

Abstract
About 250 to 350 million people worldwide are chronically infected with hepatitis B virus (HBV), and about 700000 patients per year die of HBV-related cirrhosis or hepatocellular carcinoma (HCC). Several anti-viral agents, such as interferon and nucleos(t)ide analogues (NAs), have been used to treat this disease. NAs especially have been shown to strongly suppress HBV replication, slowing the progression to cirrhosis and the development of HCC. However, reactivation of HBV replication often occurs after cessation of treatment, because NAs alone cannot completely remove covalently-closed circular DNA (cccDNA), the template of HBV replication, from the nuclei of hepatocytes. Anti-HBV immune responses, in conjunction with interferon-γ and tumor necrosis factor-α, were found to eliminate cccDNA, but complete eradication of cccDNA by immune response alone is difficult, as shown in patients who recover from acute HBV infection but often show long-term persistence of small amounts of HBV-DNA in the blood. Several new drugs interfering with the life cycle of HBV in hepatocytes have been developed, with drugs targeting cccDNA theoretically the most effective for radical cure of chronic HBV infection. However, the safety of these drugs should be extensively examined before application to patients, and combinations of several approaches may be necessary for radical cure of chronic HBV infection.
Keywords: Covalently-closed circular DNA, Genome editing technology, Immune response, Immunotherapy, Program death-1, Interferon-γ, Tumor necrosis factor-α
Core tip: Among the agents used to treat chronic hepatitis B virus (HBV) infection are nucleos(t)ide analogues, which have been shown to strongly suppress HBV replication. HBV replication, however, may be reactivated after cessation of treatment, because complete removal of covalently-closed circular DNA (cccDNA) from hepatocyte nuclei is extremely difficult. Immune responses have been shown to destroy cccDNA, but immune response alone is insufficient for complete eradication of template DNA. Several drugs were recently developed to block the HBV life cycle in hepatocytes, with drugs targeting cccDNA being, at least theoretically, the most effective for radical cure of chronic HBV infection. The safety of these agents should be extensively examined before their use in patients. Combinations of two or more classes of agent may be necessary for radical cure of chronic HBV infection.
INTRODUCTION
About 250 to 350 million people worldwide are chronically infected with hepatitis B virus (HBV)[1,2], with about 700000 patients per year dying from HBV-related cirrhosis or hepatocellular carcinoma (HCC)[3]. Several anti-viral agents, including interferons and nucleos(t)ide analogues (NAs), have been shown effective, with NA-based treatment strongly suppressing the replication of HBV-DNA and normalizing serum alanine aminotransferase activity, resulting in little or no progression of liver disease[4-6]. NAs target the viral reverse transcriptase, effectively reducing serum HBV-DNA concentrations. However, intrahepatic HBV-DNA, such as converted covalently closed circular DNA (cccDNA), is not a direct target of NAs. cccDNA is a template for all viral RNAs and HBV-DNA replication can be induced to start from residual cccDNA after cessation of treatment with NAs[7]. Small amounts of HBV-DNA can be found in serum long after patients recover from acute HBV infection, suggesting that cccDNA may persist for decades[8]. Thus, cccDNA is difficult to eradicate once infection is established, and should be the main target for the complete eradication of HBV infection. However, measuring intrahepatic cccDNA concentrations is difficult in a clinical setting[9]. The cccDNA levels in HBV-infected human hepatocytes are low, ranging from 1 to 50 copies per hepatocyte[10]. Real-time polymerase chain reaction (PCR) amplification with specific primers for cccDNA or Southern blotting can be used for the detection. However, PCR amplification may be hampered by other co-extracted viral DNA and Southern blotting needs much time and effort. Moreover, the form of cccDNA may be changed during the DNA extraction procedure. Therefore, further investigation should be required to establish the precise evaluation of intrahepatic cccDNA. As an alternative, the reduction in HBV surface antigen (HBsAg) concentration has been reported to partly reflect the decrease in intrahepatic cccDNA, with the goal of treatment for chronic HBV infection being the complete disappearance of HBsAg[11]. Fewer than 10% of patients receiving interferon-based therapy[4-6], and few patients treated with NAs[12,13], achieve complete loss of HBsAg. Various trials have tested agents targeting the life cycle of HBV in hepatocytes, including the elimination of cccDNA. This review summarizes and discusses the radical cure (Table 1) of chronic HBV infection, mainly focusing on the elimination of cccDNA.
Table 1.
Cure status of hepatitis B virus infection
| Serum HBV-DNA | Serum HBsAg | Intraheptic cccDNA | HBV-DNA-intergrated hepatocytes | |
| Functional cure (clinical cure) | Low | (-)-(++) | (+) | (-)-(+) |
| Radical cure (virological cure) | (-) | (-) | (-) | (-) |
HBV: Hepatitis B virus; cccDNA: Covalently-closed circular DNA; HBsAg: Hepatitis B virus surface antigen.
HBV REPLICATION CYCLE AND THE PRODUCTION OF HBV-RELATED PROTEINS
HBV replication cycle
HBV is a DNA virus that belongs to the family Hepadnaviridae, with a 3.2 kb-long partially double-stranded relaxed circular DNA (rcDNA) genome[14]. The life cycle of HBV is shown in Figure 1. HBV virions are thought to enter hepatocytes through a high-affinity interaction between the myristoylated preS1 region of HBV and the surface structures of hepatocytes, including sodium taurocholate cotransporting polypeptide (NTCP)[15-17]. After entry into hepatocytes, uncoated rcDNA is released into the cytoplasm and then enters the nucleus, where it is converted to cccDNA. The cccDNA remains for a long time in the nucleus, where it serves as a template for the transcription of viral mRNA[17,18]. All viral RNAs, pregenomic RNAs (pgRNA) and RNAs encoding the surface proteins, precore and HBx of HBV, are transcribed from cccDNA, with efficient transcription regulated by liver-specific transcription factors[19] and the HBx protein itself[20]. Epigenetic control of cccDNA transcriptional activity, such as acetylation, methylation or phosphorylation, appears to occur[21]. Cytoplasmic pgRNA and polymerase protein are subsequently packaged into envelope proteins, with rcDNA produced from the reverse transcription of pgRNA. Nucleocapsids packaging rcDNA are encapsulated by HBsAg as the envelope protein and released from hepatocytes as virions. The precise understanding of these processes is important for the development of new strategies for the radical cure of chronic HBV infection.
Figure 1.
