

Abstract
Next-generation sequencing technology has facilitated the discovery of millions of variants in human genomes. A sizeable fraction of these alleles are thought to be deleterious. We review the pattern of deleterious alleles as ascertained in genomic data and ask whether human populations differ in their predicted burden of deleterious alleles, a phenomenon known as “mutation load.” We discuss three demographic models that are predicted to affect mutation load and relate these models to the evidence (or the lack thereof) for variation in the efficacy of purifying selection in diverse human genomes. We also discuss why accurate estimation of mutation load depends on assumptions regarding the distribution of dominance and selection coefficients, quantities that are poorly characterized for current genomic datasets.
Introduction
The process of mutation constantly creates deleterious variation in a population. These mutations can persist for some time, depending on the intensity of drift and purifying selection. The burden of deleterious variants carried by a population was the subject of classical work in population genetics during the mid-20th century and termed “mutation load” [sidebar]. This mathematical theory described the expected mutational load under idealized genetic models whereby deleterious mutations reduce the reproductive success of carriers compared to a hypothetical genotype with no such deleterious variation. As mutations occur over time, populations accumulate a “mutational load” compared to a hypothetical population with only the fittest genotypes. A key finding was that very deleterious variants, despite their large potential for damage, tend to be quickly eliminated and rarely rise to high frequencies. By contrast, variants of weaker effect may reach appreciable frequencies due to random drift, and can contribute significantly to mutational load because they affect more individuals in the population1–3.
The role of genetic drift in these models raises the possibility that human populations may vary in their mutational burden, given the varied patterns of population growth and decline that have characterized different human groups since their initial divergence more than 100,000 years ago4–7. Although the theory of genetic load generated strong interest in the 1950s–60s, there has been limited opportunity to test these models in the context of human genomics.
The aim of this review is to synthesize recent work on the frequency of deleterious variants in the human genome and the behavior of these variants under different demographic models. What are the characteristics of deleterious variants that have been discovered in large-scale sequencing experiments? Do demographic simulations predict differences in mutational load among populations, and how realistic are these models of human demographic history? What other important parameters, such as dominance, epistasis, or interaction with the environment, should be considered when calculating the burden of deleterious alleles in each population? While there have been significant advances in quantifying how mutational load may vary among human populations, a complete understanding will remain elusive until we can better characterize the relative roles of local adaption and purifying selection in the diversification of human populations.
Models of Mutational Load
Neutral theory [sidebar] emerged in the context of empirical and theoretical work on genetic load in the mid-20th century. At that time, genetic polymorphisms were typically considered to be functional. However, as new protein polymorphism data were generated, much more genetic variation was discovered within and among species than had been previously appreciated. The estimated rate of amino acid substitutions across species phylogenies, estimated at one substitution per genome every two years in mammals, was deemed too rapid for plausible models of selective evolution8: such a rapid rate of adaptation could only be accomplished through the selective deaths of an exceedingly high number of less fit individuals [cite Haldane’s The cost of Natural selection?]. This substitution load [sidebar] would lead to population decline. Motoo Kimura recognized the significance of the estimated evolutionary rates for genetic loci and instead proposed that the vast majority of polymorphisms were in fact neutral with regard to fitness1–3. This shift in worldview, to one where neutrality is the dominant factor driving allele frequency change, recast population genetic models in terms of two evolutionary forces: the neutral mutation rate and genetic drift. In this setting, genetic polymorphism is simply “a transient phase of molecular evolution” [Kimura]. Since purifying selection eliminates highly deleterious mutations quickly, rates of heterozygosity, q, simply reflect the product of mutation rate, μ, the fraction of mutations that are neutral, f, and the effective population size, Ne: i.e., θ = 4 Ne μ f. This result only hold if positive natural selection or weak negative selection is rare. Kimura’s focus on genetic drift in a finite population led to an examination of the interaction between genetic drift, natural selection and mutation in determining the accumulation of deleterious alleles in a population.
Tomoko Ohta and Kimura extended the principles of the neutral theory to argue that mutations with very small fitness effects behave effectively as if they were neutral. If a mutation induces a fractional change s in the expected number of offspring of carriers, it is effectively neutral if |s|≪1/Ne. The evolution of such loci can be accurately modeled using equations involving only drift and mutation. Nearly neutral mutations [sidebar] were defined as a related class of loci with selection coefficients s approximately equal to 1/Ne. A given mutation with fitness coefficient s that is effectively neutral in a small population can be nearly neutral in an intermediate-size population and even considered a large effect variant in a large population, where drift is much reduced. Its effect on average fitness therefore depends on the population demography. Genetic drift occasionally drives nearly neutral mutations to fixation9, leading to a decrease in fitness of the entire population (sometimes referred to as “drift load”10).
To compare the overall effect of demography on fitness across human populations, we turn to the definition of genetic load, the reduction in fitness in a population or species due to presence of alleles that are detrimental, in comparison to the genotype with the maximum fitness (Box 1):
where Wmax is the maximum possible fitness, and Wmean is the average fitness of all genotypes in the population. Wmax is often assigned a value of 1 for algebraic convenience. This definition applies wherever genotypic fitness can be measured or inferred. However, most theoretical results are established under simplifying assumptions of time-independent fitness across generations11, environmental uniformity, and of additive or multiplicative effects across loci. Even though the maximum fitness Wmax is easy to identify in idealized models, it is much more challenging to arrive at meaningful empirical estimates in real populations.
Box 1. Summary Statistics for Mutational Load.
A variety of summary statistics have been used to quantify differences in mutational load between human populations. Some studies estimate mutational load by comparing the estimated load per individual in a population. The total number of derived deleterious alleles present in a single individual’s genome is a straightforward statistic if an unbiased genome is available. In this metric, derived homozygotes are counted twice and heterozygotes are counted once. Under neutrality, each individual is expected to carry the same number of mutations, with some stochasticity due to the finite genome. There is little evidence of substantive differences between populations in the mean number of deleterious alleles per individual48,50.
There are a number of alternative approaches that consider more general statistics of the allele frequency distribution, such as the average frequency among all deleterious alleles, or the proportion of nonsynonymous to synonymous varitions. By contrast with the genetic load, it is rather straightforward to identify differences across populations in these more general statistics. For example, Figure 2a shows the SFS for variants predicted to have a large deleterious effect in 4 populations: the western African Yoruba (YRI) have a notable excess of low frequency variants, whereas populations with Out-of-Africa ancestry like the Japanese (JPT), Tuscans (TSI) and Mexicans (MXL) have an excess of fixed variants. These statistics measure the interaction between selective forces and drift, and show that the frequency distributions of deleterious variants are different across populations. This has important consequences for the achitecture of disease across populations, but does not imply that selection was more efficient, in an absolute sense [cite?], in some populations compared to others.
Strikingly, we find no published estimates of the mutational load LT as it is classically defined2 using human genomic data (see above); we do so here for four populations (Figure 5). Only slight differences are detected across human populations when we consider an additive model (h=0.5). As reflected in the SFS, there are more deleterious variants in the YRI population compared to the JPT (20,672 vs. 13,392, respectively), but the contribution of large effect variants to mutational load is slightly higher in the Out-of-Africa population (2.40 in JPT vs. 2.37 in YRI) because more of these variants are at higher frequencies in the Japanese. [We assume all of the large effect mutations (GERP 4:6) to be equally damaging.] However, strong differences emerge under a recessive model (h=0) (Figure 5). In summary, if the mutation load is calculated according to classical models and a DFE, then differences depend largely on dominance mode. These calculations are laden with many assumptions (see Other Considerations in text), and may have little bearing on the public health of the populations.
