

Sir,
We recently reported in Brain a large multi-centre study suggesting that truncating SYNE1 mutations are a recurrent cause of recessive ataxia also outside Quebec (23/434 = 5.3% of patients with unexplained early-onset ataxia) (Synofzik et al., 2016). Moreover, this study indicated that SYNE1 ataxia might commonly present with complex multisystemic phenotypes rather than pure cerebellar ataxia, including in particular motor neuron and brainstem dysfunction (Synofzik et al., 2016). However, confirmation of both the frequency estimate and the complex phenotypic spectrum is still lacking, raising the question whether these findings indeed represent systematic results rather than just exceptional or coincidental associations.
Here, we now report the mutational and phenotypic findings on SYNE1 from a second, independent ataxia series of 116 patients. These findings not only confirm the high frequency of SYNE1 ataxia and extend both the mutational spectrum (seven novel index patients, 12 novel SYNE1 mutations) and the multisystemic phenotypic spectrum, including amyotrophic lateral sclerosis (ALS)-like motor neuron features, they also indicate that muscle immunohistochemistry might provide a valuable diagnostic biomarker for clarifying the pathogenic contribution of SYNE1 missense variants. This observation may have consequences for clinical SYNE1 diagnostics, as diagnostic tests are urgently needed for clarifying the role of the ubiquitous SYNE1 missense variants with unknown clinical significance (VUS), which are frequently found in neurological and non-neurological patients and controls (Synofzik et al., 2016).
Index subjects (n = 116) with unexplained degenerative ataxia compatible with autosomal recessive inheritance (no ataxia in the parental generation) and negative for trinucleotide repeat expansions causing Friedreich’s ataxia (FRDA) were compiled from three sources: the Early Onset Ataxia Consortium (n = 88), the ataxia centre Antwerp, Belgium (n = 9), and the ataxia centre Milano, Italy (n = 19). All subjects originated from European, Middle East or Mediterranean countries. This series was sequenced after and independent from the cohort of the previous SYNE1 study (Synofzik et al., 2016). None of the subjects had been part of the previous screening cohort. Subjects were screened for SYNE1 mutations by one of the following three next-generation sequencing methods: (i) a high coverage HaloPlex gene panel kit (Agilent) including >120 known ataxia genes (n = 88); (ii) targeted exon-capture sequencing strategy (Illumina Nextera Rapid Capture Custom kit) including 107 known ataxia genes (n = 19); or (iii) whole-exome sequencing using the SureSelect Human All Exon 50 Mb kit (Agilent) (n = 9) (for technical details, filter settings, and criteria for inclusion of SYNE1 missense variants, see the online Supplementary material). All index patients carrying two pathogenic SYNE1 alleles and their affected siblings received a systematic clinical assessment, as described in detail in the previous study (Synofzik et al., 2016) (Table 1).
Table 1.
Clinical, imaging and electrophysiological features of SYNE1 patients
| Patient ID | Phenotype category | Genotype (cDNA) | Origin/ gender | SARA score | SDFS | Age of onset | First clinical symptom | Age at last examin ation (years) | Cerebellar ataxia | Dysfunction upper motor neuron | Dysfunction lower motor neuron | Additional clinical features | Cerebellar atrophy (MRI) | Peripheral neuropathy (NCS) | EMG changes (muscle) | Abnormal MEPs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1-1 | Ataxia MND | c.18583C>T; c.18583C>T; | Turkey/m | 10 | 2 | 17 | Fasciculations face | 25 | +; onset 20 yrs | + signs; no spasticity | Fasciculations face | + | n | Anterior tibial and deltoid muscle: acute and chronic denervation | Prolonged UL + LL | |
