

Synopsis
Many dementia subtypes have more shared signs and symptoms than defining ones. We shall review eight cases with four overlapping syndromes and demonstrate how to distinguish the cases. These include focal cortical presentations of Alzheimer’s Disease(AD) (Posterior Cortical Atrophy and Corticobasal Syndrome), fluent aphasia (Semantic Dementia and Logopenic Aphasia), late-onset slowly progressive dementia (Hippocampal Sclerosis and Limbic Predominant AD) and rapidly progressive dementia (Creutzfeldt-Jakob Disease and Limbic Encephalitis). Recognizing the different syndromes described in this paper can help the clinician improve their diagnostic skills leading to improved patient outcomes by early and accurate diagnosis, prompt treatment, appropriate counseling and guidance.
Keywords: atypical Alzheimer’s, fluent aphasia, late-onset dementia, rapid dementia
1. Introduction
Many dementia subtypes have more shared signs and symptoms than defining ones. We shall review eight cases with four overlapping syndromes and demonstrate how to distinguish the cases. These include focal cortical presentations of Alzheimer’s Disease(AD) (Posterior Cortical Atrophy and Corticobasal Syndrome), fluent aphasia (Semantic Dementia and Logopenic Aphasia), late-onset slowly progressive dementia (Hippocampal Sclerosis and Limbic Predominant AD) and rapidly progressive dementia (Creutzfeldt-Jakob Disease and Limbic Encephalitis). We shall illustrate each syndrome with a case and then describe the characteristic clinical features, commonly associated pathophysiology and main differential diagnoses.
2. Focal Cortical Presentations of Alzheimer’s Disease(AD)
2.1 Posterior Cortical Atrophy (PCA)
2.1.i Clinical Case
74 year-old right-handed woman with a PhD in English was evaluated for memory impairment with visuospatial difficulties. The first symptom she noticed 10 years previously was being unable to follow the correct line when reading a book and used a magnification device to follow the right line. She had difficulty finding objects in her refrigerator and failed to recognize objects. She had difficulty attending to stimuli on the left. Memory difficulties began two years previously. She had to stop driving due to her visual impairment and was functioning well otherwise. She was referred to the behavioral neurology clinic by her ophthalmologist, who had ruled out primary ocular abnormality. Her Kokmen Short Test of Mental Status[1] score was 28 of 38. She registered four words over one trial but did not recall any with a delay. Her constructional abilities were severely impaired, and she had a tendency to neglect the left visual field. She was able to name primary colors but had difficulty discriminating visually between like colors. She was unable to see numbers on the Ishihara plates and had prominent simultagnosia as well as ocular apraxia. No significant Parkinsonism, dyspraxia, dysphasia, cortical sensory loss or surface dyslexia were noted. Her brain magnetic resonance imaging(MRI) showed bilateral occipitoparietal atrophy (Figure 1). 18F-fluorodeoxyglucose positron emission tomography scan (FDG-PET) (Figure 2) revealed severe right more than left occipital hypometabolism.
Figure 1.

MRI brain showing bilateral occipitoparietal atrophy in Posterior Cortical Atrophy (PCA).
Figure 2.

Corresponding FDG-PET in PCA revealing severe right more than left occipital hypometabolism.
2.1.ii Clinical features
Early age of onset compared to typical AD
Relative preservation of orientation and episodic memory early in the disorder
Visual field deficits
Simultanagnosia
Acalculia
Alexia
Anomia
2.1.iii Pathophysiology
The term PCA was first used by Benson and colleagues[2] to describe a syndrome characterized by progressive decline in higher order visual processing with relatively intact memory and language in the early stages along with posterior brain atrophy. Hof and colleagues[3] were the first to report that the majority of these cases had Alzheimer pathology with a high tangle count in the posterior cortical areas. In a large series by Tang-Wai and colleagues[4] described the frequent associated symptoms and confirmed the neuritic pathology in the posterior cortical area and hippocampal sparing which is related to the early preservation of anterograde memory. The typical presentation as in this case is that patient goes to the ophthalmologist who does not find an ocular cause for the visual impairment and then is referred to neurology. Because there is relative preservation of anterograde memory doctors may not recognize that AD is the most frequent cause. Common clinical features are early age of onset (mean age 60.5 in the Tan-Wai series), simultanagnosia (inability to see all the details of a picture quickly and efficiently), visual field deficits, acalculia, alexia and anomia. Because of the preserved anterograde memory, good insight is often preserved. Other diseases that should be included in the differential diagnosis include Corticobasal Degeneration (2 of Tang-Wai cases had this), Lewy Body Disease (these patients often have visuospatial difficulty) and Creutzfeld-Jakob Disease(which can present with visual difficulty). Amyloid PET scan is abnormal in PCA patients with AD but the distribution is hard to distinguish from AD[5] however tau PET scan shows disproportionate tau deposits in the visual areas[6]. Recently Carrasquillo and colleagues[7] found ApoE to be related to PCA and indicated that, CLU, BIN1 and ABCA7 may be related but further study is needed.
2.2 Corticobasal Syndrome (CBS)
2.2.i Clinical Case
A 55-year-old right-handed woman was evaluated for one year history of progressive memory impairment, limb apraxia, bradykinesia and gait instability. Her husband noted that she also developed difficulty understanding spatial relations. She was still able to perform her job as a nurse and drive without accidents. Neurological examination revealed ideomotor and limb-kinetic apraxia, focal dystonia of the right hand, asymmetric (right>left) Parkinsonism, and decreased graphesthesia bilaterally. No myoclonus, alien limb phenomena or eye movement abnormalities were noted. Detailed neuropsychological assessment revealed marked impairment in memory, language and visuospatial skills. MRI brain showed diffuse mild cortical atrophy, more prominent bifrontally. Comprehensive laboratory workup, CSF studies and EEG were unremarkable. The working diagnosis was Corticobasal Syndrome (CBS). Medical disability process was initiated and she graciously stopped driving. She was seen yearly and progressive cognitive impairment continued. She was placed on donepezil and memantine eventually as atypical Alzheimer Disease was a consideration. She died seven years after the initial assessment. The autopsy revealed advanced[8] (Braak stage VI) Alzheimer’s Disease with relatively less neuronal loss and neurofibrillary degeneration in hippocampus compared to the cortex (Hippocampal Sparing AD) (Figure 3).