Mutational load is the component of genetic load attributable to the reduction in fitness caused by new and recent deleterious mutations. Other components of genetic load include: the segregation load, the inbreeding load, and the transitory load. Segregation load occurs when a heterozygous genotype has a higher fitness than either of the homozygotes (i.e. a heterozygote advantage or overdominance). Inbreeding load occurs when recessive deleterious alleles are found in excess homozygous state relative to Hardy-Weinberg equilibrium (HWE) due to inbreeding12. (Ignoring selection, the probability of homozygosity is Pr(AA) = Fp + (1-F)pˆ2 under inbreeding vs. Pr(AA) = pˆ2 under HWE, where p is allele frequency and F is the rate of autozygosity.). Transitory load occurs while populations adapt to a new fitness landscape and the previous optimal genotype is now suboptimal. Multiple classes of effects potentially contribute to genetic load in humans, but we focus here on the mutational load and the inbreeding load13. In an infinite population, the classical mutation-selection balance [sidebar] tells us that the expected mutational load at a single site is bounded between μ and 2μ, depending on the level of dominance at that site. Importantly, it does not depend on the selection coefficient at that site: the increased cost per allele of damaging alleles is cancelled exactly by the reduction in frequency due to selection. Most populations, including human populations, however are neither infinite nor in mutation-selection balance. The equilibrium results still hold approximately true in finite populations for very deleterious variants, for which mutation and selection are the largest effects. For weakly deleterious (i.e., nearly neutral) variants, however, drift becomes more significant and can act to increase the average load.
Dominance
The proportion of deleterious mutations that are recessive, additive or dominant is an open question in human genetics. Answering is important to both evolutionary and medical genetics. The effect of dominance on fitness is quantified by the parameter h, where the fitnesses of genotypes AA, Aa, and aa are 1, 1-hs, 1-s. Across loci, there is a distribution of h, with h=0 for recessive alleles, h=0.5 for additive alleles, and all levels of partial dominance, including outside this (0,1) range. Dominance is perhaps the most important quantity that has not been estimated from genome-wide data. For a large population at equilibrium, a classical prediction is that the load per new deleterious mutation is greater under an additive than under a recessive model. The load is approximately 2u when h = 0.5 and u when h = 02. This is because, by definition, additive mutations exhibit some penetrance while recessive mutations do not. Dominance can lead to substantial differences in load across populations, because of differences in population history can have a strong impact on the proportion of homozygotes14,15. By contrast, the effect of drift on load under an additive model is much weaker. The effect of dominance on load also depends on the frequency of deleterious variants (Figure 1). New variants are almost exclusively found in heterozygous form, so rare recessive mutations have little impact on load.
Figure 1. Proportion of deleterious variants found in an individual’s genome classified by their frequency in the population (common vs. rare).
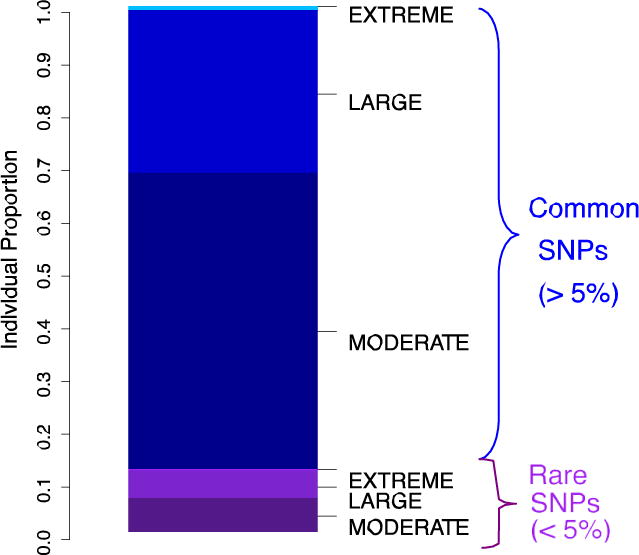
We wanted to ascertain whether the burden of the deleterious portion of an individual’s genome is mostly represented by rare or by common variants. For the 1000 Genomes Yoruba (YRI) population, variants were assigned to three selection regimes (moderate, large, extreme), according to GERP score categories in increasing order of phylogenetic conservation 2:4, 4:6, >6. The more conserved a site is, the more likely a new allele is to be deleterious (Box 2). Deleterious variants with a derived allele frequency lower than 5% within the population (purple) are classified as “rare”, and the rest as “common” (blue). Almost 70% of the deleterious variants found in an individual genome are common, and most of them have small predicted effect (“moderate”). Half of the rare variants also have a moderate effect, and half of them have a large effect, demonstrating how low frequency, large effect variants have not yet been purged by purifying selection.
Hints about the distribution of dominance come from a diversity of experimental systems. Mutation-accumulation experiments in model organisms indicate that there is an inverse relationship between dominance and the severity of mutations16–18: the more severe a mutation, the lower its dominance coefficient h (i.e., the more recessive it is). The average dominance of mildly deleterious mutations across a variety of studied non-human organisms is partially recessive ~0.2519. We also know that there are many recessive mutations of strong effect in humans, particularly evident in consanguineous unions or endogamous populations20–22, sometimes referred to as inbreeding depression13. A recent population genetic approach by Szpiech et al.23 considered long runs of homozygosity (ROH) across human populations and looked at the enrichment of deleterious variants in runs of different length. Long runs of homozygosity contain more deleterious variants on average than short ROH or homozygotes not in ROH, indicating that the deleterious variants are more likely to lie on long, recent haplotypes. They suggest that long ROH represent recent inbreeding, demonstrating how recent non-random mating can exacerbate deleterious effects for recessive loci.
Even mutations of weak effect demonstrate inbreeding depression for human height in European populations24, indicative of a recessive or partially recessive model for the majority of deleterious mutations. Well-validated recessive disease mutations, such as those used in newborn disease screening panels, have been shown to be relatively common in standard sequencing data sets; as many as 45% of individuals in a recent exome study carried at least one recessive allele of strong effect25,26. The X chromosome carries proportionally more rare deleterious variation than the autosomes, potentially because recessive alleles are exposed in the hemizygous male and, thus, reach lower frequency, on average, than recessive alleles in the autosome27. In an attempt to quantify recessive versus dominant diseases, Erickson and Mitchison28 surveyed 14 well-characterized diseases and found that autosomal recessive genes were more common than autosomal dominant ones, but they had a smaller relative contribution to human disease.
Rare Variants, Deleterious Variants
The rise of low-cost, large-scale next-generation sequencing has empowered the study of human genetic variation in ever larger samples at a whole genome scale. Newly discovered genomic variants are found at low frequency in human populations (i.e. present in less than one per thousand people). These rare variants tend to be geographically restricted29,30 or even private to an individual or family (Box 2). Rare variants also tend to be younger than common variants31. Compared to common variants, these rare variants are more likely to affect protein composition, to do so in a more disruptive manner, and to occur at predicted functional sites27,32–37. Furthermore, the lower the frequency of a variant in a sample, the more likely it is to be annotated as deleterious using a variety of variant effect prediction algorithms (Box S1).
Box 2. Properties of Private versus Shared Variants.
