

Abstract
Studying the environmental occurrence of parasites of concern for humans and animals based on coprosamples is an expanding field of work in epidemiology and the ecology of health. Detecting and quantifying Toxocara spp. and Echinococcus multilocularis, two predominant zoonotic helminths circulating in European carnivores, in feces may help to better target measures for prevention. A rapid, sensitive, and one-step quantitative PCR (qPCR) allowing detection of E. multilocularis and Toxocara spp. was developed in the present study, combined with a host fecal test based on the identification of three carnivores (red fox, dog, and cat) involved in the life cycles of these parasites. A total of 68 coprosamples were collected from identified specimens from Vulpes vulpes, Canis lupus familiaris, Canis lupus, Felis silvestris catus, Meles meles, Martes foina, and Martes martes. With DNA coprosamples, real-time PCR was performed in duplex with a qPCR inhibitor control specifically designed for this study. All the coprosample host identifications were confirmed by qPCR combined with sequencing, and parasites were detected and confirmed (E. multilocularis in red foxes and Toxocara cati in cats; 16% of samples presented inhibition). By combining parasite detection and quantification, the host fecal test, and a new qPCR inhibitor control, we created a technique with a high sensitivity that may considerably improve environmental studies of pathogens.
INTRODUCTION
For detection of infectious agents in the environment, which involves both wild and domestic animals, coprosamples are noninvasive and valuable isolates, as opposed to necropsy, which is time-consuming, logistically onerous, and often considered unethical by the public. In this field of work, Echinococcus multilocularis is the causative agent of alveolar echinococcosis, one of the most worrying zoonoses in the Northern Hemisphere (1), and is in constant progression (2–4). Toxocara spp. are the causative agents of toxocariasis, a widespread disease that is still described in known areas of endemicity despite relevant anthelminthic programs (5). Both E. multilocularis and Toxocara spp. are zoonotic agents involved in a fecal-oral transmission cycle from carnivores to humans, with no vectors or intermediate actors (5). These helminth parasites are both found in at least one wild carnivore (red fox) and two domestic carnivore (dog and cat) host species. While E. multilocularis is a common tapeworm of the red fox, low parasite prevalences have often been described for European domestic host species (cat and dog) (6–8). E. multilocularis eggs are highly resistant to environments with low temperatures and high percentages of relative humidity. Under controlled laboratory conditions, the eggs were found to survive in water for 478 days at 4°C. Under these experimental conditions, eggs remained infectious for rodents (9). Carnivores become infected after predation of contaminated rodents and release eggs into the environment via their feces. Alveolar echinococcosis in humans remains a lethal disease in untreated cases (10). In contrast, Toxocara cati is found mainly in cats, and Toxocara canis is found in dogs. The role of the fox in spreading this parasite was previously thought to be minor in comparison to that of the dog. However, in light of recent evidence, the fox's role should now be reconsidered (11). It takes Toxocara eggs between 3 weeks and several months to reach the infective stage in the environment, and they can remain infectious for several years (12). Carnivores can be infected by predation on contaminated small mammals (rodents or rabbits) or by transplacental and transmammary routes to puppies and kittens. Humans can be contaminated by ingesting eggs on raw vegetables, by having contact with soil and failing to wash their hands, or through larvae in undercooked meat (13). Both Toxocara species can be involved in the disease affecting humans (13). Although it can resolve spontaneously, some rare complications can develop from ocular larva migrans, which may lead to irreversible blindness, neurological symptoms (14), and related allergies and asthma (15, 16). Because of the time necessary for Toxocara eggs to mature outside the definitive host and the fact that carnivore stools remain in the environment for extended periods, this source of contamination for humans and animals is clearly of greater concern than contact with contaminated fur (5).
The use of coprosamples to study the environmental occurrence of these parasites of concern for humans and animals is an expanding field of work in epidemiology and health ecology. This noninvasive method is based on collection of feces. Small carnivore feces can easily be discriminated from larger carnivore feces by size, shape, and content, but correctly identifying the species often requires the opinion of specialists (17). Even experts may find it difficult to visually differentiate between species and therefore misidentify them, especially when feces come from species having similar morphological characteristics (18). Correct identification is particularly important when it is used to diagnose pathogen infection in order to avoid drawing inappropriate epidemiological conclusions. In order to confirm the morphological identification of host fecal samples, PCR techniques based on the amplification and sequencing of mitochondrial DNA (mtDNA) were developed (19, 20). Total DNA extracted from stools is analyzed by PCR or real-time quantitative PCR (qPCR). Thanks to these advanced techniques, zoonotic parasites such as Toxocara spp. (21) and Echinococcus multilocularis (22, 23) can be detected and quantified in stools. Real-time nested PCR using fluorescence resonance energy transfer (FRET) probes was designed to detect and discriminate between different host species by melting curve analysis for field studies focusing on Echinococcus multilocularis investigations and the carnivore hosts Vulpes vulpes (red fox), Vulpes ferrilata (Tibetan fox), and Canis lupus familiaris/Canis lupus (domestic dog/wolf) (22). However, in field studies dealing with helminth parasite detection and quantification, a larger panel of wild and domestic carnivores needs to be taken into account.
E. multilocularis DNAs in fox stools were quantified by a qPCR targeting a mitochondrial marker (23). However, to date, very few sensitive tools have been developed to describe environmental contamination by Toxocara spp. (21) and to assess the respective roles of their definitive hosts in spreading these organisms.
The aim of the present study was to develop a rapid, sensitive, and one-step qPCR combining carnivore feces identification and E. multilocularis and Toxocara species detection. We first developed a Toxocara species-specific qPCR assay (Toxocara qPCR) and improved a previously developed E. multilocularis qPCR, and then we combined these two qPCR assays with a new host fecal test based on the identification of three carnivores involved in the life cycles of these parasites (red fox, dog, and cat). All these tools used quantitative real-time PCR technologies with hydrolysis.
MATERIALS AND METHODS
Collection of host and parasite samples.
The molecular identification of host feces first focused on the three species involved in the life cycles of E. multilocularis and Toxocara spp. (Vulpes vulpes, C. l. familiaris, and Felis silvestris catus) and then on two carnivores for differential diagnosis (Meles meles and Martes foina [badger and stone marten, respectively]). A collection of tissue samples and coprosamples was created from captive animals (CA) and road kill animals (animals killed in road accidents; RA).
The collection of tissue samples included samples from Vulpes vulpes-CA (muscle), C. l. familiaris-CA (testis), F. s. catus-CA (ear tip), Meles meles-RA (liver), and Martes foina-RA (liver), which were maintained in 70% (vol/vol) ethanol before use. DNAs were extracted with a High Pure PCR template preparation kit (Roche Diagnostics, Penzberg, Germany) as recommended by the manufacturer and were maintained at −20°C until use.
