

Abstract
Lack of molecular biology evidence has led clinical success of alternative and complementary medicine (CAM) to be marginalized. In turn, a large portion of life Science researchers could not communicate and help to develop therapeutic potential laid in these therapeutic approaches. In this study, we began to quantify descriptive classification theory in one of the CAM branches i.e. Iranian traditional medicine (ITM). Using proteomic tools and network analysis, the expressed proteins and their relationships were studied in mitochondrial lysate isolated from PBMCs from two different temperaments i.e. Hot-wet (HW) and Cold-dry (CD). The 82% of the identified proteins are over- or under-represented in distinct temperaments. Also, our result showed the different protein-protein interaction networks (PPIN) represented in these two temperaments using centrality and module finding analysis. Following the gene ontology and pathway enrichment analysis, we have found enriched biological terms in each group which are in conformity with the physiologically known evidence in ITM. In conclusion, we argued that the network biology which naturally consider life at the system level along with the different omics data will pave the way toward explicit delineation of the CAM activities.
From a mechanistic biology point of view, complementary and alternative medicine (CAM) have long been struggled with a lack of molecular biology evidence. Technological advances accompanying with new multi-disciplinary integrative approaches in biomedical studies help to bridge this gap1. It is expected to emerge correlations between molecular and CAM clinical evidence which are functionally connected. Among other fields of CAM, Traditional Chinese medicine (TCM) is the most fortunate to be expressed in the language of modern molecular biology (1–9). Although the new findings based on omics technology, network analysis, and modeling in TCM clarify the similar basis of much traditional ethnic medicines due to humorism as common theory, there are still a lot of unanswered questions in this era.
The common dominator between the systems’ level understanding of biological systems, called systems biology, and the various branches of CAM is the holistic approach or considering the whole system consisting subjects and interactions1,2. This aforementioned approach could cause to mine emerging properties of biological phenomena. For instance, Ma et al. underlined the genetic background of Cold syndrome using the microarray expression profiling and systems biology methods. Their study revealed that genes related to energy metabolism, neurotransmitter, hormones and cytokines are differentially expressed and significantly enriched in the Cold Syndrome based on protein-protein interaction network (PPIN) analysis1. Also, Aiping Lu’s research group confirmed Hot and Cold classification in rheumatoid arthritis patients using gene expression and metabolite profiles following by the network analysis methods applied in systems biology. In the underlying biological network, they found the enrichment of G protein signaling pathway, oxidation-reduction in fatty acid metabolism and T cell proliferation pathways in the highly connected clusters of PPIN involved in rheumatoid arthritis patients3,4,5,6. There are several other studies which prompted to pave the way for the development of TCM into a new historical period, mainly divided into three categories; TCM-based disease classification3,4,6,7,8,9, TCM network pharmacology10,11,12 and biological investigations on manual therapy such as acupuncture and cupping13.
In the Iranian traditional medicine (ITM), the human body categorized into some classes or temperaments based on signs and symptoms similar to TCM14,15,16,17. Although several forenamed omics-based and non-omics-based researches have been performed on describing some of these temperaments, there are still many undefined points in the theory of ITM and TCM. In this study, the proteomic profiles of two types of temperaments were compared to each other in order to describe molecular foundation of ITM human body classification theory and candidate molecular biomarkers in these temperaments. It should be noted that in the present study, the people who represent Hot-Wet (HW) and Cold-Dry (CD) temperaments were compared. These categories are two of four combinatory temperaments, produced by two humors; Sanguine and Hypochondria respectively, expressed in ITM, TCM and Ayurveda11,18,19,20.
Results
The identified proteins in HW group in comparison with CD is presented in Fig. 1. Some of the identified proteins are specifically related to each group and some of them are differentially overexpressed in specified group. As it shown in Fig. 1, the most of the differentially identified proteins (72%) are related to the CD group which is indicated the level of complexity in this group. Previously, some DEGs were introduced in similar work focused on Hot and Cold syndrome in TCM conducted by Ma et al. and Chen et al. in healthy and patient individuals1,3. They revealed the genetic background of these syndrome using mRNA microarray technology. Although grouping in our study is somewhat different with above-mentioned studies and we analyzed differential expression at the protein levels, some proteins expression are in conformity with their results including FLNB, FLNA, SRC, YWHAZ, UBQLN4, HSPA1A, H2AFX, and MYO5B.