In general, rare variants tend to be private to a population, and common variants tend to be shared across populations (inset). For example, Gravel et al.55 show that variants below 5% allele frequency show little sharing across continental human population. The great number of shared common variants is expected from the relatively low degree of genetic differentiation seen among human populations. The large number of private and rare variants can be largely explained by neutral forces: Rare variants are likely to have occurred during or after population divergence. These variants will be population specific and found at very low frequencies. This can be compounded by the effect of natural selection, which tends to keep deleterious variants at low frequency and may act differentially across populations. Lohmueller et al.47 and Peischl et al.64 show that the proportion of deleterious alleles is higher among rare variants than among common variants, and that rare variants that are also population-specific are even more likely to be deleterious. [Cite Marth, Gabor T., et al. “The functional spectrum of low-frequency coding variation.” Genome biology 12.9 (2011): R84., who shows reduced sharing among more deleterious variants]
Shared variants are typically older, and therefore are more likely to be found at higher frequencies at a global scale. Again, this pattern can be largely explained by neutral forces: If these mutations are benign or neutral, they can be maintained over long periods of time and in multiple populations. If these mutations are slightly or even moderately deleterious but have been driven to higher frequencies and spread across populations due to the increased effect of genetic drift during range expansions in very early human dispersals. Peischl et al.64 have performed simulations that show an increase in frequency of deleterious mutations under a range expansion model, and have observed that 10% of common variants shared between Africans and non-Africans are predicted to be deleterious. A similar proportion (14%) of large effect variants is found to be shared between the eastern African Luhya population and the Finns from northern Europe in the 1000 Genomes Phase 1 data. More interestingly, though, these variants are generally found at very high frequencies, and actually represent 86% of the total number of large effect variants in the dataset. This scenario is in agreement with most of the large effect variants being private and found at low frequencies, and a smaller proportion of variants being shared but common, compatible with the range expansion model. However, large effect variants that are also shared can also be driven to high frequencies in cases where they have a beneficial effect to the fitness of the population.
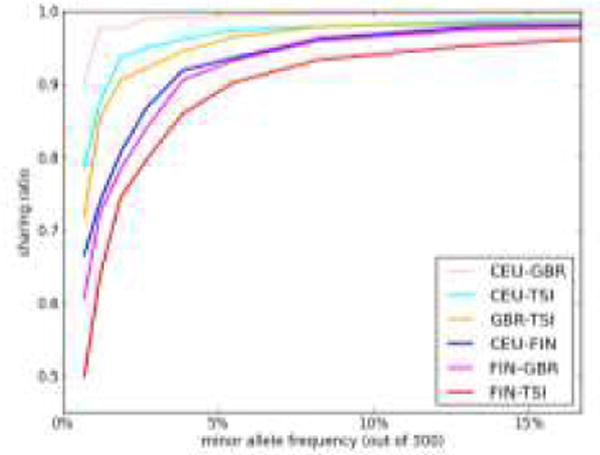
Allele sharing versus allele frequency among European populations: the sharing ratio is the probability that two minor alleles drawn randomly in a pooled sample come from different populations, relative to the panmictic expectation55. A panmictic population has a ratio of 1, and completely diverged populations have a ratio of 0. GBR: Great Britain; CEU: Central European from Utah; TSI: Tuscan; FIN:Finnish.
Variant effect prediction algorithms attempt to combine available information to predict the effect of a mutation on function (impact on protein structure/function, expression, degradation, etc.) or evolutionary fitness (i.e., expected number of offspring that a carrier leaves). These effects are distinct but sometimes related: Some mutations are strongly evolutionarily deleterious precisely because they impact protein structure or function of important genes. For example, loss-of-function mutations32,33,38 disrupt the generation of a fully functional protein either by introduction of a stop codon or by truncating the reading frame of the protein, and are thus selected against if the gene product is essential. Other mutations impact non-essential genes or only slightly alter the protein or impact gene expression. These mutations may be evolutionarily weakly deleterious if the expected number of offspring left by carriers is only slightly lower than wildtype. Rare, deleterious variants may lead to early onset diseases, and may also inflate an individual’s susceptibility to common, complex diseases36,39–42. For example, approximately 70% of nonsynonymous variants in two hundred drug target genes were estimated to have sufficiently large negative selection coefficients such that these variants would be unlikely to reach even 5% frequency within the current European population2,35,43. [This may reflect partial ascertainment bias, because drug target genes appear to be under stronger purifying selection than the average gene]. However, even in full exome data, it is predicted that 47% of single nucleotide variants detected are deleterious in a sample of size […]36,44,45. The concordance of predictions of deleterious effects across leading effect prediction algorithms is modest (Box S1), and thus there is still substantial uncertainty in the true number of functional or deleterious variants2,32,36. Despite this uncertainty, variants of large effect are enriched among rare variants in several populations from the 1000 Genomes dataset, a conclusion that is independent of the prediction algorithm (Figure 2).
Figure 2. Differences in the site frequency spectrum across populations for neutral and deleterious variants.
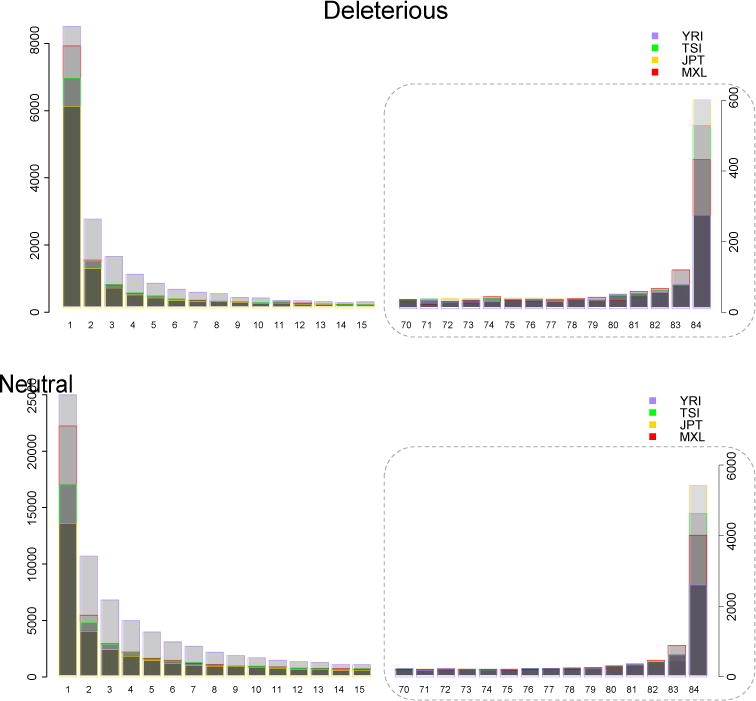
The site frequency spectrum (SFS) can be a powerful method of summarizing genomic data. We show the SFS for four populations focusing on both low frequency variants (<15%, left panel) and nearly fixed variants (>90%, right panel). Using 1000 Genomes Phase 1 exome data33, we sampled 42 individuals from the Yoruba (YRI, Nigeria), Mexicans (MXL, Mexico), Tuscans (TSI, Italy) and Japanese (JPT, Japan) populations. [Only individuals on the same Agilent exome platform were compared here to avoid biases in target capture between platforms.] Derived variants were annotated with GERP (Box S1) and we plot variants predicted to have a “large” deleterious effect (GERP>4) (top panels) and “neutral” effect (GERP<2) (bottom panels). Demography generates different SFS for each population. Neutral variants provide a null demographic model. The African Yoruba have the greatest number of rare deleterious variants, though the Japanese and Tuscans have many more deleterious fixed variants, likely due to ancient founder effects resulting in the fixation by strong drift (also noted in47). By comparing the difference between the neutral and deleterious SFS (Figure S1), one can infer the impact of purifying selection. For example, non-African populations have a larger proportion of deleterious variants that are fixed, compared to what is seen neutrally.