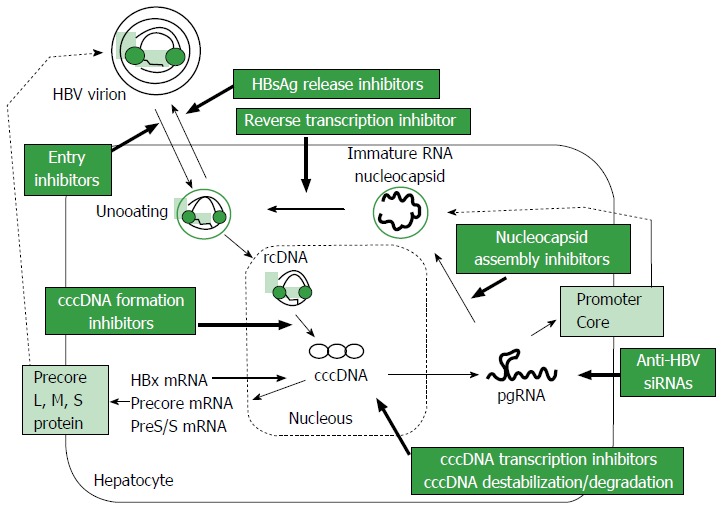
Simplified schema of the hepatitis B virus life cycle and possible targets of therapy. HBV: Hepatitis B virus; cccDNA: Covalently-closed circular DNA; HBsAg: Hepatitis B virus surface antigen; rcDNA: Relaxed circular DNA; siRNAs: Small interfering RNAs; pgRNA: Pregenomic RNAs.
HBV-related proteins and their roles in hepatocarcinogenesis
The HBV-related proteins translated from cccDNA consist not only of the envelope, core and polymerase proteins of HBV, but may play a role in hepatocarcinogenesis itself.
Studies analyzing the role of HBx proteins in hepatocellular transformation and HCC progression have found that low levels of HBx protein are present in non-tumor tissues of HBV-infected liver, whereas high levels of HBx protein are present in HCCs arising in HBV infected individuals, suggesting that this protein has an oncogenic function[22,23]. Moreover, HBx transgenic mice often develop liver cancer[24,25], and HBx protein has been found to accumulate in hepatocytes, affecting the expression of genes associated with signal transduction, cell cycle control, transcription, and immune response[23,26]. Expression of genes on the X-chromosome is regulated epigenetically, including by DNA and histone methyltransferases[27,28], and by microRNAs[29,30].
HBx is not only involved in carcinogenesis but in the progression of HCC. HBx has been shown to increase beta-catenin signaling through epigenetic control or microRNA[31,32] and to be an independent predictor of survival after HCC resection[33].
HBsAg is also involved in hepatocarcinogenesis. The ground glass appearance of hepatocytes was shown to be a typical histological finding in HBV-infected livers, with this ground glass appearance resulting from the accumulation of HBsAg with preS mutations[34-36]. PreS-mutated HBsAg, especially large HBsAg, was found to accumulate in cytoplasm, leading to the induction of ER stress and oxidative DNA damage[35-37]. Furthermore preS mutations upregulated intracellular signaling via hepatocyte proliferation[35,38]. High serum HBsAg levels showed a definite correlation with HCC development in patients with controlled HBV-DNA[39-41]. Like HBV-related proteins, spliced HBV proteins were found to activate intracellular signaling via hepatocyte proliferation[42,43]. These findings suggest that not only HBV replication, but the production of HBV-related proteins, should be suppressed to efficiently prevent hepatocarcinogenesis.
IMMUNE RESPONSE AGAINST HBV INFECTION
Immune responses against HBV are involved in both the pathogenesis and control of HBV infection[44-47]. Therefore, understanding the immune response against HBV may result in better control of HBV infection.
Acute infection
Analysis of immune responses that occur during acute HBV infection may provide valuable information on strategies by which immune responses control HBV infection.
A mouse model of acute viral hepatitis B was established by injecting HBsAg-specific T-cell clones into HBV transgenic mice[48]. Although HBsAg-specific T-cells were found to kill small numbers of HBV-replicating hepatocytes, these T cell clones destroyed intracellular HBV-RNA and HBV-DNA in most infected hepatocytes without killing these cells. This effect was found to be due to interferon (IFN)-γ and tumor necrosis factor (TNF)-α[40,49-51]. Because HBV transgenic mice do not have cccDNA[52], the effects of these cytokines on cccDNA were unclear. In cccDNA-expressing cultured cells, however, IFN-γ and TNF-α inhibited HBV replication and reduced cccDNA in an additive manner[53]. Moreover, the decay of cccDNA was found to require activation of APOBEC3 deaminases[53], which are expressed in liver tissues of individuals with acute, but not chronic, HBV infection. These observations indicate that HBV-specific T-cell activation followed by treatment with anti-viral cytokines, such as IFN-γ and TNF-α, could eradicate HBV without cytolysis.
In a chimpanzee model, cccDNA was found to disappear during the course of acute hepatitis B, and HBV-DNA was found to be susceptible to noncytolytic control by cytokines[54]. Moreover, HBV-DNA titers in these livers were reduced before T-cell influx, suggesting that non-T-cells, possibly natural killer cells, may have an important role in the noncytolytic destruction of HBV-DNA in liver during early phases of acute HBV infection[54].
Broad and vigorous CD4+ and CD8+ T-cell responses have been reported in patients with acute hepatitis B[55]. Moreover, HBV-specific T-cell responses were observed during the incubation period of acute hepatitis, with HBV-DNA reduced before alanine aminotransferase concentration peaked, indicating that noncytolytic eradication of HBV also occurs in acute hepatitis B in humans[56]. However, recovery from acute hepatitis B does not imply complete eradication of HBV, as small amounts of HBV-DNA can be detected in the blood for a long time after resolution of acute hepatitis B[8]. T-cell responses are therefore not sufficient to completely eradicate cccDNA from infected livers, even in acute hepatitis B.
Chronic infection
Immune responses in patients chronically infected with HBV were found to consist of four phases: The immunotolerant, immune-active, inactive carrier, and reactivation phases[57]. Although the exact mechanism by which HBV induces immune tolerance is unclear, it may arise from central deletion or peripheral non-recognition of HBV-specific T-cells[58]. Immune tolerance may be broken after several decades by as yet undetermined mechanisms, but these may involve the maturation of dendritic cell (DC) function[59]. Breaking immune tolerance to HBV can lead to the immune-active phase, resulting in some degree of hepatitis. During this phase, suppression of HBV replication is observed in 85% to 90% of patients, leading to an inactive carrier state. Most patients in an inactive carrier state do not need antiviral treatments, but cccDNA may be present in their livers. The cccDNA persisting in inactive carriers may be a template for reactivation of HBV replication. The 10% to 15% of patients who remain in the immune-active phase continue to experience liver inflammation with active replication of HBV, and may be at high risk for progression to liver cirrhosis and the development of HCC. The number of HBV-specific CD8+ T-cells was found to be the same in livers with low HBV replication and little hepatitis and in livers with high HBV replication and severe hepatitis[60]. These findings suggest that HBV replication is suppressed by immune surveillance of HBV-specific T-cells in the liver and that these T-cells are important in controlling HBV replication in a noncytolytic manner in inactive carriers. In contrast, HBV-specific immune responses are thought to be dysregulated in livers with active hepatitis, and several possible mechanisms have been proposed.