In order to test the usefulness of the qPCRs in the field, a collection of 68 coprosamples was used and was made up of 13 Vulpes vulpes-CA, 13 Canis lupus familiaris-CA, 13 C. lupus-CA, 17 F. s. catus-CA, 9 Meles meles-CA, 2 Martes foina-RA, and 1 Martes martes-RA (pine marten) sample (Table 1). The samples were placed at −80°C for 7 days for parasite inactivation and then maintained at −20°C until use. Total DNA was extracted with a QIAamp Fast DNA stool minikit (Qiagen, Hilden, Germany) as recommended by the manufacturer. Modifications were made as previously described by Knapp et al. (23), using 500 mg of each coprosample. Briefly, proteins were digested, DNA was bound to a QIAamp silica membrane, and impurities were washed away by using InhibitEX solution. Elution was performed in 200 μl of elution buffer, and extracts were maintained at −20°C until use.
TABLE 1.
Carnivore coprosample collection and detection of host DNA and Toxocara sp. and E. multilocularis parasites by qPCRe
| Carnivore species | Sample code | Other animal(s) presentb |
Cq value |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V. vulpes | C. lupus | F. catus | M. meles | M. foina | Toxocara | E. multilocularis | Toxo/Alea | Em/Alea | |||
| Vulpes vulpes | Em2736 | None | 25.8 | ND | ND | ND | ND | ND | 35.6 | 31.6 | 34.0 |
| Em2711a | None | 24.5 | ND | ND | ND | ND | ND | 26.3 | 32.1 | 34.1 | |
| Em2724 | None | 28.8 | ND | ND | ND | ND | ND | 39.5 | 33.4 | 34.1 | |
| Em2713 | None | 27.7 | 38.3 | 39.6c | ND | ND | ND | ND | 31.6 | 33.5 | |
| Em2603a | None | 18.1 | ND | ND | ND | ND | ND | 30.8 | 32.0 | 33.5 | |
| Em2416 | None | 17.9 | ND | ND | ND | ND | ND | 33.6 | 32.0 | 33.7 | |
| Em2714 | None | 25.5 | ND | ND | ND | ND | ND | 40.0 | 36.9 | 34.5 | |
| Em2738 | None | 25.4 | ND | ND | ND | ND | ND | 32.7 | 32.1 | 33.9 | |
| Em2692 | None | 24.9 | ND | ND | ND | ND | ND | 34.6 | 32.0 | 32.9 | |
| Em2888 | None | 21.4 | ND | ND | ND | ND | ND | ND | 32.5 | 33.4 | |
| Em2254 | None | 16.3 | ND | ND | ND | ND | ND | 31.5 | 33.6 | 35.0 | |
| Em2257 | None | 26.9 | ND | ND | ND | ND | ND | ND | 32.7 | 33.8 | |
| Em2562 | None | 17.2 | ND | ND | ND | ND | ND | 33.9 | 31.8 | 32.8 | |
| Canis lupus familiaris | YAYa | Cat/dog | ND | 22.0 | 33.3 | ND | ND | ND | ND | 32.9 | 35.0 |
| TIT | Cat/dog | ND | 20.2 | 37.7 | ND | ND | ND | ND | 34.3 | 36.2 | |
| IZIa | Cat | ND | 26.8 | ND | ND | ND | ND | ND | 35.3 | 37.8 | |
| 5108 | 2 dogs | ND | 23.3 | ND | ND | ND | ND | ND | 32.7 | 36.3 | |
| 5101 | None | ND | 21.6 | ND | ND | ND | ND | ND | 31.8 | 33.8 | |
| 5111 | Dog/cat | ND | 25.2 | ND | ND | ND | ND | ND | 34.4 | 37.6 | |
| 5065 | None | ND | 17.9 | ND | ND | ND | ND | ND | 32.2 | 34.4 | |
| 5077 | 3 dogs | ND | 20.2 | ND | ND | ND | ND | ND | 32.9 | 34.0 | |
| 5107 | 2 dogs | 42.0c | 24.0 | ND | ND | ND | ND | ND | 33.8 | 35.5 | |
| 5114 | None | ND | 20.9 | ND | ND | ND | ND | ND | 32.1 | 34.0 | |
| 5104 | None | ND | 22.0 | ND | ND | ND | ND | ND | 35.0 | 35.0 | |
| 5069 | None | ND | 25.4 | ND | ND | ND | ND | ND | 33.6 | 34.3 | |
| 5110 | None | ND | 26.2 | ND | ND | ND | ND | ND | 33.1 | 34.4 | |
| Canis lupus | 6850 | None | ND | 27.1 | ND | ND | ND | ND | ND | 33.9 | 35.2 |
| 6851 | None | ND | 25.1 | ND | ND | ND | ND | ND | 34.3 | 36.4 | |
| 6852a | None | ND | 21.4 | ND | ND | ND | ND | ND | 32.5 | 34.0 | |
| 6853 | None | ND | 27.9 | ND | ND | ND | ND | ND | 34.1 | 37.7 | |
| 6854 | None | ND | 26.5 | ND | ND | ND | ND | ND | 34.3 | 39.0 | |
| 6855 | None | ND | 27.8 | ND | ND | ND | ND | ND | 35.4 | 38.2 | |
| 6856 | None | ND | 26.4 | ND | ND | ND | ND | ND | 34.6 | 36.9 | |
| 6857 | None | ND | 28.7 | ND | ND | ND | ND | ND | 34.3 | 37.9 | |
| 6858d | None | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| 6865 | None | ND | 24.6 | ND | ND | ND | ND | ND | 32.9 | 34.2 | |
| 5670 | None | ND | 23.6 | ND | ND | ND | ND | ND | 33.1 | 34.9 | |
| 4861 | None | ND | 27.4 | ND | ND | ND | ND | ND | 35.7 | 37.9 | |
| 5667 | None | ND | 22.6 | ND | ND | ND | ND | ND | 32.6 | 34.1 | |
| Felis silvestris catus | POUa | Dog | ND | 33.6 | 25.3 | ND | ND | ND | ND | 36.6 | 37.8 |
| ESTa | None | ND | ND | 22.6 | ND | ND | ND | ND | 32.2 | 34.2 | |
| NOIa | Dog | ND | 34.4 | 23.5 | ND | ND | ND | ND | 33.3 | 34.2 | |
| ZIZa | None | ND | 34.1 | 22.4 | ND | ND | ND | ND | 33.3 | 35.5 | |
| GAM | ND | ND | 21.6 | ND | ND | ND | ND | 32.7 | 32.5 | ||
| PRI | ND | ND | 24.4 | ND | ND | 32.2 | ND | 33.7 | 35.6 | ||
| OSI | ND | 37.5c | 23.1 | ND | ND | 32.9 | ND | 31.6 | 33.0 | ||
| MIN | ND | ND | 21.7 | ND | ND | 32.1 | ND | 31.8 | 34.0 | ||
| EM5621 | None | ND | ND | 31.8 | ND | ND | ND | ND | 35.3 | 38.1 | |
| EM5212 | None | ND | ND | 27.9 | ND | ND | ND | ND | 35.4 | 37.8 | |
| EM5131 | None | ND | ND | 31.0 | ND | ND | ND | ND | 36.8 | 34.4 | |
| EM5191 | None | ND | ND | 30.4 | ND | ND | ND | ND | 35.2 | 35.0 | |
| EM5368 | None | ND | ND | 25.1 | ND | ND | ND | 40.2 | 35.5 | 37.6 | |
| EM5540 | Cat | ND | ND | 28.7 | ND | ND | ND | ND | 32.3 | 34.5 | |
| EM5536 | None | ND | ND | 21.0 | ND | ND | ND | ND | 34.1 | 36.5 | |
| EM5072 | Cat | ND | ND | 25.9 | ND | ND | ND | ND | 32.2 | 34.9 | |
| EM5018 | None | ND | 37.0c | 26.8 | ND | ND | ND | ND | 31.8 | 32.9 | |
| Meles meles | B1 | None | ND | ND | ND | 27.9 | ND | ND | ND | 31.7 | 33.7 |