Empirical Estimates of Load In Humans
The distribution and evolution of deleterious mutations is fundamental to understanding the genetic architecture of human disease. Are more diseases caused by common variants shared across populations, or rare variants specific to a population or family (Box 2)35,41? The relative proportion of rare vs. common disease variants may in fact differ by population given unique historical modes of population growth or bottlenecks36,46. Mutation load provides a framework for quantifying and summarizing the overall effect of population-specific history on deleterious variation. Perhaps more importantly, the models of mutation load illuminate the complexity of factors involved in understanding genetic disease risk from genomic data alone.
Every human individual carries many deleterious mutations (Figure 1). New mutations that enter the gene pool have widely varying impacts on fitness, and we can think of them as being drawn from an underlying distribution of fitness effects (DFE)45 [sidebar]. A very early paper by Morton and colleagues20 attempted to measure the total mutational damage in humans by considering consanguineous marriages [sidebar], where inbreeding load would be revealed by the expression of recessive homozygotes. Total mutational damage was defined as the average number of lethal mutations that would result when occurring in a homozygous state. They estimated that there are between 3–5 lethal equivalent mutations per zygote, and that this number was likely an underestimate given that inference relied only on stillbirths and other major pre-reproductive abnormalities and not, for example, on infertility.
Recently, with access to full human genomes to assay the number of deleterious mutations, interest in mutational load has been revived. However, empirical work has been limited and has primarily considered populations of European and African-American ancestry25,26,47–49. In one of the first studies to revisit this topic, Lohmueller et al.47 aimed to address whether human populations carried different numbers of deleterious mutations. Using a set of ~10,000 genes, Lohmueller et al.47 quantified the total number of damaging SNPs in two population samples (n=15 African-Americans, n=20 European-Americans) and also the per-genome rate in heterozygous vs. homozygous state. They found a significantly higher proportion of putatively deleterious alleles in the European-American sample as compared to the African-American sample. Among SNPs that were private to each population (i.e. only segregating in Europeans or only in African-Americans), the proportion of predicted damaging mutations was significantly higher in Europeans (16%) vs. African-Americans (12%). However, the total number of deleterious variants was greater in African-Americans. African-Americans carried vastly more neutral variants as well. Under a strong Out-of-Africa bottleneck (discussed in the Models section below), Lohmueller and colleagues showed via forward simulations that a severe bottleneck coupled with subsequent population growth could result in the dramatic differences in the proportion of deleterious mutations.
Whereas Lohmueller et al.47 hypothesized that the result might be due to reduced efficacy of selection (Box 3) after the Out-of-Africa bottleneck, this view has been recently contested50. Simons et al.48 revisited the question using a larger dataset of recently generated European and African-American exomes51. They contrasted the number of deleterious alleles per individual in each population (regardless of zygosity), annotated either as nonsynonymous or predicted to be damaging by Polyphen (Box S1). Under this summary of the data, European and African-American individuals carried, on average, similar numbers of deleterious mutations, and there was no significant difference in the average frequency of deleterious mutations. Simons et al. concluded that the differences observed by Lohmueller et al.47 did not indicate differences in mutation load. Rather, they suggested it can be explained by assuming that each population has the same amount of deleterious variation, but that populations differ in how many of these deleterious variants are common, and how many are rare. These results were replicated in a smaller sample from the 1000 Genomes Phase 1 data, Yoruba (Nigeria) and European-Americans (CEU from Utah)33 demonstrating that the lack of difference was not due to recent European admixture in African-Americans. The difference between Lohmueller at al.’s original paper and Simons et al.48 is largely one of interpretation, as the datasets in broad agreement52; the disagreement is about whether these are informative about the efficacy of selection [doi: http://dx.doi.org/10.1101/010934?]. However, another recent paper, by Fu and colleagues52, re-analyzed the same exome data as Simons et al., but annotating deleterious variants with a PhyloP, a conservation based algorithm (Box S1). They found significantly more deleterious alleles in Europeans than in African-Americans; these mutations were typically mildly deleterious and many of them were fixed in the European population. Thus, the choice of an effect prediction algorithm can have a large impact on the final interpreatation. Upcoming functional characterization of variants through high-throughput mutagenesis, and the resulting improvement of functional algorithms, will be a huge asset in resolving these long-standing questions about human evolution.
Box 3. Efficacy of Purifying Selection.
Lohmueller et al.47 proposed that differing patterns of deleterious variation across populations might be due to differences in the efficacy of selection (specifically, the higher proportion of nonsynonymous to synonymous variants among Europeans). But how can selection be more efficient if the mean number of deleterious mutations per individual, such as between Europeans and African-Americans, is not different (see also Lohmueller76)? Lohmueller et al.47 estimate the efficacy of selection by comparing it to the effect of drift at a given locus. Given s, negative selection is more efficient in larger populations because drift is reduced. This definition is inspired by nearly neutral theory [sidebar], wherein the fixation of deleterious alleles depends crucially on the ability of drift to overcome negative selection at individual sites. However, for rare variation and over short periods of time, this efficacy may have little to do with mean fitness decrease in a population: copies of a recent deleterious allele evolve almost independently from each other and of the population size. If we define the efficiency of selection as its ability to purge deleterious alleles globally, we may not see any appreciable difference between human populations: for short time scales, we can have an equal number of deleterious alleles across populations, but differences in drift (i.e., in the changes in frequency of these variants). Measuring the efficacy of selection by its effect on load, and by its relative strength vs drift, can lead to dramatically different conclusions [cite? doi: http://dx.doi.org/10.1101/010934?]].
Recent population history also demonstrates how some deleterious alleles can reach high frequency following a severe bottleneck. In an empirical study, Casals et al.49 examined the effect of a strong bottleneck in a French-Canadian population descended from French migrants who settled in the Quebec region beginning in 17th century. Using over 100 exome sequences, Casals et al.49 show a strong decrease in heterozygosity in French-Canadians compared to the source French population, 19% relative to 12.5%, as expected under a founder effect. French-Canadians also carry proportionally more missense alleles at both low frequencies and fixed, while the French carry proportionally more missense alleles at intermediate frequencies. Missense and nonsense mutations in the French-Canadians are more damaging than their counterparts in the French population, as measured by having larger GERP scores (Box 1); or, in other words, the deleterious variants observed in the French-Canadians have, on average, more negative selection coefficients. Similarly, the Finnish population also carries a higher proportion of low frequency loss-of-function variants (expected to be highly damaging) than their counterparts in other European countries due to a recent population bottleneck53,54. However, there is no evidence so far that these populations carry a larger genetic load.
Demographic Simulations of Mutation Load
Empirical data from human genomes appears to support discordant theories of genetic load. Do human populations differ in their levels of mutation load? Has purifying selection acted more efficiently in some human populations compared to others? Some of this confusion is due to different reported summary statistics (Box 1), but the debate also centers on simulated results obtained under several idealized demographic models. Multiple simulation efforts have considered three major demographic effects: an Out-of-Africa bottleneck dramatically reducing variation in non-African populations, serial founder effects across a geographic range whereby drift is increased during the founding of new populations, and rapid population growth due to recent agricultural and technological changes (Figure 4). These models are by no means mutually exclusive, and many simulations include bottleneck and growth periods. However, these idealized models can help us build intuition about the effect of different events on patterns of diversity and examine their effect over time. As we are dealing with a dynamic system rather than a population at equilibrium, some effects are short-lived and others take many generations to evolve before a strong difference is detectable.
Figure 4. Schematic of different demographic models for the Out-of-Africa dispersal.