| 1-2 | Ataxia MND | c.18583C > T; c.18583C > T; | Turkey/m | 23 | 3 | 11 | Speech disturbance | 28 | +; onset 17 yrs | + signs; no spasticity | + thenar and hypothenar atrophy; fibrillation tongue | CK elevation (257U/l), pes equinovarus, | + | Tibial CMAP amplitude reduced | Anterior tibial and deltoid muscle: acute and chronic denervation | Prolonged UL + LL |
| 2-1 | Ataxia MND | c.23554_23555delCA; c.4732C > T | Belgium/f | 14 | 5 | 6 | Mild spasticity LL | 28 | +; onset 15yrs | + signs; no spasticity | + thenar atrophy (4-/5), mainly proximal in LL (4/5) | Unilateral diaphragm paralysis left with restrictive lung function, CK elevation (341 U/L), | n (age 22 years) | Reduced CMAP amplitudes LL; normal SNAP | Chronic denervation proximal and distal PSW, fibrillation potentials | n.d. |
| 3-1 | Ataxia MND | c.24814C > T c.13567_13570delGAAG | Turkey/f | 19 | 6 | 28 | Gait disturbance | 37 | +; onset 28yrs | + signs; mild spasticity | - | Downbeat nystagmus | + | n | n | Normal UL, LL |
| 4-1 | Ataxia cognition | c.15029_15032delAGGA; c.15438 + 2T > A | Germany/f | 14.5 | 4 | 42 | Gait disturbance | 49 | +; onset 42yrs | - | - | Cognitive impairment: attention, memory, executive function | + | n | n | Normal UL, LL |
| 5-1 | Ataxia MND | c.10379_10380delAT; c.10996_ 11003delCTGGACGA | Germany/m | 13 | 3 | 23 | Gait disturbance | 44 | +; onset 23yrs | + spasticity | fasciculations and atrophy tongue | Writer's cramp, polyneuropathy | + | Reduced sural SNAP and SNCV, normal tibial NCS | n.d. | n.d. |
| 6-1 | Ataxia MND cognition | c.1903dupA; c.1903dupA | Italy/m | 9,5 | 2 | 7 | Cognitive deficits; gait disturbance | 35 | +; onset 7 yrs | + signs; spasticity | - | Cognitive impairment: attention, memory, executive function; impaired colour vision; urinary dysfunction; depression | + | n.d. | n.d. | n |
| 7-1 | Ataxia MND | c.25655delG; c.21971C > A | Italy/m | 20 | 3 | 6 | Speech and gait disturbance | 27 | +; onset 6 yrs | + signs; spasticity | distal atrophy UL and LL | Hammer-toes, motor polyneuropathy | + | Reduced CMAP, normal, SNAP | Acute denervation LL and UL | n.d. |
Patient ID = family number_individual number; ataxia MND = cerebellar ataxia plus motor neuron disease involving upper and/or lower neuron damage; m = male; f = female; + = present; - = absent; SARA = scale for the assessment and rating of ataxia; SDFS = spinocerebellar degeneration functional score, assessing disability stage from 1 to 7 (0: no functional handicap; 1: no functional handicap but signs at examination; 2: mild, able to run, walking unlimited; 3: moderate, unable to run, limited walking without help; 4: severe, walking with one stick; 5: walking with two sticks; 6: unable to walk, requiring wheelchair; 7: confined to the bed); upper motor neuron signs = extensor plantar response positive and/or hyperreflexia of muscle tendon reflexes; UL = upper limb, LL = lower limb; CK = creatine kinase; NCS = nerve conduction studies; CMAP = compound muscle action potential; SNAP = sensory nerve action potential; SNCV = sensory nerve conduction velocity; EMG = electromyography; MEP = motor evoked potentials; PSW = positive sharp waves; n.d. = not done; n = normal.
We identified six index patients carrying two truncating SYNE1 alleles and one index patient carrying one truncating plus one missense SYNE1 allele, thus yielding a total of seven index patients out of 116 in the total ataxia cohort (6%). In these seven index patients, we observed a total of 12 different mutations, consisting of seven frameshift, three nonsense, one splice site, and one missense mutation in the actin binding domain (Table 2 and Fig. 1A). All 12 mutations, each confirmed by Sanger sequencing, have not yet been reported in association with human disease. For all families where DNA of at least one parent was available (5/7 families), we were able to show that the respective parent carried only one of the two corresponding SYNE1 variants, supporting a biallelic localization of the variants in the index child. In one of the remaining two families, consanguinity was also suggestive of a biallelic location of the observed homozygous mutations.