| B2a | None | ND | ND | ND | 24.5 | ND | ND | ND | 35.6 | 34.8 | |
| B3d | None | ND | ND | ND | 32.4 | ND | ND | ND | 37.2 | ND | |
| B4 | None | ND | ND | ND | 23.5 | ND | ND | ND | 32.6 | 34.5 | |
| B5 | None | ND | ND | ND | 23.0 | ND | ND | ND | 30.9 | 33.1 | |
| B6 | None | ND | ND | ND | 24.1 | ND | ND | ND | 36.0 | 37.9 | |
| B7 | None | ND | ND | ND | 25.7 | ND | ND | ND | 36.1 | 38.1 | |
| B8 | None | ND | ND | ND | 23.0 | ND | ND | ND | 31.5 | 34.0 | |
| B9 | None | ND | ND | ND | 27.0 | ND | ND | ND | 35.8 | 35.9 | |
| Martes foina | F1a | None | ND | ND | ND | ND | 22.5 | ND | ND | 32.5 | 35.3 |
| F2a | Cat/dog | ND | 40.1c | 39.7c | ND | 30.0 | ND | 30.2 | 33.9 | 34.2 | |
| Martes martes | M1a | None | ND | ND | 37.5c | ND | 41.8 | ND | ND | 32.0 | 34.0 |
Specimens sequenced by SEQ-PCR for host identification.
Other animals present are described for domestic animals living with a different domestic animal(s) in their neighborhood.
qPCR was positive once for qPCR performed in duplicate.
Samples were removed from the interpretation of the analyses.
ND, no amplification was obtained or more than 45 cycles were required. Underlining indicates samples with another carnivore DNA identified in the extract by sequencing.
In order to test the specificity of the Toxocara qPCR, a collection of DNAs from parasites present in canines, felids, both, or neither was used and was made up of 2 nematodes (2 specimens of Toxocara cati and 9 specimens of Toxocara canis), 10 cestodes (E. multilocularis, Echinococcus granulosus sensu stricto, Taenia hydatigena, Taenia crassiceps, Taenia serialis, Taenia pisiformis, Taenia multiceps, Taenia polyacantha, Hydatigera taeniaeformis, and Mesocestoides sp.), and 1 trematode (Alaria sp.). DNAs were extracted from adult worms as described above by use of a High Pure PCR template preparation kit (Roche Diagnostics, Penzberg, Germany), and all specimens were tested by qPCR at a DNA concentration of 5 ng/μl.
Internal control for copro-qPCR.
To identify the presence of PCR inhibitors, an internal control was developed. A nucleotide sequence of 167 bp, named Alea, was randomly designed by use of a random DNA sequence generator program available online (http://www.faculty.ucr.edu/∼mmaduro/random.htm) (Table 2). A comparison was made by using online genetic databases and the Basic Local Alignment Search Tool (BLAST) on the NCBI website to confirm that this nucleotide sequence was not in the NCBI genetic database. The hydrolysis probe and primers were designed by use of Primer Express 3.0 and were designed to specifically amplify the sequence by real-time PCR (Table 2). The nucleotide sequence was generated by GeneCust Services (Dudelange, Luxembourg). The sequence was inserted into plasmid pET-11aH6 (24) and transformed into Escherichia coli DH5α. Plasmids were purified using a QIAfilter Plasmid Midi kit (Qiagen, Hilden, Germany) as recommended by the manufacturer. The elution step was performed with Tris-EDTA buffer, pH 8.0 (Sigma-Aldrich, St. Louis, MO). A nanophotometer apparatus (Implen, Munich, Germany) was used to measure DNA concentration. The Alea internal control was calibrated in terms of plasmid number in order to obtain the best concentration of plasmids to detect PCR inhibitors with good repeatability. The plasmid DNA range was set from 100 copies/μl (6 × 10−7 ng/μl) to 100,000 copies/μl (6 × 10−4 ng/μl). For each plasmid concentration point, qPCR was performed seven times in order to check the repeatability.
TABLE 2.
Sequences of PCR inhibitor internal control, primers, and hydrolysis probe for qPCR
| Component | Nucleotide sequence (5′–3′) |
|---|---|
| Alea sequence | GTTCCATGGAAATGCCACCCCGAAGAAACCGCCTAAAAATGTCTATGATTGGTCCACTAAAGTTGATTAAATCAACTCCTAAATCCGCGCGATAGGGCATTAGAGGTTTAATTTTGTATGGCAAGGTACTCCCGATCTTAATGAATGGCCGGAAGTGGTGGATCCTT |
| Alea forward primer | CCTAAAAATGTCTATGATTGGTCCACTA |
| Alea reverse primer | GGGAGTACCTTGCCATACAAAATT |
| Alea probe | VIC-TTAAATCAACTCCTAAATCCGCGCGATAGG-TAMRAa |
TAMRA, 6-carboxytetramethylrhodamine.
qPCR assays for parasite detection and host identification.
qPCR was used to detect and quantify Toxocara sp. and E. multilocularis parasites and their hosts. All primers and hydrolysis probes were designed using Primer Express 3.0 and the target gene sequences (Tables 3 and 4). Duplex qPCRs were designed to optimize the analysis yield. The duplexes were defined as follows: Em/Alea, E. multilocularis and Alea; Toxo/Alea, Toxocara spp. and Alea; Vv/Cf, V. vulpes and C. l. familiaris; Fc/Alea, F. s. catus and Alea; and Mf/Mm, M. foina and M. meles. Each PCR assay was performed in a final volume of 20 μl, containing 10 μl of 2× TaqMan Gene Expression master mix (Life Technologies, Foster City, CA), 5 pmol of each primer, 0.4 pmol of the hydrolysis probes with compatible fluorochromes, and 0.2 μl of water to obtain 15 μl of mixture volume, with 5 μl of total extracted copro-DNA added to this mixture. The qPCR was run on a model 7500 Fast real-time PCR system (Life Technologies, Foster City, CA). The PCR program was the same as that used to detect E. multilocularis in fox stools by qPCR with 45 cycles, designed by Knapp and coworkers (23). All PCRs were performed in duplicate, and results were expressed as quantitative cycle (Cq) numbers.