Figure 1. The identified proteins in temperaments.
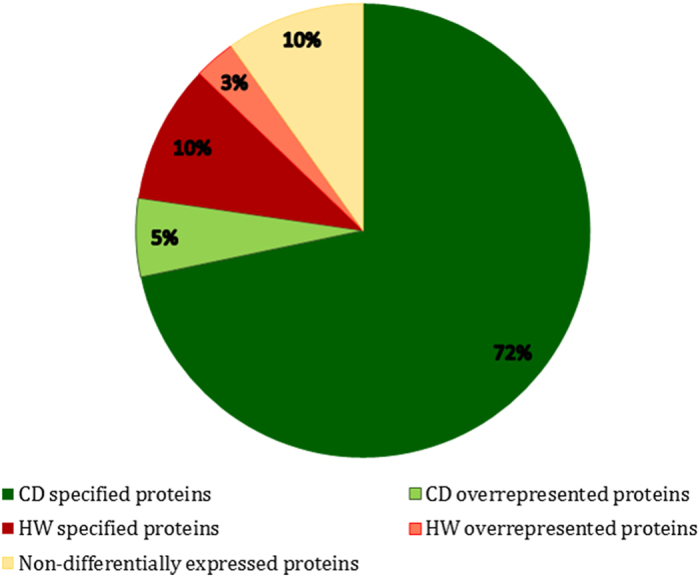
The ratio of the specified and differentially expressed gene products are presented in each temperament i.e. cold-dry and hot-wet.
Then, focusing on the pathway enrichment analysis, 17, 6, and 4 pathways are listed based on KEGG, Panther and Reactome databases, respectively. These pathways are significantly enriched in each identified protein set (adjusted p-value < 0.05) are presented in Table 1. Some of the pathways are uniquely disturbed among each group such as dilated cardiomyopathy, Endocytosis, Hypertrophic cardiomyopathy (HCM), Leukocyte trans-endothelial migration, Muscle contraction and Nicotinic acetylcholine receptor signaling pathway in CD individuals. The pathways such as ECM-receptor interaction, Hematopoietic cell lineage and vascular smooth muscle contraction were enriched only in HW individuals.
Table 1. The enriched biological pathways.
| Database | Term | Cold-Dry | Hot-Wet | ||
|---|---|---|---|---|---|
| KEGG | Frequency | P-value | Frequency | P-value | |
| Antigen processing and presentation | 9% | 3.68E-03 | 13% | 1.39E-03 | |
| Dilated cardiomyopathy | 7% | 5.72E-03 | |||
| ECM-receptor interaction | 9% | 2.08E-02 | |||
| Endocytosis | 10% | 1.38E-03 | |||
| Focal adhesion | 17% | 1.21E-07 | 9% | 1.26E-02 | |
| Gap junction | 10% | 1.27E-05 | 9% | 2.20E-02 | |
| Hematopoietic cell lineage | 9% | 2.70E-02 | |||
| Hypertrophic cardiomyopathy (HCM) | 7% | 4.08E-03 | |||
| Leukocyte transendothelial migration | 9% | 6.30E-04 | |||
| Malaria | 9% | 7.87E-03 | |||
| Pathogenic E. coli infection | 13% | 6.04E-11 | |||
| Phagosome | 14% | 6.08E-06 | |||
| Regulation of actin cytoskeleton | 17% | 3.84E-08 | |||
| Tight junction | 15% | 6.88E-09 | |||
| Vascular smooth muscle contraction | 9% | 3.24E-02 | |||
| Viral myocarditis | 13% | 1.22E-08 | |||
| PANTHER | |||||
| Blood coagulation | 8% | 1.03E-04 | |||
| Cytoskeletal regulation by Rho GTPase | 16% | 8.94E-11 | 17% | 9.94E-04 | |
| Hedgehog signaling pathway | 5% | 2.52E-03 | |||
| Integrin signaling pathway | 12% | 7.70E-04 | |||
| Nicotinic acetylcholine receptor signaling pathway | 9% | 1.85E-04 | |||
| Parkinson disease | 9% | 4.98E-04 | |||
| REACTOME | |||||
| Hemostasis | 24% | 2.67E-13 | 22% | 2.13E-03 | |
| Integrin cell surface interactions | 7% | 4.33E-03 | |||
| Metabolism of proteins | 11% | 2.06E-03 | |||
| Muscle contraction | 4% | 8.01E-03 | |||
The statistically significant enriched pathways are listed based on KEGG, Panther and Reactome databases in each temperament. Some of them are presented in both temperaments with different frequencies.