Impairment of innate immune response
Innate immune system such as pattern recognition receptors, macrophages, DCs, natural killer cells or natural killer T cells are involved in the pathogenesis of HBV infection especially at an early stage of infection[61,62]. HBV has been shown to alter the function of macrophages by modulating the secretion of cytokines[63,64] or type-1 IFN gene expression[64]. Hepatitis B e antigen was shown to directly suppress toll-like receptor (TLR) signaling via interaction with Toll/IL-1 receptor-containing proteins such as TRAM and Mal[65]. HBV has been shown to downregulate TLR-2 expression in patients with chronic HBV infection[66]. Thus, innate immunity alteration plays a role, at least in part, in the pathogenesis of chronic HBV infection and TLR-7 agonists have been applied as immune-modulatory components[67,68]. On the other hand, the effect of IFN-α on intrahepatic cccDNA has been recently explored[69], and IFN-α in addition to lymphotoxin-β receptor (LTβR) activation has been shown to induce cccDNA degradation through upregulation of nuclear APOBEC3 deaminases[70]. APOBEC3 can deaminate double-stranded DNA cytidines to uridines[71] and induce cccDNA degradation. IFN-γ and TNF-α produced form T-cells can induce deamination of cccDNA without cytolysis, supporting the essential role of APOBEC3 in reduction of cccDNA[53]. Collectively, type-1 IFN-mediated effects, especially APOBEC3 upregulation, will be a key subject for development of new therapeutics.
Dysfunction of dendritic cells
DCs are the most potent antigen-presenting cells, stimulating both T- and B-cells. In patients with chronic hepatitis, the cytokine-induced maturation of circulating myeloid DCs is impaired, possibly by exposure to high amounts of HBV or HBsAg[72,73]. Dysfunctional DCs may act as tolerogenic antigen-presenting cells, resulting in a failure to induce HBV-specific immune responses.
Alteration of the hierarchy of epitope-specific CD8+ T-cell responses
In acute hepatitis B, the CD8+ T-cell response to the immunogenic epitope HBc18-27 (HLA-A2 restricted epitope) is dominant. In contrast, HBc18-27-specific CD8+ T-cell responses are low and CD8+ T-cell responses against less immunogenic envelope (183-191) are dominant in chronic hepatitis B[74]. Although the mechanisms underlying changes in the major epitope to CD8+ T-cell response are not yet known, they may account, at least in part, for the different CD8+ T-cell responses observed in patients with acute and chronic hepatitis.
Regulatory T-cells
Regulatory T-cells (Tregs) expressing the forkhead family transcription factor, Foxp3, are specialized cells that have a major role in the maintenance of immunological self-tolerance by suppressing self-reactive cells[75]. Tregs express CD25 [interleukin (IL)-2 receptor α-chain] and/or cytotoxic T-lymphocyte antigen-4 (CTLA-4), which are excellent inhibitors of IL-2 production or downregulation of CD80 and CD86 on DCs by a CTLA-4-dependent mechanism[76].
The numbers of CD4+CD25+FoxP3+ Tregs were higher in the livers of patients with chronic hepatitis B, suggesting that these cells suppress intrahepatic HBV-specific T-cell responses, leading to insufficient immune control of HBV replication in the liver[77].
Inhibitory receptors
Program death (PD)-1 is a surface receptor critical for the regulation of T-cell function[78,79]. Binding of the ligand PD-L1 to PD-1 on T-cells results in the antigen-specific inhibition of T-cell proliferation, with a molecule related to T-cell exhaustion found in the livers of patients with chronic hepatitis B. T-cell exhaustion is characterized by poor cytotoxic activity and cytokine production, as well as by the expression of inhibitory receptors, including not only PD-1 but lymphocyte activation gene-3, CTLA-4, T-cell immunoglobulin domain and mucin domain-3, and CD244[66]. These inhibitory receptors are thought to be induced by persistent exposure of intrahepatic T-cells to HBV or HBV-related proteins[80]. Exhaustion of T-cells could also account for impaired T-cell responses in the livers of patients with chronic hepatitis B, and blockade of these receptors could be therapeutic.
Patients with high serum HBV-DNA concentration have been reported likely to progress to cirrhosis and eventually HCC[81]. Transition of immune-active patients to an inactive state with low HBV-DNA replication by the direct stimulation of HBV-specific T-cells or removal of immunosuppressive factors, may be sufficient to inhibit progression to cirrhosis or HCC. Inactive HBV carriers may not require specific treatment, because spontaneous HBsAg develops at a rate of 1% to 1.9%/year in these patients, making the development of HCC rare[82]. Therefore, an inactive HBV carrier may be regarded as in a state of functional cure (Table 1). However, HBV replication may be reactivated, either spontaneously or during treatment with an immunosuppressive or anticancer agent, resulting in a higher risk of hepatocarcinogenesis than in the general population[83]. The rate of HCC development was recently reported to be greater in patients with high than with low serum HBsAg concentrations, even in inactive HBV carriers with low serum HBV-DNA concentrations[36,37].
Collectively, these results suggest that induction of immune control against HBV infection may result in functional cure of HBV infection. Functional cure, however, may be an unstable condition, allowing progression to cirrhosis or HCC under various conditions. Although radical cure (Table 1) is desirable, it is problematic because of the difficulty in eliminating HBV cccDNA from the liver.
THERAPEUTIC STRATEGIES FOR HBV INFECTION
Immunotherapy
Radical cure of HBV infection could be achieved by both the elimination of cccDNA in the liver and the destruction of HBV-DNA-integrated hepatocytes. The primary goals of immunotherapy in HBV-infected individuals include the induction or stimulation of HBV-specific immune responses, leading to the killing of infected cells or the degradation of HBV-RNA and HBV-DNA in a noncytolytic manner, inhibiting progression to liver cirrhosis and hepatocarcinogenesis. Although immune responses involving cytokines such as IFN-γ and TNF-α can eliminate cccDNA[50,53], cccDNA is not completely eliminated even after resolution of acute hepatitis B[8], suggesting that immune responses alone may be insufficient to achieve radical cure of HBV infection.
Induction or stimulation of HBV-specific immune responses
Efforts to stimulate HBV-specific T-cells have included immunizations with HBV-peptides, viral proteins, DCs, and DNA, as well as treatment with cytokines[84]. Because HBV-specific T-cells in patients with chronic hepatitis B are exhausted by long-term exposure to high levels of HBV-related antigens, activation of those cells by immunization would be ineffective without functional restoration of the cells by blocking the inhibitory signals responsible for T-cell exhaustion. Blockade of PD-1, CTLA-4 or Tim-3 has been shown to restore exhausted HBV-specific T-cells[80], suggesting that the combination of immunization and blockade of inhibitory signals would be effective in activating HBV-specific T-cells.
