

Resurgent sodium currents arise from channel reopening during repolarisation, and are predicted to increase neuronal excitability. Patel et al. show that epilepsy-associated mutations in the voltage-gated sodium channel Nav1.6, but not Nav1.1, upregulate resurgent currents. Cannabidiol preferentially targets these currents, suggesting a strategy for reducing neuronal hyperexcitability associated with epilepsy.
Keywords: cannabidiol, Dravet syndrome, GEFS+, resurgent current, VGSC

Resurgent sodium currents arise from channel reopening during repolarisation, and are predicted to increase neuronal excitability. Patel et al. show that epilepsy-associated mutations in the voltage-gated sodium channel Nav1.6, but not Nav1.1, upregulate resurgent currents. Cannabidiol preferentially targets these currents, suggesting a strategy for reducing neuronal hyperexcitability associated with epilepsy.
Abstract
Mutations in brain isoforms of voltage-gated sodium channels have been identified in patients with distinct epileptic phenotypes. Clinically, these patients often do not respond well to classic anti-epileptics and many remain refractory to treatment. Exogenous as well as endogenous cannabinoids have been shown to target voltage-gated sodium channels and cannabidiol has recently received attention for its potential efficacy in the treatment of childhood epilepsies. In this study, we further investigated the ability of cannabinoids to modulate sodium currents from wild-type and epilepsy-associated mutant voltage-gated sodium channels. We first determined the biophysical consequences of epilepsy-associated missense mutations in both Nav1.1 (arginine 1648 to histidine and asparagine 1788 to lysine) and Nav1.6 (asparagine 1768 to aspartic acid and leucine 1331 to valine) by obtaining whole-cell patch clamp recordings in human embryonic kidney 293T cells with 200 μM Navβ4 peptide in the pipette solution to induce resurgent sodium currents. Resurgent sodium current is an atypical near threshold current predicted to increase neuronal excitability and has been implicated in multiple disorders of excitability. We found that both mutations in Nav1.6 dramatically increased resurgent currents while mutations in Nav1.1 did not. We then examined the effects of anandamide and cannabidiol on peak transient and resurgent currents from wild-type and mutant channels. Interestingly, we found that cannabidiol can preferentially target resurgent sodium currents over peak transient currents generated by wild-type Nav1.6 as well as the aberrant resurgent and persistent current generated by Nav1.6 mutant channels. To further validate our findings, we examined the effects of cannabidiol on endogenous sodium currents from striatal neurons, and similarly we found an inhibition of resurgent and persistent current by cannabidiol. Moreover, current clamp recordings show that cannabidiol reduces overall action potential firing of striatal neurons. These findings suggest that cannabidiol could be exerting its anticonvulsant effects, at least in part, through its actions on voltage-gated sodium channels, and resurgent current may be a promising therapeutic target for the treatment of epilepsy syndromes.
Introduction
There are over 700 mutations identified in brain isoforms of voltage-gated sodium channels (VGSCs) in patients with distinct epileptic phenotypes. The majority of these mutations occur in the gene (SCN1A) encoding Nav1.1 and result in a phenotype known as generalized epilepsy with febrile seizures plus or a more severe form of this termed Dravet syndrome. These mutations lead to mostly protein truncation and amino acid substitutions that occur throughout the channel protein. Functionally, they are primarily thought to cause loss of channel activity. In accordance with the prominent role of Nav1.1 in parvalbumin-positive GABAergic neurons, the prevailing hypothesized mechanism underlying Dravet syndrome is the loss of Nav1.1 channel activity leading to decreased excitability of GABAergic neurons and consequently an increase in circuit excitability (Yu et al., 2006; Ogiwara et al., 2007; Cheah et al., 2012; Dutton et al., 2013; Hedrich et al., 2014). Indeed, selective deletion of Nav1.1 in parvalbumin-positive GABAergic neurons can lead to seizures and mimic a Dravet syndrome phenotype (Cheah et al., 2012; Han et al., 2012; Kalume et al., 2013; Ogiwara et al., 2013). Moreover, Higurashi et al. (2013) have observed reduced excitability of GABAergic neurons differentiated from pluripotent stem cells derived from a human patient. However, it is important to note that recent data from human patient-derived induced pluripotent stem cells challenge this hypothesis and suggests that rather there is an overall upregulation of VGSC activity leading to increased excitability of both excitatory and inhibitory neurons (Liu et al., 2013; Chopra and Isom, 2014). Recently, there have been mutations identified in SCN8A (coding for Nav1.6) in patients with a severe early infantile epileptic encephalopathy (Veeramah et al., 2012; Carvill et al., 2013; O'Brien and Meisler, 2013; Blanchard et al., 2015; Kong et al., 2015; Larsen et al., 2015). Of the Nav1.6 mutations characterized in heterologous expression systems, it has been found that most of these mutations result in gain-of-function in channel properties (Veeramah et al., 2012; Estacion et al., 2014), although putative loss-of-function mutations have also been reported (de Kovel et al., 2014; Blanchard et al., 2015). Mutations in these two isoforms cause phenotypically distinct syndromes and understanding the functional consequences of these mutations can provide invaluable insight into the potential role of these channels in physiological and pathophysiological conditions.
Approximately 30% of patients with epilepsy are refractory to treatment; therefore there is a great need for the development of alternative anti-epileptic medications. Cannabidiol has recently received attention for its potential efficacy in the treatment of childhood epilepsies (Devinsky et al., 2016) although more studies are needed to confirm this finding. Intriguingly, endogenous, exogenous and synthetic cannabinoids have been shown to target peak transient currents generated by VGSCs (Turkanis et al., 1991; Theile and Cummins, 2011; Foadi et al., 2014; Hill et al., 2014; Okura et al., 2014). In addition, our laboratory has previously shown that with Nav1.7, which is present in dorsal root ganglion neurons but not expressed in the brain, anandamide can selectively inhibit resurgent current over peak transient current (Theile and Cummins, 2011). Furthermore, Foadi et al. (2014) found that a synthetic derivative of Δ9-tetrahydrocannabinol, ajulemic acid, can inhibit resurgent currents generated by Nav1.5, although this isoform is predominantly expressed in the heart. Resurgent current is an atypical current predicted to enhance neuronal excitability (Khaliq et al., 2003; Cruz et al., 2011). Mechanistically, these currents arise from channel re-opening during the repolarization phase of the action potential due to unbinding of an open-channel blocker (Lewis and Raman, 2014). Therefore, these currents provide a depolarizing drive to approach threshold for firing additional action potentials. These currents have been observed to be dysregulated in both acquired and inherited disorders of excitability. Resurgent currents are increased in a kindling model of temporal lobe epilepsy as well as by mutations in VGSCs associated with pain, myotonia and cardiac arrhythmias (Jarecki et al., 2010; Hargus et al., 2011, 2013; Sittl et al., 2012). Targeting resurgent sodium current is a potentially novel therapeutic strategy for the treatment of multiple epilepsy syndromes.
Here we focused on mutations in Nav1.1 and Nav1.6 due to the severity of the clinical phenotypes associated with these mutations (Oliva et al., 2012). We asked whether epilepsy-associated mutations in these channel isoforms alter resurgent sodium current generation, and whether we can preferentially inhibit resurgent sodium current over peak transient current generated by these two channel isoforms. We found that epilepsy-associated mutations in Nav1.6 dramatically enhanced resurgent current generation while mutations in Nav1.1 did not, suggesting that mutations in these channel isoforms are acting by distinct mechanisms to induce epileptogenesis. Moreover, cannabidiol can selectively inhibit resurgent current over peak transient current generated by wild-type Nav1.6 as well as aberrant resurgent and persistent current generated by Nav1.6 mutant channels. We validated our findings using endogenous sodium currents from striatal neurons and found that indeed cannabidiol inhibits endogenous resurgent and persistent current in neurons. Furthermore, we found that excitability of striatal neurons is reduced in the presence of cannabidiol. Therefore, cannabidiol could be exerting its anti-epileptic effects, at least in part, through its action on aberrant activity in VGSCs.
Materials and methods
Complementary DNA constructs
Optimized human constructs for Nav1.1 and Nav1.6 (hNav1.1 and hNav1.6) were designed in-house and purchased from Genscript. cDNA constructs for wild-type Nav1.1 and Nav1.6 channels encode for amino acid sequences corresponding to the accession numbers BAC21102.1 and NP_055006.1 in the NCBI database, respectively. Mutations were introduced into wild-type cDNA constructs (hNav1.1 R1648H, hNav1.1 N1788K, hNav1.1 N1788D, hNav1.6 L1331V, hNav1.6 N1768D, and hNav1.6 N1768K) using the QuikChange® II XL site-directed mutagenesis kit from Agilent Technologies according to the manufacturer’s instructions. Mutant channel constructs were fully sequenced (ACGT, Inc.) to confirm the presence of the correct mutation and absence of additional mutations.