Figure 3.

This patient presented with corticobasal syndrome, but upon autopsy was found to have neuropathologic changes consistent with Alzheimer’s disease. (A) Parasagittal atrophy was noted when examining the fixed tissue. Immunohistochemical evaluation with a tau antibody revealed (B) a paucity of neurofibrillary tangles, (C) relative to the abundance of mature and extracellular tangles noted in the parietal lobe. A 1X magnification of the (D) parietal cortex shows an appreciably higher tau burden compared to the (E) hippocampus. Scale bar at 50 μm for 20x images.
2.2.ii Clinical features
Progressive Cognitive Impairment
Apraxia
Asymmetric Parkinsonism and Dystonia
Decreased Graphestesia bilaterally
Visuospatial Impairment
2.1.iii Pathophysiology
The clinical picture of CBS may have many pathological causes in addition to corticobasal degeneration such as in this case which had AD[9]. Other causes include Progressive Supranuclear Palsy, Picks disease and Creutzfeldt-Jakob disease. Although hippocampal atrophy rates have been used to monitor progression of AD and to predict cognitive decline [10–12], a subset of AD cases has less hippocampal atrophy relative to the predominance of cortical atrophy[13, 14], which challenges the Braak staging scheme of neurofibrillary changes[8] and results in symptomatic heterogeneity, as in our case. Murray and colleagues[14] looked into a large cohort (N=889) of autopsy- proven AD cases and showed that AD has distinct clinicopathologic subtypes: typical, hippocampal sparing and limbic-predominant. They pointed out that compared with the other subtypes, hippocampal sparing cases were mostly presented as focal cortical clinical syndromes, such as CBS, behavioral variant FTD, progressive aphasia, or posterior cortical atrophy (PCA). Hippocampal sparing cases were mostly men and were younger at death, with early onset and rapid progression. They had higher neurofibrillary tangle densities in the association cortices and the motor cortex, with significantly lower H1H1 MAPT genotype. Murray and colleagues [14]added that atypical AD (hippocampal sparing and limbic-predominant) might account 25% of the cases, and so should be considered in diagnostic evaluation and treatment studies. In a recent study by Paterson and colleagues[15], the typical and atypical AD groups were matched for age, sex, rate and severity of cognitive decline and had similar biomarker profiles, both different from controls. The atypical AD group appeared to be an heterogeneous entity with evidence for subtle differences in amyloid processing and neurodegeneration between subsets. For example they showed that PCA patients had the lowest cerebrospinal fluid total tau(T-tau), P-tau T-tau/Ab1-42 ratio and the slowest rate of cognitive decline as opposed to frontal variant group which had the highest T-tau, P-tau T-tau/Ab1-42 ratio and fastest rate of cognitive decline. Overall, these findings may have practical implications for CSF biomarker interpretation.
3. Fluent Aphasia
3.1 Semantic Dementia (SD)
3.1.i Clinical Case
A 72-year-old right-handed woman with a Master’s Degree in Speech Pathology presented with five years history of progressive word finding problems followed by difficulty recognizing faces even of familiar people. She was functioning well otherwise, was able to manage her finances and drive without getting lost. On examination, she scored 26/30 on the Folstein Mini Mental Status Examination (MMSE)[16], 8 of 10 on the orientation and 1 of 3 on the memory items. Her score was 8/20 on the first 20 words of the Boston Naming Test. On the famous faces test, she scored 0/20. On naming the last three presidents and their wives, first names, she scored 1/6. Her speech was fluent without any articulation difficulty. Her sentence repetition was intact. She had difficulty understanding sentences in the passive voice. Neuropsychological assessment revealed impaired semantic retrieval, including confrontation naming and speeded semantic fluency, with intact phonemic fluency, visual spatial skills, cognitive speed, executive function, and immediate attention. Brain MRI showed severe left more than right temporal lobe atrophy(Figure 4). Autopsy was not available.
Figure 4.
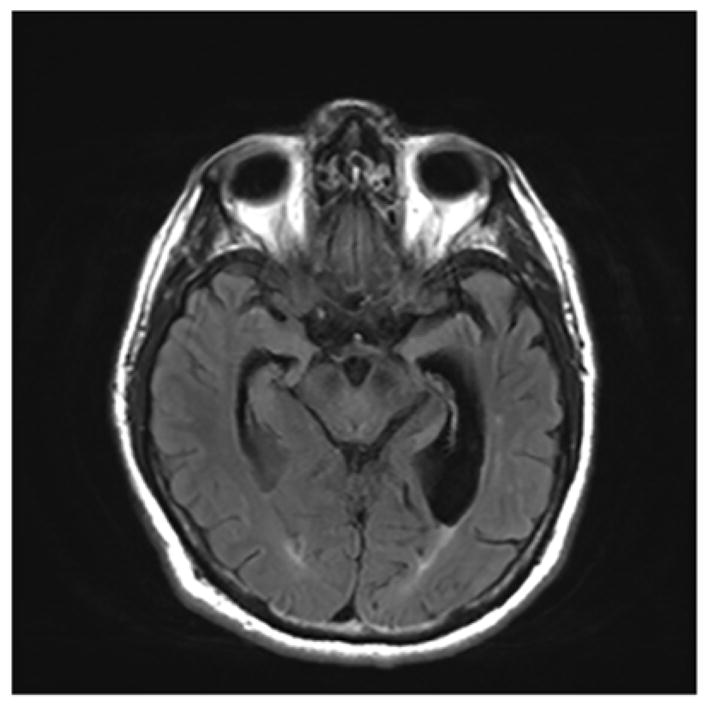
MRI brain showing left temporal lobe atrophy in a patient with Semantic Dementia (SD).