Other immunotherapeutic approaches to HBV infection include administration of cytokines, such as IFN-γ, IL-6, IL-1β, LTβR-agonists and/or TLR-7 agonist, as well as IFN-γ and TNF-α which were shown to cause silencing or degradation of cccDNA[67]. This strategy may be more effective in the complete eradication of HBV infection than strategies involving the activation of HBV-specific cells, suggesting that only cytokine administration results in the elimination of cccDNA.
Elimination of HBV-infected hepatocytes by a novel approach
A novel approach to eliminate HBV-core containing hepatocytes[85] was based on findings showing that elimination of HBV is impaired by cellular inhibitor of apoptosis proteins (cIAPs), which inhibit the TNF-α-mediated death of HBV-infected cells[86]. This led to testing the effects of inhibitors of cIAPs, including birinapant and other Smac mimetics, on HBV-infected hepatocytes. These inhibitors of cIAPs resulted in the rapid reduction in serum HBV-DNA and HBsAg concentrations, possibly by eliminating HBV-core containing hepatocytes. However, the effects of those drugs on cccDNA are unclear.
Immunotherapeutic strategies for HBV-DNA-integrated hepatocytes
Three main mechanisms are responsible for hepatocarcinogenesis: (1) the oncogenic potential of the HBV-related proteins, HBsAg and HBx; (2) HBV-DNA integration into the host genome, dysregulating the cell cycle by the introduction of deletions, cis/trans-activations, and/or translocations, and/or inducing generalized genomic instability; and (3) persistent inflammation in the liver causing rapid turnover of hepatocyte regeneration, enhancing the instability and/or mutagenesis of host genomes.
Therefore, if future advances in therapeutic modalities result in the complete elimination of cccDNA, hepatocarcinogenesis resulting from HBV-DNA integration into the host genome should be addressed. HBV-DNA integration into the hepatocyte genome has been observed in 86.4% of HBV-related HCCs and in 30.7% of adjacent liver tissue[87]. Integration of HBV-DNA into areas of the host genome encoding genes that regulate cellular proliferation, such as telomerase or proliferation signal transduction genes, may lead to cis-/trans-activation, inducing malignant transformation[88]. Furthermore, integration of HBV-DNA may induce genetic instability by altering the expression of oncogenes, tumor suppressor genes and microRNAs[87,89]. In addition, a viral-human chimeric transcript was reported to function as a noncoding RNA and promote hepatocarcinogenesis[90]. Integration of HBV-DNA into the host hepatocyte genome of transiently infected individuals has been reported to be a rare event, occurring in 0.01%-0.1% of hepatocytes[91]. Further investigations are needed to determine the mechanism by which HBV-DNA integration into the host genome induces carcinogenesis. The immune cytolysis of cells expressing HBV-related peptides may be the only strategy that effectively eliminates HBV-DNA-integrated hepatocytes. However, if non-immunogenic regions of HBV-DNA are integrated, elimination of those cells by immune attack would be impossible.
Taken together, these findings indicate that immunotherapy against HBV can control viral replication and reduce cccDNA, but may not be sufficient to completely eradicate HBV-infected or -integrated hepatocytes.
Inhibition of HBV replication
Currently available NAs can efficiently reduce viremia but cannot eliminate intracellular cccDNA. However, complete suppression of HBV polymerase can result in the complete elimination of cccDNA through the death of cccDNA-containing hepatocytes after one natural lifespan of these cells[92]. Among the agents being tested are prodrugs of HBV polymerase inhibitors[93]. These include prodrugs of tenofovir, such as AGX1009 (Agenix) and TAF (GS-7340, Gilead Sciences), which have been evaluated in phase 3 trials[93,94], and CMX157, a lipid conjugate of tenofovir, which has been evaluated in phase 1/2 trials[93,95]. RNase H inhibitors are also being tested, based on the specificity of HBV replication, which depends on the RNase H activity of HBV polymerase to degrade pgRNA[10]. Evaluations of selective inhibitors of HBV polymerase RNase H activity[96] suggest that they might be more effective when combined with NAs[93].
Destruction of cccDNA
Eradication of cccDNA in hepatocytes is essential to achieve radical cure of established HBV infection. Several trials have targeted cccDNA. For example, gene silencing techniques, such as small interfering RNAs (siRNAs) or antisense oligonucleotides (ASOs), have been evaluated for their ability to reduce viremia and cccDNA. Although siRNAs may have promising activity, methods to effectively deliver them to hepatocytes have not been determined[97]. RNAi can inhibit all steps of HBV replication, and ARC-520 has been tested in a phase 2 trial in patients with chronic hepatitis B[95]. In contrast, a single injection of ASO, consisting of liver-targeted peptides, into a mouse model of chronic HBV infection was shown to reduce HBV-RNA, proteins and HBV-DNA for a long time, suggesting that ASO may become a promising treatment in patients with chronic HBV[98]. Furthermore, disubstituted sulfonamide was shown to selectively inhibit the formation of cccDNA[99].
In addition, several genome editing technologies have been developed to silence sequence-specific cleavage of cccDNA. These include zinc finger nucleases (ZFNs)[100,101], transcription activator-like effector nucleases (TALENs)[102,103], and the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR associated system (Cas). These sequence-specific genome editing technologies could induce double-stranded breaks at certain DNA sites. ZFNs consist of a zinc finger domain, which contains a sequence-specific binding site, and a FokI nuclease domain. ZFNs form heterodimers and induce double-stranded breaks at targeted sites. These breaks are subsequently repaired by homology-directed repair or non-homologous end joining. The specificity of ZFNs may be context-dependent, resulting from interactions between DNA binding domains and neighboring zinc fingers[104]. TALENs have transcription activator-like effector specific DNA binding activity, with DNA-binding sites more specific than those of ZFNs[105]. However, both ZFNs and TALENs require pairs of site-specific nucleases for each target to produce customized proteins[9]. In contrast, CRISPR/Cas technology is a novel genome-editing method, which is more useful than ZFNs or TALENs[106]. CRISPR/Cas loci encode RNA guided endonucleases, which are induced by immune responses against foreign genetic elements such as bacteriophages and plasmids[107]. The type 2 CRISPR/Cas system from Streptococcus pyogenes is a chimeric single-guide RNA with Cas9 protein[108]. The CRISPR/Cas9 system was shown to suppress HBV replication in cultured cells and in mouse models[109-117], reducing both HBsAg[109,110,112-116] and cccDNA[110,113-115,117]. These findings suggest that genome editing technology, such as a CRISPR/Cas system, may be a potential therapeutic option for the complete eradication of HBV infection in future. However, cleavage of cccDNA and subsequent DNA repair may introduce mutations into the host genome. These mutations may be harmful to the host, resulting in the possible development of malignancy[9,118,119], suggesting the need for further improvements in efficacy and safety prior to the therapeutic use of these systems.