TABLE 3.
Toxocara sp. and Echinococcus multilocularis primers and hydrolysis probes for qPCR, designed for a partial sequence of the cox1 gene of Toxocara cati and for the rrnL gene of E. multilocularis
| Host species | Primer or probe | Oligonucleotide sequence (5′–3′) | Reference |
|---|---|---|---|
| Toxocara spp. | Tox forward primer | AAAATAGCCAAATCCACACTACTACCA | This study |
| Tox reverse primer | GGTGTGGGACTAGTTGAACTGTGTA | This study | |
| Tox probe | FAM-CCCCATAGTCCTCAAAG-MGB | This study | |
| Tox forward primer (SEQ-PCR) | TGGATGTTACCTTTGATGTTGGG | This study | |
| E. multilocularis | rrn-Em forward primer | CTGTGATCTTGGTGTAGTAGTTGAGATTT | 23 |
| rrn-Em reverse primer | GGCTTACGCCGGTCTTAACTC | 23 | |
| rrn-Em probe | FAM-TGGTCTGTTCGACCTTTTTAGCCTCCAT-TAMRA | 23 | |
| rrn-Em reverse primer (SEQ-PCR) | GGGGTCAATCACAACAACCC | This study |
TABLE 4.
Host fecal test primers and probes for qPCR identification of red fox (Vulpes vulpes), domestic dog (Canis lupus familiaris), and domestic cat (Felis silvestris catus), designed for a fragment of the cytB gene, and for qPCR identification of stone marten (Martes foina) and badger (Meles meles), designed for a fragment of the cox1 gene
| Host species | Primer or probe | Oligonucleotide sequence (5′–3′) |
|---|---|---|
| Vulpes vulpes | Vv forward primer | ACCTTCCCGCACCATCAAA |
| Vv reverse primer | TGTTGCAATCTGTAGAATAAGGCATA | |
| Vv probe | FAM-CTGCCTGATGGAACTTCGGGTCCC-TAMRA | |
| Vv reverse primer (SEQ-PCR) | GCCATAGTTAACGTCTCGGC | |
| Canis lupus familiaris | Cf forward primer | CCACCCACTAGCCAAAATTGTT |
| Cf reverse primer | AAGTTCCATCAAGCAGAGATGTTAGA | |
| Cf probe | VIC-ATAACTCATTCATTGACCTCCCAGCGCC-TAMRA | |
| Cf reverse primer (SEQ-PCR) | TGTGGCTGTGTCCGATGTAT | |
| Felis silvestris catus | Fc forward primer | CCCTTCTAGGAGTCTGCCTAATCTT |
| Fc reverse primer | CGGTTATTGTGTCTGATGTGTAGTGT | |
| Fc probe | FAM-AAATCCTCACCGGCCTCTTTTTGGC-TAMRA | |
| Fc reverse primer (SEQ-PCR) | TTCCCCGTCCCACATGTATG | |
| Martes foina | Mf forward primer | CCTCAACATCATCACCTTTCAAAA |
| Mf reverse primer | GCGCTTTCATTGTAGGTTTATTGTC | |
| Mf probe | FAM-TAACAAGCAGTCAATAGCT-MGB | |
| Mf reverse primer (SEQ-PCR) | TGCTGTTGGTTGTGGGATTG | |
| Meles meles | Mm forward primer | CCTCAACATCATCTCCCTTCAAG |
| Mm reverse primer | GAGCTTTTGTTGTTGGTTTATTGTCT | |
| Mm probe | VIC-ATAGCAAGCCATCAAT-MGB | |
| Mm reverse primer (SEQ-PCR) | GCTGTTGGTTGTGGGATTGT |
The Alea internal control allowed the detection of PCR inhibitors. Because of the different fluorochromes used, duplex PCRs were possible between the Alea control (probe labeled with VIC dye) and the parasite (probe labeled with 6-carboxyfluorescein [FAM] dye) or carnivore. For this duplex qPCR, water in the mixture preparation was replaced by an Alea solution to obtain 15 μl of mixture volume. The Alea qPCR products were 102 bp long.
Primers and a hydrolysis probe were designed from the complete mitochondrial genome of Toxocara cati (GenBank accession number AM411622.1) and from part of the mitochondrial cytochrome c oxidase subunit 1 (cox1) gene (Table 3). Primer specificity was tested by using the NCBI Primer BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). The qPCR products were 79 bp long. A minor groove binder (MGB) was coupled with the hydrolysis probe in order to increase the melting temperature (Tm) of a shorter sequence, thus improving specificity. For qPCR calibration, total DNA was extracted from adult T. cati parasites. The DNA concentration was checked using a nanophotometer (Implen, Munich, Germany). In order to test the method detection limits (MDLs) or the probability of successfully detecting n positive results out of N trials for Toxocara spp. with the Toxo/Alea qPCR, a dilution series was made to obtain a DNA range with 10 concentration points, from 5 to 5 × 10−9 ng/μl. Technical limits were assessed by performing the PCR on each point of the DNA range seven times. The last point of the DNA range presenting seven positive PCRs in seven trials was considered to be the MDL for T. cati DNA detection (25). The sensitivity of Toxocara qPCR was tested under a microscope by use of a range of isolated eggs from a T. cati adult female worm. DNAs were extracted from isolated eggs (3 extracts of 1, 5, and 10 eggs) as described above by use of a High Pure PCR template preparation kit (Roche Diagnostics, Penzberg, Germany). DNA was eluted in 100 μl of the elution buffer provided in the kit. Toxocara qPCR was performed in triplicate on each egg DNA extract.
The previously developed qPCR for E. multilocularis detection and quantification (23) was tested in this study in duplex with the Alea target (Em/Alea qPCR). The marker was designed from part of the mitochondrial rrnL gene. In order to test the MDLs of the qPCR performed in duplex, DNA was obtained from an in vitro culture of E. multilocularis, and a dilution series was performed to obtain a DNA range with seven concentration points, from 5 to 5 × 10−6 ng/μl. Technical limits (MDLs) were assessed by performing the PCR on each point of the DNA range seven times. The last point of the DNA range presenting seven positive PCR results was considered the MDL for E. multilocularis DNA analysis (25). The primers and hydrolysis probe employed in the present study were described previously (23) and are reported in Table 3.
To check the morphological identification of host feces determined in the field, qPCR was performed. For V. vulpes, C. l. familiaris, and F. s. catus, primers and TaqMan probes were designed from part of the mitochondrial cytochrome b gene (cytB), and for M. meles and M. foina, primers and probes were designed from part of the cox1 gene (Table 4). The qPCR products were 83 bp long for V. vulpes, 78 bp for C. l. familiaris, 81 bp for F. s. catus, 72 bp for M. meles, and 72 bp for M. foina. Primer specificity was tested using the NCBI Primer BLAST tool.