3.1.ii Clinical features[17]
Impaired confrontation naming
Impaired single-word comprehension
Impaired object/person knowledge
Intact repetition
Intact motor speech
Surface dyslexia and/or dysgraphia
3.1.iii Pathophysiology
Arnold Pick [18] was first to describe the clinical syndrome in late 19th century, long before Mesulam[19] reawakened the interest in 1982, and the term “Semantic Dementia” was coined by Snowden[20] and colleagues. The underlying pathology is usually TDP-43, characterized by dystrophic neurites[21], categorized as FTLD-TDP Type. Hodges and Patterson[22] reported autopsy-proven case series of 20 patients: 14 with TDP-43, 4 with Pick’s disease, and 2 with AD pathology. Neurodegeneration in SD appears to begin in the anterior temporal lobe, more left than right, then spreads to entorhinal cortex, hippocampus, inferior, middle and superior (anterior part) of temporal gyrus. Hodges and Patterson[22] have suggested that sparing of the cingulate in SD might explain intact orientation and ability to learn new information despite severe degeneration of the hippocampal and entorhinal cortices which leads to disorientation and poor anterograde memory in AD. Using lesion method and positron emission tomography, Damasio and colleagues[23] pointed out that different parts of the left temporal lobe take an intermediary role in naming and organization of conceptual knowledge of different categories; faces in the anterior temporal lobe, animals in the inferior temporal lobe and tools in the posterolateral temporal lobe. Gefen and colleagues[24] demonstrated that impaired face naming was correlated with atrophy of the left anterior temporal lobe whereas impaired face recognition was correlated with bilateral anterior temporal lobe atrophy, as in this case.
3.2 Logopenic Aphasia
3.2.i Clinical Case
72-year-old right-handed man with 18 years of formal education presented with a 3-year history of progressively worsening memory and naming difficulties that had not interfered with his daily functioning. He noted no difficulty carrying out his volunteer job and was able to drive without getting lost. Although he was able to manage his household finances, it was taking him longer to complete the task and his wife started double checking after several errors were noted. Family history was significant for his mother developing dementia in her early 70s. On examination his Folstein MMSE[16] was 22/30; he scored 8/10 on the orientation items, 0/3 on the memory items and his sentence repetition was impaired. He named 12 out of first 15 items on Boston Naming Test. Although his speech was fluent his rate was slow. He made a few phonemic paraphasic errors. Grammar and articulation were preserved. Brain MRI showed diffuse cortical atrophy, more in the left temporoparietal region.
3.2.ii Clinical Features [17]
Impaired word finding
Impaired sentence repetition
Preserved single word comprehension
Phonetic paraphasic errors
No agrammatism
Normal motor speech
3.2.iii Pathophysiology
Gorno-Tempini and colleagues[17] described that impaired single-word retrieval and sentence repetition are main features of logopenic aphasia. Phonological paraphasic errors may also be seen. Object knowledge, single-word comprehension, motor speech and grammar are typically preserved. They have suggested phonological loop deficit as core mechanism[25]. Neuropathological and biomarker studies[26]have linked logopenic aphasia to Alzheimer’s disease (AD). Mesulam and colleagues[27] described that memory is preserved early in the disease due to relative sparing of the medial temporal lobe, which was shown in a pathological comparison of a patient with logopenic aphasia and a patient with typical Alzheimer’s disease (AD). In the patient with logopenic aphasia there were more tangles in temporoparietal junction and less tangles in the entorhinal area and the reversal of the pattern was seen in the patient with typical AD. Neuroimaging studies revealed prominent atrophy in the left posterior temporal and inferior parietal regions in logopenic aphasia.
4. Late-onset Dementia
4.1 Hippocampal Sclerosis (HpScl)
4.1.i Clinical Case
An 85-year-old right-handed man presented with six months of subtle memory difficulties. His wife noted that he had started to repeat his stories and was having difficulty remembering his fellow golfers’ names. He was functioning very well otherwise. He was able to drive without problems, perform his activities of daily living and play golf three times a week. He scored 26/30 on the Folstein MMSE[16], scoring 9 of 10 on the orientation and 0 of 3 on the memory items. Neuropsychological assessment was suggestive of amnestic Mild Cognitive Impairment(MCI) with high-average performance across all domains, except inefficient learning and poor retention of information over time. Brain MRI was unremarkable, showing diffuse mild cortical atrophy, compatible with his age. He was seen three years after the initial evaluation. Although he reported progressive memory difficulties, he had continued to function well, was still driving, managing his finances, and was playing golf once a week. His performance on Folstein MMSE was similar (25/30) to his initial evaluation. Neuropsychological assessment revealed relatively isolated memory dysfunction, remained to be consistent with amnestic Mild Cognitive Impairment (MCI), although his performance had declined comparing to his baseline evaluation (Dementia Rating Scale, DRS, scores of 109/144 and 129/144 respectively). He died at age 92, and the autopsy revealed moderate temporal lobe atrophy. The histology showed Hippocampal Sclerosis (HpScl) with TDP-43 pathology limited to medial temporal and limbic lobes with predominant neuronal cytoplasmic inclusions (NCI) along with tangle dominant (Braak stage IV) Alzheimer’s type pathology (Figure 5).
Figure 5.

This patient presented with a long-standing amnestic syndrome. Upon autopsy (A) no obvious cortical atrophy was noted when examining the fixed tissue. (B) When the hippocampus was observed, however, focal discoloration of the subiculum was noted in the fixed tissue and (c) upon examination with H&E without observable senile plaques. Immunohistochemical evaluation with a TDP-43 antibody revealed (D) fine neurites consistent with hippocampal sclerosis of a neurodegenerative etiology. (E) By definition, there was insufficient tau pathology to account for the (F) extensive neuronal loss (inset: pyknotic neuron). Scale bar at 100 μm for 20x images and 50 μm for inset.
4.1.ii Clinical Features [28]
(Comparing to typical AD)
Older age of onset
Slower rate of cognitive decline
Stronger family history of dementia
4.2. Limbic Predominant Alzheimer’s Disease (LP-AD)
4.2.i Clinical Case
A 74-year-old left-handed woman presented with three years history of progressive memory difficulties. She was able to perform her activities of daily living, run her house and drive without problems. She scored 28/30 on the Folstein MMSE[16], scoring 1 of 3 on the memory items. Comprehensive neuropsychological assessment was suggestive of amnestic Mild Cognitive Impairment(MCI) with isolated memory difficulty, particularly list learning and logical memory difficulties, despite above-average performance in most areas. Brain MRI was unremarkable. She was seen two years after the initial presentation and was placed on donepezil by her primary care physician in the interim. She stopped driving and was functioning well otherwise. She scored 25/30 on Folstein MMSE and her neuropsychological testing showed some deterioration in memory and language domains. She continued to follow up yearly. Progressive cognitive deterioration was noted to the point that she was unable to perform her activities of daily living and eventually moved into a retirement home at age 83 with her husband. She died when she was 87. The autopsy revealed Limbic Predominant AD (LP-AD) with predominance of neurofibrillary degeneration in the medial temporal lobe structures without any significant atrophy(Figure 6).