Future perspectives on radical cure of chronic HBV infection
Various trials have assessed agents that can terminate the HBV life cycle in hepatocytes, including inhibitors of HBV-DNA polymerase, virus entry, core assembly and HBsAg secretion (Table 2)[93,95,120,121]. Especially Myrcludex B, a synthetic lipopeptide that targets NTCP, has been shown to efficiently prevent viral spread and has been applied in clinical trials[15,17,122,123]. These agents, including Myrcludex, are not themselves sufficient to eliminate HBV from chronically infected hepatocytes, as shown by the remaining cccDNA in the nuclei and HBV-DNA-integrated hepatocytes. Immunotherapy may potentially eliminate both cccDNA and HBV-DNA-integrated hepatocytes, but its effects would be limited. Although drugs targeting cccDNA in hepatocytes are theoretically ideal for complete eradication of HBV, no single drug or strategy, whether currently available or under development, has shown the ability to completely eliminate HBV with established safety and efficacy. Future trials, testing combination of different agents or strategies, will be necessary.
Table 2.
Tgerapectic agents against hepatitis B virus currently in clinical development
| Mode of actions | Target | Stage of development | Ref. |
| Entry inhibitions | NTCP | Myrcludex in phage 2 | [14,123] |
| cccDNA | |||
| Formation inhibitions | DSS | Preclinical | [99] |
| Transcription inhibitions | ASO | IONIS-HBVRx in phase 1 | [98] |
| Destabilization/degradation | ZFN | Preclinical | [100,101] |
| TALEN | Preclinical | [102,103] | |
| CRISPR/Cas9 | Preclinical | [109-117] | |
| SiRNA | PgRNA | ARC-520 in phase 2 | [95] |
| Nucleocapsid assembly inhibitions | Capsid formation | BAY4109 in phase 1 | [93,95] |
| NV1221 in phase 1 | [93,95] | ||
| Reverse transcription inhibitions | Polymerase | TAF in phase 3 | [93,94] |
| Cmx157 in phase 1/2 | [93,95] | ||
| HBsAg release inhibitions | HBsAg secretion | Preclinical | [109] |
| HBsAg secretion | Rep2139 in phase 1/2 | [110] | |
| Immune modulating | TLR-7 agonist | GS-9620 in phase 2 | [67,68] |
| HBV-specific | Preclinical | [84] | |
| cIAPS | Preclinical | [86] |
cccDNA: Covalently-closed circular DNA; ZFN: Zinc finger nuclease; TALEN: Transcription activator-like effector nuclease; CRISPR: Clustered regularly interspaced short palindromic repeat; Cas: CRISPR associated system; pgRNA: Pregenomic RNAs; HBsAg: Hepatitis B virus surface antigen; TLR: Toll-like receptor; HBV: Hepatitis B virus; cIAPS: Cellular inhibitor of apoptosis proteins; DSS: Disubstituted sulfonamide; ASO: Antisense oligonucleotides; TAF: Tenofovir Alafenamide; NTCP: Na+/taurocholate cotransporting polypeptide.
Footnotes
Conflict-of-interest statement: Tajiri K and Shimizu Y declare there is no conflict of interest related to this publication.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Specialty Type: Gastroenterology and Hepatology
Country of Origin: Japan
Peer-Review Report Classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): D
Grade E (Poor): 0
Peer-review started: March 28, 2016
First decision: May 17, 2016
Article in press: June 29, 2016
P- Reviewer: Bowden S, Gong ZJ, Jin DY S- Editor: Qi Y L- Editor: A E- Editor: Li D
References
- 1.Liaw YF, Chu CM. Hepatitis B virus infection. Lancet. 2009;373:582–592. doi: 10.1016/S0140-6736(09)60207-5. [DOI] [PubMed] [Google Scholar]
- 2.Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 3.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trépo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 5.Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, Gane E, Fried MW, Chow WC, Paik SW, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352:2682–2695. doi: 10.1056/NEJMoa043470. [DOI] [PubMed] [Google Scholar]
- 6.Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, Simon C, So TM, Gerken G, de Man RA, et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet. 2005;365:123–129. doi: 10.1016/S0140-6736(05)17701-0. [DOI] [PubMed] [Google Scholar]
- 7.Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 8.Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat Med. 1996;2:1104–1108. doi: 10.1038/nm1096-1104. [DOI] [PubMed] [Google Scholar]
- 9.Ohno M, Otsuka M, Kishikawa T, Yoshikawa T, Takata A, Koike K. Novel therapeutic approaches for hepatitis B virus covalently closed circular DNA. World J Gastroenterol. 2015;21:7084–7088. doi: 10.3748/wjg.v21.i23.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lampertico P, Maini M, Papatheodoridis G. Optimal management of hepatitis B virus infection - EASL Special Conference. J Hepatol. 2015;63:1238–1253. doi: 10.1016/j.jhep.2015.06.026. [DOI] [PubMed] [Google Scholar]
- 12.Lai CL, Gane E, Liaw YF, Hsu CW, Thongsawat S, Wang Y, Chen Y, Heathcote EJ, Rasenack J, Bzowej N, et al. Telbivudine versus lamivudine in patients with chronic hepatitis B. N Engl J Med. 2007;357:2576–2588. doi: 10.1056/NEJMoa066422. [DOI] [PubMed] [Google Scholar]
- 13.Chang TT, Gish RG, de Man R, Gadano A, Sollano J, Chao YC, Lok AS, Han KH, Goodman Z, Zhu J, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med. 2006;354:1001–1010. doi: 10.1056/NEJMoa051285. [DOI] [PubMed] [Google Scholar]
- 14.Summers J, O’Connell A, Millman I. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc Natl Acad Sci USA. 1975;72:4597–4601. doi: 10.1073/pnas.72.11.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gripon P, Cannie I, Urban S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J Virol. 2005;79:1613–1622. doi: 10.1128/JVI.79.3.1613-1622.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Fälth M, Stindt J, Königer C, Nassal M, Kubitz R, et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology. 2014;146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Brahmania M, Feld J, Arif A, Janssen HL. New therapeutic agents for chronic hepatitis B. Lancet Infect Dis. 2016;16:e10–e21. doi: 10.1016/S1473-3099(15)00436-3. [DOI] [PubMed] [Google Scholar]
- 18.Summers J, Mason WS. Replication of the genome of a hepatitis B--like virus by reverse transcription of an RNA intermediate. Cell. 1982;29:403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- 19.Tang H, McLachlan A. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci USA. 2001;98:1841–1846. doi: 10.1073/pnas.041479698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, Zoulim F, Hantz O, Protzer U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55:996–1003. doi: 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 21.Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 22.Paterlini P, Poussin K, Kew M, Franco D, Brechot C. Selective accumulation of the X transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology. 1995;21:313–321. [PubMed] [Google Scholar]