Sequencing for confirmation of host and parasite identifications.
To confirm the molecular screening for host identification and the presence of the parasite(s) targeted, sequencing was performed on qPCR products. For this purpose, sequencing-qPCR (SEQ-PCR) was carried out. The forward primer and the probe designed for the screening were used with a new reverse primer for each target, except for Toxocara spp. (a new forward primer combined with the qPCR reverse primer was used in this case) (Tables 3 and 4). SEQ-PCR was performed to obtain longer qPCR products. The SEQ-PCR product size was 202 bp for E. multilocularis (Em-SEQ-PCR), 200 bp for Toxocara spp. (Toxo-SEQ-PCR), 170 bp for V. vulpes (Vv-SEQ-PCR), 163 bp for C. l. familiaris (Cf-SEQ-PCR), 201 bp for F. s. catus (Fc-SEQ-PCR), 232 bp for M. meles (Mm-SEQ-PCR), and 233 bp for M. foina (Mf-SEQ-PCR). Forward and reverse strands of qPCR products were sequenced using the Sanger method (26), and detection was performed on a model 3130 Applied Biosystems genetic analyzer (Life Technologies, Carlsbad, CA). Sequences were aligned using BioEdit 7.0.9 (27) and nucleotide databases for species identification.
RESULTS
Internal control Alea.
Alea qPCR was tested in simplex over a plasmid DNA range with four concentration points from 6 × 10−4 to 6 × 10−7 ng/μl, ranging from 100,000 down to 100 plasmids/μl. The average Cq for each point of the Alea DNA range performed seven times was obtained (for 6 × 10−4 ng/μl, Cq = 28.23 [standard deviation {SD} = 0.17]; for 6 × 10−5 ng/μl, Cq = 31.27 [SD = 0.21]; for 6 × 10−6 ng/μl, Cq = 34.18 [SD = 0.06]; and for 6 × 10−7 ng/μl, Cq = 35.28 [SD = 0.44]). A suitable balance was found between the use of an inhibition control with an acceptable variation coefficient and the detection of an early decrease in PCR efficacy. The point of 6 × 10−6 ng/μl (1,000 plasmids/μl), with a Cq of 35, was thus retained for testing of the inhibition control. The control was tested on the panel of 68 coprosamples, and the average Cq for Alea at 6 × 10−6 ng/μl mixed with copro-DNA extracts was 33.5 (SD = 1.6) for Toxo/Alea qPCR and 35.1 (SD = 1.7) for in Em/Alea qPCR. For the Alea marker, a linear correlation coefficient (r) was calculated for the results of the qPCR duplexes Toxo/Alea and Em/Alea obtained with the sample panel. The r value was 0.83. The threshold of detection for PCR inhibitor presence was therefore a Cq of 35 for Toxo/Alea qPCR and a Cq of 37 for Em/Alea qPCR. Based on these thresholds, 20 samples presented inhibition, with 11 showing inhibition in both the Toxo/Alea and Em/Alea qPCRs (Table 1).
Toxocara sp. detection by qPCR.
With Toxocara qPCR, no amplification was observed from available parasite DNAs in the laboratory, except for the T. canis and T. cati specimens. The specimens of the two species (9 specimens of T. canis, with an average Cq for Toxocara qPCR of 24.1 [SD = 3.2], and 2 specimens of T. cati, with Cq values of 18.0 and 22.2) were sequenced by using the SEQ-PCR products (Fig. 1). Six mutations allowed us to discriminate T. cati from T. canis in a 111-bp aligned sequence. Polymorphisms were found among the two species and reference sequences. Primer specificity was tested online, and the primer set was described only for T. cati for the combined forward and reverse primer couple. In comparison to T. canis, one mutation was found in the forward primers (for qPCR and SEQ-PCR) and three were found in the reverse primer, whereas all T. canis adult specimens were amplified.
FIG 1.

Sequence alignment (partial cox1 gene) of 111-bp sequences from Toxocara cati and T. canis reference specimens, adult worms from the laboratory collection (T. canis, 9 specimens; and T. cati, 2 specimens), and three positive samples from the present coprosample collection.
The average Cq for each point of the T. cati DNA range tested seven times in duplex (Toxo/Alea qPCR) was obtained for the seven points from 5 to 5 × 10−6 ng/μl (for 5 ng/μl, Cq = 17.52 [SD = 0.09]; for 0.5 ng/μl, Cq = 21.13 [0.14]; for 0.05 ng/μl, Cq = 24.83 [0.18]; for 5 × 10−3 ng/μl, Cq = 28.04 [0.13]; for 5 × 10−4 ng/μl, Cq = 31.48 [0.07]; for 5 × 10−5 ng/μl, Cq = 35.26 [0.23]; and for 5 × 10−6 ng/μl, Cq = 39.14 [0.58]). The points of the DNA range of 5 × 10−7 and 5 × 10−8 ng/μl were amplified in 2/7 and 1/7 PCRs, respectively, and the point of 5 × 10−9 ng/μl was negative for the seven repetitions. For the DNA range data, the standard curve y intercept was 19.89, and the slope was −1.526. The correlation coefficient (r) of this curve was −0.99 (r2 = 0.98). The MDL of the Toxocara qPCR tested on T. cati corresponded to 39 cycles. Beyond this limit, DNA is considered to be detectable but not quantifiable.
From this calibration curve, the average Cq obtained for three PCRs in triplicate (Fig. 2) with one egg DNA extract (3 × 1 egg extracted; each tested 3 times by PCR) was 40.29 (SD = 2.29). This result corresponded to a DNA concentration of 2.9 fg/μl in the extract. For five eggs (3 × 5 eggs extracted), the average Cq obtained for three PCRs in triplicate was 37.5 (SD = 1.84), corresponding to a DNA concentration of 16.6 fg/μl. For 10 eggs (3 × 10 eggs extracted), the average Cq was 34.45 (SD = 0.39), corresponding to a DNA concentration of 0.11 pg/μl.
FIG 2.