Cell cultures and transfections
The use of human embryonic kidney 293T (HEK293T) cells (Dubridge et al., 1987) was approved by the Institutional Biosafety Committee and followed the ethical guidelines for the National Institutes of Health for the use of human-derived cell lines. HEK293T cells were grown under standard tissue culture conditions. HEK293T cells were transiently transfected using the calcium phosphate precipitation method. Briefly, calcium phosphate-DNA mixture [4.5 μg channel construct and 0.5 μg enhanced green fluorescent protein (EGFP)] was added to cells in serum-free media for 4 to 5 h, after which it was replaced with normal media. Twelve to twenty-four hours post-transfection, cells were split onto laminin-coated glass coverslips. Cells were identified by expression of EGFP using a fluorescent microscope and whole-cell patch clamp recordings were obtained 36–72 h post-transfection. Wild-type and mutant Nav1.6 channels were incubated at 30°C overnight to increase protein surface expression.
Striatal neuron cultures
Procedures with mice were performed in accordance with the Institutional Animal Care and Use Committee approved protocol. FVB/NJ mice were purchased from Jackson Laboratories and breeding colonies were established. The mice were housed under standard conditions with free access to water and food. Striatal neuronal cultures were prepared from individual striata of postnatal Day 1 wild-type FVB/NJ mice of both sexes as previously described (Dubinsky, 1993). We used neuronal–glial co-cultures derived from postnatal Day 1 mouse pups because it is more physiologically relevant and allows for the study of more mature, better developed cells than pure neuronal cultures derived from embryonic animals. For all platings, 35 mg/ml uridine plus 15 mg/ml 5-fluoro-20-deoxyuridine were added 24 h after plating to inhibit proliferation of non-neuronal cells. Neurons were cultured in a 5% CO2 atmosphere at 37°C in Neurobasal® medium with B27 supplement (Life Technologies). Cultures were used for electrophysiological recordings at 7–12 days.
Chemicals and solutions
Anandamide and cannabidiol were purchased from Sigma Aldrich and Cayman Chemical Company, respectively. Anandamide was dissolved in ethanol to a stock concentration of 138.3 mM, aliquoted and stored at −20°C. Cannabidiol was dissolved in methanol to a stock concentration of 31.8 mM, aliquoted into tubes topped will argon gas and stored at −80°C. A fresh aliquot of drug was used and diluted with the extracellular patch clamp solution to desired concentration for each experiment. Control data were collected in the presence of the corresponding vehicle used to dissolve the drug.
Whole-cell patch clamp recordings
All whole-cell patch clamp recordings were obtained at room temperature (∼23°C) using a HEKA EPC-10 amplifier and the Pulse program (v 8.80, HEKA Electronic) was used for data acquisition. For experiments with HEK293T cells, electrodes were fabricated from 1.7 mm capillary glass and fire-polished to a resistance of 0.9–1.3 MΩ using a Sutter P-97 puller (Sutter Instrument Company). All voltage protocols were started 5 min after obtaining a GΩ seal and entering the whole-cell configuration, which controlled for time-dependent shifts in channel properties and allowed time for diffusion of the Navβ4 peptide. Voltage errors were minimized to <5 mV using series resistance compensation and passive leak currents were cancelled by P/−5 subtraction. The bath solution contained (in mM): 140 NaCl, 1 MgCl2, 3 KCl, 1 CaCl2, and 10 HEPES, adjusted to a pH of 7.30 with NaOH. The pipette solution contained (in mM): 140 CsF, 10 NaCl, 1.1 EGTA, and 10 HEPES, adjusted to a pH of 7.30 with CsOH. To induce resurgent currents in HEK293T cells, 200 μM Navβ4 peptide (KKLITFILKKTREK-OH) (Biopeptide Co), a peptide that corresponds to the sequence of part of the C-terminal tail of the full-length Navβ4 subunit, was included in the pipette solution.
For recordings obtained from striatal neurons, pipettes were fabricated from 1.5 mm borosilicate glass and fire-polished to a resistance of 2–4 MΩ. Series resistance was compensated by 70 to 85%. For voltage clamp recordings, the striatal neuron bath solution contained (in mM): 130 NaCl, 30 TEA-Cl, 3 KCl, 1 MgCl2, 1 CaCl2, 0.05 CdCl2, 5 4-aminopyridine and 10 HEPES, adjusted to a pH of 7.3 with NaOH. The pipette solution was the same CsF containing solution mentioned above but did not contain the Navβ4 peptide. For current clamp recordings, the bath solution contained (in mM): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2 and 10 HEPES adjusted to a pH of 7.3 with NaOH. The pipette solution contained (in mM): 140 KCl, 0.5 EGTA, 5 HEPES and 3 Mg-ATP adjusted to a pH of 7.3 with KOH. Recordings were all started 2 min after establishing the whole-cell configuration. Only cells with a stable resting membrane potential more negative than −35 mV were used. To eliminate variation between cells, cells were held at −60 mV or −80 mV for action potential activity recordings.
For all experiments involving drugs, recordings were made in the presence of extracellular bath solution containing either the drug or vehicle control. Each coverslip was recorded from for up to 1.5 h before discarding.
Data analysis
Electrophysiological data were analysed using Pulsefit (v 8.67 HEKA Electronic), Microsoft Excel, Origin (v 8.0, OriginLab), and Prism (v 6.0, Graphpad Software). Steady-state activation and inactivation curves were fit to a Boltzmann function to obtain midpoint (V1/2) and slope values. Time constants for recovery were obtained by fitting data from each cell to a first order exponential function and averaging time constants across cells. Input resistance (R) was calculated from the change in voltage (V) at the end of a 200 ms, −200 pA current (I) stimulus using the equation V = I / R. All data points are presented as mean ± standard error of the mean (SEM) and n is the number of experimental cells from which recordings were obtained. Statistical significance was assessed using an unpaired t-test, one-way ANOVA with Dunnett post hoc test or a two-way ANOVA with Bonferroni post hoc test.
Results
Epilepsy-associated mutations in Nav1.1 did not alter peak resurgent current
We examined an arginine to histidine mutation at position 1648 in Nav1.1, which is located in the S4 segment of domain IV, identified in patients with generalized epilepsy with febrile seizures plus (Fig. 1A) (Escayg et al., 2000). This mutation has been previously characterized in both heterologous expression systems and mouse models and from the reported biophysical defects that this mutation confers we predicted it would alter resurgent current generation (Spampanato et al., 2001; Kahlig et al., 2006; Vanoye et al., 2006; Tang et al., 2009; Martin et al., 2010). Additionally, we examined an asparagine to lysine mutation at position 1788 in Nav1.1, which is located at the end of the S6 segment of domain IV, identified in a patient with Dravet syndrome (Fig. 1A). Coincidentally, this mutation occurs at the exact same position as the first epilepsy-associated mutation identified in human Nav1.6 (Veeramah et al., 2012) and has not been previously characterized (Fig. 3A). We characterized the biophysical properties of the wild-type and mutant hNav1.1 channels with whole-cell patch clamp recordings from transiently transfected HEK293T cells. Representative families of current traces from wild-type and mutant channels, elicited by applying depolarizing step pulses ranging from −80 mV to +80 mV for 50 ms from a holding potential of −100 mV, are shown in Fig. 1B. Current density curves were obtained by normalizing peak current at each voltage by the cell capacitance. hNav1.1 R1648H and hNav1.1 N1788K have a significantly (P < 0.05; two-way ANOVA) decreased current density compared to hNav1.1 wild-type (Fig. 1C). We next examined the voltage dependence of channel activation and steady-state inactivation (Fig. 1D and Supplementary Table 1). hNav1.1 N1788K has a significantly (P < 0.05; one-way ANOVA) more depolarized V1/2 of channel activation and steady-state inactivation compared to hNav1.1 wild-type (Supplementary Table 1). hNav1.1 R1648H did not show significant changes in channel activation but has a significantly (P < 0.001; one-way ANOVA) hyperpolarized voltage dependence of steady-state inactivation (−73.0 ± 2.2 mV; n = 10) compared to wild-type hNav1.1 (−61.6 ± 1.7 mV; n = 20). We also examined persistent current generation by wild-type and mutant channels using a protocol in which incremental step pulses from −80 mV to + 30 mV were applied from a holding potential of −100 mV for 200 ms (Fig. 1E inset). Persistent current is a non-inactivating (or very slowly-inactivating) current and was measured at the end of a long (200 ms) depolarizing step pulse. These currents can amplify subthreshold currents and facilitate repetitive firing (Crill, 1996; Kiss, 2008). We found that neither the hNav1.1 R1648H nor the hNav1.1 N1788K mutant channel significantly altered persistent current (Fig. 1E). Additionally, we examined steady-state slow inactivation, recovery from slow inactivation as well as development of slow inactivation. We found no significant effects of hNav1.1 mutant channels on steady-state slow inactivation or development of slow inactivation. hNav1.1 N1788K significantly (P < 0.0001; one-way ANOVA) slowed recovery from slow inactivation compared to wild-type hNav1.1 (Supplementary Fig. 1A–C and Supplementary Table 2). From these findings, it appears that both hNav1.1 R1648H and hNav1.1 N1788K mutants are loss-of-function.