- 23.Zhang XD, Wang Y, Ye LH. Hepatitis B virus X protein accelerates the development of hepatoma. Cancer Biol Med. 2014;11:182–190. doi: 10.7497/j.issn.2095-3941.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 25.Yu DY, Moon HB, Son JK, Jeong S, Yu SL, Yoon H, Han YM, Lee CS, Park JS, Lee CH, et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J Hepatol. 1999;31:123–132. doi: 10.1016/s0168-8278(99)80172-x. [DOI] [PubMed] [Google Scholar]
- 26.Qiu X, Dong S, Qiao F, Lu S, Song Y, Lao Y, Li Y, Zeng T, Hu J, Zhang L, et al. HBx-mediated miR-21 upregulation represses tumor-suppressor function of PDCD4 in hepatocellular carcinoma. Oncogene. 2013;32:3296–3305. doi: 10.1038/onc.2013.150. [DOI] [PubMed] [Google Scholar]
- 27.Jung JK, Arora P, Pagano JS, Jang KL. Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1 pathway. Cancer Res. 2007;67:5771–5778. doi: 10.1158/0008-5472.CAN-07-0529. [DOI] [PubMed] [Google Scholar]
- 28.Rivière L, Gerossier L, Ducroux A, Dion S, Deng Q, Michel ML, Buendia MA, Hantz O, Neuveut C. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol. 2015;63:1093–1102. doi: 10.1016/j.jhep.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 29.Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13:123–135. doi: 10.1038/nrc3449. [DOI] [PubMed] [Google Scholar]
- 30.Yuan K, Lian Z, Sun B, Clayton MM, Ng IO, Feitelson MA. Role of miR-148a in hepatitis B associated hepatocellular carcinoma. PLoS One. 2012;7:e35331. doi: 10.1371/journal.pone.0035331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JO, Kwun HJ, Jung JK, Choi KH, Min DS, Jang KL. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene. 2005;24:6617–6625. doi: 10.1038/sj.onc.1208827. [DOI] [PubMed] [Google Scholar]
- 32.Arzumanyan A, Friedman T, Kotei E, Ng IO, Lian Z, Feitelson MA. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene. 2012;31:563–572. doi: 10.1038/onc.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie Y, Liu S, Zhao Y, Guo Z, Xu J. X protein mutations in hepatitis B virus DNA predict postoperative survival in hepatocellular carcinoma. Tumour Biol. 2014;35:10325–10331. doi: 10.1007/s13277-014-2331-0. [DOI] [PubMed] [Google Scholar]
- 34.Su IJ, Wang HC, Wu HC, Huang WY. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J Gastroenterol Hepatol. 2008;23:1169–1174. doi: 10.1111/j.1440-1746.2008.05348.x. [DOI] [PubMed] [Google Scholar]
- 35.Pollicino T, Cacciola I, Saffioti F, Raimondo G. Hepatitis B virus PreS/S gene variants: pathobiology and clinical implications. J Hepatol. 2014;61:408–417. doi: 10.1016/j.jhep.2014.04.041. [DOI] [PubMed] [Google Scholar]
- 36.Wu HC, Tsai HW, Teng CF, Hsieh WC, Lin YJ, Wang LH, Yuan Q, Su IJ. Ground-glass hepatocytes co-expressing hepatitis B virus X protein and surface antigens exhibit enhanced oncogenic effects and tumorigenesis. Hum Pathol. 2014;45:1294–1301. doi: 10.1016/j.humpath.2013.10.039. [DOI] [PubMed] [Google Scholar]
- 37.Na B, Huang Z, Wang Q, Qi Z, Tian Y, Lu CC, Yu J, Hanes MA, Kakar S, Huang EJ, et al. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PLoS One. 2011;6:e26240. doi: 10.1371/journal.pone.0026240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hildt E, Munz B, Saher G, Reifenberg K, Hofschneider PH. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002;21:525–535. doi: 10.1093/emboj/21.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawanaka M, Nishino K, Nakamura J, Oka T, Urata N, Goto D, Suehiro M, Kawamoto H, Kudo M, Yamada G. Quantitative Levels of Hepatitis B Virus DNA and Surface Antigen and the Risk of Hepatocellular Carcinoma in Patients with Hepatitis B Receiving Long-Term Nucleos(t)ide Analogue Therapy. Liver Cancer. 2014;3:41–52. doi: 10.1159/000343857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tseng TC, Liu CJ, Yang HC, Su TH, Wang CC, Chen CL, Hsu CA, Kuo SF, Liu CH, Chen PJ, et al. Serum hepatitis B surface antigen levels help predict disease progression in patients with low hepatitis B virus loads. Hepatology. 2013;57:441–450. doi: 10.1002/hep.26041. [DOI] [PubMed] [Google Scholar]
- 41.Liu J, Yang HI, Lee MH, Lu SN, Jen CL, Batrla-Utermann R, Wang LY, You SL, Hsiao CK, Chen PJ, et al. Spontaneous seroclearance of hepatitis B seromarkers and subsequent risk of hepatocellular carcinoma. Gut. 2014;63:1648–1657. doi: 10.1136/gutjnl-2013-305785. [DOI] [PubMed] [Google Scholar]
- 42.Voehringer D, Blaser C, Grawitz AB, Chisari FV, Buerki K, Pircher H. Break of T cell ignorance to a viral antigen in the liver induces hepatitis. J Immunol. 2000;165:2415–2422. doi: 10.4049/jimmunol.165.5.2415. [DOI] [PubMed] [Google Scholar]
- 43.Chen WN, Chen JY, Jiao BY, Lin WS, Wu YL, Liu LL, Lin X. Interaction of the hepatitis B spliced protein with cathepsin B promotes hepatoma cell migration and invasion. J Virol. 2012;86:13533–13541. doi: 10.1128/JVI.02095-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan AT, Koh S, Goh V, Bertoletti A. Understanding the immunopathogenesis of chronic hepatitis B virus: an Asian prospective. J Gastroenterol Hepatol. 2008;23:833–843. doi: 10.1111/j.1440-1746.2008.05385.x. [DOI] [PubMed] [Google Scholar]
- 45.Chisari FV, Isogawa M, Wieland SF. Pathogenesis of hepatitis B virus infection. Pathol Biol (Paris) 2010;58:258–266. doi: 10.1016/j.patbio.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertoletti A, Wang FS. Overview of the special issue on HBV immunity. Cell Mol Immunol. 2015;12:253–254. doi: 10.1038/cmi.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guidotti LG, Isogawa M, Chisari FV. Host-virus interactions in hepatitis B virus infection. Curr Opin Immunol. 2015;36:61–66. doi: 10.1016/j.coi.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moriyama T, Guilhot S, Klopchin K, Moss B, Pinkert CA, Palmiter RD, Brinster RL, Kanagawa O, Chisari FV. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science. 1990;248:361–364. doi: 10.1126/science.1691527. [DOI] [PubMed] [Google Scholar]
- 49.Chisari FV. Cytotoxic T cells and viral hepatitis. J Clin Invest. 1997;99:1472–1477. doi: 10.1172/JCI119308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 51.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 52.Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. J Virol. 1995;69:6158–6169. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia Y, Stadler D, Lucifora J, Reisinger F, Webb D, Hösel M, Michler T, Wisskirchen K, Cheng X, Zhang K, et al. Interferon-γ and Tumor Necrosis Factor-α Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology. 2016;150:194–205. doi: 10.1053/j.gastro.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 54.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 55.Rehermann B, Fowler P, Sidney J, Person J, Redeker A, Brown M, Moss B, Sette A, Chisari FV. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J Exp Med. 1995;181:1047–1058. doi: 10.1084/jem.181.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Webster GJ, Reignat S, Maini MK, Whalley SA, Ogg GS, King A, Brown D, Amlot PL, Williams R, Vergani D, et al. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology. 2000;32:1117–1124. doi: 10.1053/jhep.2000.19324. [DOI] [PubMed] [Google Scholar]