Table 2.
SYNE1 mutations identified in this study
| Variant ID | Family ID | Phenotype | Genomic variant | cDNA variant | Protein change | Variant type | Zygosity | PhyloP | CADD score | MAF 1000 genomes | MAF ExAC | MAF EVS6500 | in HGMD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | #1 | Ataxia MND | chr6:152590412G > A | c.18583C > T | p.Q6195* | Stopgain SNV | Homo | 8.90 | 56 | 0 | 0 | 0 | absent |
| 2 | #2 | Ataxia MND | chr6:152485320_ 152485321delTG | c.23767_23768delCA | p.Q7923Efs*4 | Frameshift deletion | Het | NA | 43 | 0 | 0 | 0 | absent |
| 3 | #2 | Ataxia MND | chr6:152751303G > A | c.4732C > T | p.P1578S | Missense | Het | 6.03 | 24.2 | 0 | 2.26 × 10−4 | 7.70 × 10−5 | absent |
| 4 | #3 | Ataxia MND | chr6:152469342G > A | c.24814C > T | p.R8272* | Stopgain SNV | Het | 1.93 | 53 | 0 | 8.25 × 10−6 | 0 | absent |
| 5 | #3 | Ataxia MND | chr6:152652250_ 152652253delCTTC | c.13567_13570delGAAG | p.E4523Sfs*34 | Frameshift | Het | NA | 35 | 0 | 0 | 0 | absent |
| 6 | #4 | Ataxia cognition | chr6:152647692_ 152647695delTCCT | c.15029_15032delAGGA | p.E5010Vfs*13 | Frameshift | Het | NA | 37 | 0 | 0 | 0 | absent |
| 7 | #4 | Ataxia cognition | chr6:152647091A > T | c.15438 + 2T > A | ? | Splice donor | Het | 5.79 | 24.1 | 0 | 0 | 0 | absent |
| 8 | #5 | Ataxia MND | chr6:152680513_ 152680514delAT | c.10379_10380delAT | p.Y3460Cfs*28 | Frameshift | Het | NA | 36 | 0 | 0 | 0 | absent |
| 9 | #5 | Ataxia MND | chr6:152674803_ 152674810del TCGTCCAG | c.10996_11003delCT GGACGA | p.L3666Efs*51 | Frameshift | Het | NA | 37 | 0 | 0 | 0 | absent |
| 10 | #6 | Ataxia MND | chr6:152786422dup | c.1903dupA | p.M635Nfs*35 | Frameshift | Homo | NA | 33 | 0 | 0 | 0 | absent |
| 11 | #7 | Ataxia MND | chr6:152457757delC | c.25655delG | p.C8552Sfs*7 | Frameshift | Het | NA | 46 | 0 | 0 | 0 | absent |
| 12 | #7 | Ataxia MND | chr6:152540211G > T | c.21971C > A | p.S7324* | Stopgain SNV | Het | 7.44 | 59 | 0 | 8.24 × 10−6 | 0 | absent |
Genomic positions of the variants according to genome build hg19.
DNA changes according to NM_182961.3. Variant type and protein changes according to GVS function based on NP_149062. MND = motor neuron disease; zygosity = homozygous (homo) or heterozygous (het); PhyloP = PhyloP conservation score based on base-wise conservation across 100 vertebrates; CADD score = scaled Combined Annotation Dependent Depletion score, integrating many diverse annotations into a single measure (C score) for each variant. The predicted pathogenicity of each variant is scored and ranked relative to all ∼8.6 billion single nucleotide variants of the GRCh37/hg19 reference. A scaled CADD score of 20 indicates variants at the top 1%, a CADD score of 30 indicates variants at the top 0.1%, etc. (Kircher et al., 2014). MAF = minor allele frequency; ExAC = Exome Aggregation Consortium; EVS = Exome Variant Server 6500 exomes all from the NHLBI GO Exome Sequencing Project; HGMD = Human Gene Mutation Database; NA = not available.
Figure 1.