Figure 1.

Biophysical characterization of hNav1.1 wild-type, R1648H and N1788K mutant channels. (A) Linear schematic of the structure of the VGSC α-subunit depicting the locations of the hNav1.1 R1648H (green circle) and hNav1.1 N1788K (orange circle) mutations. (B) Representative family of current traces generated by hNav1.1 wild-type (WT), R1648H and N1788K expressing HEK293T cells. Currents were elicited with step depolarizations ranging from −80 mV to +80 mV for 50 ms from a holding potential of −100 mV. Peak current traces are in bold. (C) Current density–voltage curve for hNav1.1 wild-type (blue squares), R1648H (green circles) and N1788K (orange triangles). Current density values were calculated by normalizing the peak sodium current at each voltage to the cell capacitance and subsequently averaged across cells. (D) Voltage dependence of steady-state activation and inactivation curves fit with a Boltzmann function. Steady-state inactivation was measured using a protocol in which cells were held at a series of voltages ranging from −120 mV to +30 mV for 500 ms followed by a 20-ms step pulse to +10 mV to measure channel availability (inset). (E) Persistent current amplitude plotted as a function of voltage. Persistent current was measured at 180 ms into current traces elicited by 200-ms step depolarizations ranging from −80 mV to +30 mV from a holding potential of −100 mV (inset). (F) Resurgent currents were elicited with a step depolarization from −100 mV to +60 mV for 20 ms to open channels allowing them to undergo open-channel block and subsequently repolarizing to a series of potentials ranging from +25 mV to −80 mV for 50 ms to allow the blocker to unbind. (G) Representative family of resurgent current traces generated by hNav1.1 wild-type (top), R1648H (middle) and N1788K (bottom). Peak resurgent current traces are bolded. (H) Per cent resurgent current plotted as a function of the repolarization voltage for hNav1.1 wild-type (blue squares; n = 29), R1648H (green circles; n = 17) and N1788K (orange triangles; n = 11). Per cent resurgent current was calculated by normalizing peak resurgent current at each voltage to peak transient current measured with a single step pulse from −120 mV to +10 mV. *P < 0.05; two-way ANOVA.
Figure 3.
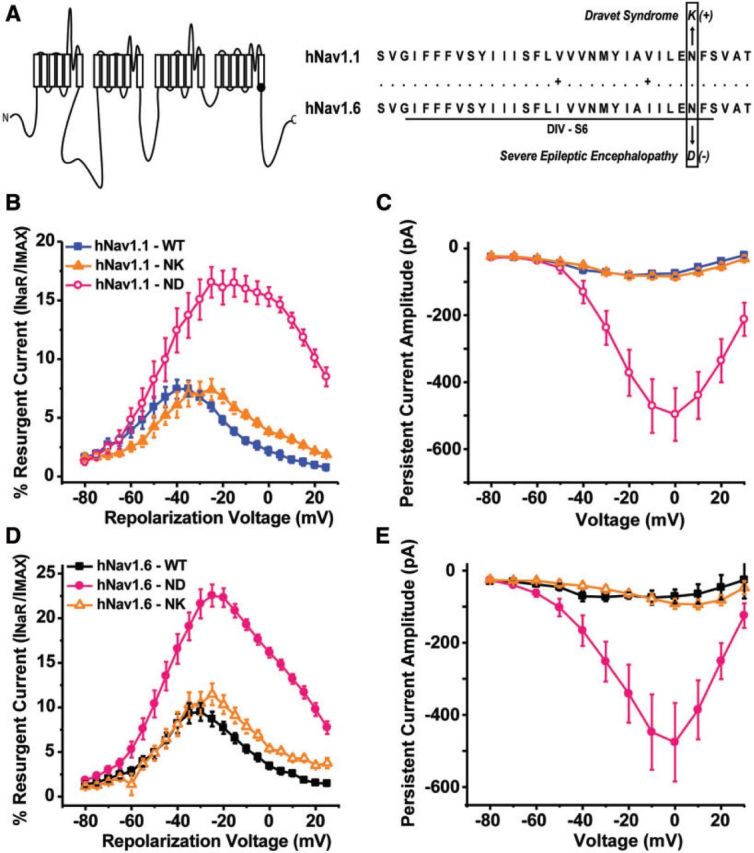
Resurgent and persistent current generation by reciprocal epilepsy-associated mutation in non-native channel isoform. (A) Position of epilepsy-associated mutations in the linear schematic of the VGSC α- subunit and in the sequence alignment of hNav1.1 and hNav1.6. (B and C) Per cent resurgent current and peak persistent current amplitude generated by hNav1.1 wild-type (WT) (blue squares), hNav1.1 N1788K (orange triangles) and the reciprocal N1788D mutation in hNav1.1 (open pink circles). (D and E) Per cent resurgent current and peak persistent current amplitude generated by hNav1.6 wild-type (black squares), hNav1.6 N1768D (pink circles) and the reciprocal N1768K mutation in hNav1.6 (open orange triangles).
We next asked whether epilepsy-associated mutations in hNav1.1 alter resurgent sodium current generation. Resurgent currents are predicted to increase neuronal excitability and therefore alterations in these currents could potentially underlie the hyperexcitability seen in epilepsy. Resurgent currents were observed by applying an initial depolarizing step to +60 mV from −100 mV and subsequently repolarizing incrementally from +25 mV to −80 mV (Fig. 1F). Representative families of resurgent current traces from hNav1.1 wild-type and mutant channels can be seen in Fig. 1G. Peak resurgent current was measured after 1.5 ms into the repolarization step to bypass fast tail currents. The per cent resurgent current was reported after normalizing the peak resurgent current at each repolarizing voltage step to a single measure of the peak transient current amplitude in each cell. The estimate of peak transient current amplitude in each cell was obtained from a depolarization step to +10 mV from −120 mV. This provides a consistent and reliable peak transient current value for each cell, and normalizing the peak resurgent current values to this measure of peak transient current greatly helps to control for variability in channel expression in HEK293T cells. We found that there was no significant difference in the normalized magnitude of the peak resurgent current generated by mutant hNav1.1 channels compared to wild-type hNav1.1 (Fig. 1H). Although there appears to be a shift in the voltage-dependence of resurgent current for mutant hNav1.1 channels, these shifts correspond with shifts in channel activation seen in Fig. 1D. Therefore, it is plausible that these shifts are a consequence of alterations in channel activation rather than direct effects of the mutation on the channels’ ability to generate resurgent current. It was surprising that the hNav1.1 R1648H mutant channel did not alter peak resurgent current since it demonstrated a faster rate of inactivation (data not shown). However, from these data it appears that resurgent currents are not altered by epilepsy-associated mutant Nav1.1 R1648H and N1788K channels.