Conspicuously, these enriched pathways are in harmony with the physiological signs and symptoms stated in the ITM theory. In addition to human body classification including HW, CD, Hot-Dry (HD) and Cold-Wet (CW), the human body organs are also classified using these temperaments which are the main constituent of that organ based on ITM theory21,22,23. For instance, the heart’s temperament is HW and all diseases which functionally impaired toward decreasing cardiac efficiency are diseases related to CD temperament. Therefore, significancy of the enriched terms like Viral Myocarditis, Dilated cardiomyopathy and Hypertrophic cardiomyopathy (HCM) in CD temperaments is in accordance with the biomedical significancy prevalent in ITM. This consistency is occurred in several other cases such as Leukocyte Transendothelial migration and Integrin signaling pathway in CD group which conforms to diverse chronic autoimmune and inflammatory disease are common in people with advanced CD symptoms (CD Syndrome); or Parkinson pathway and Blood coagulation which typically classified in CD signs and symptom. Considering the discussed conformity about CD temperaments, in addition, Hematopoietic cell lineage and Vascular Smooth muscle contraction pathways are significantly enriched in HW temperaments as expected based on ITM explanation for HW sings and symptom.
In the next step, the gene ontology related to the identified proteins were explored based on enrichment analysis (Fig. 2). Diverse similar or unique biological processes (BPs) and molecular function (MFs) were distributed among the both groups. The HW specified BPs were generally related to DNA packaging and protein-DNA complex assembly while the CD related BPs were cellular component morphogenesis, movement and transport. The binding to GTP, ATP, Actin and other cytoskeletal constitutes versus the binding to ADP and extracellular matrix is another difference between CD and HW individuals. Interestingly, some of the enriched terms are previously reported in the gene ontology analysis of DEGs found by microarray3 including response to stress (GO:0006950), response to wounding (GO:009611), protein localization to cell surface (GO: 0034394), regulation of apoptotic process (GO: 0042981), activation of immune response (GO:0002253), generation of precursor metabolites and energy (GO:0006091), meiotic cell cycle process (GO:1903046), small GTPase mediated signal transduction (GO:0007264), Rho protein signal transduction (GO:0007266), intracellular signal transduction (GO:0035556) and defense response (GO:0006952). The conformity of the gene ontology enrichment analysis is improved in the next step when the PPIN structure is considered to evoke functional associations between differentially expressed proteins.
Figure 2. The enriched biological processes (BP) and molecular functions (MF).

The statistically significant enriched BP (A) and MF (B) are listed based on gene ontology database in each temperament. Some of them are presented in both temperaments with different frequencies. The p-values related to each term are listed in Supplementary file 1.
To more clarify protein functions, the protein-protein interactions of each specified groups were extracted via STRING database. As it is shown in Fig. 3, the protein-protein interaction network (PPIN) associated with each temperament are represented with sorted node size based on degree centrality. In each PPIN, the modular structures were represented by different color. The high degree proteins in each module are listed in Table 2. The high degree proteins were mostly related to cytoskeletal proteins in both groups. The actin and actin-associated proteins were dominants in the CD while other structural proteins such as tubulin beta and myosin constituted the central nodes in HW PPIN. Interestingly, the pyruvate kinase PKM was overrepresented in HW group while the polyubiquitin-C was overexpressed in CD group. It indicates the role of energy metabolism in HW versus protein degradation in CD individuals along with the complex involvement of cytoskeletal and cell to cell signaling apparatus. The overall BPs in the modules of each network are represented in the figure according to their significant level. The enriched terms related to mitochondria permeabilization and organization accompanying with different processes related to metabolic precursor generation and energy consumption conformed the dissimilar basal metabolism in two studied ITM patterns. Additionally, the repeated processes related to the actin polymerization are also the indication of cytoskeletal organization, cell movement and connection, and signal transduction to discriminate between CD and HW pattern (Fig. 3A). On the other hand, the specified PPIN module related to chromatin assembly bold the importance of gene expression regulation in HW pattern. The gas transport and post-translational folding processes observed in this network discriminate the HW and CD patterns too (Fig. 3B). The proteins related to muscle cell development and contraction, actin filament-based movement and cell-to-cell junction processes also divide the network into distinct modules in HW similar to CD patterns to highlight the importance of signal transduction in developing these patterns.