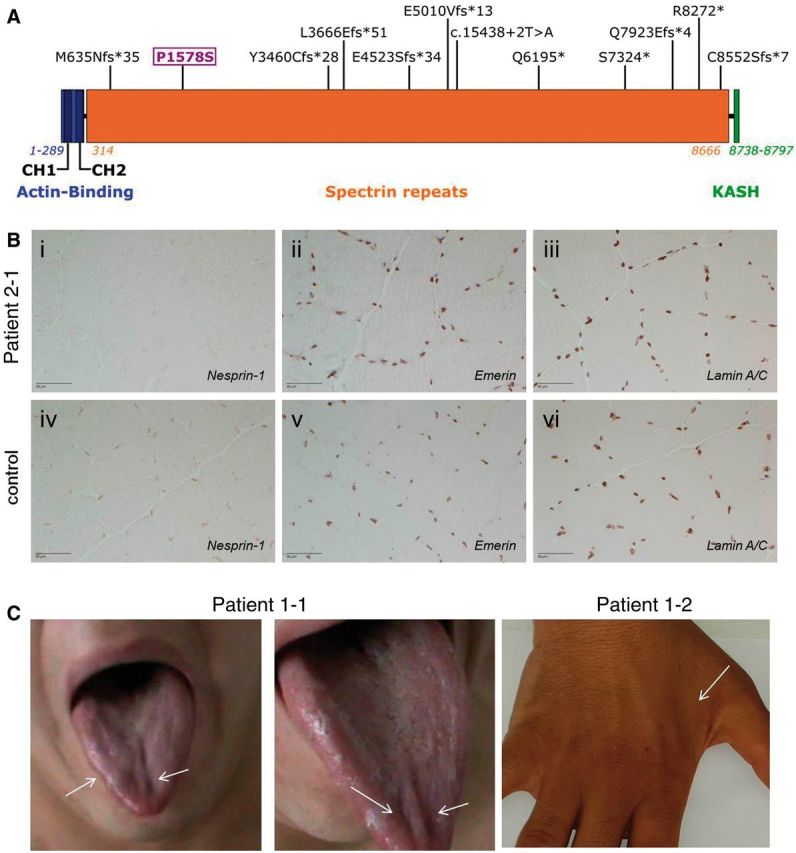
Mutational spectrum, muscle immunolabelling, and atrophy findings in SYNE1 patients. (A) Graphical overview of the mutations found in this study in relation to the SYNE1 domains. The mutations identified in this study are indicated at their respective position in the SYNE1 gene. The N-terminal actin-binding domain (in blue) contains two calponin homology domains (CH1 and CH2); spectrin repeats (orange) contain all the mutations detected in this study; the C-terminal KASH domain (Klarsicht/ANC-1/Syne homology domain) is coloured in green. The missense variant investigated in this study is marked in purple. (B) Absent SYNE1 staining in muscle tissue in a patient carrying a missense plus a truncating SYNE1 mutation. After immunolabelling of nesprin-1, staining of the nuclear envelope was absent in Patient 2-1 carrying a missense (c.4732C > T; p.P1578S) plus a truncating SYNE1 mutation (i), but present in quadriceps muscle of a healthy control (iv) (peroxidase-antiperoxidase technique). Immunolabelling of emerin and lamin A/C at the inner nuclear membrane were normal in patient and control (ii/iii and v/vi, respectively) (avidin-biotin complex technique), thus ruling out an unspecific lack of staining in this subject. Scale bar = 50 µm. (C) Tongue and interosseous muscle atrophy illustrating lower motor neuron degeneration in SYNE1 disease. Photographs of the tongue of Patient 1-1 (left) and of the hand of Patient 1-2 (right) illustrating atrophy of the right lateral and medial tongue and of the first dorsal interosseous muscle, respectively, illustrating affection of bulbar and cervical motor neurons in SYNE1 disease. The tongue also showed generalized fasciculations (not seen on the static photographic image).