Epilepsy-associated mutations in Nav1.6 increase peak resurgent current
Epilepsy-associated mutations in Nav1.6 are phenotypically distinct from those in Nav1.1 (Oliva et al., 2012; Wagnon and Meisler, 2015). Therefore we examined how epilepsy-associated mutations in Nav1.6 alter biophysical characteristics of the channel. We examined a leucine to valine mutation at position 1331 in hNav1.6, which is located in the S4-S5 linker in domain III, identified in a patient with an early infantile epileptic encephalopathy (Fig. 2A) (Carvill et al., 2013). This mutation is in a region of the channel important for coordinating the binding site for the intrinsic fast inactivation particle of the channel (Smith and Goldin, 1997; Catterall, 2000). Although this mutation has not yet been characterized, we predicted it would alter resurgent current generation based on its location. Additionally, we examined an asparagine to aspartic acid mutation at position 1768 in hNav1.6, which is located at the end of the S6 segment of domain IV, identified in a patient with a severe epileptic encephalopathy (Fig. 2A) (Veeramah et al., 2012). As mentioned previously, this mutation (hNav1.6 N1768D) occurs at the same position as the Dravet syndrome-associated hNav1.1 N1788K mutation (Fig. 3A). Representative families of peak transient current traces from hNav1.6 wild-type and mutant channels can be seen in Fig. 2B. We did not observe significant differences in peak current density or the V1/2 of channel activation of mutant channels compared to hNav1.6 wild-type (Fig. 2C and D; Supplementary Table 1). Peak current density was measured from hNav1.6 wild-type and mutant channels from HEK293T cells that had been cultured at 30°C overnight prior to obtaining patch clamp recordings. This procedure can boost channel expression at the cell plasma membrane, but as has been reported by others, might help rescue expression of mutant channels with folding or trafficking defects (Rusconi et al., 2007, 2009; Sharkey et al., 2009; Cestele et al., 2013b). While we do not know how specific disease mutations might alter functional channel density in patients, it is important to note that this methodological procedure does not affect the voltage-dependence of channel activation or inactivation (de Kovel et al., 2014). The voltage dependence of steady-state inactivation is significantly (P < 0.001; one-way ANOVA) depolarized for hNav1.6 N1768D (−58.5 ± 2.7 mV; n = 14) compared to wild-type hNav1.6 (−71.1 ± 1.8 mV; n = 18). Both mutations in hNav1.6 impaired fast inactivation as evidenced by incomplete inactivation in the steady-state inactivation curve (Fig. 2D, inset). We next examined persistent current and found that hNav1.6 N1768D produces a very large persistent current while hNav1.6 L1331V has similar persistent current amplitudes as wild-type (Fig. 2E). hNav1.6 mutant channels had opposing effects on slow inactivation (Supplementary Fig. 1 D–F and Supplementary Table 2). hNav1.6 L1331V had a significantly (P < 0.05; one-way ANOVA) more depolarized V1/2 of slow inactivation and was slower to develop slow inactivation compared to wild-type hNav1.6. hNav1.6 N1768D had a significantly (P < 0.05; one-way ANOVA) more hyperpolarized V1/2 of slow inactivation, was slower to recover from slow inactivation and faster to develop slow inactivation. It is not clear how these effects on slow inactivation would impact excitability as the prolonged depolarizations that are needed to induce slow inactivation may not occur under normal physiological conditions and the temperature sensitivity of slow inactivation is unclear. However, slow inactivation could be a factor during seizure activity. Our findings on hNav1.6 N1768D mutant channels are consistent with previous reports and clearly demonstrate that this mutation results in gain-of-function (Veeramah et al., 2012). In contrast, biophysical defects conferred by the hNav1.6 L1331V mutation are more subtle but (at this point in the analysis) could be characterized as loss-of-function.
Figure 2.

Biophysical characterization of hNav1.6 wild-type, L1331V and N1768D mutant channels. (A) Linear schematic of the structure of the VGSC α-subunit depicting the locations of the hNav1.6 L1331V (purple circle) and N1768D (pink circle) mutations. (B) Representative family of current traces generated by HEK293T cells expressing hNav1.6 wild-type (WT), L1331V and N1768D channels. Peak current traces are in bold. (C) Plot of current density versus voltage. (D) Steady-state inactivation and activation curves fit with a Boltzmann function. Inset: Magnification of the voltage-dependence of inactivation curve between −20 mV to +30 mV depicting incomplete inactivation of mutant channels. (E) Peak persistent current amplitude plotted as a function of voltage. (F) Representative family of resurgent current traces generated by hNav1.6 wild-type (top), L1331V (middle) and N1768D (bottom). Peak resurgent current traces are in bold. (G) Per cent resurgent current plotted as a function of the repolarization voltage for hNav1.6 wild-type (black squares; n = 20), L1331V (purple triangles; n = 11) and N1768D (pink circles; n = 14). *P < 0.05; two-way ANOVA.
To further explore the biophysical consequences of the hNav1.6 epilepsy-associated mutations, we examined resurgent sodium current generation. Representative families of resurgent current traces generated by wild-type and mutant channels can be seen in Fig. 2F. We found that both hNav1.6 L1331V and hNav1.6 N1768D have a greater propensity to generate resurgent sodium current compared to wild-type hNav1.6. The peak resurgent current amplitude generated by hNav1.6 L1331V and hNav1.6 N1768D is nearly tripled and doubled, respectively, compared to wild-type hNav1.6 (Fig. 2G). This finding clearly demonstrates that both mutations in hNav1.6 are gain-of-function, which is in contrast to the mutations we examined in hNav1.1, suggesting that epilepsy-associated mutations in these two channel isoforms act by distinct mechanisms and consequently result in different phenotypes.
Alterations in peak resurgent and persistent current conferred by the mutation are independent of channel isoform
Nav1.6 N1768D was the first human epilepsy-associated mutation to be reported in the Nav1.6 isoform and coincidentally occurs at the same position as a previously identified Dravet syndrome-associated mutation in Nav1.1 (Nav1.1 N1788K) (Fig. 3A) (Depienne et al., 2009; Veeramah et al., 2012). This gave us a unique opportunity to ask whether disease mutations confer the same biophysical consequences to different channel isoforms. To address this we created the reciprocal mutations in the non-native channel isoform (hNav1.1 N1788D and hNav1.6 N1768K). We measured resurgent and persistent sodium current generation by these mutant channels since we observed dramatic changes in these properties with the epilepsy-associated mutant channels. It is noteworthy that these disease mutations result in oppositely charged residue substitutions. We found similar alterations in resurgent and persistent current for the reciprocal mutations in the non-native channel isoform, although the magnitudes of the changes varied. In hNav1.1 the N1788D mutation increases both resurgent and persistent current, similar to the epilepsy-associated hNav1.6 N1768D mutant (Fig. 3B and C). In hNav1.6 the N1768K mutation did not alter peak resurgent current or persistent current amplitude, similar to the epilepsy-associated hNav1.1 N1788K mutant (Fig. 3D and E). These data show that epilepsy-associated mutations cause similar alterations in channel properties independent of channel isoform and that different amino acid substitutions at the same position in the channel can cause significantly different effects on channel properties. This suggests that distinct phenotypes likely arise from not only differences in the expression pattern and regulation of the channel isoform in which the mutation occurs but is also dependent on the amino acid residue itself.
Cannabidiol can selectively inhibit peak resurgent sodium current generated by wild-type Nav1.6
Targeting resurgent currents is a novel strategy for the treatment of epilepsy. However, there are currently no selective pharmacological inhibitors of this current for brain isoforms of VGSCs. It has been shown that anandamide and ajulemic acid can inhibit resurgent current generated by Nav1.7 and Nav1.5 (Theile and Cummins, 2011; Foadi et al., 2014), respectively, but these isoforms are not present at significant levels in the brain. We therefore asked whether anandamide could similarly inhibit resurgent current over peak transient current generated by hNav1.1 and hNav1.6. Additionally, we examined the effects of cannabidiol due to its potential efficacy in the treatment of paediatric epilepsies (Devinsky et al., 2016) and similarity in structure to ajulemic acid. We found that neither 5 μM anandamide nor 1 μM cannabidiol had any significant effect on peak current density or peak resurgent current generated by hNav1.1 (Fig. 4A and B). We also did not observe significant effects of cannabidiol on the voltage-dependence of activation or inactivation, recovery, or persistent current generated by hNav1.1 (data not shown). Peak current density of hNav1.6 was significantly (P < 0.05; unpaired t-test) inhibited by 5 μM anandamide, but peak resurgent current was not preferentially altered by 5 μM anandamide in our preparation (Fig. 4C). Remarkably, 1 μM cannabidiol significantly (P < 0.01; unpaired test) inhibited the peak resurgent current generated by hNav1.6 (measured as a percentage of the peak transient current in each cell) while having no significant effect on peak current density (Fig. 4C and D). This finding identifies a novel target (i.e. resurgent sodium current) and mechanism underlying the anti-convulsant properties of cannabidiol.
Figure 4.

Effects of anandamide and cannabidiol on peak current density and resurgent current generated by wild-type hNav1.1 and hNav1.6 channels. (A and B) Effects of 5 μM anandamide (AEA) (n = 14) and 1μM cannabidiol (CBD) (n = 26–28) on peak current density and peak resurgent current generated by wild-type hNav1.1. (C and D) Effects of 5 μM AEA (n = 12–13) and 1 μM CBD (n = 26–30) on peak current density and peak resurgent current generated by hNav1.6. *P < 0.05, **P < 0.01; unpaired t-test.