Figure 6.
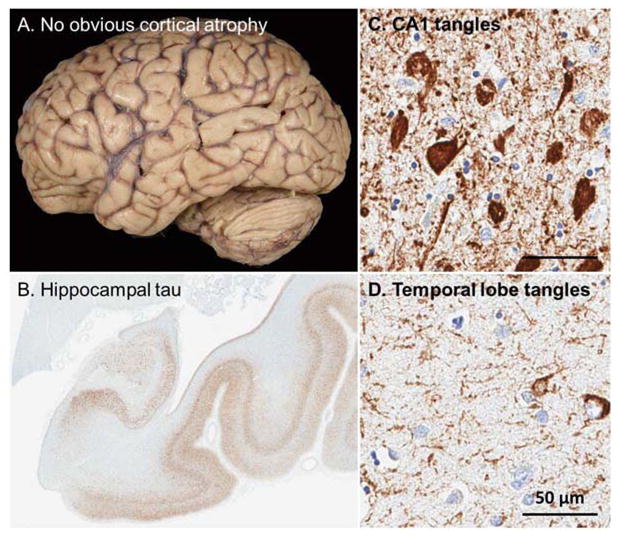
This patient presented with a long-standing amnestic syndrome. Upon autopsy (A) no obvious cortical atrophy was noted when examining the fixed tissue. (B) The hippocampus, however, was greatly atrophied with extensive tau pathology noted. (C) Frequent neurofibrillary tangles were noted with extensive neuronal loss in CA1 and subiculum of the hippocampus. (D) Consistent with the limbic predominant form of Alzheimer’s disease, sparse neurofibrillary tangles were noted in the neocortex as illustrated by the temporal lobe. Scale bar at 50 μm for 20x images.
4.2.ii Clinical Features
Age of onset and cognitive decline are similar to typical AD
4.2.iii Pathophysiology
Differentiating late-onset amnestic dementias can be difficult as clinical presentations are similar and multiple pathologies can co-exist [29]. Pathological subtypes of AD are described based on the relative density of NFTs in the hippocampus and cortex: typical AD, hippocampal-sparing AD, and limbic-predominant AD (LP-AD) [14, 30]. While typical AD follows the Braak staging [8] LP-AD has disproportionately greater NFTs in the hippocampus than in the neocortex. HpScl is characterized by severe hippocampal volume loss and neuronal loss in CA1 and the subiculum which is disproportionate to the number of NFTs [31]. Considering the medial temporal lobe involvement in both HpScl and LP-AD, neuroimaging and pathologic analysis can be limited in distinguishing [32]. Concomitant HpScl and AD can further blur the picture. Murray and colleagues [28] examined a large cohort of autopsy-confirmed cases of typical AD, LP-AD, HpScl and HpScl-AD to identify distinct clinical, genetic, and pathologic characteristics. They showed that existence of AD pathology (Typical AD, LP-AD, HpScl-AD) dominates the phenotype, while “pure HpScl” phenotype is distinct with significantly older age of onset (79 in comparison to 71 in typical AD) and slower cognitive decline. They pointed out that AD groups were more likely to be carriers of APOE ε4, the strongest genetic risk factor for AD, while HpScl groups (HpScl and HpScl-AD) were more likely to exhibit genetic variants in GRN and TMEM106B that are associated with frontotemporal lobar degeneration [33–35] and APOE status was independent of risk for HpScl [34, 36, 37] which may have diagnostic value along with a novel polymorphism that was recently described in the ATP-binding cassette, sub-family C member 9 (ABCC9) in HpScl [38]. Murray and colleagues also showed that HpScl groups had a high frequency of TDP-43 pathology that was most often Type A morphology and distribution[33], while AD groups had a significantly lower frequency of TDP-43 pathology that was most often Type B[14, 30]. Taken together, it is suggested that HpScl and AD are pathologically and genetically distinct and non-synergistic neurodegenerative processes despite similar clinical presentations [28, 39, 40].
5. Rapidly Progressive Dementia (RPD)
5.1 Creutzfeldt- Jakob Disease (CJD)
5.1.i Clinical Case
82-year-old, right-handed otherwise healthy woman presented with one year history of rapidly progressive cognitive impairment. Despite her short-term memory problems and word-finding difficulties, she was functioning well. She was able to live alone and drive without getting lost. She reported some difficulty standing from a sitting position. She was feeling increasingly unsteady and most recently started using a cane to ambulate. No falls or abnormal movements were reported. She scored 26/30 on Folstein MMSE[16] examination, 2/10 on orientation and 1/3 on the memory items. She scored 14/20 on the first 20 items of Boston Naming Test. She had significant simultanagnosia. Examination did not reveal ataxia, myoclonus or Parkinsonism. Serum and CSF paraneoplastic panels were unremarkable. CSF 14-3-3 protein was positive and T-tau level was high (3615 pg/mL). Diffusion-weighted MRI brain images showed cortical ribboning in the occipital and temporal lobes bilaterally, left more than right (Figure 7). EEG was not performed. The patient died within two months of her evaluation. Autopsy was not performed.
Figure 7.

Diffusion weighted imaging (DWI) shows cortical ribboning in the occipital and temporal lobes bilaterally, left more than right.