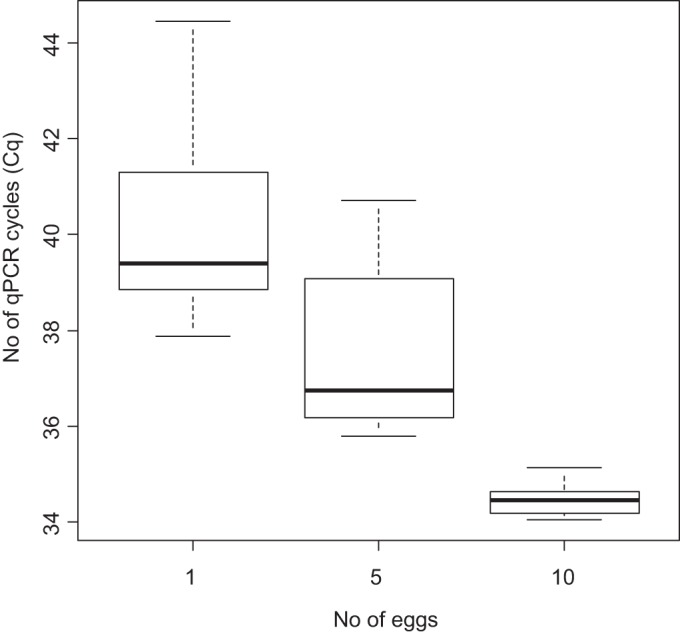
Box plots representing the results of Toxocara qPCR performed on DNA extracted from isolated T. cati eggs. Results are expressed as numbers of quantitative cycles (Cq); all qPCRs were performed in triplicate.
Toxocara qPCR was tested on the 68 coprosamples. All samples were negative, except for three cat fecal samples which were found to be positive, with Cq values ranging from 32.06 (corresponding to 0.5 pg/μl of DNA) to 32.87 (0.3 pg/μl of DNA) (Table 1). After sequencing of the SEQ-PCR products, T. cati was identified for the three cats (Fig. 1).
E. multilocularis detection by qPCR.
The average Cq for each point of the E. multilocularis DNA range, performed seven times in duplex with the Alea target, was obtained for six points from 5 to 5 × 10−5 ng/μl (for 5 ng/μl, Cq = 19.94 [SD = 0.05]; for 0.5 ng/μl, Cq = 23.77 [SD = 0.18]; for 0.05 ng/μl, Cq = 27.11 [SD = 0.06]; for 5 × 10−3 ng/μl, Cq = 30.57 [SD = 0.21]; for 5 × 10−4 ng/μl, Cq = 33.76 [SD = 0.24]; and for 5 × 10−5 ng/μl, Cq = 37.21 [SD = 0.45]). The point of the DNA range of 5 × 10−6 ng/μl was amplified in 6/7 PCRs. For the DNA range data, the standard curve y intercept was 22.56, and the slope was −1.487. The correlation coefficient (r) of this curve was −0.99 (r2 = 0.98). Based on the point of 5 × 10−5 ng/μl, the MDL of the E. multilocularis rrnL qPCR tested on E. multilocularis DNA corresponded to 38 cycles. Beyond this limit, DNA is considered to be detectable but not quantifiable.
Em/Alea qPCR was performed on the coprosample panel. Ten fecal samples from foxes were positive (Cq values of 26.26 [83 pg/μl] to 39.99 [8 fg/μl]), as well as that of one cat (Cq = 40.2 [7 fg/μl]) (Table 1), according to the calibration curve. The Em-SEQ-PCR was positive for 8/10 positive foxes, and the PCR products were sequenced. All the isolates were identified as E. multilocularis. The three negative samples in Em-SEQ-PCR presented Cq values for E. multilocularis qPCR of >39.9 (Table 1).
Host identification.
The specificities of the primers for amplification of only V. vulpes, C. l. familiaris, F. s. catus, M. foina, and M. meles DNAs were tested online. For the five carnivores targeted, primer homology was 100%. With the F. s. catus primers, Felis silvestris silvestris targets could be amplified (100% identity in the sequencing primers), and with the C. l. familiaris primers, C. lupus targets could be amplified (100% identity in the sequencing primers).
The qPCR primers and probes were first tested on the tissue DNAs of the five species. Each point of these DNA ranges was tested in 5 independent runs, and Cq averages were obtained (Table 5).
TABLE 5.
Average Cq values for qPCRs performed 5 times independently on each point of the DNA ranges for the 5 carnivores studieda
| Host species | Mean (SD) Cq at indicated DNA concn (ng/μl) | ||
|---|---|---|---|
| 50 ng/µl | 5 ng/µl | 0.5 ng/µl | |
| Vulpes vulpes | 16.9 (1.41) | 20.4 (1.01) | 24.5 (1.46) |
| Canis lupus familiaris | 13.9 (1.02) | 17.6 (1.85) | 21.0 (0.73) |
| Felis silvestris catus | 15.8 (0.78) | 20.1 (0.33) | 24.0 (0.77) |
| 40 ng/µl | 4 ng/µl | 0.4 ng/µl | |
| Martes foina | 12.4 (2.47) | 16.3 (2.69) | 21.4 (3.53) |
| 8 ng/µl | 0.8 ng/µl | 0.08 ng/µl | |
| Meles meles | 12.4 (0.43) | 16.2 (0.45) | 21.0 (0.55) |
All qPCRs were performed in duplicate.
Carnivore qPCR screening allowed us to validate the morphological identification of all 68 stools collected (Tables 1 and 6). Cross-reactions were observed among the tested coprosamples, with gaps of 7.8 to 18.9 cycles depending on the targeted species (Tables 1 and 6).
TABLE 6.
Average, minimum, and maximum Cq values for qPCRs for five targeted carnivore species, with cross-reaction data (all qPCRs performed in duplicate)
| Carnivore species | Avg Cq | Minimum Cq | Maximum Cq | Minimum–maximum Cq for cross-reaction (no. of animals with cross-reaction) |
|---|---|---|---|---|
| Vulpes vulpes | 23.1 | 16.3 | 28.8 | 42.0 (1) |
| Canis lupus familiaris | 22.8 | 17.9 | 26.8 | 33.6–40.1 (7) |
| Canis lupus | 25.8 | 21.4 | 28.7 | |
| Felis silvestris catus | 25.5 | 21.0 | 31.8 | 33.3–39.7 (5) |
| Meles meles | 25.7 | 23.0 | 32.4 | (0) |
| Martes foinaa | 26.25 | 22.5 | 30.0 | 41.8 (1) |
Two specimens were available.
The SEQ-PCRs were tested on the 5 species by using extracted DNAs from coprosamples (Table 1). The products were sequenced, and the identity of the host was confirmed for all tested samples (Table 1). For one canine stool (YAY), a cytB sequence from Fc-SEQ-PCR was obtained after sequencing and showed 100% identity to F. s. catus; for three cat stools (POU, NOI, and ZIZ), a cytB sequence from Cf-SEQ-PCR was obtained after sequencing and revealed 100% identity to C. l. familiaris. For the M. martes stool (M1), a cytB sequence from Mf-SEQ-PCR was obtained after sequencing and showed 99% identity to the M. martes reference sequence (GenBank accession number KC660129.1).