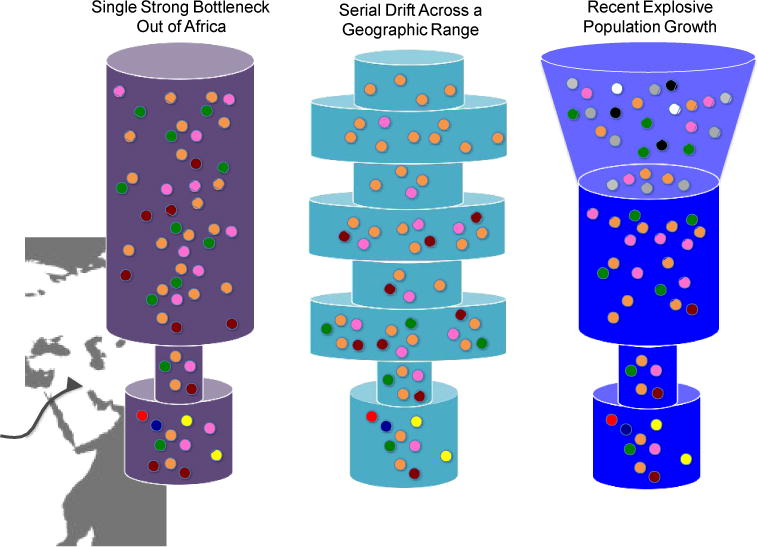
Three demographic models have been discussed in the context of changes in genetic load due to extreme genetic drift across different human populations. All three models allow for a severe Out-of-Africa bottleneck and recovery but with varying degrees of subsequent population size changes. Colored dots indicate allelic diversity; width of the column is proportional the effective population size, Ne. The bottom tube represents the ancestral African population size, with later events occurring in temporal sequence towards the top of the figure.
Bottleneck
A scenario that is commonly simulated is a classical bottleneck, in which a population experiences a drastic reduction in size before recovering. The most readily observed effect of a bottleneck is an increase in genetic drift, which in turn reduces heterozygosity. The amount of drift depends on the bottleneck intensity, I=T/(2NB), where T is the duration of the bottleneck and NB the effective population size during the bottleneck. A single Out-of-Africa bottleneck model captures the reduction in genomic diversity in populations currently residing outside of Africa15, and is arguably the most noticeable genomic consequence of varying demographic histories in human populations15,55. Many studies support a severe bottleneck during the initial colonization of the Eurasian continents, reducing the effective population size of the founders to only <1,000–2,500 individuals4,5,35,46. Recent coalescent analysis56,57 based on whole genome sequences also supports an Out-of-Africa bottleneck with a nearly fifteen-fold reduction in Ne to only about 1,000 individuals, leading to a higher proportion of recent common ancestry among non-African individuals4,58. Interestingly, the coalescent method also suggests a bottleneck of varying magnitudes in African populations at approximately the same time as the Out-of-Africa dispersal. Whether this is due to reduction in sub-structure across African populations, parallel bottlenecks due to the MIS 4 and 5 glacial periods, founder effects during the expansion throughout Africa59 or other demographic possibilities has not yet been determined.
Here we consider in detail published simulations of the Out-of-Africa (OOA) bottleneck on load. The single bottleneck models simulated by Lohmueller47,60 and Simons et al.48 vary in the length of bottleneck they consider (T), from instantaneous to 7,700 generations. The OOA bottleneck length inferred from genetic data varies from a few hundred generations to 50,000 years55,58. [[Immediately following a deep bottleneck, the number of deleterious polymorphisms decreases –that’s not true as written, I tried to guess what was meant see comment]], reflecting the fixation of low frequency variants. But as the population recovers, the now larger population accumulates rare deleterious variants whose distribution of fitness effects is more similar to that of new mutations. Compared to the variants in the pre-bottleneck population, these new variants will tend to have more deleterious effects. Importantly, during the bottleneck, the reduced population size inflates the role of random genetic drift and allows some deleterious mutations to drift to intermediate and high frequencies. This ultimately leads to more high frequency and fixed deleterious variants in a population that has undergone a bottleneck (Figure 2), however this can be a slow process. The Simons et al.48 simulations under a single-step bottleneck model illustrate how these two opposing factors can interact; the decrease in the number of deleterious variants is balanced by the increased frequency of the remaining variants. This balance is exact right after the bottleneck, and is maintained over time for very deleterious variants (as simulated in Simons et al.), or very weakly deleterious ones, but more moderately deleterious alleles can be more influenced by differential purifying selection (see below, [doi: http://dx.doi.org/10.1101/010934?]). Assuming weaker overall variant effects, Fu et al.52 found that a tenfold bottleneck increased mutation load by 4.5% relative to a constant size population, primarily due to common and fixed variants of weak effect.
Serial Founder Effects/Range Expansion
The second strong signature in genetic data from non-African populations is the continuous trend of decreasing genetic diversity (e.g. heterozygosity) with distance from eastern Africa61,62. This observation can be modeled by serial founder effects in which a small founder population buds off from the ancestral group and contains only a subset of the original diversity as it colonizes a new uninhabited geographic area32,34. When described using geographically explicit simulation models, the effect of genetic drift is further exacerbated by the sampling of demes from the wave front of the population as it expands in space (see Moreau et al.63 for a historically documented example). For populations toward the end of the range expansion (e.g. Native Americans), demographic history is characterized by many, perhaps hundreds, of bottlenecks followed by population recoveries. While computationally intensive to simulate, the serial founder effect model probably best describes the long period of human population history after the dramatic Out-of-Africa event32.
Simulations using the serial founder effect show that varying demographic details can result in large differences in genetic load. Peischl et al.64 used spatially explicit forward simulations to examine the effect of extreme drift at the expansion wave front on the pattern of deleterious alleles. These wave front expansions will affect both new mutations and standing variation where drift is especially extreme and alleles can ‘surf’ to high frequencies rapidly43. A range expansion can thus increase the mutational load of a population at the edge of an expansion relative to one in the geographic center, assuming similar environments and selection coefficients. This effect is particularly pronounced for small selection coefficients and mutations that newly occur either during or after the Out-of-Africa bottleneck. Expansion load in these simulations was particularly sensitive to the carrying capacity (K, the population size reached before founding a new deme and allowing for migration), with large carrying capacities reducing the probability of fixation44. Simulations of the Out-of-Africa serial founder effect identify K≈1,000 and moderate rates of migration as a good fit to current human heterozygosity32. These results would involve a high probability of local fixation for new deleterious mutations, even with selection coefficients as strong as −0.01.
Population growth
We know that the global human population has grown at a prodigious rate – this has been well documented from historical and archeological records from the past few thousand years (Figure 3). What has been appreciated only recently is the magnitude of the impact of this recent growth on the pattern of genetic variation in humans. It is only with large samples sizes that surveys of population variation reveal this impact. The studies of Coventry et al.65, Nelson et al.35, Tennessen et al.36 and Fu et al.51 examined samples of fully-sequenced genes or exomes in samples of thousands, and give a consistent picture – larger sample sizes reveal many more rare variants than expected under a constant population model. This excess of rare variation reflected by the nearly five-fold excess of singletons in the 10,000+ sample of Coventry and colleagues, is consistent with realistic models of recent population growth. Demographers report global human growth rates on the order of 1%–2% per year for the last century or more, and although a somewhat lower rate is obtained from genetic data, this is likely because genetic growth rates reflect the effective population size.
Figure 3. Demographic history based on the site frequency spectrum and sharing of rare alleles.