Figure 3. The protein-protein interaction network.

The PPIN involved in the cold-dry (A) and hot-wet (B) temperaments are presented separately. The proteins are sorted by node size based on degree centrality in ascending order and colored differently based on interconnectedness known as graph modules.
Table 2. The important proteins in both groups based on PPIN analysis.
| Protein Name | Group | UniProt ID | Description |
|---|---|---|---|
| UBC | CD | P0CG48 | Polyubiquitin-C |
| ACTG1 | CD | P63261 | Actin, cytoplasmic 2 |
| SRC | CD | P12931 | Proto-oncogene tyrosine-protein kinase Src |
| ACTA1 | CD | P68133 | Actin, alpha skeletal muscle |
| ITGA2B | CD | P08514 | Integrin alpha-IIb |
| CFL1 | CD | P23528 | Cofilin-1 |
| MYH11 | CD | P35749 | Myosin-11 |
| TLN1 | CD | Q9Y490 | Talin-1 |
| YWHAZ | HW | D0PNI1 | Epididymis luminal protein 4 |
| MSN | HW | P26038 | Moesin-1 |
| MYH9 | HW | P35579 | Myosin-9 |
| PKM2 | HW | P14618 | Pyruvate kinase PKM |
| MYL6 | HW | P60660 | Myosin light polypeptide 6 |
| H2AFV | HW | Q71UI9 | Histone H2A.V |
| TAGLN2 | HW | P37802 | Transgelin-2 |
| TUBB2A | HW | Q13885 | Tubulin beta-2A chain |
The CD and HW high degree proteins are listed respectively.
The activity of proteins is totally affected by different kinds of post-translational modifications situate on protein sequence. These effects could be considered as activation-inactivation or high-low activity which enable signal transduction pathways triggered toward contradictory sides24,25. In order to discover post-translational modification distribution, the PEIMAN software was used in both groups and compare the results. As it is shown in Fig. 4, the statistically significant enriched PTMs are varied in the many cases. Overally, the various types of phosphorylation, lipoprotein, myristate, Cycteinesulfunic acid, deamidated asparagine, cycteinepersulfide, glycation, and ADP-ribosylcysteine are dominant in CD pattern while in the HW groups other kinds of PTMs are dominantly disturbed such as various types of acetylation, methylation, hydroxylation and citrullination.
Figure 4. The post-translational modification enrichment analysis.

The ratio of the chemical modifications on proteins in both groups are compared together. These terms are statistically significant in enrichment analysis using PEIMAN software.
Discussion
The four humors i.e. Sanguine, Hypochondria, Bile and Phlegm, which constitute nine various temperaments or patterns with specified signs and symptom are the key terms in ITM theory. With this classification, the many physiological and pathological events could be categorized and consequently a distinct therapeutic strategy could be identified. The emphasis of these classifications when it emerged that we face to heterogeneous diseases such as rheumatoid arthritis or migraine considering the necessity of personalized medicine. Additional efforts have been projected to elucidate the biomedical foundation of traditional ethnic medicine classification of live phenomena. These efforts include a wide range of basic researches from biothermodynamics to molecular biology investigations14,26,27. In conclusion, it seems that the network biology which naturally consider life at the system level will pave the way toward this goal.
Material and Methods
Materials, volunteers and sample preparation procedure
Materials and methods used in this project are reported earlier in detail16,28. In a nutshell, normal volunteers (aged from 18 to 24 years) were classified in two temperaments according to a questionnaire and physical examination which are commonly used by ITM physicians: Hot-Wet (4 cases), and Cold-Dry (6 cases). The nine indicators were used in order to determine temperaments in ITM; touch, body features, hair features, background color of the body, sleep-wake pattern, waste material (feces, urine and sweat), organ size, temperamental manifestation of warmth, coldness, dryness and moisture on function and behavior, mental states and mood.
Then, peripheral blood mononuclear cells (PBMCs) were extracted from 5 ml of human blood by Ficoll28. In the next step, the mitochondrial lysate isolated from PBMCs to extract their related proteins. Detergents and salts were removed by buffer exchange methods, and proteins were solved in 50% acetonitrile and stored at −80 °C. These are subjected to a previously described tryptic digestion protocol29,30 so that they are reduced, alkylated, and finally subjected to tryptic digestion.
