Google Scholar]
- 28.Zhong H, Wu Y, Belardinelli L, Zeng D. A2B adenosine receptors induce IL-19 from bronchial epithelial cells, resulting in TNF-alpha increase. Am J Respir Cell Mol Biol. 2006;35:587–92. doi: 10.1165/rcmb.2005-0476OC. [DOI] [PubMed] [Google Scholar]
- 29.Anvari F, Sharma AK, Fernandez LG, et al. Tissue-derived proinflammatory effect of adenosine A2B receptor in lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2010;140:871–7. doi: 10.1016/j.jtcvs.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stone ML, Sharma AK, Mas VR, et al. Ex vivo perfusion with adenosine A2A receptor agonist enhances rehabilitation of murine donor lungs after circulatory death. Transplantation. 2015;99:2494–503. doi: 10.1097/TP.0000000000000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Z, Sharma AK, Linden J, Kron IL, Laubach VE. CD4+ T lymphocytes mediate acute pulmonary ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2009;137:695–702. doi: 10.1016/j.jtcvs.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer. 1976;17:62–70. doi: 10.1002/ijc.2910170110. [DOI] [PubMed] [Google Scholar]
- 33.Sharma AK, Fernandez LG, Awad AS, Kron IL, Laubach VE. Proinflammatory response of alveolar epithelial cells is enhanced by alveolar macrophage-produced TNF-alpha during pulmonary ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol. 2007;293:L105–13. doi: 10.1152/ajplung.00470.2006. [DOI] [PubMed] [Google Scholar]
- 34.Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar Epithelial A2B Adenosine Receptors in Pulmonary Protection during Acute Lung Injury. J Immunol. 2015;195:1815–24. doi: 10.4049/jimmunol.1401957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y, Schneider DJ, Morschl E, et al. Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J Immunol. 2011;186:1097–106. doi: 10.4049/jimmunol.1002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feoktistov I, Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest. 1995;96:1979–86. doi: 10.1172/JCI118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D. A(2B) adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol. 2004;30:118–25. doi: 10.1165/rcmb.2003-0118OC. [DOI] [PubMed] [Google Scholar]
- 38.Feoktistov I, Goldstein AE, Ryzhov S, et al. Differential expression of adenosine receptors in human endothelial cells: role of A2B receptors in angiogenic factor regulation. Circ Res. 2002;90:531–8. doi: 10.1161/01.res.0000012203.21416.14. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Kolachala V, Walia B, et al. Agonist-induced polarized trafficking and surface expression of the adenosine 2b receptor in intestinal epithelial cells: role of SNARE proteins. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1100–7. doi: 10.1152/ajpgi.00164.2004. [DOI] [PubMed] [Google Scholar]
- 40.Karmouty-Quintana H, Philip K, Acero LF, et al. Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J. 2015;29:50–60. doi: 10.1096/fj.14-260182. [DOI] [PMC free article] [PubMed] [Google Scholar]