Cannabidiol can inhibit aberrant resurgent and persistent current generated by mutant Nav1.6 channels
One of the major biophysical defects we observed with epilepsy-associated mutations in hNav1.6 was increased resurgent current; we therefore asked if we could target this aberrant activity with cannabidiol. We first examined the effects of 1 μM cannabidiol on hNav1.6 L1331V generated currents. Cannabidiol did not significantly inhibit peak current density at this concentration (Fig. 5A and B). However, peak resurgent current was significantly (P < 0.05; two-way ANOVA) inhibited by cannabidiol, similar to cannabidiol effects on wild-type hNav1.6 (Fig. 5C and D). We also examined gating properties of hNav1.6 L1331V in the presence of cannabidiol and found a slight but significant (P < 0.05; unpaired t-test) depolarizing shift in channel activation (−28.6 ± 1.4 mV; n = 10) compared to hNav1.6 L1331V in the presence of vehicle (−33.8 ± 1.7 mV; n = 9) (Fig. 5E). Additionally, we measured channel recovery from inactivation at −80 mV (Fig. 5F). To do this, we used a voltage command protocol in which we assessed the peak current by an initial depolarizing step to 0 mV and allowed channels to recover from inactivation for increasing durations at −80 mV before measuring channel availability with a test pulse to 0 mV (Fig. 5F, inset). Unexpectedly, cannabidiol significantly (P < 0.001; unpaired t-test) slowed hNav1.6 L1331V (τ = 32.1 ± 2.5 ms) recovery from inactivation compared to vehicle (τ = 16.8 ± 1.6 ms), which was not observed with wild-type hNav1.6 (data not shown). We also examined persistent current and found no difference in the presence of cannabidiol (Fig. 5G).
Figure 5.

Effects of 1 µM cannabidiol on hNav1.6 L1331V generated currents. (A) Representative family of current traces generated by hNav1.6 L1331V in presence of vehicle (left) and 1 μM cannabidiol (CBD) (right). (B) Current density curve showing no statistical difference in the peak current density between vehicle (purple triangles; n = 9) and CBD (black triangles; n = 10). (C) Representative family of resurgent current traces generated by hNav1.6 L1331V in presence of vehicle (left) and 1 μM CBD (right). Peak resurgent current traces are bolded. (D) Per cent resurgent current plotted against voltage. (E) Steady-state activation and inactivation curves fit with a Boltzmann function. (F) Normalized available current plotted against recovery duration and fit with an exponential function. Recovery from fast inactivation was measured by applying an initial depolarizing step to 0 mV to assess the peak current and then repolarizing to −80 mV for increasing durations followed by a final test pulse 0 mV to measure channel availability (inset). (G) Persistent current amplitude plotted versus voltage. *P < 0.05; two-way ANOVA.
We next examined cannabidiol effects on hNav1.6 N1768D generated currents. Again, we found no significant difference in peak current density in the presence of 1 μM cannabidiol (Fig. 6A and B). Cannabidiol did significantly (P < 0.05; two-way ANOVA) inhibit peak resurgent current generated by the hNNav1.6 N1768D mutant (Fig. 6C and D). It is important to note that resurgent current is measured relative to the peak transient current in each cell. Thus while there is a slight, but non-significant, decrease in peak transient currents, the reported decrease in resurgent current is in addition to this effect of cannabidiol. Unlike with the hNav1.6 L1331V mutant, cannabidiol did not alter the voltage dependence of activation of hNav1.6 N1768D channels (Fig. 6E). However, recovery from inactivation at −80 mV was significantly (P < 0.05; unpaired t-test) slowed in the presence of cannabidiol (τ = 9.7 ± 1.0 ms; n = 10) compared to vehicle (τ = 6.5 ± 0.9 ms; n = 9) (Fig. 6F). Surprisingly, we found that cannabidiol inhibited peak persistent current generated by the hNav1.6 N1768D mutant. The inhibition of persistent current occurs within the same voltage range as the inhibition of resurgent current and is likely contributing to the reduction in resurgent current. Overall, cannabidiol alters multiple biophysical properties of Nav1.6 mutant channels in ways that are all consistent with decreasing channel activity.
Figure 6.

Effects of 1 µM cannabidiol on hNav1.6 N1768D generated currents. (A) Representative family of current traces generated by hNav1.6 N1768D in the presence of vehicle (left) and cannabidiol (CBD) (right). (B) Current density curves for hNav1.6 N1768D in the presence of vehicle (pink circles; n = 20) and the presence of CBD (black circles; n = 19). (C) Representative family of resurgent current traces generated by hNav1.6 N1768D in the presence of vehicle (left) and CBD (right). Peak resurgent current traces are in bold. (D) Summary data showing the per cent resurgent current versus the repolarization voltage. (E) Steady-state activation and inactivation curves fit with a Boltzmann function. (F) Normalized available current plotted versus recovery duration and fit with an exponential function. (G) Persistent current amplitude plotted against voltage. *P < 0.05, two-way ANOVA.
Cannabidiol can inhibit endogenous resurgent and persistent current and decrease excitability of striatal neurons
Cannabidiol inhibited Nav1.6 resurgent currents in HEK293T cells. However, these currents were elicited with the aid of the Navβ4 peptide, not full-length Navβ4. Therefore to further verify our findings, we examined the effects of cannabidiol on endogenous sodium currents from striatal neurons. Striatal neurons have a very high expression of Navβ4 and generate resurgent sodium current (Miyazaki et al., 2014). This allows us to determine if cannabidiol can influence resurgent currents dependent on full-length Navβ4. As we observed in HEK293T cells expressing hNav1.6, 1μM cannabidiol did not have a significant effect on peak transient current, estimated with a test pulse to +10 mV from a holding potential of −80 mV (Fig. 7A and Supplementary Fig. 2A). We next examined resurgent currents using a standard protocol (Fig. 7B, inset). Representative resurgent current traces can be seen in Fig. 7B. Cannabidiol significantly (P < 0.05; two-way ANOVA) decreased peak resurgent current in striatal neurons (Fig. 7C). Additionally, we found that the presence of 1 μM cannabidiol slightly but significantly (P < 0.05; unpaired t-test) shifted the V1/2 of steady-state inactivation to more hyperpolarizing potentials (−52.5 ± 1.14 mV; n = 23) compared to vehicle control (−48.4 ± 1.1; n = 24), but did not alter steady-state activation or the rate of fast inactivation (Fig. 7D, Supplementary Fig. 2B and Supplementary Data). However, we examined recovery from fast inactivation at −80 mV and found that indeed cannabidiol significantly (P < 0.05; unpaired t-test) slows recovery (τ = 7.5 ± 1.0 ms; n = 22) compared to vehicle control (τ = 4.4 ± 0.3 ms; n = 24) (Fig. 7E). Moreover, persistent current was also significantly (P < 0.05; two-way ANOVA) reduced by cannabidiol (Fig. 7F).
Figure 7.

Effects of 1 µM cannabidiol on endogenous sodium currents recorded from striatal neurons. (A) Peak transient current for vehicle (red) and 1 μM cannabidiol (CBD) (black). Peak transient current was measured with a step pulse from −80 mV to +10 mV from the steady-state inactivation protocol. (B) Representative family of resurgent current traces from striatal neurons in the presence of vehicle (left) and 1 μM cannabidiol (right). Peak resurgent current traces are in bold and magnified below. To elicit resurgent current in striatal neurons an initial prepulse to +30 mV for 20 ms was applied and followed by repolarizing steps ranging from +25 mV to −90 mV for 200 ms (inset). (C) Percent resurgent current plotted versus the repolarization voltage. (D) Steady-state inactivation curve fit with a Boltzmann function. Steady-state inactivation was measured using a prepulse ranging from −80 mV to +30 mV for 500 ms followed by a test pulse to +10 mV to measure channel availability (inset). (E) Normalized available current plotted versus recovery duration and fit with an exponential function. (F) Persistent current amplitude plotted as a function of voltage. *P < 0.05; two-way ANOVA.