All of the materials and reagents were supplied from Sigma-Aldrich including EDTA, SDS, TEMED, TCA, PMSF, CHAPS, DTT, Bis Acrylamide, Acrylamide and etc. Protein ladder was provided from Fermentase (Page ruler unstained ladder). The central apparatuses used in this study are itemized below: sonicator (Hielscher), refrigerated centrifuge (Kendro, D37520), electrophoresis tools (PayaPajoohesh), speed vacuum (Scanvac), a nano-LC column (obtained from Phoenix S&T, Chester, PA, USA), Easy-nLC system (Thermo Fisher) and a LTQ-Orbitrap mass spectrometry system (Thermo Fisher).
Study procedure was approved by the institutional Ethics Committee of the Pasteur Institute of Iran, and all experiments were performed in accordance with relevant guidelines and regulations issued by the Committee. Informed consent was obtained from all the individuals.
Proteome analysis
An Easy-nLC system (Thermo Fisher) equipped with a capillary column (150 × 0.075 mm) was used for LC– MS/MS analysis of the tryptic samples. The column obtained from Phoenix S&T (Chester, PA, USA), and the slurry was packed in-house using a 5-μm, 100-Å pore size Magic C18 stationary phase resin (MichromBioResources, Auburn, CA, USA). The chromatography gradient using mobile phase A (0.1% formic acid in deionized water) and the mobile phase B (0.1% formic acid in acetonitrile) was designed for a linear increase from 0 to 8% B in 5 min, 5 to 25% B in 100 min, 25 to 45% B in 10 min, and 45 to 60% B in 10 min. Then an LTQ-Orbitrap mass spectrometry system (Thermo Fisher) was used for identification of peptides from digested proteins. The Xcalibur system (version 2.1; Thermo Fisher) was used to generate peak lists. The optimized parameters were as follow: Orbitrap full MS scans acquired from m/z 350 to 1500 at a resolution of 15 000 (at m/z 400) using an automatic gain control (AGC) value of 2 × 105, the minimum threshold was set to 100 000 ion counts, parent ions fragmented using the LTQ (isolation width of 2 m/z units) with a maximum injection time of 100 ms combined with an AGC value of 1 × 104 using three fragmentation modes such as collision-induced dissociation (CID) alone, electron-transfer dissociation (ETD) alone, and decision tree-based CID/ETD. For ETD MS/MS, the reagent ion source emission current, reagent ion electron energy, and reagent ion source chemical ionization pressure were set to 35 mA, 70 V, and 26 psi, respectively. The activation time and dynamic exclusion time was set to 100 ms and 30 s, respectively. Internal calibration was performed using the background polysiloxane ion signal at m/z 445.120025 as the calibrant. The Agilent 6530 Accurate-Mass Q-TOF combined with the nano chip HPLC system (Agilent, Wilmington, DE, USA) was employed for peptide identification31.
Enrichment and network analysis
Visualization of networks and the global network properties analysis were implemented in Gephi32. Given a network, nodes and edges are representative of proteins and pairwise interactions extracted from STRING 10 database33. Network clustering was implemented using the fast unfolding clustering algorithm to identify network modules34.
Enrichr was used to recognize overrepresented terms in the annotations of HW and CD specified proteins before and after network module finding analysis35. This tool was applied to scrutinize whether the protein set were involved in common biological processes (BP), molecular functions (MF), KEGG, Reactome and Panther pathways. These terms in Enrichr output were designated and filtered with adjusted P-value < 0.05. Additionally, Post Translational Modification Enrichment, Integration and Matching Analysis software (PEIMAN 1.0) was used to compare enriched PTMs in two temperaments36. Using hypergeometric test, the enriched PTMs were selected considering adjusted P-value < 0.05.
Additional Information
How to cite this article: Rezadoost, H. et al. Proteomics of hot-wet and cold-dry temperaments proposed in Iranian traditional medicine: a Network-based Study. Sci. Rep. 6, 30133; doi: 10.1038/srep30133 (2016).
Supplementary Material
Acknowledgments
We would like to thank Prof. Alireza Ghassempour, Dr. Mahvash Khodabandeh and Dr. Mostafa Rezaie-Tavirani for the valuable comments and aids during the project. This work is in part supported by a grant from INSF project (no. 89000833).