The missense variant c.4732C>T; p.P1578S (observed in Patient 2-1) (i) segregated in trans with the frameshift deletion c.23767_23768delCA; p.Q7923Efs*4; (ii) had a very low minor allele frequency in ExAc (2.26E-04) and EVS6500 (7.70E-05); (iii) predicted to be damaging by three out of three in silico algorithms (Mutation Taster) (Schwarz et al., 2010; Wang et al., 2010); PolyPhen-2 HDIV (Adzhubei et al., 2010), and Likelihood Ratio Test (LRT) (Chun and Fay, 2009); (iv) highly evolutionary conserved with scores PhyloP 100way = 6.03 and PhastCons 100way = 1.0 (Pollard et al., 2010); and (v) ranked among the top 1% of all 8.6 billion single nucleotide variants in the GRCh37/hg19 (CADD score: 24.2) (Kircher et al., 2014). However, given that even rare, well-conserved missense SYNE1 variants have been shown to present a ubiquitous finding in control subjects (Synofzik et al., 2016), additional functional evidence is needed to demonstrate a pathogenic contribution. Immunohistochemistry assessment of muscle tissue in Patient 2-1 showed severely reduced SYNE1 staining (Fig. 1B; for methodological details see Supplementary material), in line with the findings seen in patients with two truncating SYNE1 mutations (Synofzik et al., 2016). This suggests that the p.P1578S missense mutation, in combination with another truncating SYNE1 variant, leads to loss of SYNE1 protein.
Clinical data were available for all eight affected subjects belonging to the seven index families. Age of disease onset was variable, ranging from 6 to 42 years (median onset: 14 years). Disease started in 4/8 patients (50%) with non-ataxia features, namely facial muscle fasciculations, speech disturbances, spasticity and cognitive deficits, respectively. At last examination (median age: 35 years), all 8/8 patients showed a ‘cerebellar ataxia plus’ phenotype, i.e. none of them showed the classical SYNE1 phenotype of pure cerebellar ataxia. Seven of eight subjects (88%) exhibited ataxia plus motor neuron disease, which involved both upper motor neuron dysfunction (bilateral positive extensor plantar reflex and/or spasticity) and lower motor neuron dysfunction muscle atrophy, including bulbar muscles (Fig. 1C) combined with reduced reflexes; fasciculations clinically or on EMG; acute denervation in EMG] in 5/8 patients (63%), and only upper motor dysfunction in 2/8 patients (25%). In 2/8 patients (one also with motor neuron disease), ataxia was complicated by additional moderate-to-severe cognitive impairment across manifold neuropsychological domains, affecting in particular processing speed, attention, memory, and executive functions (for detailed neuropsychological test results of Patient 4-1, see Supplementary material). One index patient (Patient 2-1) showed a severe and complex multisystemic phenotype, comprising of very early onset (6 years of age) ataxia, upper and lower motor neuron damage, including acute neurogenic changes with creatine kinase elevation, and unilateral diaphragm paralysis with restrictive lung function. This finding confirms that the complex early-onset phenotypes reported in three subjects in the previous report (Synofzik et al., 2016) are a recurrent manifestation of SYNE1 disease.
These findings provide evidence from an independent, second series that SYNE1 deficiency is indeed a relatively common cause of non-FRDA recessive ataxia also outside Quebec, with a frequency of 5% (Synofzik et al., 2016) to 6% (this report). Moreover, these findings further extend the mutational spectrum of SYNE1 disease by 12 novel mutations spread throughout the gene (yet sparing the KASH-domain, Fig. 1A), demonstrating the need to sequence not only particular ‘hot spot’ regions, but indeed the whole giant 146-exon gene.
These findings also help to explicate the multisystemic spectrum and disease course of SYNE1 disease. They show that multisystemic ataxia plus syndromes are not the exception, but the rule, with up to 100% of SYNE1 patients outside Canada presenting with ataxia plus syndromes, in particular complicated by upper and/or lower motor neuron disease. In up to 50% of patients, SYNE1 disease even starts with non-cerebellar features. The combination of upper plus lower motor neuron disease (seen in 63% of patients), which can include fasciculations, acute denervation and damage of bulbar motor neurons (see tongue atrophy, Fig. 1C), resembles ALS-like motor neuron features. Our findings demonstrate that such complex early-onset syndromes with upper and lower motor neuron disease and respiratory features are not a coincidental finding, but a recurrent manifestation of truncating SYNE1 mutations (Izumi et al., 2013; Synofzik et al., 2016). This contrasts the view on SYNE1 ataxia as relatively benign, slowly progressive ataxia, which was largely traced from French-Canadian SYNE1 patients (Dupre et al., 1993/2012, 2007).