To further explore the implications of cannabidiol actions on excitability, we obtained current clamp recordings from striatal neurons. Striatal neurons were not spontaneously active and therefore evoked activity was measured. We observed no differences in the resting membrane potential or input resistance from striatal neurons in the presence of vehicle or cannabidiol (Fig. 8B and C). With a holding potential of −60 mV, we found that the number of action potentials fired during a 200 ms stimulus with increasing intensity was significantly (P < 0.05; two-way ANOVA) reduced in the presence of cannabidiol (Fig. 8A and D). Action potential peak amplitude and current threshold for action potential firing from a holding potential of −60 mV was measured with a 1 ms stimulus increasing incrementally from 0 pA to 1nA in 20 pA steps. Cannabidiol reduced (P < 0.0001; unpaired t-test) the action potential peak (20.9 mV ± 3.6; n = 12) compared to vehicle control (48.7 mV ± 2.8; n = 14) (Fig. 8E). Moreover, cannabidiol significantly (P < 0.01; unpaired t-test) increases the threshold current needed to elicit an action potential compared to vehicle (from 587.7 ± 48.2 pA to 776 ± 31 pA) (Fig. 8F). To determine if the effects on action potential number, peak amplitude and current threshold were due to the hyperpolarizing shift in the voltage-dependence of inactivation (Fig. 7D), we additionally examined the effects of cannabidiol on striatal neuron excitability with a holding potential of −80 mV. We found that the number of action potentials fired with increasing stimulus intensity was still substantially reduced in the presence of cannabidiol with a holding potential of −80 mV (Fig. 8A and G). We then measured peak action potential amplitude and current threshold from a holding potential of −80 mV with a 1 ms stimulus increasing incrementally from 0 pA to 2 nA in 40 pA steps. Cannabidiol did not significantly (n = 33) change peak action potential amplitude or current threshold with this holding potential (Fig. 8H and I). Overall, our data show that 1 μM cannabidiol reduces overall excitability of striatal neurons.
Figure 8.

Effects of 1 µM cannabidiol on excitability of striatal neurons. (A) Representative traces of activity evoked with a 200 ms stimulus of 100 pA and 200 pA for vehicle (top) and 1 μM cannabidiol (CBD) (bottom) from a holding potential of −60 mV (grey and black traces) and −80 mV (red and black traces). (B) Resting membrane potential of striatal neurons in the presence of vehicle (open, red) and 1 μM cannabidiol (CBD) (open, black) from all recorded cells (n = 47 and 45, respectively). (C) Input resistance calculated from the change in voltage with a 200 ms, −200 pA stimulus according to V = IR for vehicle (open, red) and 1 μM cannabidiol (open, black). (D) Number of action potentials elicited by a 200 ms stimulus of increasing intensity from 20 pA to 200 pA in 20 pA steps from a holding potential of −60 mV for vehicle (grey circles; n = 14) and cannabidiol (black squares; n = 12). (E) Action potential peak was measured at the current threshold with a holding potential of −60 mV for vehicle (grey) and 1 μM cannabidiol (black). (F) Current threshold measured using a 1 ms stimulus increasing incrementally from 0 pA to 1 nA in 20 pA steps with a holding potential of −60 mV. (G) Number of action potentials elicited by a 200 ms stimulus of increasing intensity from 20 pA to 200 pA in 20 pA steps from a holding potential of −80 mV for vehicle (red circles; n = 33) and cannabidiol (black squares; n = 33). (H) Action potential peak was measured at the current threshold with a holding potential of −80 mV for vehicle (red) and 1 μM cannabidiol (black). (I) Current threshold measured using a 1 ms stimulus increasing incrementally from 0 pA to 2 nA in 40 pA steps with a holding potential of −80 mV. *P < 0.05, **P < 0.01, ##P < 0.0001; unpaired t-test and two-way ANOVA.
Discussion
In this study, we asked how epilepsy-associated mutations in Nav1.1 and Nav1.6 alter biophysical properties of these channels. Although several biophysical properties were altered by the disease mutations, we found differential effects on resurgent current generation, which suggests a divergence in the mechanism by which mutations in these two channel isoforms induce epileptogenesis and consequently result in different phenotypes. We found that mutations in Nav1.1 (R1648H and N1788K) that result in generalized epilepsy with febrile seizures plus or Dravet syndrome, did not alter peak resurgent current. In contrast, mutations in Nav1.6 (L1331V and N1768D), which result in a severe infantile epileptic encephalopathy, dramatically increased peak resurgent sodium current. These findings are consistent with the hypothesis that epilepsy-associated mutations in Nav1.1 are predominantly loss-of-function while mutations in Nav1.6 are primarily gain-of-function. Moreover, we found that cannabidiol can preferentially inhibit Nav1.6 generated resurgent current over peak transient current. This led us to ask whether cannabidiol could inhibit the aberrant activity generated by Nav1.6 mutant channels. We found that, indeed, a low concentration of cannabidiol can reduce mutant channel activity by shifting the voltage-dependence of activation to more depolarizing potentials, slowing channel recovery from fast inactivation and inhibiting resurgent and persistent current. We further confirmed our findings using endogenous sodium currents from striatal neurons, again demonstrating that cannabidiol can inhibit endogenous resurgent and persistent current. Overall, cannabidiol inhibits voltage-gated sodium channel activity contributing to a decrease in neuronal excitability.
Although brain isoforms of VGSCs have a similar functional role, that is mediating the inward depolarizing current underlying the upstroke of the action potential, each isoform is crucial for normal excitability. This is evidenced by the fact that knockout of brain isoforms of VGSCs in mice results in premature lethality suggesting that other isoforms cannot compensate for the lost activity (Sprunger et al., 1999; Planells-Cases et al., 2000; Ogiwara et al., 2007; Cheah et al., 2012). In this study, we focused on Nav1.1 and Nav1.6 due to the phenotypic severity of epilepsy-associated mutations identified in these two channel isoforms. Nav1.1 and Nav1.6 only exhibit 75% sequence identity and have distinct expression patterns in the brain. Specifically, Nav1.1 is expressed in parvalbumin positive GABAergic neurons in somato-dendritic compartments as well as the proximal axon initial segment, while Nav1.6 is found ubiquitously but is highly concentrated in the distal axon initial segment (Yu et al., 2006; Ogiwara et al., 2007; Lorincz and Nusser, 2010). The expression pattern of these two channel isoforms is critical to understanding the potential mechanisms by which mutations in these channels lead to pathological hyperexcitability. Our findings show that, despite having only 75% sequence identity, reciprocal disease mutations conferred similar biophysical defects to both channel isoforms; at least for this position, the specific amino acid substitution itself is critical to the biophysical consequences. Understanding the biophysical consequence of epilepsy-associated mutations can give us insight into the potential mechanism by which different phenotypes arise.
Voltage-gated sodium channels are highly regulated proteins that can be modulated by many post-translational processes, and therefore the cellular background in which they are expressed can greatly influence the biophysical properties they exhibit. This caveat is a limitation of all studies of mutant channels in expression systems and is exemplified by the Nav1.1 R1648H mutant. While we replicated the impairment in inactivation and decreased current density, we did not observe an increase in persistent current or enhancement of slow inactivation as seen by some others (Alekov et al., 2000; Spampanato et al., 2001; Lossin et al., 2002; Tang et al., 2009; Martin et al., 2010; Hedrich et al., 2014). This could be due to the lack of co-expressing full-length auxiliary beta subunits in our study. Interestingly, enhanced persistent currents were not observed in interneurons from transgenic mice expressing the R1648H mutation (Lossin et al., 2002; Tang et al., 2009; Martin et al., 2010; Hedrich et al., 2014), but the possibility that Nav1.1 R1648H enhances persistent currents in specific neuronal populations cannot be ruled out. However, our study replicated all the biophysical defects previously characterized with the Nav1.6 N1768D mutant including: increased persistent current, incomplete inactivation, and a depolarizing shift in steady-state inactivation (Veeramah et al., 2012). Heterologous expression systems and animal models are useful, but have limitations due to extensive modulation of sodium channels and therefore mutant channels may exhibit distinct biophysical properties in human neuronal subtypes.
