

Abstract
Interleukin-17A is a member of the IL-17 family, and is known as CTLA8 in the mouse. It is produced by T lymphocytes and NK cells and has proinflammatory roles, inducing cytokine and chemokine production. However, its role in tumor biology remains controversial. We investigated the effects of locally produced IL-17A by transferring the gene encoding it into CT26 colon cancer cells, either in a secretory or a membrane-bound form. Expression of the membrane-bound form on CT26 cells dramatically enhanced their proliferation in vitro. The enhanced growth was shown to be due to an increased rate of cell cycle progression: after synchronizing cells by adding and withdrawing colcemid, the rate of cell cycle progression in the cells expressing the membrane-bound form of IL-17A was much faster than that of the control cells. Both secretory and membrane-bound IL-17A induced the expression of Sca-1 in the cancer cells. When tumor clones were grafted into syngeneic BALB/c mice, the tumor clones expressing the membrane-bound form IL-17A grew rapidly; those expressing the secretory form also grew faster than the wild type CT26 cells, but slower than the clones expressing the membrane-bound form. These results indicate that IL-17A promotes tumorigenicity by enhancing cell cycle progression. This finding should be considered in treating tumors and immune-related diseases.
Keywords: colon cancer, interleukin 17A, membrane-bound cytokine, pro-tumor, sca-1
INTRODUCTION
Th17 lymphocytes are a recently identified subset of CD4+ T lymphocytes, which develop in response to IL-6, TGF-β and IL-23. They are typically characterized by the secretion of the cytokine IL-17 (Weaver et al., 2006; 2007). IL-17 was first cloned and referred to as cytotoxic T lymphocyte antigen 8 (CTLA-8), and exists in the form of subtypes IL-17A-F (Rouvier et al., 1993; Yao et al., 1996). Of these, IL-17A and IL-17F have the highest degree of homology and are produced mainly by activated T lymphocytes (Moseley et al., 2003). IL-17 is known as a proinflammatory cytokine that promotes the secretion of IL-6, IL-1β and chemokines from macrophages and fibroblasts, via the NF-κB, ERK, JNK and p38 MAPK intracellular signaling pathways. It can also cause autoimmune disease by inducing excessive inflammatory responses. Since IL-17 receptors are ubiquitously expressed on immune cells and various epithelial tissues (Moseley et al., 2003), IL-17 signaling may affect cellular homeostasis beyond the immune system.
Cytokines play an essential role in the immune response for regulating tumorigenicity. The functional role of IL-17 has also been studied in tumor biology. In brief, some tumors such as those of the prostate and ovary have been found to generate Th17 cells (Miyahara et al., 2008; Sfanos et al., 2008). However, the ectopic expression of the secreted form of IL-17 in most types of cancer did not significantly affect their in vitro growth (Hirahara et al., 2001; Numasaki et al., 2003; Tartour et al., 1999). Grafting IL-17-producing tumors into syngeneic immunocompetent mice has yielded contradictory results; the ectopic expression of IL-17 in some hematopoietic cancer cells and fibrosarcomas inhibited their growth after transplantation (Benchetrit et al., 2002; Hirahara et al., 2001), whereas the growth of some other types of cancer was stimulated (Numasaki et al., 2003; Tartour et al., 1999). Blocking endogenous IL-17 expression also increased or decreased tumor growth depending on the type of cancer and system (Hyun et al., 2012; Kryczek et al., 2009; Wang et al., 2009; 2010).
Although certain cytokines have potent antitumor effects, systemic administration of cytokines has significant toxicity that limits their clinical use (Atkins et al., 1997; Car et al., 1999; Leonard et al., 1997). To avoid this problem gene transfer strategies that achieve localized cytokine production have been tried. In many cases, expression of a cytokine in membrane-bound form increased the immunogenicity of tumor cells and stimulated antitumor immunity more effectively than expression in the secreted form (Ji et al., 2002; Kim et al., 2000; Marr et al., 1997; Soo Hoo et al., 1999). Previously we showed that tumor cells expressing membrane-bound IL-12p35 had reduced tumorigenicity, and led to CD8+ T cell-dependent antitumor immunity of MethA cells (Lim et al., 2010). In present study we investigated whether expression of the membrane-bound form of IL-17A on CT26 colon cancer cells promotes or inhibits their growth as tumors, and found, for the first time, that membrane-bound IL-17A greatly increased their proliferation and tumorigenicity both in vitro and in vivo.
MATERIALS AND METHODS
Mice and tumor cell lines
Female BALB/c mice 6 to 8 weeks old were obtained from the Korea Research Institute of Chemical Technology (Korea). All animal procedures were approved and guided by the committee for experimental animal care of Chungnam National University. The murine colon tumor cell line CT26 originating from BALB/c mice was propagated and maintained in RPMI-1640 (GIBCO BRL, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; GIBCO-BRL), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma St, Louis, MO) in humidified 5% CO2 at 37°C. G-418 (Santa Cruz, US) at 0.2 μg/ml was used as selective agent for transfections. Cells were incubated for 24 hours and then exposed to medium containing colcemid or IL-17A.