Determining the pathogenicity of SYNE1 missense mutations will present a complex challenge in future clinical diagnostics, given that even rare, well conserved missense variants can be found in 5.6% of controls with unrelated disease conditions or phenotypes (Synofzik et al., 2016). Here we show that SYNE1 muscle staining might help to show a loss of SYNE1 protein in subjects carrying a SYNE1 missense variant. Muscle immunohistochemistry would thus help to corroborate the pathogenicity of at least some well selected, rare, highly conserved SYNE1 missense variants, in particular if located in trans with another truncating variant.
Supplementary Material
Acknowledgements
We are grateful to J. Reichbauer (Hertie-Institute for Clinical Brain Research, Tübingen) and Inge Bats (University of Antwerpen) for technical support.
Funding
The sequencing work of this study was supported, in part, by Actelion pharmaceuticals. This company was not involved in any parts of data acquisition, data analysis or data interpretation. This work was moreover supported by the Else Kröner-Fresenius-Stiftung (award to M.S.), the European Union [grant F5-2012-305121 ‘NEUROMICS’ to L.S., P.B., T.K., and grant PIOF-GA-2012-326681 ‘HSP/CMT genetics’ and ‘NEUROLIPID’ (01GM1408B) to R.S.], E-RARE grants of the respective national research ministries to the EUROSCAR project (to L.S., P.B., and F.T.) (grant 01GM1206), grant RF-2011-02351165 from the Italian Ministry of Health to F.T., the EUROSPA project (grant 01GM0807) (to L.S. and P.B.), and the National Institute of Health (NIH) (grants 5R01NS072248, 1R01NS075764, 5R01NS054132, 2U54NS065712 all to S.Z.). This work was supported by the Association Belge contre les Maladies Neuromusculaires (ABMM). I.M. is supported by a PhD fellowship of the agency for Innovation by Science and Technology (IWT). J.B. is supported by a Senior Clinical Researcher mandate of the Research Fund - Flanders (FWO).
Conflicts of interest: Dr Bauer is Chief Operating Officer at Centogene AG, Rostock, since January 2016. This company has no direct market-related interests in this study and was not involved in any parts of this study. Dr Synofzik received consulting fees from Actelion Pharmaceuticals Ltd. The remaining authors report no conflicts of interest.
Supplementary material
Supplementary material is available at Brain online.
Appendix 1
We acknowledge the following members of the Early Onset Ataxia (EOA) consortium for their contribution of ataxia patients to the screening cohort: Thomas Klopstock, Claudia Stendel (Department of Neurology with Friedrich-Baur-Institute, Ludwig-Maximilians-University, Munich, Germany); Christoph Kamm, Ales Dudesek (Department of Neurology, University of Rostock); Stefan Vielhaber, Dorothea Henkel (Department of Neurology, University of Magdeburg); Janina Gburek-Augustat (Tübingen).
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res 2009; 19: 1553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre N, Gros-Louis F, Bouchard JP, Noreau A, Rouleau GA. SYNE1-related autosomal recessive cerebellar ataxia. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. Seattle, WA: University of Washington: 1993/2012. Accessed on March 26th, 2016. [Google Scholar]
- Dupre N, Gros-Louis F, Chrestian N, Verreault S, Brunet D, de Verteuil D, et al. Clinical and genetic study of autosomal recessive cerebellar ataxia type 1. Ann Neurol. 2007Jul;62(1):93-8. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Miyamoto R, Morino H, Yoshizawa A, Nishinaka K, Udaka F, et al. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology 2013; 80: 600–1. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010; 20: 110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–6. [DOI] [PubMed] [Google Scholar]
- Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014; 137(Pt 1): 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmüller C, Baets J, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: findings from a large-scale multi-center screening. Brain; 2016; 139: 1378–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.