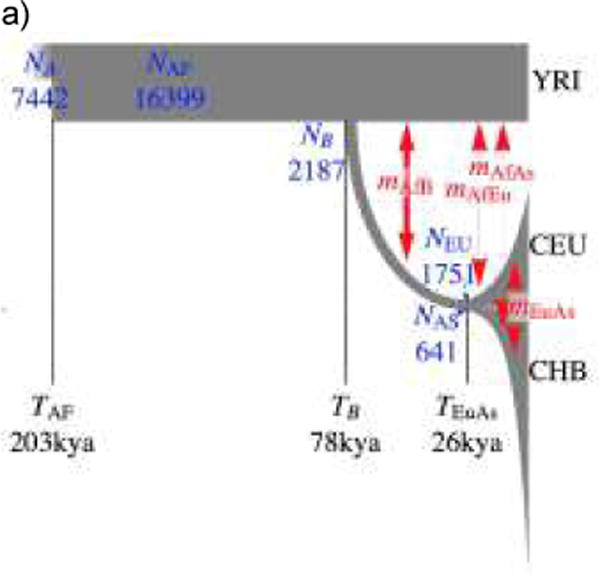
a) Updated three-population demographic model based on synonymous sites from 1000 Genomes Phase 1 data33, assuming a mutation rate of 2.36×10−8/bp/g and a generation time of 25 years (for ease of comparison with Gravel et al.55 and Tennessen et al.36). Estimated times and population sizes are inversely proportional to the assumed mutation rate.
Population growth stretches back more than just a couple thousand years in many regions of the world. Analysis of mtDNA lineages shows a strong increase in female effective populations size (Nef) during the Holocene in Africa and Eurasia66,67, compared to Upper Paleolithic lineages. Zheng et al. analyzed over 300 East Asian mitochondrial genomes and find that major mtDNA lineages underwent expansions starting around 13 kya and lasting until 4 kya, with changes in Ne from a few hundreds of thousands to millions. Western African populations, far from being the constant population size often portrayed in simulations, have experienced tremendous growth, particularly over the past 5,000 years as Bantu-speaking populations and other groups adopted agriculture4,36,66. Europeans also likely experienced a notable bottleneck during the Last Glacial Period beginning 21 kya as northern and central Europe became glaciated68, but populations recovered after 15 kya just before the widespread adoption of agriculture resulted in sustained growth.
Population expansion has a complex effect on the fate of deleterious variation. For example, growth increases the mean survival time of all new mutations in the population, including deleterious ones. However, a longer survival time does not necessarily mean a larger effect on genetic load—variants survive longer largely because they are allowed to exist at lower frequency after the population has grown. Similarly, population expansions increase the proportion of recent (and rare) mutations in a population, and these rare variants tend to be more deleterious than common variants because of differences in the efficacy of purifying selection between rare and common variants. However, this may not have a large effect on the genetic load, because rare variants make up a small fraction of an individual’s overall mutational load48,69 (Figure 1).
Other Considerations And Future Directions
With a few exceptions, genetic load differences across human populations are expected to have little bearing on the health and reproduction of present-day populations—the current fitness/cultural landscape is very different from what it was over the last hundred thousand years. Exceptions include populations that have undergone particularly strong bottlenecks and experience a temporary increase in recessive deleterious variants. Models of mutation load that attempt to accurately quantify differences across populations or to test the predictions of specific theoretical models should consider several other inter-connected issues: variant prediction accuracy, spatial variation in selection coefficients and local adaptation.
As noted in Box S1, almost all the variants considered in these studies are predicted to be deleterious, and most estimates rely on bioinformatic heuristics that are informative but far from perfect. Substantial discrepancy exists among methods. More generally, methods that attempt to estimate mutational load share many assumptions11 that are debatable. One potential issue is the radical mis-assignment of selection coefficients for adaptive variants. For example, the EDARV370A missense mutation is computationally predicted to have a strong effect on a downstream signal transducer and yet this mutation may be locally adaptive in East Asia for an increased number of eccrine sweat glands70. Even if local adaptation is not considered to be a pervasive force in recent human evolution71, small numbers of adaptive alleles, under a selective sweep model, will reach high frequency in the population and contribute significantly to the mutational load if erroneously annotated as deleterious. One potential way to overcome local adaptation would be to disregard alleles found at high frequencies, though that would not be appropriate for all non-African populations, as severe genetic drift will lead to bona fide high-frequency deleterious variants49 (Figure 2). Even for alleles that have a more global distribution, the notion that alleles can be assigned an absolute fitness coefficient shared among all human groups is untenable. Alleles that are deleterious in some human groups have been shown in some cases to be beneficial in others. As an example, it has been shown that children with anemia have a fourfold increase in risk for pneumonia at high altitudes compared to lower altitudes72. G6PD deficiency alleles may have a negative consequence of hemolytic anemia but nevertheless carry a fitness advantage in areas where malaria is endemic. The genome-wide dependence of fitness coefficients on time and place is largely unknown.
Finally, the above models assume that fitness effects are additive over all loci in the genome, which means they ignore the possibility of epistatic interactions, despite the fact that experimental mutation-accumulation experiments with model organisms often see diminishing returns (negative) epistasis73,74 Direct tests of the importance of epistasis in humans have been badly underpowered by the explosion in the number of tests (there are n(n-1)/2 pairwise epistasis tests across n SNPs), and by the fact that these tests require contrasts of phenotypes across multiple genotypic classes, requiring very large sample sizes to recover rare double homozygous genotypes. In short, the absence of large numbers of reports of epistasis in humans is not evidence for a lack of epistasis.
There may not be enough information in the genomes of all human individuals to accurately infer all the unknown parameters governing human evolution. To answer these questions, we will need to complement genomic data with direct high-throughput experiments measuring the cellular impact of mutations through saturation mutagenesis [], and experimental evolution in model organisms[].
Conclusions
Current human genomic datasets and genomic simulations provide conflicting evidence for differences in mutation load among human populations. A variety of statistics have been used to summarize the distribution of deleterious polymorphisms within populations, and this has contributed to the confusion. While recent work has emphasized an abundance of deleterious rare variants, rare variants have only a small effect on differences in mutation load between populations.
Rather than a focus on rare variants, we believe that more realistic assessment and simulation of different dominance models are key to understanding the distribution of genetic load across populations. If only a fraction of deleterious alleles are recessive, as predicted from disease and model organism studies, then Out-of-Africa bottlenecked populations are expected to see an increase in load due to intermediate-effect variants compared to an additive model (Figure 5). Moreover a large number of deleterious mutations may also exist in the non-coding portion of the genome75, meaning that studies focusing on exomes have only studied a small portion of the mutational load that may exist in the human genome37.
Figure 5. Mutational load under an additive and a recessive model.
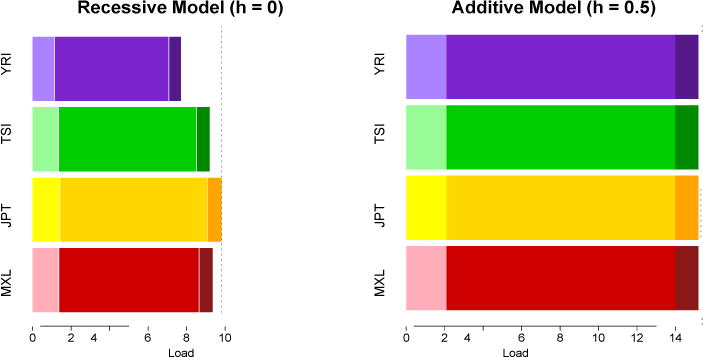
Using the same dataset as in Figure 2, we computed the total mutation load2 for each population. GERP scores were annotated in whole-exome data. Variants were grouped in three categories according their GERP score (2:4, 4:6, >6), corresponding to different biological functional effects. The more phylogenetically conserved a site is, the more likely a new allele is to be deleterious and have a high GERP score (Box S1). Within each category, three selection coefficients were assigned: (s= −4.5 × 10−4), (s= −4.5 × 10−3) and (s= −1 × 10−2), using the inferred s coefficients in Boyko et al.45. Total mutational load is the sum of load for each locus2. Mutational load under an additive model is higher than mutational load under a recessive model because the phenotypic effect of a variant is masked in the recessive homozygous state. While only slight differences exist between populations for an additive model of dominance (~1.5%), strong differences occur under a recessive model because of the differential number of derived homozygotes among populations.