Antibodies and reagents
Anti-IL-17A polyclonal antibody (ab79056) and FITC-conjugated anti-MHC Class 1 (ab25056) were purchased from Abcam (USA). Anti-Sca1 antibody was kindly given by Dr. Alfred L. M. Bothwell (Yale University, USA). FITC-conjugated goat anti-mouse IgG and Cy3-conjugated donkey anti-rabbit IgG (711-165-152) were purchased from Sigma (USA). Goat anti-mouse (sc-2005) and goat anti-rabbit (sc-1004) HRP- conjugated secondary antibodies were from Santa Cruz Biotechnology (USA).
Plasmid construction and transfection
Mouse splenocyte cDNA was used as template to amplify S-IL-17A and mb-IL-17 cDNAs. Mouse TNF-α cDNA was obtained from Dr. Sang Young Chun (Chonnam National University, Korea). To construct pcDNA3.1/S-IL-17A and chimeric pcDNA3.1/mb-IL-17Aplasmids, primers specific for S-IL-17A (Forward 5′ TCA GGATCCGCAAACATGAGTCCAGGG 3′, Reverse 5′ GGT CTCGAGTTAGGCTGCCTG GCGGAC 3′), mb-IL-17A (Forward 5′ AGTGGATCCAAGCAGCGATCATCC CTC 3′, Reverse 5′ GGTCTCGAGTTAGGCTGCCTGGCGGAC 3′) and TNF-α (Forward 5′ TCAAGCTTATGAGCACAGAAAGC 3′, Reverse 5′ ATTGGATCCCTCCGGCCA TAGA 3′) were used to amplify the respective cDNA fragments. The S-IL-17A (501bps) and the Mb-IL-17A (410 bps) PCR fragments were digested with BamHI /XhoI. A 240 bp TNF-α cDNA fragment encoding the transmembrane region (from −79 to −45), cytoplasmic region (from −44 to −24), and extracellular region (from −23 to −5) was digested with HindIII/BamHI and ligated to the Mb-IL-17A expression plasmid. The chimeric cDNAs were then subcloned into the pcDNA3.1(+) expression vector. The constructs were confirmed by DNA sequencing (Genotech, Korea). CT26 colon cancer cells were transfected with these constructs or empty vector using BioT Transfection Reagent (Morganville Scientific, USA). After 48 h, the cells were plated in G418 (1 mg/ml) containing medium. Drug-resistant colonies were usually visible 2–3 weeks after transfection.
RT-PCR
To examine the expression of S-IL-17A and Mb-IL-17A mRNAs, total RNA was extracted using a Hybrid-R kit (GeneAll, Korea). 2 μg of total RNA was reverse transcribed using oligo (dT) primers and M-MLV reverse transcriptase (Affymetrix, USA) at 42°C for 1 h. Semi-quantitative PCR was performed using HSTag premix (Geneall, Korea) on a DNA thermal Cycler (Biorad; USA).
ELISA for IL-17A
An enzyme-linked immunosorbent assay (ELISA) was used to measure the concentration of bioactive IL-17A in culture supernatants. 1 × 106 cells were incubated in 1 ml of culture medium in 24-well culture plates at 37°C for 24 h. IL-17A in the supernatants was measured with an enzyme-amplified sensitive immunoassay kit (KOMA Biotech Inc., Korea).
Flow cytometric analysis
Cells were incubated with primary antibodies diluted in staining buffer (1× PBS containing 0.02% sodium azide and 2% FBS) for 1 h at 4°C. After washing off unbound antibodies with the staining buffer, the cells were stained with suitable secondary antibodies for 45 min at 4°C in the dark. After further washing they were analyzed with a flow cytometer (FACSCalibur, Becton Dickinson, USA). For BrdU staining, cells were incubated with 50 μM BrdU for the final 12 h of culture. After harvesting, they were fixed in ice-cold 70% ethanol and permeabilized with 2 M HCl. Incorporation of BrdU was determined by flow cytometry after staining with anti-BrdU (Santa Cruz) and FITC-conjugated anti-mouse IgG antibodies.
In vitro cell proliferation assay
1 × 104 cells were plated on 96-well plates. The cells were cultured for 72 h and their proliferation was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.
Tumor challenge
Five syngeneic BALB/c mice were challenged with each CT26 clone. The tumor cells were washed twice with PBS and suspended in PBS. The 1 × 106 tumor cells in 100 μl PBS were injected subcutaneously into the left lower back quadrants of the mice with a 1 ml disposable syringe. Tumor size was measured serially with calipers and calculated according to the formula: 0.52 × S2 × L, where L is length and S is the width of the tumor. As controls, 5 BALB/c mice each were challenged with either CT26 wild-type or mock-transfected CT26 cells. At the end of the experiment, the animals were killed in a CO2-containing chamber, and the tumors were collected.
Analysis of cell cycle progression rate
The Stathmokinetic method is a metaphase-arrest technique that provides the rate of entry to mitosis or the cell birthrate (Puck and Steffen, 1963). In this study, the Stathmokinetic agent, colcemid, was used to analyze cell proliferation. Cells were incubated with 0.5 μg/ml colcemid (Gibco) for 20 h and samples were collected periodically after colcemid removal. The samples were fixed with 70% ice-cold ethanol and analyzed for the DNA content with a FACSCalibur (Becton Dickinson, USA). Nuclear DNA was stained with 50 μg/ml propidium iodide after digesting cellular RNA with RNAase.
Statistical analysis
All data are presented as means ± SEMs (error bars). One way or two way analysis of variance (ANOVA) and Student’s t-tests were used to analyze differences between groups, as indicated in the figure legends. A P-value less than 0.05 was considered significant.