Footnotes
Author Contributions H.R., M.K. and M.J. participated in the design of the study. H.R. helped experimental proteomic analysis and in the writing of the draft. M.K. participated in preparation of the questionnaire, physical examination, clinical interpretation of the data and revising the manuscript critically. M.J. carried out acquisition, computational analysis and interpretation of the data and drafted the manuscript. All authors read and approved the final manuscript.
References
- Ma T. et al. Bridging the gap between traditional Chinese medicine and systems biology: the connection of Cold Syndrome and NEI network. Molecular bioSystems 6, 613–619, 10.1039/b914024g (2010). [DOI] [PubMed] [Google Scholar]
- Li Q.-y. & Su S.-b. Application of Systems Biology in Traditional Chinese Medicine Research. World Science and Technology 10, 1–6, 10.1016/S1876-3553(09)60017-X (2008). [DOI] [Google Scholar]
- Chen G. et al. A network-based analysis of traditional Chinese medicine cold and hot patterns in rheumatoid arthritis. Complementary therapies in medicine 20, 23–30, 10.1016/j.ctim.2011.10.005 (2012). [DOI] [PubMed] [Google Scholar]
- Cheng L. et al. Biological Basis of Cold and Heat Pattern of Rheumatoid Arthritis in Traditional Chinese Medicine. World Science and Technology 12, 814–817, 10.1016/s1876-3553(11)60028-8 (2010). [DOI] [Google Scholar]
- Lu A., Jiang M., Zhang C. & Chan K. An integrative approach of linking traditional Chinese medicine pattern classification and biomedicine diagnosis. Journal of Ethnopharmacology 141, 549–556, http://dx.doi.org/10.1016/j.jep.2011.08.045 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang C., Jiang M., Chen G. & Lu A. Incorporation of traditional Chinese medicine pattern diagnosis in the management of rheumatoid arthritis. European Journal of Integrative Medicine 4, e245–e254, http://dx.doi.org/10.1016/j.eujim.2012.02.004 (2012). [Google Scholar]
- Lu A. P., Jia H. W., Xiao C. & Lu Q. P. Theory of traditional chinese medicine and therapeutic method of diseases. World Journal of Gastroenterology 10, 1854–1856 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L. H., Xiao C. & Yan X. P. Correlation between heat or cold syndrome and cytokine, and laboratory index in women with early rheumatoid arthritis. Acta Univ Trad Med Sin Pharm Shanghai 2006 20, 21–24 (2006). [Google Scholar]
- van Wietmarschen H. et al. Systems biology guided by Chinese medicine reveals new markers for sub-typing rheumatoid arthritis patients. Journal of clinical rheumatology: practical reports on rheumatic & musculoskeletal diseases 15, 330–337, 10.1097/RHU.0b013e3181ba3926 (2009). [DOI] [PubMed] [Google Scholar]
- Xu Y., Zhang Z. & Sun Z. Drug resistance to Mycobacterium tuberculosis: From the traditional Chinese view to modern systems biology. Critical reviews in microbiology 7828, 1–12, 10.3109/1040841X.2013.860948 (2014). [DOI] [PubMed] [Google Scholar]
- Gorji A. Pharmacological treatment of headache using traditional Persian medicine. Trends in pharmacological sciences 24, 331–334, 10.1016/s0165-6147(03)00164-0 (2003). [DOI] [PubMed] [Google Scholar]
- Fu Y., Wang Y. & Zhang B. Systems pharmacology for traditional Chinese medicine with application to cardio-cerebrovascular diseases. Journal of Traditional Chinese Medical Sciences 1, 84–91, 10.1016/j.jtcms.2014.09.006 (2014). [DOI] [Google Scholar]
- Zhang A., Sun H., Yan G., Cheng W. & Wang X. Systems biology approach opens door to essence of acupuncture. Complementary Therapies in Medicine 21, 253–259, 10.1016/j.ctim.2013.03.002 (2013). [DOI] [PubMed] [Google Scholar]
- Shahabi S. et al. Hot and Cold natures and some parameters of neuroendocrine and immune systems in traditional Iranian medicine: a preliminary study. Journal of alternative and complementary medicine 14, 147–156, 10.1089/acm.2007.0693 (2008). [DOI] [PubMed] [Google Scholar]
- Naseri M. & MRS A. The school of traditional Iranian medicine, the definition, origin and advantages. J Int Soc History Islamic Med 3, 17–21 (2004). [Google Scholar]