- 57.Wu JF, Chang MH. Natural history of chronic hepatitis B virus infection from infancy to adult life - the mechanism of inflammation triggering and long-term impacts. J Biomed Sci. 2015;22:92. doi: 10.1186/s12929-015-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mamun-Al-Mahtab SM, Uddin H, Khan SI, Rahman S. Early termination of immune tolerance state of hepatitis B virus infection explains liver damage. World J Hepatol. 2014;6:621–625. doi: 10.4254/wjh.v6.i8.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shimizu Y, Guidotti LG, Fowler P, Chisari FV. Dendritic cell immunization breaks cytotoxic T lymphocyte tolerance in hepatitis B virus transgenic mice. J Immunol. 1998;161:4520–4529. [PubMed] [Google Scholar]
- 60.Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, King AS, Herberg J, Gilson R, Alisa A, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med. 2000;191:1269–1280. doi: 10.1084/jem.191.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fisicaro P, Valdatta C, Boni C, Massari M, Mori C, Zerbini A, Orlandini A, Sacchelli L, Missale G, Ferrari C. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut. 2009;58:974–982. doi: 10.1136/gut.2008.163600. [DOI] [PubMed] [Google Scholar]
- 62.Busca A, Kumar A. Innate immune responses in hepatitis B virus (HBV) infection. Virol J. 2014;11:22. doi: 10.1186/1743-422X-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu L, Yin W, Sun R, Wei H, Tian Z. Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatology. 2014;59:443–452. doi: 10.1002/hep.26668. [DOI] [PubMed] [Google Scholar]
- 64.Zeisel MB, Lucifora J, Mason WS, Sureau C, Beck J, Levrero M, Kann M, Knolle PA, Benkirane M, Durantel D, et al. Towards an HBV cure: state-of-the-art and unresolved questions--report of the ANRS workshop on HBV cure. Gut. 2015;64:1314–1326. doi: 10.1136/gutjnl-2014-308943. [DOI] [PubMed] [Google Scholar]
- 65.Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J Hepatol. 2011;55:762–769. doi: 10.1016/j.jhep.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 66.Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, Rodgers S, Kurtovic J, Chang J, Lewin S, et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology. 2007;45:102–110. doi: 10.1002/hep.21482. [DOI] [PubMed] [Google Scholar]
- 67.Isorce N, Lucifora J, Zoulim F, Durantel D. Immune-modulators to combat hepatitis B virus infection: From IFN-α to novel investigational immunotherapeutic strategies. Antiviral Res. 2015;122:69–81. doi: 10.1016/j.antiviral.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 68.Gane EJ, Lim YS, Gordon SC, Visvanathan K, Sicard E, Fedorak RN, Roberts S, Massetto B, Ye Z, Pflanz S, et al. The oral toll-like receptor-7 agonist GS-9620 in patients with chronic hepatitis B virus infection. J Hepatol. 2015;63:320–328. doi: 10.1016/j.jhep.2015.02.037. [DOI] [PubMed] [Google Scholar]
- 69.Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49:S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 70.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol. 2010;17:222–229. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang FS, Xing LH, Liu MX, Zhu CL, Liu HG, Wang HF, Lei ZY. Dysfunction of peripheral blood dendritic cells from patients with chronic hepatitis B virus infection. World J Gastroenterol. 2001;7:537–541. doi: 10.3748/wjg.v7.i4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Op den Brouw ML, Binda RS, van Roosmalen MH, Protzer U, Janssen HL, van der Molen RG, Woltman AM. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: a possible immune escape mechanism of hepatitis B virus. Immunology. 2009;126:280–289. doi: 10.1111/j.1365-2567.2008.02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Webster G, Bertoletti A. Quantity and quality of virus-specific CD8 cell response: relevance to the design of a therapeutic vaccine for chronic HBV infection. Mol Immunol. 2001;38:467–473. doi: 10.1016/s0161-5890(01)00082-7. [DOI] [PubMed] [Google Scholar]
- 75.Miyara M, Sakaguchi S. Human FoxP3(+)CD4(+) regulatory T cells: their knowns and unknowns. Immunol Cell Biol. 2011;89:346–351. doi: 10.1038/icb.2010.137. [DOI] [PubMed] [Google Scholar]
- 76.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 77.Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, Zhao JM, Zhang B, Shi M, Ding X, et al. Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis B. J Immunol. 2006;177:739–747. doi: 10.4049/jimmunol.177.1.739. [DOI] [PubMed] [Google Scholar]
- 78.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fife BT, Pauken KE. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N Y Acad Sci. 2011;1217:45–59. doi: 10.1111/j.1749-6632.2010.05919.x. [DOI] [PubMed] [Google Scholar]
- 80.Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. 2015;6:e1694. doi: 10.1038/cddis.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen CJ, Yang HI, Iloeje UH. Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology. 2009;49:S72–S84. doi: 10.1002/hep.22884. [DOI] [PubMed] [Google Scholar]
- 82.Invernizzi F, Viganò M, Grossi G, Lampertico P. The prognosis and management of inactive HBV carriers. Liver Int. 2016;36 Suppl 1:100–104. doi: 10.1111/liv.13006. [DOI] [PubMed] [Google Scholar]
- 83.Tong MJ, Trieu J. Hepatitis B inactive carriers: clinical course and outcomes. J Dig Dis. 2013;14:311–317. doi: 10.1111/1751-2980.12051. [DOI] [PubMed] [Google Scholar]
- 84.Shimizu Y. T cell immunopathogenesis and immunotherapeutic strategies for chronic hepatitis B virus infection. World J Gastroenterol. 2012;18:2443–2451. doi: 10.3748/wjg.v18.i20.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ebert G, Allison C, Preston S, Cooney J, Toe JG, Stutz MD, Ojaimi S, Baschuk N, Nachbur U, Torresi J, et al. Eliminating hepatitis B by antagonizing cellular inhibitors of apoptosis. Proc Natl Acad Sci USA. 2015;112:5803–5808. doi: 10.1073/pnas.1502400112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ebert G, Preston S, Allison C, Cooney J, Toe JG, Stutz MD, Ojaimi S, Scott HW, Baschuk N, Nachbur U, et al. Cellular inhibitor of apoptosis proteins prevent clearance of hepatitis B virus. Proc Natl Acad Sci USA. 2015;112:5797–5802. doi: 10.1073/pnas.1502390112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 88.Paterlini-Bréchot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, Lagorce D, Bréchot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–3916. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 89.Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007;252:157–170. doi: 10.1016/j.canlet.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 90.Lau CC, Sun T, Ching AK, He M, Li JW, Wong AM, Co NN, Chan AW, Li PS, Lung RW, et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell. 2014;25:335–349. doi: 10.1016/j.ccr.2014.01.030. [DOI] [PubMed] [Google Scholar]