RESULTS
Expression of secretory and membrane-bound IL-17A in CT26 colon cancer cells
To define the role of IL-17A in cancer cells we used murine CT26 colorectal carcinoma cells, which are derived from BALB/c mice. The cells were transfected with control vector (pcDNA3.1+), or vectors encoding secretory IL-17A (S-IL-17A, 1–158 a.a. of IL-17A) or membrane-bound IL-17A (Mb-IL-17A). Mb-IL-17A is a chimeric form of the transmembrane/cytoplasmic region of TNF-α and 26–158 a.a. of IL-17A (Fig. 1A). The TNF-α-converting enzyme (TACE) cutting site of cytoplasmic region of TNF-α was excluded to create a stable membrane-bound form of the chimeric protein (Horiuchi et al., 2010). Wild type CT26 cells were shown not to express endogenous IL-17A (Fig. 1B). Stably transfected cell lines were selected with G418, and expression of S-IL-17A and Mb-IL-17A mRNAs was measured by RT-PCR (Fig. 1C). Two clones each of S-IL-17A- and Mb-IL-17A-expressing cells were chosen for further experiments and designated S11/S15 and Mb6/Mb10, respectively. IL-17A protein was measured by ELISA in culture supernatants of the CT26 sublines. It was detected in the culture supernatant of S-IL-17A transfectants, S11 and S15, but not in those of the Mb-IL-17A transfectants (Fig. 1D). Surface expression of IL-17A was analyzed by flow cytometry, and a significant increase of expression in the Mb-IL-17A clones compared with the basal level of wild type CT26 cells (P) and mock-transfected cells (M) was detected in repeated experiments (Fig. 1E).
Fig. 1.
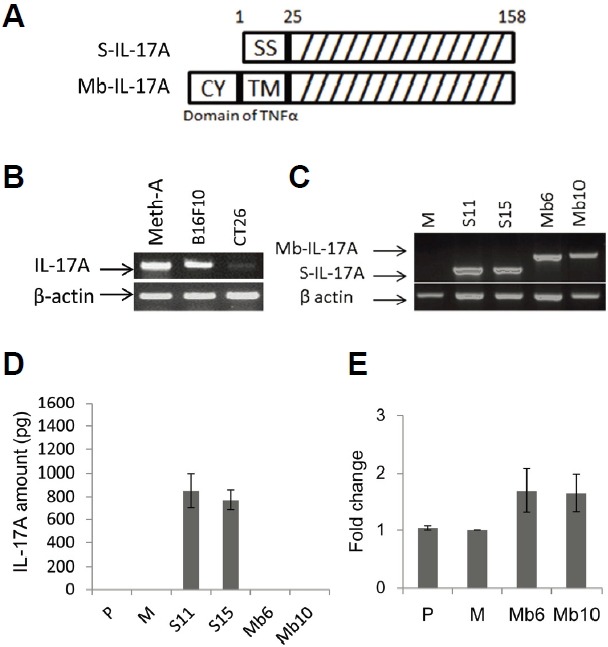
Generation of CT26 clones stably expressing S-IL-17A or Mb-IL-17A. (A) The construct of the secretory form of IL-17A (S-IL-17A) was composed of signal sequence (1–25 a.a.) and IL-17A domain (26–158 a.a.). That of the membrane-bound form (Mb-IL-17A) was composed of the cytoplasmic domain (from −75 to −45, CY), transmembrane fragment (from −44 to −24, TM) and 19 extracellular amino acids (from −23 to −5) of murine TNF-α and the IL-17A domain (26–158 a.a.). (B) Several tumor types were tested for endogenous expression of IL-17A. Meth-A; Meth-A fibrosarcoma, B16F10; B16 melanoma and CT26; CT26 colon cancer. (C) S-IL-17A transfectants (S11, S15) and Mb-IL-17A transfectants (Mb6, Mb10) that strongly expressed IL-17A at the mRNA level, were used in following experiments. M; a mock-vector transfected CT26 clone. (D) IL-17A expression in the culture supernatant of CT26 clones was analyzed by ELISA. Wild-type CT26 cells (P) and a mock-vector transfected CT26 clone (M) were used as negative controls. (E) Membrane-bound IL-17A was stained with polyclonal anti-mouse IL-17A antibody and analyzed by flow cytometry. Each transfectant was also stained with the isotype control and Cy-3-conjugated secondary antibody. Mean florescence intensity was determined by subtracting the basal level from each sample. Data are shown as fold change relative to the mock control (M). Data are means ± SEM of three independent experiments.
Expression of Mb-IL-17A on colon cancer cells promotes tumor cell growth
The S-IL-17A and Mb-IL-17A clonal transfectants were cultured in conventional culture medium and their proliferation were analyzed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays. After 48 hours the numbers of cells of both of the Mb-IL-17A clones were ∼2.3 fold higher than those of the wild-type or mock-transfected cells, whereas growth of the S-IL-17A transfectants was similar to that of the control CT26 cells (Fig. 2A).
Fig. 2.