- Jafari M. et al. Proteomics and traditional medicine: new aspect in explanation of temperaments. Forschende Komplementärmedizin/Research in Complementary Medicine 4, Accepted-Accepted (2014). [DOI] [PubMed] [Google Scholar]
- Ardekani M. R. S., Rahimi R., Javadi B., Abdi L. & Khanavi M. Relationship between Temperaments of Medicinal Plants and Their Major Chemical Compounds. Journal of Traditional Chinese Medicine 31, 27–31, http://dx.doi.org/10.1016/S0254-6272 (11)60006-X (2011). [DOI] [PubMed] [Google Scholar]
- Gorji A. & Khaleghi Ghadiri M. History of headache in medieval Persian medicine. Lancet neurology 1, 510–515 (2002). [DOI] [PubMed] [Google Scholar]
- Tadjbakhsh H. Sayyed Esma ‘il Jorjani, founder of Persian medicine. Journal of Veterinary Research 62, 131–140 (2007). [Google Scholar]
- Jiang W. Therapeutic wisdom in traditional Chinese medicine: a perspective from modern science. Trends in pharmacological sciences 26, 558–563, 10.1016/j.tips.2005.09.006 (2005). [DOI] [PubMed] [Google Scholar]
- Avicenna. The Cannon of Medicine. (Sorush Publisher, 2004).
- Abduvaliev A. A. Modern views on the theory of nature (mizadj) by Ibn Sina in medicine. Likars’ka sprava 3, 102–105 (2003). [PubMed] [Google Scholar]
- Abduvaliev A. A. Etiological and pathogenetic aspects of joint diseases in Abu Ali Ibn Sina’s papers and their modern significance. Likars’ka sprava/Ministerstvo okhorony zdorov’ia Ukraïny, 103–108 (2006). [PubMed] [Google Scholar]
- Jordan J. D., Landau E. M. & Iyengar R. Signaling networks: the origins of cellular multitasking. Cell 103, 193–200 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. & Snyder M. ‘Omic’ approaches for unraveling signaling networks. Current Opinion in Cell Biology 14, 173–179, 10.1016/S0955-0674(02)00315-0 (2002). [DOI] [PubMed] [Google Scholar]
- Kang G. L., Li S. & Zhang J. F. Entropy-based model for interpreting life systems in traditional Chinese medicine. Evidence-based Complementary and Alternative Medicine 5, 273–279, 10.1093/ecam/nem026 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Q. et al. Understanding ZHENG in traditional Chinese medicine in the context of neuro-endocrine-immune network. IET Systems Biology 1, 51–60, 10.1049/iet-syb:20060032 (2007). [DOI] [PubMed] [Google Scholar]
- Pooreydy B. et al. Organelle Isolation for Proteomics: Mitochondria from Peripheral Blood Mononuclear Cells. Journal of Paramedical Sciences (JPS) 4, 79–86 (2013). [Google Scholar]
- Lee H.-J. et al. Application of a peptide-based PF2D platform for quantitative proteomics in disease biomarker discovery. Proteomics 8, 3371–3381, 10.1002/pmic.200800111 (2008). [DOI] [PubMed] [Google Scholar]
- Jafari M. et al. Comparison of in-gel protein separation techniques commonly used for fractionation in mass spectrometry-based proteomic profiling. Electrophoresis 33, 2516–2526, 10.1002/elps.201200031 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-J. et al. Comprehensive genome-wide proteomic analysis of human placental tissue for the Chromosome-Centric Human Proteome Project. Journal of proteome research 12, 2458–2466, 10.1021/pr301040g (2013). [DOI] [PubMed] [Google Scholar]
- Bastian M., Heymann S. & Jacomy M. Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media (2009).
- Szklarczyk D. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research, 1–6, 10.1093/nar/gku1003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel V. D., Guillaume J.-L., Lambiotte R. & Lefebvre E. Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment 2008, 6–6, 10.1088/1742-5468/2008/10/P10008 (2008). [DOI] [Google Scholar]
- Chen E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC bioinformatics 14, 128–128, 10.1186/1471-2105-14-128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickchi P., Jafari M. & Kalantari S. PEIMAN 1.0: Post-translational modification Enrichment, Integration and Matching ANalysis. Database 2015, bav037–bav037, 10.1093/database/bav037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.