5.1.ii Clinical Features [41]
Progressive dementia
Visual disturbances
Myoclonus
Cerebellar, pyramidal/extrapyramidal findings
Akinetic mutism
CSF 14-3-3 positivity
Periodic Sharp Wave Complexes (PSWCs) in EEG
5.1.iii Pathophysiology
Considering the diagnosis of CJD can be delayed irrespective of the age of onset[42], older age can further complicate the picture. The World Health Organization (WHO) criteria for sCJD[41] require either a typical EEG (PSWCs) or a positive CSF 14-3-3 along with progressive dementia resulting in fatality within two years. Diagnostic accuracy of protein 14-3-3 has been questioned as approximation of its sensitivity differs anywhere between 50% to 92% with an approximate specificity of 80% [43, 44] comparing to T-Tau with sensitivity and specificity higher than 90%[45, 46]. Dr. Forner and colleagues[47] recently looked into diagnostic tools for sporadic CJD, and noted that diffusion-weighted MRI had a higher diagnostic accuracy (97%) than CSF biomarkers (14-3-3, neuron-specific enolase, NES, T-Tau) among which T-Tau had the highest diagnostic accuracy (79.6%). The newer RT-QuIC (real-time quaking-induced conversion) may also be helpful[48]. Despite its high diagnostic accuracy for sCJD, similar diffusion-weighted MRI hyperintensity patterns can be seen in nonprionRPD (npRPD). Several other conditions, particularly autoimmune encephalitis and rapidly progressive neurodegenerative diseases, can mimic CJD[49]. In a case series of 15 subjects who had an initial diagnosis of CJD but eventually confirmed to have Voltage-Gated Potassium Channel (VGKC) autoantibody–associated encephalopathy, Geschwind and colleagues[50] noted that more than half (60%) of the subjects satisfied World Health Organization diagnostic criteria for CJD; with rapidly progressive dementia, elevated CSF 14-3-3 protein or NES. The T2-weighted and FLAIR hyperintensities of the medial temporal regions were noted in 5 subjects, which is classic for autoimmune limbic encephalitis but can also be seen in CJD. EEG showed no PSWCs despite diffuse slowing along with frontal intermittent rhythmic delta activity and focal epileptogenic activity. Hyponatremia was common (60%) and was thought to have an important diagnostic value favoring an autoimmune etiology. Neoplasia was confirmed histologically in 5 patients (33%) and was suspected in another 5. Most patients(92%) improved after immunomodulatory therapy. Rapidly Progressive Alzheimer’s Disease (rpAD) is another consideration, in a cohort (N=89) of autopsy-confirmed rpAD, Schmidt and colleagues[51] showed that rpAD is a different clinical syndrome than typical AD, at least in the early stages, and is similar to CJD symptomatology. Fast decline appears to be associated with certain symptoms, such as motor signs. Although CSF AD biomarkers did not differ from that of typical AD pattern, protein 14-3-3 was frequently detected (42%) in rpAD.
5.2 Limbic Encephalitis
5.2.i Clinical Case
A 71-year-old right-handed engineer presented with one-year history of seizures and cognitive impairment. After being evaluated by multiple providers he was referred to Behavioral Neurology by an epileptologist. His initial symptom was a sudden onset of focal amnesia, followed by persistent memory impairment and paroxysmal episodes of olfactory hallucinations, diaphoresis, oral automatisms and dysarthria lasting for several minutes. He was placed on memantine and donepezil without any significant benefit. Oxcarbazepine had diminished the frequency and severity of the seizures, however they persisted with intermittent generalization. Medical and family history indicated no autoimmunity. He scored 19/30 on Folstein MMSE. Neurological examination was otherwise unremarkable. Comprehensive neuropsychological assessment revealed profound memory impairment with mild frontal-subcortical involvement. Laboratory workup showed hyponatremia (125 mmol/L) and voltage-gated potassium channel antibodies (VGKC-Abs) in the serum at a level of 0.59 nmol/L (normal level <0.02 nmol/L). CSF studies were unremarkable. EEG revealed subclinical electrographic seizure activity arising from bilateral temporal lobes in the sleep states. MRI-brain was unremarkable with no evidence for encephalopathy (Figure 8). Full body FDG-PET scan revealed no abnormality. Diagnosis of Limbic Encephalitis with VGKC-Abs was considered and the patient was placed on IV methylprednisolone (1 g IV daily for 3 days then weekly for 6 weeks and then biweekly for 6 weeks). Blood count, liver and kidney functions were monitored closely. He was placed on vitamin D and calcium replacement along with trimethoprim/sulfamethoxazole (1 double-strength tablet three times a week) for Pneumocystis carinii pneumonia. His return visit was twelve weeks after the initiation of immunotherapy. He reported significant cognitive improvement, which was also noted by his wife with particular improvement in days immediately after infusions, then slow gradual decline until the following infusion. His seizures were resolved. Detailed neuropsychological assessment confirmed notable improvement across domains relative to his initial evaluation, except persistent memory impairment. His blood sodium was normalized (135 mmol/L). He was switched to oral prednisolone (60mg daily for 3 months) and azathioprine was started (50mg twice a day) simultaneously with an aim to increase his MCV by 5 points. Thiopurine methyltransferase (TPMT) enzyme activity was checked and was normal prior to initiation of azathioprine[52]. He was seen 12 weeks after initiation of azathioprine. Neuropsychological assessment revealed stable cognitive function, although the patient and his wife reported cognitive improvement. Azathioprine was titrated (100mg twice a day) while oral prednisolone was continued at the dose of 60mg daily for 12 weeks, until his return visit. Neuropsychological assessment revealed stable cognitive function, with subjective report of continuous cognitive improvement. MCV had increased by 10, Na:139 mmol/L, liver and kidney functions were normal. Steroid taper was initiated (10mg per month until 10mg daily followed by 1mg/day/month decrements until completion). He was seen upon completion of steroid taper. He reported stable cognitive function which was confirmed by the Neuropsychological assessment. Azathioprine (100mg twice a day) was continued.
Figure 8.
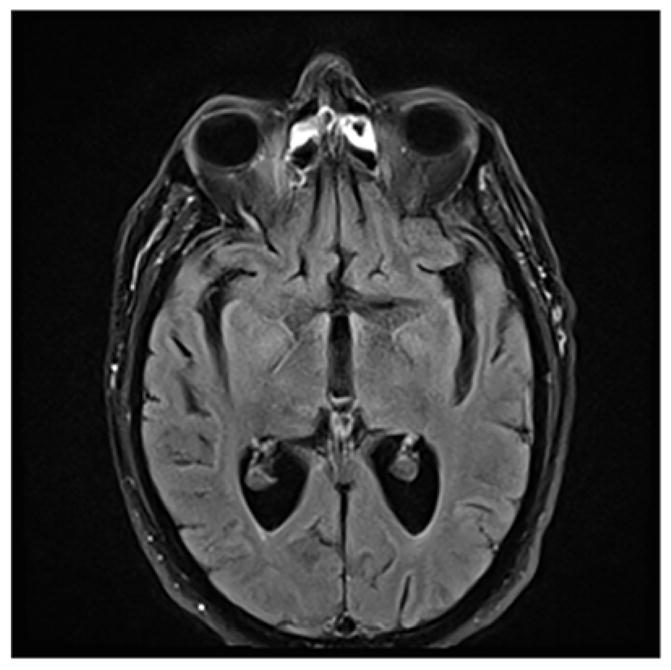
MRI brain can be negative in limbic encephalitis.