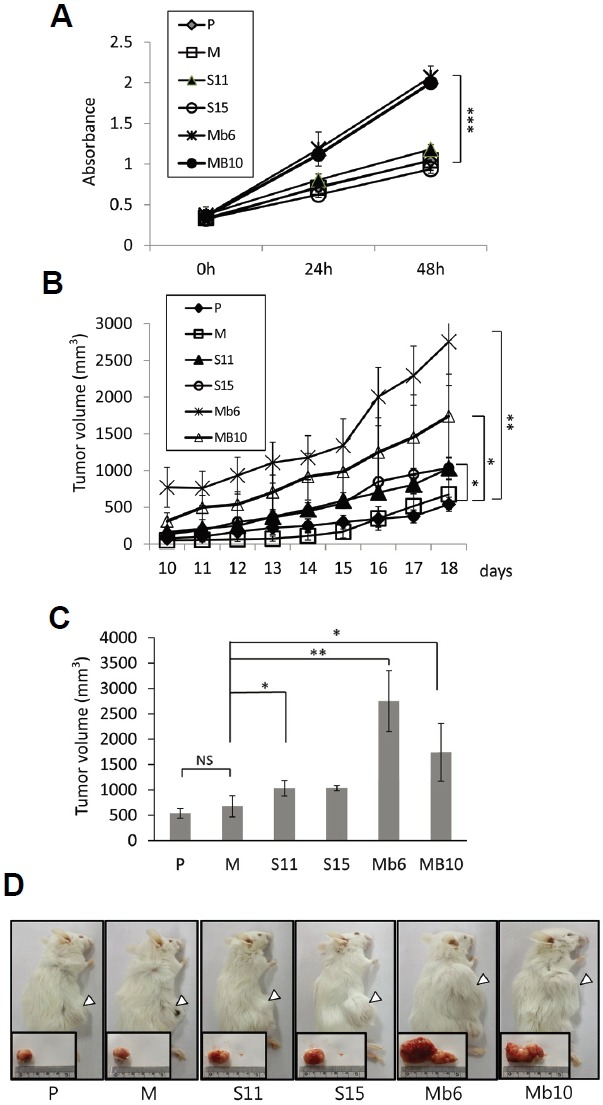
Mb-IL-17A enhances the growth rate and tumorigenicity of CT26 cells. (A) The 1 × 104 cells of the wild-type CT26 cells (P) and the indicated transfectants were seeded in 96-well plates. Their growth rates were analyzed by MTT assay over 48 h. Data represent means ± SEM of three independent experiments. The statistical significance of the differences between Mb6 and MB10 vs. M were analyzed by one way analysis of variance (ANOVA). ***P < 0.001. (B) Aliquots of 1 × 106 cells of each subline were injected into mice subcutaneously. Tumor size was measured with a Vernier caliper and calculated as described in “Materials and Methods”. (C) Tumor volumes on day 18 after tumor clone implantation. The differences between S11, Mb6 and MB10 vs. M were analyzed by one way ANOVA. *P < 0.05; **P < 0.01; NS, not significant. (D) The animals were killed on day 25 after tumor clone implantation, and tumor tissues were collected. Representative photos of the animals and their tumors in the various groups are shown.
To see whether the difference in in vitro proliferation rate was reflected in the tumorigenicity of the colon cancer cells, IL-17A expressing and control CT26 cells were implanted subcutaneously into syngeneic BALB/c mice. Each of five mice per group was challenged with 1 × 106 clonal cells, and tumor growth was monitored for 1 month, during which all the IL-17A expressing and control CT26 cells formed solid tumors. S11 and S15 grew faster than the controls, but slower than Mb6 and Mb10 (Fig. 2B). The tumor volumes formed by the S11 and S15 transfectants was ∼1.5 fold greater than those formed by the mock-transfected cells on day 18 (1030 mm3 and 1034 mm3 vs. 675 mm3) (Fig. 2C). The volumes of the tumors formed by Mb6 and Mb10 were ∼3.9 and ∼2.6 fold greater, respectively, than those of the controls on day 18 (2754 mm3 and 1742 mm3 vs. 675 mm3). This result is confirmed by the photographs of the tumors made on day 25 (Fig. 2D). This finding demonstrates that the ectopic expression of IL-17A increases the tumorigenicity of CT26 cells, and especially of the cells expressing the membrane-bound form of IL-17A.
Mb-IL-17A expression on cancer cells promotes cell cycle progression
To analyze how the expression of Mb-IL-17A affects cell proliferation in vitro we used colcemid, a reagent inducing cell cycle arrest at metaphase (Kung et al., 1990; Rieder and Palazzo, 1992). We assumed that if two different cell populations have different rates of cell cycling, the fraction of cells arrested at metaphase by colcemid over a given period of time will be different. Therefore, mock-control and IL-17A-expressing CT26 tumor clones were analyzed with propidium iodide, and the fraction of cells in each phase was measured. Without colcemid treatment, the transfectants did not differ in the fraction of cells in each phase (Fig. 3A). After treatment with 0.1 μg/ml colcemid, samples were collected at two-hour intervals and stained with propidium iodide, and Mb6 and Mb10 were found to accumulate more rapidly in the G2/M phase than the other cell lines (Fig. 3B). In both Mb6 and Mb10, more than 60% of cells had entered G2/M phase by 8 h, whereas S11 and S15 accumulated in G2/M phase more slowly and the maximal fraction of cells accumulating did not exceed 50%. These data show that the cell cycle progression is more rapid in the Mb6 and Mb10 cells than in the S11 and S15 cells or the control cells.
Fig. 3.