DISCUSSION
Molecular analyses based on environmental material are often affected by inhibitor agents, such as humic acids, calcium, or tannic acids (28), which can lead to false-negative PCR results or underestimation of DNA loads. Adding an integrated control to duplex or multiplex qPCR mixtures is thus mandatory for screening feces for parasites. In the present study, we developed an internal control with a random sequence inserted into a plasmid and used for duplex qPCR. For the panel of 68 samples used, PCR inhibitors were detected in some samples. The expected Cq value for the Alea target was set at 35 to 37 cycles for Toxo/Alea and Em/Alea qPCRs, which was a compromise to avoid premature signal detection but was close to the values obtained in the field. For samples with inhibition, diluting copro-DNA to reduce the effects of inhibitors (23) or reextracting DNA from the coprosamples may be recommended. Because this is an independent target, it could be implemented in other studies for working on environmental samples with qPCR technology. A similar internal amplification control, obtained by artificial construction but amplified with the same primers as those for the targeted DNA, was also used to obtain a comparable amplification efficacy (29) and to diagnose E. multilocularis in feces (8). In the present study, we chose a random sequence as an internal control in order to check independently for the presence of inhibitors.
qPCR screening for pathogens in carnivore stools was improved in our study. First, we developed qPCR detection and quantification of Toxocara sp. parasites. DNA from a single egg was detected for T. cati, though it was close to the MDL. With the qPCR designed for T. cati, both Toxocara species were amplified by Toxocara qPCR for all available adult worms and for T. cati in three domestic cat feces. Toxocara species discrimination and identification were made possible by the development of SEQ-PCR, which enabled us to sequence fragments of about 200 bp. Similar Cq values were obtained for the same DNA concentration for T. cati and T. canis adult worms, despite mutations present in the forward and reverse primers. However, further studies with T. canis eggs are necessary to optimize the Toxocara qPCR, with the aim of better evaluating the presence of this parasite in the environment. Duplex qPCR with primers designed for T. canis coupled with T. cati primers may help to better evaluate the presence of each species in the environment. A rapid diagnostic test that is able to quantify each species is needed to assess the relevance of control strategies in both domestic and wild reservoirs. Indeed, although parasite control campaigns do exist, these parasites are still very prevalent in Europe (30).
With SEQ-PCR, the number of steps required before sequencing was reduced in comparison to classical PCR (which involves agarose gel loading with PCR products and electrophoresis to check correct amplification). In the present study, the amplification curves obtained during qPCR were used as controls before sequencing of high-quality PCR products. Alternatively, pyrosequencing was used by Umhang and coworkers to confirm the presence of E. multilocularis in feces (8). Another target was amplified with this technique to avoid problems of contamination by previous qPCR products. However, SEQ-PCR is the easiest way to obtain confirmation by use of the same equipment and by checking the size (in base pairs) of the SEQ-PCR products compared to qPCR products.
E. multilocularis detection in coprosamples was further developed here, with inhibition control performed in a duplex qPCR. The Cq results for the Em/Alea duplex qPCR were very similar to those for the previously developed E. multilocularis rrn simplex qPCR (23). With an additional sequencing step, we were able to confirm the parasite identity. SEQ-PCR results could be considered a confirmation test of specificity. This confirmation step may prove to be essential in studies performed on domestic carnivore coprosamples for both prevention and deworming.
Confirming the morphological identification of carnivore feces is a critical step toward avoiding substantial bias in field studies (18). We chose qPCR technology because it is a highly sensitive, one-step reaction process. The method provides quick results and makes it possible to combine analyses with pathogen detection. In the present study, host identification was validated by SEQ-PCR. Cross-reactions were observed in a few cases, but the nontargeted isolates always had Cq values of >7 cycles, in contrast to the values obtained for the targeted species. Over and above the expected values, confirmation by sequencing of SEQ-PCR products must be performed. Additional carnivore DNA was detected in the feces of three pets living with another pet (dog or cat). These results can be explained by the ingestion of cells from animals sharing the same home. It is noteworthy that one domestic cat living alone presented canine DNA in its stool. Contamination in the laboratory, e.g., by handling errors, cannot be ruled out, but the Cq difference in qPCR screening provides the first confirmation of correct identification. In the present study, duplex qPCR was developed to reduce the cost of the analysis, and it could be improved by multiplexing after fluorochrome compatibility was tested, combining detection of the host, helminth parasites, and the PCR inhibition control.
For parasite detection involving wild and domestic animals, the use of qPCR coupled with SEQ-PCR allows the screening of large panels of coprosamples. The role of domestic animals in environmental contamination by helminths may thus be better understood, and public health prevention and control programs implemented, based on a rapid, available, and ethical protocol.
ACKNOWLEDGMENTS
We are very grateful to Peter Deplazes, Marie-Hélène Guislain, Francis Raoul, Patrick Giraudoux, Steffi Rocchi, Claudine Rosinek, Coralie Barrera, and Guillaume Halliez for providing samples for the design and assessment of techniques. We give many thanks to Pamela Albert for language editing.
This work was supported by a Projet Interdisciplinaire Ecologie de la Santé 2014 grant from the CNRS-INEE and by funds from the InVS National Reference Center for Alveolar Echinococcosis.
REFERENCES
- 1.Torgerson PR, Keller K, Magnotta M, Ragland N. 2010. The global burden of alveolar echinococcosis. PLoS Negl Trop Dis 4:e722. doi: 10.1371/journal.pntd.0000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jenkins EJ, Peregrine AS, Hill JE, Somers C, Gesy K, Barnes B, Gottstein B, Polley L. 2012. Detection of European strain of Echinococcus multilocularis in North America. Emerg Infect Dis 18:1010–1012. doi: 10.3201/eid1806.111420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schweiger A, Ammann RW, Candinas D, Clavien P-A, Eckert J, Gottstein B, Halkic N, Muellhaupt B, Prinz BM, Reichen J, Tarr PE, Torgerson PR, Deplazes P. 2007. Human alveolar echinococcosis after fox population increase, Switzerland. Emerg Infect Dis 13:878–882. doi: 10.3201/eid1306.061074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takumi K, Hegglin D, Deplazes P, Gottstein B, Teunis P, van der Giessen J. 2012. Mapping the increasing risk of human alveolar echinococcosis in Limburg, The Netherlands. Epidemiol Infect 140:867–871. doi: 10.1017/S0950268811001221. [DOI] [PubMed] [Google Scholar]