Overall, our data suggest that mutations in Nav1.1 and Nav1.6 are acting by distinct mechanisms to induce epileptogenesis. Although the two Nav1.1 mutations that we characterized were selected from among hundreds of Nav1.1 mutations associated with epilepsy, most Nav1.1 mutations are believed to be loss-of-function. There will likely be exceptions to this paradigm. Several mutations in SCN1A have been identified in patients with migraine and some of these appear to be gain-of-function (Cestele et al., 2013a). By contrast, far fewer SCN8A epilepsy mutations have been identified and characterized (Wagnon and Meisler, 2015). The two Nav1.6 mutations that we characterized, and an additional Nav1.6 I1327V mutant that we characterized but is not included in this study, had direct enhancing effects on resurgent currents. It is probable that not all gain-of-function mutations in Nav1.6 will enhance resurgent current activity. Indeed, we previously found that painful Nav1.7 mutations could be divided into one of two groups (Theile et al., 2011). Paroxysmal extreme pain disorder mutations impairing inactivation enhanced resurgent currents, while primary erythromelalgia mutations enhancing activation did not alter resurgent currents. Mutations in domains III and IV or the C-terminus of the channel Nav1.6 most likely impair inactivation and enhance resurgent currents, while mutations that enhance activation such as the Nav1.6 N984K mutant (Blanchard et al., 2015) may not directly enhance resurgent current. A few apparent loss-of-function mutations have also been reported in Nav1.6 (de Kovel et al., 2014; Blanchard et al., 2015). It is not entirely clear how these mutations lead to epilepsy, although as Nav1.6 is expressed in both excitatory and inhibitory neurons, there could be important reductions in inhibitory tone with loss-of-function Nav1.6 mutations. Interestingly, enhanced Nav1.6 resurgent currents have been observed in an animal model of induced temporal lobe epilepsy (Hargus et al., 2011, 2013). This raises the intriguing possibility that Nav1.6 resurgent currents may be increased in epilepsy syndromes caused by brain injury and of other aetiologies.
It is likely that the clinical phenotypes manifested by mutations in Nav1.1 and Nav1.6 will require tailored treatment strategies to achieve maximal seizure control in different patients. It has been observed that many VGSC blockers exacerbate seizure severity in patients with Dravet syndrome while they may be efficacious in patients with mutations in Nav1.6 (Guerrini et al., 1998; Liao et al., 2010; Carvill et al., 2013; Boerma et al., 2015; Kong et al., 2015). This can be expected due to the primary role of Nav1.1 in inhibitory neurons and the loss-of-function effects of Dravet syndrome-associated mutations in Nav1.1. Further inhibition of inhibitory neuron excitability by non-selective VGSC blockers would not be expected to restore inhibitory-excitatory balance. In contrast mutations in Nav1.6 appear to be primarily gain-of-function and would lead to an overall increase in excitability of all neurons due to Nav1.6’s ubiquitous expression; broadly inhibiting VGSC activity in this case could help dampen the overall increase in excitability. In this study, we found that cannabidiol can inhibit VGSC activity; however, cannabidiol has shown some efficacy in the treatment of Dravet syndrome as well as other epilepsy syndromes (Porter and Jacobson, 2013; Szaflarski and Bebin, 2014; Devinsky et al., 2016). This may seem contradictory to our previous discussion, but we posit that cannabidiol’s efficacy lies in its specificity. At low concentrations, cannabidiol appears to be specific for Nav1.6 over Nav1.1 generated currents. More specifically, cannabidiol is selective for resurgent sodium current over peak transient currents generated by Nav1.6. Resurgent sodium currents are expressed in subpopulations of neuronal subtypes, many of which are in key circuits implicated in epilepsy syndromes such as striatal medium spiny neurons, perirhinal layer II pyramidal neurons, hippocampal dentate gyrus, ventral CA1 pyramidal neurons, globus pallidus, subthalamic nuclei, and medial entorhinal cortex (Lewis and Raman, 2014). Moreover, these currents are thought to occur at the axon initial segment and therefore can directly influence action potential initiation (Castelli et al., 2007). In addition, there is the possibility that seizure activity itself can lead to increased Nav1.6 resurgent currents in excitatory neurons (Hargus et al., 2011, 2013). Thus the selective expression of this atypical current may make it an ideal target to help rebalance inhibitory-excitatory tone and/or reduce pathological hyperexcitability even if the primary defect does not directly enhance Nav1.6 resurgent current activity.
In this study, we used a low concentration (1 µM) of cannabidiol to mimic the concentrations achievable in vivo (Deiana et al., 2012; Stott et al., 2013). We also tested a 500 nM concentration of cannabidiol and found a similar level of inhibition of resurgent current generated by wild-type Nav1.6 (data not shown). Hill et al. (2014) have found that high concentrations (10–30 µM) of cannabidiol can inhibit peak transient currents generated by other VGSC isoforms including: Nav1.1, Nav1.2, and Nav1.5, but the physiological significance of those findings are unclear. It still remains unclear as to how and where cannabidiol is interacting with Nav1.6. Because cannabidiol had no significant effect on Nav1.1, it is likely not acting on a conserved region in these two isoforms i.e. the local anaesthetic site, which is consistent with findings by Foadi et al. (2014) with ajulemic acid on Nav1.5. One possibility is that cannabidiol targets the domain IV voltage-sensor, which is critical for inactivation and can be differentially targeted by toxins and small molecules (McCormack et al., 2013; Xiao et al., 2014). We note that while we observed pronounced effects of cannabidiol on resurgent currents in population studies, we did not observe significant effects on peak transient or resurgent current in perfusion experiments on cells already patch-clamped, presumably due to disruption of the cytoplasmic milleau. This could suggest a possible indirect mechanism of action. Physiologically, it is plausible that cannabidiol is working indirectly on VGSCs. Cannabidiol has been shown to inhibit the degradation of anandamide extracellularly, which we and others have found can inhibit Nav1.6 peak current density (Leweke et al., 2012; Okura et al., 2014). In cultured striatal neurons cannabidiol substantially reduced repetitive firing, which is consistent with the inhibition of resurgent and persistent currents. However, the signficant reduction in action potential amplitude from a holding potential of −60 mV suggests additional actions. Cannabidiol induced a small but significant negative shift in the voltage-dependence of steady-state inactivation of striatal sodium currents, which would lead to reduced sodium channel availability and action potential amplitude. Indeed, when a more hyperpolarized membrane potential (−80 mV) was used to measure excitability, the effect of cannabidiol on action potential amplitude and current threshold was eliminated suggesting that the reduction was due to inactivation of VGSCs at −60 mV by cannabidiol. The pronounced effect on excitability that we observed in cultured neurons may be reduced in CNS neurons in vivo, which tend to have even more negative resting membrane potentials (−90 mV) compared to our in vitro preparation (−60 mV or −80 mV) and larger overall sodium currents (Surmeier et al., 2011; Miyazaki et al., 2014). Indeed, in the limited clinical reports that are available, cannabidiol does not seem to be associated with major CNS depression (Porter and Jacobson, 2013; Devinsky et al., 2016). Importantly, a substantial decrease in action potential number was still observed with the −80 mV holding potential and this is consistent with the reduction in resurgent currents observed using a −80 mV holding potential (Fig. 7C). While our data lead us to predict that inhibition of resurgent currents by low concentrations of cannabidiol could be important in acquired and inherited epilepsies, it is likely that the polypharmacological nature of cannabidiol (Devinsky et al., 2014) also can contribute to its ability to reduce seizure activity.
In conclusion, our present findings further elucidate a potential mechanism by which epilepsy-associated mutations in Nav1.6 lead to pathological hyperexcitability, which is distinct from mutations in Nav1.1. In addition, we found that cannabidiol can inhibit resurgent sodium current generated by wild-type and mutant Nav1.6 channels. Overall, our findings suggest that cannabidiol is mechanistically acting, in part, on VGSCs to decrease seizure activity and that resurgent sodium current may be a promising therapeutic target for the treatment of epilepsy syndromes.
Funding
The research was supported by grants from the Dravet Syndrome Foundation, the Indiana Spinal Cord and Brain Injury Research Fund and National Institutes of Health grant (NIH-NS053422) to T.R.C. R.R.P. was partially supported by a Paul and Carole Stark Medical Neuroscience Graduate Student Fellowship. N.B. was supported by a National Institutes of Health fellowship grant (NIH-NS078008).