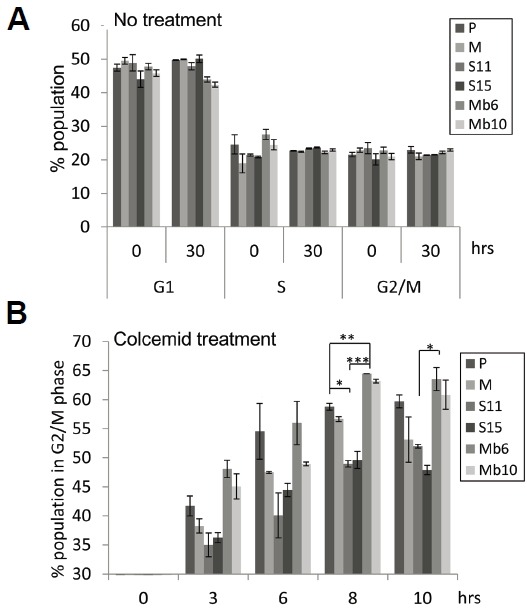
Mb-IL-17A-promoted cell cycle progression verified by colcemid-induced metaphase arrest. (A) Cells were seeded and cultured for 30 h. Each group of cells was harvested at the indicated time points and stained with propidium iodide (PI). The proportion of cells in G1, S, and G2/M phases was measured by flow cytometry. (B) The 1 × 106 cells of each transfectant type were treated with 0.1 μg/ml colcemid and the fraction of cells in G2/M phase at the indicated time points was measured by PI staining and flow cytometry. The differences between groups were analyzed using two-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001.
It is known that when a 4N cell population synchronized by colcemid is released from inhibition, a proportion of the cells produce 2N or 8N cells and the others die via apoptosis and senescence (Rieder and Maiato, 2004). We assumed that if the two different cell populations cycle at different rates, their cell cycle progression after release from metaphase block would be different. Therefore, colcemid was removed from cell cultures and the doubling time from 4N to 8N of the tumor clones was observed for 80 h. In the mock-transfected clone, and S11 and S15 cells, 8N DNA content peaked at 32 h, and then declined (Fig. 4A). However, in Mb6 and Mb10, 8N cells reached a first peak at 24 h and formed a second peak at 47 h, showing that the doubling time of 4N cells in Mb6 and Mb10 was much shorter than in the control and S-IL-17A transfectants (Fig. 4B). When the cells were monitored by optical microscopy 72 h after removal of colcemid, both of the Mb-IL-17A-expressing clones had formed larger numbers of cells than the other cell lines (Fig. 4C). Furthermore, growth of the transfectants in the presence of BrdU for 12 h resulted in significantly more BrdU incorporation into replicating DNA in the Mb-IL-17A-expressing clones than in the mock and S-IL-17A transfectants (Fig. 4D). These data taken together indicate that the more rapid proliferation of the tumor cells expressing membrane-bound IL-17A is the result of faster cell cycle progression. Further research on downstream signaling induced by the membrane-bound form of IL-17A will be helpful to dissect the mechanism of IL-17 regulation of tumor cell growth.
Fig. 4.
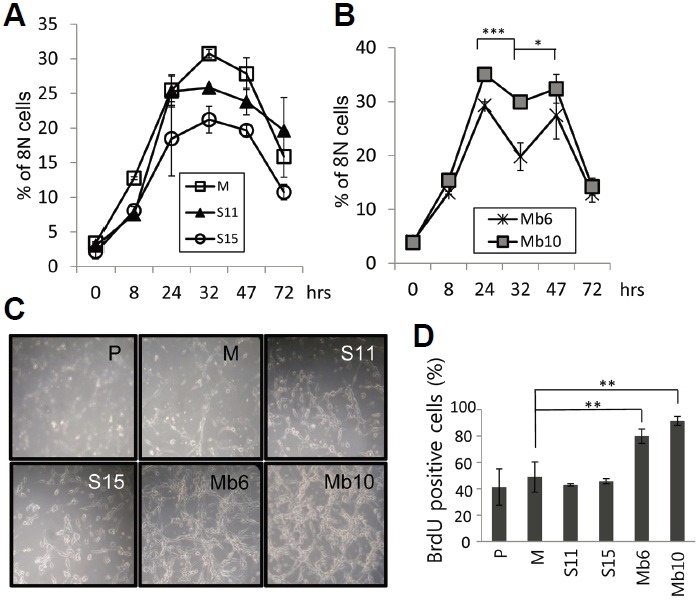
Mb-IL-17A expression enhances cell cycle progression after removal of colcemid. (A, B) The indicated transfectants were treated with 0.5 μg/ml colcemid for 20 h and the medium was replaced with fresh medium without colcemid. Cells were harvested at the indicated time points and stained with PI, and the proportion of cells containing 8N DNA was evaluated by flow cytometry. The differences between the values for MB6 at 24, 32, 47 h were analyzed using two-way ANOVA. *P < 0.05; ***P < 0.001. (C) Photographs of the indicated cells taken 3 days after colcemid removal. (D) Transfectants were pulse-labelled during exponential growth with BrdU for 12 h and the BrdU incorporation was analyzed by flow cytometry.