In addition to the question of dominance, there are three areas that require extensive research before geneticists understand the phenomenon of load. First, epistasis plays a key role in determining complex trait phenotypes in model organisms, and likely does the same in humans. At the same time, we know little of the underlying mechanisms driving epistatic interactions, especially for rare variants, or the degree to which epistasis in fitness effects impacts allele frequency dynamics. Second, the role of local adaptation in human populations from different environments and cultures must be described in order to discriminate between frequent deleterious alleles and frequent beneficial ones. Third, research should focus on integrating bioinformatic and experimental approaches to validate predicted variant effects on the phenotype. An improved partitioning of variants into a distribution of fitness effects would increase the utility of evolutionary and disease models, and is key to improving our understanding of the differences in the architecture of complex diseases across human populations.
Supplementary Material
Online Summary.
Millions of new variants have been discovered in human genomic datasets. Many of these, especially rare variants, have been annotated as deleterious.
A series of recent articles considered whether different human populations vary in their burden of deleterious alleles, a concept referred to as “mutation load”.
Several studies suggest that there is little difference in the average number of deleterious alleles per individual. These studies primarily compared genomic datasets from populations of western African and western European ancestry. However, prior studies are sensitive to annotation prediction algorithms and summary statistics leading to different, sometimes contradictory results.
These calculations also involved a number of simplifying assumptions, including: additive allelic effects, no epistasis, and simple distributions of selection coefficients across deleterious variants and across populations.
Following classical models of mutation load, we study genotype frequencies to highlight how load can change under different models of dominance.
Additionally, genetic drift has shifted the allele frequency spectrum of deleterious variants such that Out-of-Africa populations carry more common deleterious variants.
Differences in the frequency spectra across affect the genetic architecture of disease and our ability to interpret it.
References
- 1.Ohta T, Gillespie J. Development of Neutral and Nearly Neutral Theories. Theor Popul Biol. 1996;49:128–142. doi: 10.1006/tpbi.1996.0007. [DOI] [PubMed] [Google Scholar]
- 2.Kimura M, Maruyama T, Crow JF. The Mutation Load In Small Populations. Genetics. 1963;48:1303–1312. doi: 10.1093/genetics/48.10.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King JL, Jukes TH. Non-Darwinian evolution. Science. 1969;164:788–798. doi: 10.1126/science.164.3881.788. [DOI] [PubMed] [Google Scholar]
- 4.Marth GT, Czabarka E, Murvai J, Sherry ST. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–372. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laval G, Patin E, Barreiro LB, Quintana-Murci L. Formulating a Historical and Demographic Model of Recent Human Evolution Based on Resequencing Data from Noncoding Regions. PLoS ONE. 2010;5:e10284. doi: 10.1371/journal.pone.0010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. Bayesian inference of ancient human demography from individual genome sequences. Nature Genetics. 2011;43:1031–1034. doi: 10.1038/ng.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veeramah KR, et al. An Early Divergence of KhoeSan Ancestors from Those of Other Modern Humans Is Supported by an ABC-Based Analysis of Autosomal Resequencing Data. Mol Bio Evol. 2012;29:617–630. doi: 10.1093/molbev/msr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 9.Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- 10.Crow JF. Genetic Loads and the Cost of Natural Selection. Mathematical Topics in Population Genetics. 1970;1:128–177. [Google Scholar]
- 11.Agrawal AF, Whitlock MC. Mutation load: the fitness of individuals in populations where deleterious alleles are abundant. Annu Rev Ecol Evol Syst. 2012;43:115–135. [Google Scholar]
- 12.Crow JF., 2 The concept of genetic load: a reply. The American Journal of Human Genetics. 1963;15:310–315. [PMC free article] [PubMed] [Google Scholar]
- 13.Charlesworth D, Willis JH. Fundamental Concepts in Genetics: The genetics of inbreeding depression. Nature Reviews Genetics. 2009;10:783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- 14.Li JZ, et al. Worldwide Human Relationships Inferred from Genome-Wide Patterns of Variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 15.Henn BM, Cavalli-Sforza LL, Feldman MW. The great human expansion. Proceedings of the National Academy of Sciences. 2012;109:17758–17764. doi: 10.1073/pnas.1212380109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agrawal AF, Whitlock MC. Inferences About the Distribution of Dominance Drawn From Yeast Gene Knockout Data. Genetics. 2011;187:553–566. doi: 10.1534/genetics.110.124560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukai T, Chigusa SI, Mettler LE, Crow JF. Mutation rate and dominance of genes affecting viability in Drosophila melanogaster. Genetics. 1972;72:335–355. doi: 10.1093/genetics/72.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Houle D, Hughes KA, Assimacopoulos S, Charlesworth B. The effects of spontaneous mutation on quantitative traits. II. Dominance of mutations with effects on life-history traits. Genet Res. 1997;70:27–34. doi: 10.1017/s001667239700284x. [DOI] [PubMed] [Google Scholar]
- 19.Manna F, Martin G, Lenormand T. Fitness Landscapes: An Alternative Theory for the Dominance of Mutation. Genetics. 2011;189:923–937. doi: 10.1534/genetics.111.132944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morton NE, Crow JF, Muller HJ. An Estimate Of The Mutational Damage In Man From Data On Consanguienous Marriages. Proceedings of the National Academy of Sciences. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends in Genetics. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 22.Bittles AH, Black ML. Consanguinity, human evolution, and complex diseases. Proceedings of the National Academy of Sciences. 2010;107:1779–1786. doi: 10.1073/pnas.0906079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szpiech ZA, et al. Long runs of homozygosity are enriched for deleterious variation. Am J Hum Genet. 2013;93:90–102. doi: 10.1016/j.ajhg.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McQuillan R, et al. Evidence of Inbreeding Depression on Human Height. PLoS Genet. 2012;8:e1002655. doi: 10.1371/journal.pgen.1002655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tabor HK, et al. Pathogenic Variants for Mendelian and Complex Traits in Exomes of 6,517 European and African Americans: Implications for the Return of Incidental Results. The American Journal of Human Genetics. 2014;95:183–193. doi: 10.1016/j.ajhg.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue Y, et al. Deleterious- and Disease-Allele Prevalence in Healthy Individuals: Insights from Current Predictions, Mutation Databases, and Population-Scale Resequencing. The American Journal of Human Genetics. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nature Genetics. 2010:1–6. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 28.Erickson RP, Mitchison NA. The low frequency of recessive disease: insights from ENU mutagenesis, severity of disease phenotype, GWAS associations, and demography: an analytical review. J Appl Genetics. 2014;55:319–327. doi: 10.1007/s13353-014-0203-3. [DOI] [PubMed] [Google Scholar]