5.2.ii Clinical Features [53]
Rapidly progressive or fluctuating course
One or more symptoms of memory impairment, behavioral disturbance, hallucinations, focal seizures
Inflammatory markers in the CSF
Coexisting organ-specific autoimmunity (particularly thyroid)
Response to immunotherapy
5.2.iii Pathophysiology
When evaluating a patient with RPD, it is important to not to overlook potentially treatable conditions. Systematic approach is needed to ensure that all possibilities are considered. The mnemonic VITAMIN C (vascular, infectious, toxic-metabolic, autoimmune, metastatic, iatrogenic, neurodegenerative, congenital/systemic) can come handy. Flanagan and colleagues[54] looked into 72 consecutive patients received immunotherapy for suspected autoimmune dementia and showed that more than one third (35%) of the immunotherapy responders were initially diagnosed with a neurodegenerative or prion disorder. If an autoimmune dementia is suspected, several points are worth noting [53]:
The clinical manifestations are diverse and often multifocal
A personal or family history of autoimmunity/seropositivity is a risk factor
Neural-specific autoantibody positivity serves as a marker of neurologic autoimmunity
Identification of neural-specific autoantibodies may lead a targeted oncological evaluation in the context of a known cancer association (e.g. NMDAR antibody and teratoma)
Immunotherapy trial may have a “diagnostic” value
For evaluation and treatment of patients with suspected autoimmune dementia, McKeon and colleagues [53]outlined practical guidelines, starting with the exclusion of other causes of cognitive decline, followed by objective documentation of cognitive and neurologic abnormalities (comprehensive neuropsychological assessment, EEG, MRI, functional imaging) to provide a pretreatment baseline by which response to immunotherapy can be measured. CSF analysis, comprehensive antibody testing (serum and CSF), and paraneoplastic investigation (e.g. whole-body CT or PET scan) may lead to further clues about autoimmune etiology. Although the phenotype associated with any given autoantibody appears to be broader[55] there are several direct associations that are identified, such as limbic encephalitis with VGKC antibodies, ANNA-1 (antineuronal nuclear antibody/anti-Hu) and α-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antibodies[56]; fluctuating hemiplegia with complete recovery and thyroid autoantibodies[57]; and diverse psychiatric presentations early in the course with NMDA receptor antibody [58] The positive predictive values for cancer detection rates vary from 33% for VGKC anti- bodies (various cancer types)[59] to over 80% for ANNA-1 (almost always small- cell lung carcinoma)[60]. Once autoimmune dementia is suspected, immunotherapy should be initiated promptly as shorter delay to treatment is shown[61] to be one of the predictors of treatment response along with subacute onset, fluctuating course, detection of a cation channel- specific autoantibody (calcium channel, VGKC, ganglionic acetylcholine receptor) and CSF findings of elevated protein (greater than 100 mg/dL) or pleocytosis. Treatment of autoimmune dementias remains empirical. McKeon and colleagues [53] recommended starting a trial of IV corticosteroid “pulse therapy” with methylprednisolone 1 g IV daily for 3–5 days then weekly for 6–8 weeks (alternatively IVIG 0.4–1g/kg IV daily for 3–5 days then weekly for 6–8 weeks). They pointed out that plasma exchange can be tried for critically ill or when corticosteroids/IVIG is poorly tolerated. They recommended a return visit following 4-to-8-weeks of immunotherapy to investigate subjective and objective evidence of clinical improvement (reported functional/cognitive improvement, comprehensive neuropsychological assessment, neuroimaging etc). In the absence of objective evidence of improvement, a trial of IVIG can be considered; followed by reevaluations. They noted that a favorable response to immunotherapy supports the diagnosis and prompts long-term immunotherapy as relapse is common due to withdrawal of acute therapies. Extending the interval between infusions of IV corticosteroids (or IVIG) from weekly to biweekly, then every 3 weeks and then monthly, over a period of 4 to 6 months while bridging with oral steroid-sparing immunosuppressants (azathioprine 1–2 mg/kg/d in a two daily divided doses or mycophenolatemofetil 500–2000 mg/d in a two daily divided doses) to ensure reduction in corticosteroid (or IVIG) dependence. Some overlap in corticosteroid (or IVIG) and oral immunosuppressant treatment is required, approximately 12 weeks for azathioprine and 6 to 8 weeks for mycophenolate mofetil[62]. If no cognitive improvement occurs after infusion, corticosteroid (or IVIG) can be discontinued. No data are available to determine the appropriate duration of immunosuppression treatment in autoimmune dementia. While spontaneous remissions is possible, some patients are dependent lifelong on immunosuppressants. McKeon and colleagues recommend a trial of immunosuppressant medication withdrawal after 2 to 3 years [53]. Appropriate calcium (1000 mg/d – 1500 mg/d) and vitamin D (800 IU/d) intake is recommended for patients taking corticosteroids long term[63]. Prophylaxis against Pneumocystis carinii pneumonia (trimethoprim/sulfamethoxazole, 1 double-strength tablet 3 times a week) is recommended for patients receiving long term corticosteroids or a purine analog[64]. Withdrawal of chronic oral corticosteroids may lead to adrenocortical failure if not done cautiously, collaborating with an endocrinologist or internist would be beneficial. Close monitoring of blood count, liver and renal function is required for patients on immunotherapy, weekly for the first month then biweekly for two months, and monthly thereafter.
6. Discussion
The differential diagnosis of dementia can be challenging as initial clinical presentation can be variable and different neuropathologies can coexist, particularly in the elderly. It is important not to overlook focal cortical presentations of AD: visual impairment can be the only symptom early in the course leading to unfruitful ophthalmologic visits and the clinical picture of CBS may have AD as the underlying pathology. Patients with logopenic aphasia frequently have AD, while TDP-43 is the most common pathology in SD. Different neuropathologic processes can result in similar clinical presentations, as discussed in LP-AD and HpScl. Early diagnosis in RPD is of paramount importance as it can be reversible, such as autoimmune encephalitis. Recognizing the different syndromes described in this paper can help the clinician improve their diagnostic skills leading to improved patient outcomes by early and accurate diagnosis, prompt treatment, appropriate counseling and guidance.