IL-17A stimulates Sca-1 expression in CT26 cancer cells
To assess the expression of MHC class I molecules, wild type CT26 and the IL-17A-transfected clones were stained with an anti- MHC class I-Ld monoclonal antibody. Surface expression of MHC class I molecules on the Mb-IL-17A-expressing clones was greater than on the wild type CT26 or mock-vector transfected cells, but the S-IL-17A clones did not show enhanced expression of MHC class I molecules (Fig. 5). By blocking GDF10-dependent TGF-β signaling, stem cell antigen (Sca-1), a glycerophosphatidylinositol-linked surface protein, has been shown to enhance tumorigenicity (Batts et al., 2011; Upadhyay et al., 2011; Xin et al., 2005). Because IL-17A is an important TGF-β driven cytokine, the relationship between IL-17A and Sca-1 expression was tested. The expression of Sca-1 antigen on the cell surface of S-IL-17A- and Mb-IL-17A-expressing clones increased compared with wild type CT26 cells or mock-vector transfected clone (Fig. 6A). The mean values of the peaks of Sca-1 expression in the mock, S11, S15, Mb6 and Mb10 cells were 9.49, 22.7, 14.62, 27.78 and 43.53, respectively, after subtraction with that of each isotype control. In a further analysis, wild type CT26 cells were treated with the culture supernatant of mock-control and S11 cells for 72 h (Fig. 6B). The supernatant of S11 clearly increased Sca-1 expression on CT26 cells, demonstrating that IL-17A induces Sca-1 expression in the cancer cells.
Fig. 5.
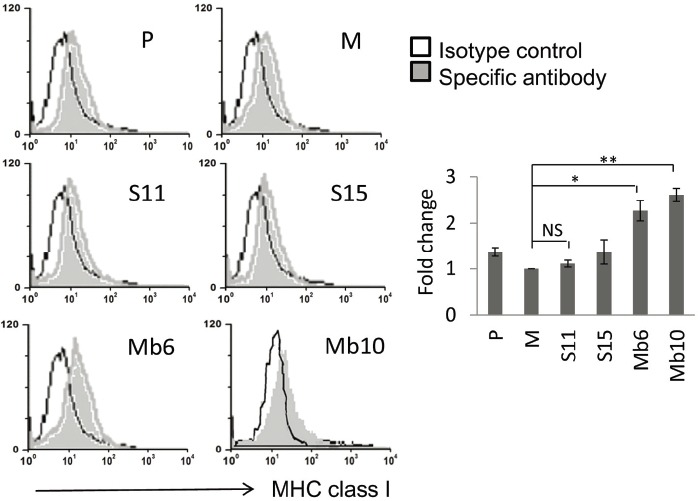
Surface MHC class I expression is induced by ectopic expression of Mb-IL-17A. MHC class I expression was analyzed in the indicated clones using anti-Ld MHC Class I monoclonal antibody (24-14-8S) and FITC-conjugated secondary anti-mouse IgG. Cells were analyzed by flow cytometry (left). The relative changes of mean values against M, measured by flow cytometry, are represented by the bar graphs. Differences between groups were analyzed using two-way ANOVA. *P < 0.05; **P < 0.01; NS, no significant (right).
Fig. 6.
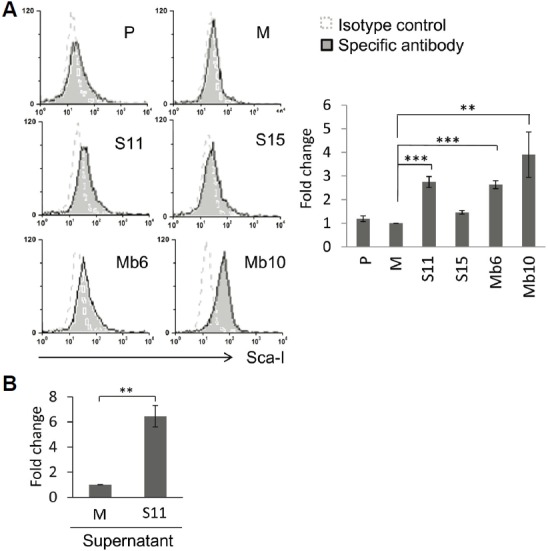
IL-17A enhances Sca-1 expression in CT26 cells. (A) Sca-1 expression was analyzed in the indicated clones using anti-Sca-1 antibody and flow cytometry (left). The relative changes of mean values against M, measured by flow cytometry, are represented by the bar graphs. Differences between groups were analyzed using two-way ANOVA. **P < 0.01; ***P < 0.001 (right). (B) The 1 × 107 cells of the indicated clones were cultured in 50 ml fresh medium for 3 days. Each culture supernatant was collected and filtered through 0.22 μm filter, and wild type CT26 cells were incubated with 1:1 diluted culture supernatants for 72 hours and Sca-1 expression was analyzed by flow cytometry. Differences between groups were analyzed using student’s t-test. **P < 0.01.