- 29.la Cruz De O, Raska P. Population structure at different minor allele frequency levels. BMC Proceedings. 2014;8:S55. doi: 10.1186/1753-6561-8-S1-S55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henn BM, Gravel S, Moreno-Estrada A, Acevedo-Acevedo S, Bustamante CD. Fine-scale population structure and the era of next-generation sequencing. Human Molecular Genetics. 2010;19:R221–R226. doi: 10.1093/hmg/ddq403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathieson I, McVean G. Demography and the Age of Rare Variants. PLoS Genet. 2014;10:e1004528. doi: 10.1371/journal.pgen.1004528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshpande O, Batzoglou S, Feldman MW, Luca Cavalli-Sforza L. A serial founder effect model for human settlement out of Africa. Proceedings of the Royal Society B: Biological Sciences. 2009;276:291–300. doi: 10.1098/rspb.2008.0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Consortium, T. 1. G. P et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2013;490:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeGiorgio M, Jakobsson M, Rosenberg NA. Explaining worldwide patterns of human genetic variation using a coalescent-based serial founder model of migration outward from Africa. Proceedings of the National Academy of Sciences. 2009;106:16057–16062. doi: 10.1073/pnas.0903341106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson MR, et al. An Abundance of Rare Functional Variants in 202 Drug Target Genes Sequenced in 14,002 People. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tennessen JA, et al. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goode DL, et al. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Research. 2010;20:301–310. doi: 10.1101/gr.102210.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacArthur DG, et al. A Systematic Survey of Loss-of-Function Variants in Human Protein-Coding Genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? The American Journal of Human Genetics. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agarwala V, Flannick J, Sunyaev S, Altshuler D. AgarwalaNatGenet2013. Nature Publishing Group. 2013;45:1418–1427. doi: 10.1038/ng.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson G. Rare and common variants: twenty arguments. Nature Reviews Genetics. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maher MC, Uricchio LH, Torgerson DG, Hernandez RD. Population Genetics of Rare Variants and Complex Diseases. Hum Hered. 2012;74:118–128. doi: 10.1159/000346826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klopfstein S. The Fate of Mutations Surfing on the Wave of a Range Expansion. Mol Bio Evol. 2005;23:482–490. doi: 10.1093/molbev/msj057. [DOI] [PubMed] [Google Scholar]
- 44.Flaxman SM. Surfing downhill: when should population range expansion be characterized by reductions in fitness? Molecular Ecology. 2013;22:5963–5965. doi: 10.1111/mec.12564. [DOI] [PubMed] [Google Scholar]
- 45.Boyko AR, et al. Assessing the Evolutionary Impact of Amino Acid Mutations in the Human Genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keinan A, Clark AG. Recent Explosive Human Population Growth Has Resulted in an Excess of Rare Genetic Variants. Science. 2012;336:740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lohmueller KE, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simons YB, Turchin MC, Pritchard JK, Sella G. The deleterious mutation load is insensitive to recent population history. Nature Genetics. 2014;46:220–224. doi: 10.1038/ng.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casals F, et al. Whole-Exome Sequencing Reveals a Rapid Change in the Frequency of Rare Functional Variants in a Founding Population of Humans. PLoS Genet. 2013;9:e1003815. doi: 10.1371/journal.pgen.1003815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Do R, et al. No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nature Publishing Group. 2015 doi: 10.1038/ng.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu W, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu W, Gittelman RM, Bamshad MJ, Akey JM. Characteristics of Neutral and Deleterious Protein-Coding Variationamong Individuals and Populations. The American Journal of Human Genetics. 2014;95:421–436. doi: 10.1016/j.ajhg.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim ET, et al. Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sajantila A, et al. Paternal and maternal DNA lineages reveal a bottleneck in the founding of the Finnish population. Proceedings of the National Academy of Sciences. 1996;93:12035–12039. doi: 10.1073/pnas.93.21.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gravel S, et al. Demographic history and rare allele sharing among human populations. Proceedings of the National Academy of Sciences. 2011;108:11983–11988. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyer M, et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the Joint Demographic History of Multiple Populations from Multidimensional SNP Frequency Data. PLoS Genet. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henn BM, et al. Hunter-gatherer genomic diversity suggests a southern African origin for modern humans. Proceedings of the National Academy of Sciences. 2011;108:5154–5162. doi: 10.1073/pnas.1017511108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lohmueller KE. The Impact of Population Demography and Selection on the Genetic Architecture of Complex Traits. PLoS Genet. 2014;10:e1004379. doi: 10.1371/journal.pgen.1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramachandran S, et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proceedings of the National Academy of Sciences. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sousa V, Peischl S, Excoffier L. Impact of range expansions on current human genomic diversity. Current Opinion in Genetics & Development. 2014;29:22–30. doi: 10.1016/j.gde.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 63.Moreau C, et al. Deep Human Genealogies Reveal a Selective Advantage to Be on an Expanding Wave Front. Science. 2011;334:1148–1150. doi: 10.1126/science.1212880. [DOI] [PubMed] [Google Scholar]
- 64.Peischl S, Dupanloup I, Kirkpatrick M, Excoffier L. On the accumulation of deleterious mutations during range expansions. Molecular Ecology. 2013;22:5972–5982. doi: 10.1111/mec.12524. [DOI] [PubMed] [Google Scholar]
- 65.Coventry A, et al. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nature Communications. 2010;1:131–6. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gignoux CR, Henn BM, Mountain JL. Rapid, global demographic expansions after the origins of agriculture. Proceedings of the National Academy of Sciences. 2011;108:6044–6049. doi: 10.1073/pnas.0914274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng HX, Yan S, Qin ZD, Jin L. MtDNA analysis of global populations support that major population expansions began before Neolithic Time. Sci Rep. 2012;2 doi: 10.1038/srep00745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forster P. Ice Ages and the mitochondrial DNA chronology of human dispersals: a review. Philosophical Transactions of the Royal Society B: Biological Sciences. 2004;359:255–264. doi: 10.1098/rstb.2003.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gazave E, Chang D, Clark AG, Keinan A. Population growth inflates the per-individual number of deleterious mutations and reduces their mean effect. Genetics. 2013 doi: 10.1534/genetics.113.153973/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kamberov YG, et al. Modeling Recent Human Evolution in Mice by Expression of a Selected EDAR Variant. Cell. 2013;152:691–702. doi: 10.1016/j.cell.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hernandez RD, et al. Classic Selective Sweeps Were Rare in Recent Human Evolution. Science. 2011;331:920–924. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moschovis PP, et al. Childhood Anemia at High Altitude: Risk Factors for Poor Outcomes in Severe Pneumonia. PEDIATRICS. 2013;132:e1156–e1162. doi: 10.1542/peds.2013-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whitlock MC, Bourguet D. Factors affecting the genetic load in Drosophila: synergistic epistasis and correlations among fitness components. Evolution. 2000;54:1654–1660. doi: 10.1111/j.0014-3820.2000.tb00709.x. [DOI] [PubMed] [Google Scholar]
- 74.Fry JD. On the rate and linearity of viability declines in Drosophila mutation-accumulation experiments: genomic mutation rates and synergistic epistasis revisited. Genetics. 2004;166:797–806. doi: 10.1534/genetics.166.2.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arbiza L, et al. Genome-wide inference of natural selection on human transcription factor binding sites. Nature Publishing Group. 2013;45:723–729. doi: 10.1038/ng.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lohmueller KE. The distribution of deleterious genetic variation in human populations. Current Opinion in Genetics & Development. 2014;29:139–146. doi: 10.1016/j.gde.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 77.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Meth. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Meth. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 79.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 81.Ng PC. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Research. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siepel A, Pollard KS, Haussler D. New methods for detecting lineage-specific selection. 2006:190–205. [Google Scholar]
- 85.Gulko B, Hubisz MJ, Gronau I, Siepel A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nature Publishing Group. 2015 doi: 10.1038/ng.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.