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviation
- VGSC =
voltage-gated sodium channel
References
- Alekov A, Rahman MM, Mitrovic N, Lehmann-Horn F, Lerche H. A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro. J Physiol 2000; 529: 533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard MG, Willemsen MH, Walker JB, Dib-Hajj SD, Waxman SG, Jongmans MC, et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet 2015; 52: 330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerma RS, Braun KP, van de Broek MP, van Berkestijn FM, Swinkels ME, Hagebeuk EO, et al. Remarkable phenytoin sensitivity in 4 children with SCN8A-related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics 2015; 13: 192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013; 45: 825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli L, Biella G, Toselli M, Magistretti J. Resurgent Na+ current in pyramidal neurones of rat perirhinal cortex: axonal location of channels and contribution to depolarizing drive during repetitive firing. J Physiol 2007; 582: 1179–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000; 26: 13–25. [DOI] [PubMed] [Google Scholar]
- Cestele S, Labate A, Rusconi R, Tarantino P, Mumoli L, Franceschetti S, et al. Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 2013a; 54: 927–35. [DOI] [PubMed] [Google Scholar]
- Cestele S, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Nonfunctional Na(V)1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc Natl Acad Sci USA 2013b; 110: 17546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 2012; 109: 14646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra R, Isom LL. Untangling the dravet syndrome seizure network: the changing face of a rare genetic epilepsy. Epilepsy Curr 2014; 14: 86–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol 1996; 58: 349–62. [DOI] [PubMed] [Google Scholar]
- Cruz JS, Silva DF, Ribeiro LA, Araujo IG, Magalhaes N, Medeiros A, et al. Resurgent Na+ current: a new avenue to neuronal excitability control. Life Sci 2011; 89: 564–9. [DOI] [PubMed] [Google Scholar]
- de Kovel CG, Meisler MH, Brilstra EH, van Berkestijn FM, van ‘t Slot R, van Lieshout S, et al. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res 2014; 108: 1511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiana S, Watanabe A, Yamasaki Y, Amada N, Arthur M, Fleming S, et al. Plasma and brain pharmacokinetic profile of cannabidiol (CBD), cannabidivarine (CBDV), Delta(9)-tetrahydrocannabivarin (THCV) and cannabigerol (CBG) in rats and mice following oral and intraperitoneal administration and CBD action on obsessive-compulsive behaviour. Psychopharmacology 2012; 219: 859–73. [DOI] [PubMed] [Google Scholar]
- Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet 2009; 46: 183–91. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol 2016; 15: 270–8. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Cilio MR, Cross H, Fernandez-Ruiz J, French J, Hill C, et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014; 55: 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neurosci 1993; 13: 623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human-cells by using an epstein-barr-virus shuttle system. Mol Cell Biol 1987; 7: 379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, et al. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis 2013; 49: 211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000; 24: 343–5. [DOI] [PubMed] [Google Scholar]
- Estacion M, O'Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, et al. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis 2014; 69: 117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foadi N, Berger C, Pilawski I, Stoetzer C, Karst M, Haeseler G, et al. Inhibition of voltage-gated Na(+) channels by the synthetic cannabinoid ajulemic acid. Anesth Analg 2014; 118: 1238–45. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998; 39: 508–12. [DOI] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012; 489: 385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Merrick EC, Nigam A, Kalmar CL, Baheti AR, Bertram EH, et al. Temporal lobe epilepsy induces intrinsic alterations in Na channel gating in layer II medial entorhinal cortex neurons. Neurobiol Dis 2011; 41: 361–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Nigam A, Bertram EH 3rd, Patel MK. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J Neurophysiol 2013; 110: 1144–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich UB, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, et al. Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J Neurosci 2014; 34: 14874–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi N, Uchida T, Lossin C, Misumi Y, Okada Y, Akamatsu W, et al. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol Brain 2013; 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, Jones NA, Smith I, Hill CL, Williams CM, Stephens GJ, et al. Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci Lett 2014; 566: 269–74. [DOI] [PubMed] [Google Scholar]
- Jarecki BW, Piekarz AD, Jackson JO, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest 2010; 120: 369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Misra SN, George AL Jr, . Impaired inactivation gate stabilization predicts increased persistent current for an epilepsy-associated SCN1A mutation. J Neurosci 2006; 26: 10958–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 2013; 123: 1798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci 2003; 23: 4899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T. Persistent Na-channels: origin and function. A review. Acta Biol Hung 2008; 59 (Suppl 1): 1–12. [DOI] [PubMed] [Google Scholar]
- Kong W, Zhang Y, Gao Y, Liu X, Gao K, Xie H, et al. SCN8A mutations in Chinese children with early onset epilepsy and intellectual disability. Epilepsia 2015; 56: 431–8. [DOI] [PubMed] [Google Scholar]
- Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015; 84: 480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2012; 2: e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Resurgent current of voltage-gated Na(+) channels. J Physiol 2014; 592: 4825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao WP, Shi YW, Long YS, Zeng Y, Li T, Yu MJ, et al. Partial epilepsy with antecedent febrile seizures and seizure aggravation by antiepileptic drugs: associated with loss of function of Na(v) 1.1. Epilepsia 2010; 51: 1669–78. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O'Malley HA, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol 2013; 74: 128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science 2010; 328: 906–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL Jr, . Molecular basis of an inherited epilepsy. Neuron 2002; 34: 877–84. [DOI] [PubMed] [Google Scholar]
- Martin MS, Dutt K, Papale LA, Dube CM, Dutton SB, de Haan G, et al. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem 2010; 285: 9823–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci USA 2013; 110: E2724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki H, Oyama F, Inoue R, Aosaki T, Abe T, Kiyonari H, et al. Singular localization of sodium channel beta4 subunit in unmyelinated fibres and its role in the striatum. Nat Commun 2014; 5: 5525. [DOI] [PubMed] [Google Scholar]
- O'Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet 2013; 4: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 2013; 22: 4784–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 2007; 27: 5903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okura D, Horishita T, Ueno S, Yanagihara N, Sudo Y, Uezono Y, et al. The endocannabinoid anandamide inhibits voltage-gated sodium channels Nav1.2, Nav1.6, Nav1.7, and Nav1.8 in Xenopus oocytes. Anesth Analg 2014; 118: 554–62. [DOI] [PubMed] [Google Scholar]
- Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia 2012; 53: 1849–59. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, et al. Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys J 2000; 78: 2878–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter BE, Jacobson C. Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav 2013; 29: 574–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi R, Combi R, Cestele S, Grioni D, Franceschetti S, Dalpra L, et al. A rescuable folding defective Nav1.1 (SCN1A) sodium channel mutant causes GEFS+: common mechanism in Nav1.1 related epilepsies? Hum Mutat 2009; 30: E747–60. [DOI] [PubMed] [Google Scholar]
- Rusconi R, Scalmani P, Cassulini RR, Giunti G, Gambardella A, Franceschetti S, et al. Modulatory proteins can rescue a trafficking defective epileptogenic Na(v)1.1 Na+ channel mutant. J Neurosci 2007; 27: 11037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey LM, Cheng X, Drews V, Buchner DA, Jones JM, Justice MJ, et al. The ataxia3 mutation in the N-terminal cytoplasmic domain of sodium channel Na(v)1.6 disrupts intracellular trafficking. J Neurosci 2009; 29: 2733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, et al. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc Natl Acad Sci USA 2012; 109: 6704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys J 1997; 73: 1885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler MH, Goldin AL. Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci 2001; 21: 7481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprunger LK, Escayg A, Tallaksen-Greene S, Albin RL, Meisler MH. Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Hum Mol Genet 1999; 8: 471–9. [DOI] [PubMed] [Google Scholar]
- Stott CG, White L, Wright S, Wilbraham D, Guy GW. A phase I study to assess the single and multiple dose pharmacokinetics of THC/CBD oromucosal spray. Eur J Clin Pharmacol 2013; 69: 1135–47. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Carrillo-Reid L, Bargas J. Dopaminergic modulation of striatal neurons, circuits, and assemblies. Neuroscience 2011; 198: 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaflarski JP, Bebin EM. Cannabis, cannabidiol, and epilepsy - From receptors to clinical response. Epilepsy Behav 2014; 41: 277–82. [DOI] [PubMed] [Google Scholar]
- Tang B, Dutt K, Papale L, Rusconi R, Shankar A, Hunter J, et al. A BAC transgenic mouse model reveals neuron subtype-specific effects of a Generalized Epilepsy with Febrile Seizures Plus (GEFS+) mutation. Neurobiol Dis 2009; 35: 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Cummins TR. Inhibition of Navbeta4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol Pharmacol 2011; 80: 724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Jarecki BW, Piekarz AD, Cummins TR. Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J Physiol 2011; 589: 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkanis SA, Partlow LM, Karler R. Delta-9-Tetrahydrocannabinol depresses inward sodium current in mouse neuroblastoma-cells. Neuropharmacology 1991; 30: 73–7. [DOI] [PubMed] [Google Scholar]
- Vanoye CG, Lossin C, Rhodes TH, George AL Jr, . Single-channel properties of human NaV1.1 and mechanism of channel dysfunction in SCN1A-associated epilepsy. J Gen Physiol 2006; 127: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012; 90: 502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Meisler MH. Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol 2015; 6: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Blumenthal K, Cummins TR. Gating-pore currents demonstrate selective and specific modulation of individual sodium channel voltage-sensors by biological toxins. Mol Pharmacol 2014; 86: 159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006; 9: 1142–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.