DISCUSSION
The role of many cytokines in promoting or inhibiting tumor immunity has been extensively studied, but the roles of several cytokines remain undefined. In this study we investigated whether the membrane-bound form of IL-17A expressed on CT26 colon cancer cells promoted or inhibited tumor growth, and we showed, for the first time, that it greatly increased cell growth and tumorigenicity in vitro and in vivo. Our finding is based on several observations. First, Mb-IL-17A clonal transfectants cultured in conventional culture medium showed greatly enhanced cellular proliferation (Fig. 2A). Second, when implanted subcutaneously into syngeneic BALB/c mice, the ectopic expression of Mb-IL-17A significantly increased tumorigenicity of CT26 (Figs. 2B–2D). Moreover the increased growth rate of transfectants with the membrane-bound form of IL-17A was shown to be due to more rapid cell cycle progression (Figs. 3 and 4). Since the membrane-bound form of IL-17A increased the growth rate in culture medium in the absence of stromal or immune cells, it is likely that Mb-IL-17A stimulates proliferation by direct cell-to-cell contact. Presumably the juxtacrine communication between Mb-IL-17A and IL-17A receptors on CT26 cells has a much more stable and stronger effect than the interaction between S-IL-17A and IL-17A receptors. Direct binding and signaling by IL-17A in cancer cells has been reported previously; endogenous secretory IL-17 was shown to directly induce IL-6 secretion by B16 melanoma and bladder carcinoma cells and thereby activate Stat3 signaling, evoking pro-survival and pro-angiogenic responses (Wang et al., 2009). In aspect of the enhanced cell cycle progression in Mb-IL-17A transfectants, our results are also consistent with several reports demonstrating that IL-17A modulates the expression of cell mitogens and cell cycle genes. In keratinocytes, secretory IL-17A directly promotes the proliferation and inhibits the differentiation of the cells by the up-regulation of cell cycle-related genes such as CCNE1, CDCA5, and CDCA25A (Chiricozzi et al., 2014). In the development of colitis-associated cancer, IL-17A knockout led to reduced expression of the key cell cycle regulators, cyclin D1, cyclin-dependent kinase 2 and cyclin E (Hyun et al., 2012). Further research on downstream signaling induced by the membrane-bound form of IL-17A should help to dissect the mechanism of IL-17 regulation of tumor cell growth. In contrast, the in vitro proliferation of CT26 transfectants producing the secreted form IL-17A did not differ from that of control CT26 cells, though they did grow faster when implanted subcutaneously into BALB/c mice (Fig. 2). These results are consistent with a previous report in which secretory IL-17 transfectants of fibrosarcoma and colon adenocarcinoma cells grew more rapidly than controls in vivo but not in vitro (Numasaki et al., 2003).
We did not observe any effect of soluble IL-17A on MHC class I expression (Fig. 5). On the other hand, Hirahara et al. reported that MHC antigen expression (class I and II) increased in soluble IL-17-transfected Meth-A cells, as did antitumor immunity mediated by CD4+ and CD8+ T cells (Hirahara et al., 2001). The discrepancy between their results and ours may result from differences between the tumor models and systems. We also tested the effect of IL-17A on the stem cell antigen-1 (Sca-1) since there is growing evidence of a role of Sca-1 in tumorigenicity (Batts et al., 2011; Upadhyay et al., 2011; Xin et al., 2005). Since Sca-1 is involved in TGF-β signaling this suggested a possible relationship between Sca-1 and IL-17A. We found that the tumor clones expressing IL-17A had higher levels of Sca-1 expression (Fig. 6). Furthermore, treatment of wild type CT26 cells with exogenous IL-17A also enhanced Sca-1 expression. This observation is the first evidence that IL-17A directly induces the expression of Sca-1. More quantitative analysis is required to confirm it in the future research. Further studies of the mechanism underlying this process should increase our understanding of the effects of cytokines on tumor immunity in general.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2013R1A1A4A 01013202), by a grant from Chungnam National University (2014), and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (HI15C1886).
REFERENCES
- Atkins M.B., Robertson M.J., Gordon M., Lotze M.T., DeCoste M., DuBois J.S., Ritz J., Sandler A.B., Edington H.D., Garzone P.D., et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin. Cancer. Res. 1997;3:409–417. [PubMed] [Google Scholar]
- Batts T.D., Machado H.L., Zhang Y., Creighton C.J., Li Y., Rosen J.M. Stem cell antigen-1 (sca-1) regulates mammary tumor development and cell migration. PLoS One. 2011;6:e27841. doi: 10.1371/journal.pone.0027841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchetrit F., Ciree A., Vives V., Warnier G., Gey A., Sautes-Fridman C., Fossiez F., Haicheur N., Fridman W.H., Tartour E. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99:2114–2121. doi: 10.1182/blood.v99.6.2114. [DOI] [PubMed] [Google Scholar]
- Car B.D., Eng V.M., Lipman J.M., Anderson T.D. The toxicology of interleukin-12: a review. Toxicol. Pathol. 1999;27:58–63. doi: 10.1177/019262339902700112. [DOI] [PubMed] [Google Scholar]