Key Points.
The differential diagnosis of dementia can be challenging as initial clinical presentation can be variable and different neuropathologies can coexist.
Visual impairment can be the only symptom early in the course of focal Alzheimer’s.
The clinical picture of Corticobasal Syndrome may have have Alzheimer’s as the underlying pathology.
Patients with logopenic aphasia frequently have Alzheimer’s pathology, while TDP-43 is the most common pathology in Semantic Dementia.
Different neuropathologic processes can result in similar clinical presentations.
It is important to not to overlook treatable causes of rapidly progressive dementia.
Footnotes
Dsclosures: The study is supported in part by Mayo Clinic Alzheimer’s Disease Research Center (ADRC) grant P50AG 16574-14(PI: Ronald Petersen, MD, PhD). The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Kokmen E, Naessens JM, Offord KP. A short test of mental status: description and preliminary results. Mayo Clin Proc. 1987;62(4):281–8. doi: 10.1016/s0025-6196(12)61905-3. [DOI] [PubMed] [Google Scholar]
- 2.Benson DF, Davis RJ, Snyder BD. Posterior cortical atrophy. Arch Neurol. 1988;45(7):789–93. doi: 10.1001/archneur.1988.00520310107024. [DOI] [PubMed] [Google Scholar]
- 3.Hof PR, et al. Atypical form of Alzheimer’s disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Res. 1997;37(24):3609–25. doi: 10.1016/S0042-6989(96)00240-4. [DOI] [PubMed] [Google Scholar]
- 4.Tang-Wai DF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004;63(7):1168–74. doi: 10.1212/01.wnl.0000140289.18472.15. [DOI] [PubMed] [Google Scholar]
- 5.Rosenbloom MH, et al. Distinct clinical and metabolic deficits in PCA and AD are not related to amyloid distribution. Neurology. 2011;76(21):1789–96. doi: 10.1212/WNL.0b013e31821cccad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ossenkoppele R, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol. 2015;77(2):338–42. doi: 10.1002/ana.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrasquillo MM, et al. Late-onset Alzheimer disease genetic variants in posterior cortical atrophy and posterior AD. Neurology. 2014;82(16):1455–62. doi: 10.1212/WNL.0000000000000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 9.Boeve BF, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999;53(4):795–800. doi: 10.1212/wnl.53.4.795. [DOI] [PubMed] [Google Scholar]
- 10.Jack CR, Jr, et al. Rate of medial temporal lobe atrophy in typical aging and Alzheimer’s disease. Neurology. 1998;51(4):993–9. doi: 10.1212/wnl.51.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frisoni GB, et al. Mapping local hippocampal changes in Alzheimer’s disease and normal ageing with MRI at 3 Tesla. Brain. 2008;131(Pt 12):3266–76. doi: 10.1093/brain/awn280. [DOI] [PubMed] [Google Scholar]
- 12.Desikan RS, et al. Selective disruption of the cerebral neocortex in Alzheimer’s disease. PLoS One. 2010;5(9):e12853. doi: 10.1371/journal.pone.0012853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giannakopoulos P, et al. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol. 2007;113(1):1–12. doi: 10.1007/s00401-006-0144-y. [DOI] [PubMed] [Google Scholar]
- 14.Murray ME, et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785–96. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paterson RW, et al. Dissecting IWG-2 typical and atypical Alzheimer’s disease: insights from cerebrospinal fluid analysis. J Neurol. 2015;262(12):2722–30. doi: 10.1007/s00415-015-7904-3. [DOI] [PubMed] [Google Scholar]
- 16.Folstein MF, Folstein SE, McHugh PR. Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 17.Gorno-Tempini ML, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–14. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pick A. Ueber die Beziehungen der senilen Hirnatrophie zur Aphasia. Prag Med Wochenschr. 1892;17(165):7. [Google Scholar]
- 19.MMM Slowly progressive aphasia without generalized dementia. Ann Neurol. 1982;11(592):8. doi: 10.1002/ana.410110607. [DOI] [PubMed] [Google Scholar]
- 20.Snowden JS, GP, Neary D. Semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol. 1989;2(167):82. [Google Scholar]
- 21.Mackenzie IR, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–3. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodges JR, Patterson K. Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol. 2007;6(11):1004–14. doi: 10.1016/S1474-4422(07)70266-1. [DOI] [PubMed] [Google Scholar]
- 23.Damasio H, et al. A neural basis for lexical retrieval. Nature. 1996;380(6574):499–505. doi: 10.1038/380499a0. [DOI] [PubMed] [Google Scholar]
- 24.Gefen T, et al. Naming vs knowing faces in primary progressive aphasia: a tale of 2 hemispheres. Neurology. 2013;81(7):658–64. doi: 10.1212/WNL.0b013e3182a08f83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorno-Tempini ML, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55(3):335–46. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teichmann M, et al. Deciphering logopenic primary progressive aphasia: a clinical, imaging and biomarker investigation. Brain. 2013;136(Pt 11):3474–88. doi: 10.1093/brain/awt266. [DOI] [PubMed] [Google Scholar]
- 27.Mesulam MM. Primary progressive aphasia and the language network: the 2013 H. Houston Merritt Lecture. Neurology. 2013;81(5):456–62. doi: 10.1212/WNL.0b013e31829d87df. [DOI] [PubMed] [Google Scholar]
- 28.Murray ME, et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol. 2014;128(3):411–21. doi: 10.1007/s00401-014-1302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jellinger KA, Attems J. Challenges of multimorbidity of the aging brain: a critical update. J Neural Transm. 2014 doi: 10.1007/s00702-014-1288-x. [DOI] [PubMed] [Google Scholar]
- 30.Janocko NJ, et al. Neuropathologically defined subtypes of Alzheimer’s disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol. 2012;124(5):681–92. doi: 10.1007/s00401-012-1044-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickson DW, et al. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol. 1994;88(3):212–21. doi: 10.1007/BF00293396. [DOI] [PubMed] [Google Scholar]