- 91.Summers J, Jilbert AR, Yang W, Aldrich CE, Saputelli J, Litwin S, Toll E, Mason WS. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc Natl Acad Sci USA. 2003;100:11652–11659. doi: 10.1073/pnas.1635109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res. 2013;98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Block TM, Rawat S, Brosgart CL. Chronic hepatitis B: A wave of new therapies on the horizon. Antiviral Res. 2015;121:69–81. doi: 10.1016/j.antiviral.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Menéndez-Arias L, Álvarez M, Pacheco B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: mechanism of action and resistance. Curr Opin Virol. 2014;8:1–9. doi: 10.1016/j.coviro.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 95.Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM, Lok AS. Present and future therapies of hepatitis B: From discovery to cure. Hepatology. 2015;62:1893–1908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tavis JE, Lomonosova E. The hepatitis B virus ribonuclease H as a drug target. Antiviral Res. 2015;118:132–138. doi: 10.1016/j.antiviral.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, Hegge JO, Klein JJ, Wakefield DH, Oropeza CE, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973–985. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Billioud G, Kruse RL, Carrillo M, Whitten-Bauer C, Gao D, Kim A, Chen L, McCaleb ML, Crosby JR, Hamatake R, et al. In vivo reduction of hepatitis B virus antigenemia and viremia by antisense oligonucleotides. J Hepatol. 2016;64:781–789. doi: 10.1016/j.jhep.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 99.Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, Mason WS, Xu X, Guo JT, Block TM, et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother. 2012;56:4277–4288. doi: 10.1128/AAC.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hoeksema KA, Tyrrell DL. Inhibition of viral transcription using designed zinc finger proteins. Methods Mol Biol. 2010;649:97–116. doi: 10.1007/978-1-60761-753-2_6. [DOI] [PubMed] [Google Scholar]
- 101.Zimmerman KA, Fischer KP, Joyce MA, Tyrrell DL. Zinc finger proteins designed to specifically target duck hepatitis B virus covalently closed circular DNA inhibit viral transcription in tissue culture. J Virol. 2008;82:8013–8021. doi: 10.1128/JVI.00366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol Ther. 2013;21:1889–1897. doi: 10.1038/mt.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen J, Zhang W, Lin J, Wang F, Wu M, Chen C, Zheng Y, Peng X, Li J, Yuan Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol Ther. 2014;22:303–311. doi: 10.1038/mt.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, Recchia A, Cathomen T, Gonçalves MA. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63. doi: 10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 108.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin SR, Yang HC, Kuo YT, Liu CJ, Yang TY, Sung KC, Lin YY, Wang HY, Wang CC, Shen YC, et al. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates In Vivo. Mol Ther Nucleic Acids. 2014;3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kennedy EM, Bassit LC, Mueller H, Kornepati AV, Bogerd HP, Nie T, Chatterjee P, Javanbakht H, Schinazi RF, Cullen BR. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology. 2015;476:196–205. doi: 10.1016/j.virol.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Seeger C, Sohn JA. Targeting Hepatitis B Virus With CRISPR/Cas9. Mol Ther Nucleic Acids. 2014;3:e216. doi: 10.1038/mtna.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu X, Hao R, Chen S, Guo D, Chen Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J Gen Virol. 2015;96:2252–2261. doi: 10.1099/vir.0.000159. [DOI] [PubMed] [Google Scholar]
- 113.Zhen S, Hua L, Liu YH, Gao LC, Fu J, Wan DY, Dong LH, Song HF, Gao X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015;22:404–412. doi: 10.1038/gt.2015.2. [DOI] [PubMed] [Google Scholar]
- 114.Dong C, Qu L, Wang H, Wei L, Dong Y, Xiong S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015;118:110–117. doi: 10.1016/j.antiviral.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 115.Ramanan V, Shlomai A, Cox DB, Schwartz RE, Michailidis E, Bhatta A, Scott DA, Zhang F, Rice CM, Bhatia SN. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci Rep. 2015;5:10833. doi: 10.1038/srep10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Karimova M, Beschorner N, Dammermann W, Chemnitz J, Indenbirken D, Bockmann JH, Grundhoff A, Lüth S, Buchholz F, Schulze zur Wiesch J, et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci Rep. 2015;5:13734. doi: 10.1038/srep13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang J, Xu ZW, Liu S, Zhang RY, Ding SL, Xie XM, Long L, Chen XM, Zhuang H, Lu FM. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J Gastroenterol. 2015;21:9554–9565. doi: 10.3748/wjg.v21.i32.9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu S, Zhang H, Gu C, Yin J, He Y, Xie J, Cao G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst. 2009;101:1066–1082. doi: 10.1093/jnci/djp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu Y, Gan Y, Gao F, Zhao Z, Jin Y, Zhu Y, Sun Z, Wu H, Chen T, Wang J, et al. Novel natural mutations in the hepatitis B virus reverse transcriptase domain associated with hepatocellular carcinoma. PLoS One. 2014;9:e94864. doi: 10.1371/journal.pone.0094864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yu W, Goddard C, Clearfield E, Mills C, Xiao T, Guo H, Morrey JD, Motter NE, Zhao K, Block TM, et al. Design, synthesis, and biological evaluation of triazolo-pyrimidine derivatives as novel inhibitors of hepatitis B virus surface antigen (HBsAg) secretion. J Med Chem. 2011;54:5660–5670. doi: 10.1021/jm200696v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Noordeen F, Vaillant A, Jilbert AR. Nucleic acid polymers inhibit duck hepatitis B virus infection in vitro. Antimicrob Agents Chemother. 2013;57:5291–5298. doi: 10.1128/AAC.01003-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang YJ, Yang L, Zuo JP. Recent developments in antivirals against hepatitis B virus. Virus Res. 2016;213:205–213. doi: 10.1016/j.virusres.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 123.Urban S, Bartenschlager R, Kubitz R, Zoulim F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology. 2014;147:48–64. doi: 10.1053/j.gastro.2014.04.030. [DOI] [PubMed] [Google Scholar]