- Chiricozzi A., Nograles K.E., Johnson-Huang L.M., Fuentes-Duculan J., Cardinale I., Bonifacio K.M., Gulati N., Mitsui H., Guttman-Yassky E., Suarez-Farinas M., et al. IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS One. 2014;(9):e90284. doi: 10.1371/journal.pone.0090284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara N., Nio Y., Sasaki S., Minari Y., Takamura M., Iguchi C., Dong M., Yamasawa K., Tamura K. Inoculation of human interleukin-17 gene-transfected Meth-A fibrosarcoma cells induces T cell-dependent tumor-specific immunity in mice. Oncology. 2001;61:79–89. doi: 10.1159/000055357. [DOI] [PubMed] [Google Scholar]
- Horiuchi T., Mitoma H., Harashima S., Tsukamoto H., Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology (Oxford) 2010;49:1215–1228. doi: 10.1093/rheumatology/keq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun Y.S., Han D.S., Lee A.R., Eun C.S., Youn J., Kim H.Y. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis. 2012;33:931–936. doi: 10.1093/carcin/bgs106. [DOI] [PubMed] [Google Scholar]
- Ji J., Li J., Holmes L.M., Burgin K.E., Yu X., Wagner T.E., Wei Y. Glycoinositol phospholipid-anchored interleukin 2 but not secreted interleukin 2 inhibits melanoma tumor growth in mice. Mol. Cancer Ther. 2002;1:1019–1024. [PubMed] [Google Scholar]
- Kim Y.S., Sonn C.H., Paik S.G., Bothwell A.L. Tumor cells expressing membrane-bound form of IL-4 induce antitumor immunity. Gene Ther. 2000;7:837–843. doi: 10.1038/sj.gt.3301175. [DOI] [PubMed] [Google Scholar]
- Kryczek I., Wei S., Szeliga W., Vatan L., Zou W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114:357–359. doi: 10.1182/blood-2008-09-177360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung A.L., Sherwood S.W., Schimke R.T. Cell line-specific differences in the control of cell cycle progression in the absence of mitosis. Proc. Natl. Acad. Sci. USA. 1990;87:9553–9557. doi: 10.1073/pnas.87.24.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J.P., Sherman M.L., Fisher G.L., Buchanan L.J., Larsen G., Atkins M.B., Sosman J.A., Dutcher J.P., Vogelzang N.J., Ryan J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–2548. [PubMed] [Google Scholar]
- Lim H.Y., Ju H.Y., Chung H.Y., Kim Y.S. Anti-tumor effects of a tumor cell vaccine expressing a membrane-bound form of the IL-12 p35 subunit. Cancer Biol. Ther. 2010;10:336–343. doi: 10.4161/cbt.10.4.12310. [DOI] [PubMed] [Google Scholar]
- Marr R.A., Addison C.L., Snider D., Muller W.J., Gauldie J., Graham F.L. Tumour immunotherapy using an adenoviral vector expressing a membrane-bound mutant of murine TNF alpha. Gene Ther. 1997;4:1181–1188. doi: 10.1038/sj.gt.3300528. [DOI] [PubMed] [Google Scholar]
- Miyahara Y., Odunsi K., Chen W., Peng G., Matsuzaki J., Wang R.F. Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc. Natl. Acad. Sci. USA. 2008;105:15505–15510. doi: 10.1073/pnas.0710686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley T.A., Haudenschild D.R., Rose L., Reddi A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- Numasaki M., Fukushi J., Ono M., Narula S.K., Zavodny P.J., Kudo T., Robbins P.D., Tahara H., Lotze M.T. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- Puck T.T., Steffen J. Life Cycle Analysis of Mammalian Cells. I. A method for localizing metabolic events within the life cycle, and its application to the action of colcemide and sublethal doses of X-irradiation. Biophys J. 1963;3:379–397. doi: 10.1016/s0006-3495(63)86828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder C.L., Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Rieder C.L., Palazzo R.E. Colcemid and the mitotic cycle. J. Cell Sci. 1992;102((Pt 3)):387–392. doi: 10.1242/jcs.102.3.387. [DOI] [PubMed] [Google Scholar]
- Rouvier E., Luciani M.F., Mattei M.G., Denizot F., Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- Sfanos K.S., Bruno T.C., Maris C.H., Xu L., Thoburn C.J., DeMarzo A.M., Meeker A.K., Isaacs W.B., Drake C.G. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin. Cancer Res. 2008;14:3254–3261. doi: 10.1158/1078-0432.CCR-07-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo Hoo W., Lundeen K.A., Kohrumel J.R., Pham N.L., Brostoff S.W., Bartholomew R.M., Carlo D.J. Tumor cell surface expression of granulocyte-macrophage colony-stimulating factor elicits antitumor immunity and protects from tumor challenge in the P815 mouse mastocytoma tumor model. J. Immunol. 1999;162:7343–7349. [PubMed] [Google Scholar]
- Tartour E., Fossiez F., Joyeux I., Galinha A., Gey A., Claret E., Sastre-Garau X., Couturier J., Mosseri V., Vives V., et al. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res. 1999;59:3698–3704. [PubMed] [Google Scholar]
- Upadhyay G., Yin Y., Yuan H., Li X., Derynck R., Glazer R.I. Stem cell antigen-1 enhances tumorigenicity by disruption of growth differentiation factor-10 (GDF10)-dependent TGF-beta signaling. Proc. Natl. Acad. Sci. USA. 2011;108:7820–7825. doi: 10.1073/pnas.1103441108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Yi T., Kortylewski M., Pardoll D.M., Zeng D., Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Yi T., Zhang W., Pardoll D.M., Yu H. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res. 2010;70:10112–10120. doi: 10.1158/0008-5472.CAN-10-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver C.T., Harrington L.E., Mangan P.R., Gavrieli M., Murphy K.M. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Weaver C.T., Hatton R.D., Mangan P.R., Harrington L.E. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Xin L., Lawson D.A., Witte O.N. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. U S A. 2005;102:6942–6947. doi: 10.1073/pnas.0502320102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z., Timour M., Painter S., Fanslow W., Spriggs M. Complete nucleotide sequence of the mouse CTLA8 gene. Gene. 1996;168:223–225. doi: 10.1016/0378-1119(95)00778-4. [DOI] [PubMed] [Google Scholar]