- 32.Dutra JR, Cortes EP, Vonsattel JP. Update on Hippocampal Sclerosis. Curr Neurol Neurosci Rep. 2015;15(10):67. doi: 10.1007/s11910-015-0592-7. [DOI] [PubMed] [Google Scholar]
- 33.Amador-Ortiz C, et al. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol. 2007;113(3):245–52. doi: 10.1007/s00401-006-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson PT, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134(Pt 5):1506–18. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson PT, et al. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol. 2013;126(2):161–77. doi: 10.1007/s00401-013-1154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Troncoso JC, et al. Lack of association of the apoE4 allele with hippocampal sclerosis dementia. Neurosci Lett. 1996;204(1–2):138–40. doi: 10.1016/0304-3940(96)12331-4. [DOI] [PubMed] [Google Scholar]
- 37.Leverenz JB, et al. Clinical and neuropathological characteristics of hippocampal sclerosis: a community-based study. Arch Neurol. 2002;59(7):1099–106. doi: 10.1001/archneur.59.7.1099. [DOI] [PubMed] [Google Scholar]
- 38.Nelson PT, et al. ABCC9/SUR2 in the brain: Implications for hippocampal sclerosis of aging and a potential therapeutic target. Ageing Res Rev. 2015;24(Pt B):111–25. doi: 10.1016/j.arr.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graff-Radford N, Murray ME, Ross OA, Duara R, Rademakers R, Whitwell JL, Dickson DW. Differentiating clinicopathologic and genetic aspects of hippocampal sclerosis in Alzheimer’s disease from limbic predominant Alzheimer’s disease and “pure” hippocampal sclerosis. Alzheimer’s Dement. 8:620–621. [Google Scholar]
- 40.Brenowitz WD, et al. Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimers Dis. 2014;39(3):691–702. doi: 10.3233/JAD-131880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.WHO. Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: Report of a WHO consultation. Geneva: 1998. [Google Scholar]
- 42.Paterson RW, et al. Differential diagnosis of Jakob-Creutzfeldt disease. Arch Neurol. 2012;69(12):1578–82. doi: 10.1001/2013.jamaneurol.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2012;79(14):1499–506. doi: 10.1212/WNL.0b013e31826d5fc3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim MO, Geschwind MD. Clinical update of Jakob-Creutzfeldt disease. Curr Opin Neurol. 2015;28(3):302–10. doi: 10.1097/WCO.0000000000000197. [DOI] [PubMed] [Google Scholar]
- 45.Coulthart MB, et al. Diagnostic accuracy of cerebrospinal fluid protein markers for sporadic Creutzfeldt-Jakob disease in Canada: a 6-year prospective study. BMC Neurol. 2011;11:133. doi: 10.1186/1471-2377-11-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Harten AC, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med. 2011;49(3):353–66. doi: 10.1515/CCLM.2011.086. [DOI] [PubMed] [Google Scholar]
- 47.Forner SA, et al. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease. Neurol Clin Pract. 2015;5(2):116–125. doi: 10.1212/CPJ.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atarashi R, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17(2):175–8. doi: 10.1038/nm.2294. [DOI] [PubMed] [Google Scholar]
- 49.Geschwind MD. Rapidly progressive dementia: prion diseases and other rapid dementias. Continuum (Minneap Minn) 2010;16(2 Dementia):31–56. doi: 10.1212/01.CON.0000368211.79211.4c. [DOI] [PubMed] [Google Scholar]
- 50.Geschwind MD, et al. Voltage-gated potassium channel autoimmunity mimicking creutzfeldt-jakob disease. Arch Neurol. 2008;65(10):1341–6. doi: 10.1001/archneur.65.10.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt C, et al. Rapidly progressive Alzheimer’s disease: a multicenter update. J Alzheimers Dis. 2012;30(4):751–6. doi: 10.3233/JAD-2012-120007. [DOI] [PubMed] [Google Scholar]
- 52.Liu YP, et al. Association between Thiopurine S-Methyltransferase Polymorphisms and Azathioprine-Induced Adverse Drug Reactions in Patients with Autoimmune Diseases: A Meta-Analysis. PLoS One. 2015;10(12):e0144234. doi: 10.1371/journal.pone.0144234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKeon A, Lennon VA, Pittock SJ. Immunotherapy-responsive dementias and encephalopathies. Continuum (Minneap Minn) 2010;16(2 Dementia):80–101. doi: 10.1212/01.CON.0000368213.63964.34. [DOI] [PubMed] [Google Scholar]
- 54.Flanagan EP, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc. 2010;85(10):881–97. doi: 10.4065/mcp.2010.0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mittal MK, et al. Autoimmune Encephalitis in the ICU: Analysis of Phenotypes, Serologic Findings, and Outcomes. Neurocrit Care. 2015 doi: 10.1007/s12028-015-0196-8. [DOI] [PubMed] [Google Scholar]
- 56.Lai M, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65(4):424–34. doi: 10.1002/ana.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaw PJ, et al. Hashimoto’s encephalopathy: a steroid-responsive disorder associated with high anti-thyroid antibody titers--report of 5 cases. Neurology. 1991;41(2 Pt 1):228–33. doi: 10.1212/wnl.41.2_part_1.228. [DOI] [PubMed] [Google Scholar]
- 58.Dalmau J, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091–8. doi: 10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan KM, et al. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008;70(20):1883–90. doi: 10.1212/01.wnl.0000312275.04260.a0. [DOI] [PubMed] [Google Scholar]
- 60.Lucchinetti CF, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology. 1998;50(3):652–7. doi: 10.1212/wnl.50.3.652. [DOI] [PubMed] [Google Scholar]
- 61.Flanagan E, MA, Lennon V. Immunotherapy responsive dementia or encephalopathy: clinical course and predictors of improvements. Ann Neurol. 2009;66:S41. [Google Scholar]
- 62.Meriggioli MN, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology. 2003;61(10):1438–40. doi: 10.1212/01.wnl.0000094122.88929.0b. [DOI] [PubMed] [Google Scholar]
- 63.American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum. 2001 doi: 10.1002/1529-0131(200107)44:7<1496::AID-ART271>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 64.Stern A, et al. Prophylaxis for Pneumocystis pneumonia (PCP) in non-HIV immunocompromised patients. Cochrane Database Syst Rev. 2014;10:CD005590. doi: 10.1002/14651858.CD005590.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]