- 5.Deplazes P, van Knapen F, Schweiger A, Overgaauw PAM. 2011. Role of pet dogs and cats in the transmission of helminthic zoonoses in Europe, with a focus on echinococcosis and toxocarosis. Vet Parasitol 182:41–53. doi: 10.1016/j.vetpar.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Dyachenko V, Pantchev N, Gawlowska S, Vrhovec MG, Bauer C. 2008. Echinococcus multilocularis infections in domestic dogs and cats from Germany and other European countries. Vet Parasitol 157:244–253. doi: 10.1016/j.vetpar.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 7.Kapel CMO, Torgerson PR, Thompson RCA, Deplazes P. 2006. Reproductive potential of Echinococcus multilocularis in experimentally infected foxes, dogs, raccoon dogs and cats. Int J Parasitol 36:79–86. doi: 10.1016/j.ijpara.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Umhang G, Forin-Wiart M-A, Hormaz V, Caillot C, Boucher J-M, Poulle M-L, Franck B. 2015. Echinococcus multilocularis detection in the intestines and feces of free-ranging domestic cats (Felis s. catus) and European wildcats (Felis s. silvestris) from northeastern France. Vet Parasitol 214:75–79. doi: 10.1016/j.vetpar.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Veit P, Bilger B, Schad V, Schäfer J, Frank W, Lucius R. 1995. Influence of environmental factors on the infectivity of Echinococcus multilocularis eggs. Parasitology 110:79–86. doi: 10.1017/S0031182000081075. [DOI] [PubMed] [Google Scholar]
- 10.Bresson-Hadni S, Blagosklonov O, Knapp J, Grenouillet F, Sako Y, Delabrousse E, Brientini M-P, Richou C, Minello A, Antonino A-T, Gillet M, Ito A, Mantion GA, Vuitton DA. 2011. Should possible recurrence of disease contraindicate liver transplantation in patients with end-stage alveolar echinococcosis? A 20-year follow-up study. Liver Transplant 17:855–865. doi: 10.1002/lt.22299. [DOI] [PubMed] [Google Scholar]
- 11.Morgan ER, Azam D, Pegler K. 2013. Quantifying sources of environmental contamination with Toxocara spp. eggs. Vet Parasitol 193:390–397. doi: 10.1016/j.vetpar.2012.12.034. [DOI] [PubMed] [Google Scholar]
- 12.Azam D, Ukpai OM, Said A, Abd-Allah GA, Morgan ER. 2012. Temperature and the development and survival of infective Toxocara canis larvae. Parasitol Res 110:649–656. doi: 10.1007/s00436-011-2536-8. [DOI] [PubMed] [Google Scholar]
- 13.Overgaauw PAM, van Knapen F. 2013. Veterinary and public health aspects of Toxocara spp. Vet Parasitol 193:398–403. doi: 10.1016/j.vetpar.2012.12.035. [DOI] [PubMed] [Google Scholar]
- 14.Moreira GMSG, Telmo PL, Mendonça M, Moreira AN, McBride AJA, Scaini CJ, Conceição FR. 2014. Human toxocariasis: current advances in diagnostics, treatment, and interventions. Trends Parasitol 30:456–464. doi: 10.1016/j.pt.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Buijs J, Borsboom G, Renting M, Hilgersom WJ, van Wieringen JC, Jansen G, Neijens J. 1997. Relationship between allergic manifestations and Toxocara seropositivity: a cross-sectional study among elementary school children. Eur Respir J 10:1467–1475. doi: 10.1183/09031936.97.10071467. [DOI] [PubMed] [Google Scholar]
- 16.Pinelli E, Aranzamendi C. 2012. Toxocara infection and its association with allergic manifestations. Endocr Metab Immune Disord Drug Targets 12:33–44. doi: 10.2174/187153012799278956. [DOI] [PubMed] [Google Scholar]
- 17.Boitani L, Powell RA. 2012. Carnivore ecology and conservation: a handbook of techniques. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 18.Monterroso P, Castro D, Silva TL, Ferreras P, Godinho R, Alves PC. 2013. Factors affecting the (in)accuracy of mammalian mesocarnivore scat identification in south-western Europe: accuracy of carnivore scat identification. J Zool 289:243–250. doi: 10.1111/jzo.12000. [DOI] [Google Scholar]
- 19.Farrell LE, Roman J, Sunquist ME. 2000. Dietary separation of sympatric carnivores identified by molecular analysis of scats. Mol Ecol 9:1583–1590. doi: 10.1046/j.1365-294x.2000.01037.x. [DOI] [PubMed] [Google Scholar]
- 20.Nonaka N, Sano T, Inoue T, Teresa Armua M, Fukui D, Katakura K, Oku Y. 2009. Multiplex PCR system for identifying the carnivore origins of faeces for an epidemiological study on Echinococcus multilocularis in Hokkaido, Japan. Parasitol Res 106:75–83. doi: 10.1007/s00436-009-1629-0. [DOI] [PubMed] [Google Scholar]
- 21.Durant J-F, Irenge LM, Fogt-Wyrwas R, Dumont C, Doucet J-P, Mignon B, Losson B, Gala J-L. 2012. Duplex quantitative real-time PCR assay for the detection and discrimination of the eggs of Toxocara canis and Toxocara cati (Nematoda, Ascaridoidea) in soil and fecal samples. Parasit Vectors 5:288. doi: 10.1186/1756-3305-5-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dinkel A, Kern S, Brinker A, Oehme R, Vaniscotte A, Giraudoux P, Mackenstedt U, Romig T. 2011. A real-time multiplex-nested PCR system for coprological diagnosis of Echinococcus multilocularis and host species. Parasitol Res 109:493–498. doi: 10.1007/s00436-011-2272-0. [DOI] [PubMed] [Google Scholar]
- 23.Knapp J, Millon L, Mouzon L, Umhang G, Raoul F, Ali ZS, Combes B, Comte S, Gbaguidi-Haore H, Grenouillet F, Giraudoux P. 2014. Real time PCR to detect the environmental faecal contamination by Echinococcus multilocularis from red fox stools. Vet Parasitol 201:40–47. doi: 10.1016/j.vetpar.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 24.Roussel S, Rognon B, Barrera C, Reboux G, Salamin K, Grenouillet F, Thaon I, Dalphin J-C, Tillie-Leblond I, Quadroni M. 2011. Immuno-reactive proteins from Mycobacterium immunogenum useful for serodiagnosis of metalworking fluid hypersensitivity pneumonitis. Int J Med Microbiol 301:150–156. doi: 10.1016/j.ijmm.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Hospodsky D, Yamamoto N, Peccia J. 2010. Accuracy, precision, and method detection limits of quantitative PCR for airborne bacteria and fungi. Appl Environ Microbiol 76:7004–7012. doi: 10.1128/AEM.01240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98. [Google Scholar]
- 28.Opel KL, Chung D, McCord BR. 2010. A study of PCR inhibition mechanisms using real time PCR. J Forensic Sci 55:25–33. doi: 10.1111/j.1556-4029.2009.01245.x. [DOI] [PubMed] [Google Scholar]
- 29.Auvray F, Lecureuil C, Dilasser F, Taché J, Derzelle S. 2009. Development of a real-time PCR assay with an internal amplification control for the screening of Shiga toxin-producing Escherichia coli in foods. Lett Appl Microbiol 48:554–559. doi: 10.1111/j.1472-765X.2009.02561.x. [DOI] [PubMed] [Google Scholar]
- 30.Otranto D, Cantacessi C, Dantas-Torres F, Brianti E, Pfeffer M, Genchi C, Guberti V, Capelli G, Deplazes P. 2015. The role of wild canids and felids in spreading parasites to dogs and cats in Europe. II. Helminths and arthropods. Vet Parasitol 213:24–37. doi: 10.1016/j.vetpar.2015.04.020. [DOI] [PubMed] [Google Scholar]