

ABSTRACT
DNA/RNA helicases, which are enzymes for eliminating hydrogen bonds between bases of DNA/DNA, DNA/RNA, and RNA/RNA using the energy of ATP hydrolysis, contribute to various biological activities. In the present study, the Euryarchaeota-specific helicase EshA (TK0566) from the hyperthermophilic archaeon Thermococcus kodakarensis (Tk-EshA) was obtained as a recombinant form, and its enzymatic properties were examined. Tk-EshA exhibited maximal ATPase activity in the presence of RNA at 80°C. Unwinding activity was evaluated with various double-stranded DNAs (forked, 5′ overhung, 3′ overhung, and blunt end) at 50°C. Tk-EshA unwound forked and 3′ overhung DNAs. These activities were expected to unwind the structured template and to peel off misannealed primers when Tk-EshA was added to a PCR mixture. To examine the effect of Tk-EshA on PCR, various target DNAs were selected, and DNA synthesis was investigated. When 16S rRNA genes were used as a template, several misamplified products (noise DNAs) were detected in the absence of Tk-EshA. In contrast, noise DNAs were eliminated in the presence of Tk-EshA. Noise reduction by Tk-EshA was also confirmed when Taq DNA polymerase (a family A DNA polymerase, PolI type) and KOD DNA polymerase (a family B DNA polymerase, α type) were used for PCR. Misamplified bands were also eliminated during toxA gene amplification from Pseudomonas aeruginosa DNA, which possesses a high GC content (69%). Tk-EshA addition was more effective than increasing the annealing temperature to reduce misamplified DNAs during toxA amplification. Tk-EshA is a useful tool to reduce noise DNAs for accurate PCR.
IMPORTANCE PCR is a technique that is useful for genetic diagnosis, genetic engineering, and detection of pathogenic microorganisms. However, troubles with nonspecific DNA amplification often occur from primer misannealing. In order to achieve a specific DNA amplification by eliminating noise DNAs derived from primer misannealing, a thermostable Euryarchaeota-specific helicase (Tk-EshA) was included in the PCR mixture. The addition of Tk-EshA has reduced noise DNAs in PCR.
INTRODUCTION
PCR is a technique for the amplification of nucleic acids invented by Mullis in 1983 (1–3). This method is generally used for cloning genes, sequencing DNA, detecting single nucleotide polymorphisms (SNPs) in genetic diagnosis, and identifying microbial infections (4–6). PCR is a simple and efficient method for DNA amplification and is now essential for molecular genetics. However, unexpected DNA sometimes appears due to primer misannealing. To avoid nonspecific amplification in PCR, optimization of the annealing temperature and Mg2+ concentration, in addition to primer redesigning, is generally attempted (7). However, these approaches are often not effective, and undesirable DNA still appears due to unfavorable primer misannealing. To reduce undesirable primer annealing, which causes nonspecific amplification, several methods were developed, such as hot start. The first method uses solid oil and is called the wax method (8). This method separates the PCR mixture into two fractions, the DNA template and DNA polymerase, by using solid oil during the first cycle. The second method uses a neutralizing monoclonal antibody directed against DNA polymerases, such as a Taq polymerase from Thermus aquaticus (9) and a KOD polymerase from Thermococcus kodakarensis (10). This method is based on the principle that the antibody inhibits polymerase activity before the onset of thermal cycling, preventing primer dimer formation and primer misannealing at various positions besides the target region. In the first denaturation step in PCR, the antibody is quickly inactivated, and PCR proceeds. The antibody-mediated hot start method is significantly more convenient than the hot start method using wax; however, hot start is not always successful, especially when long DNA and high-GC-content DNA are used as the templates. A thermostable RecA protein that is involved in DNA recombination reduces nonspecific amplification in PCR (11, 12). In addition, a technique was reported in which the mismatch-recognizing protein MutS from a thermophilic bacterium was added to the PCR mixture for accurate DNA amplification (13). MutS is an initiator of the DNA mismatch repair pathway and is conserved in a variety of thermophilic bacteria and in a very few archaea (14). MutS binds to a mismatched primer-template complex, thereby preventing the approach of the DNA polymerase to the 3′ end of the primer.
In the present study, we tested a novel approach to reduce unexpected misannealing and increase correct annealing of primers in order to efficiently reduce misamplified products. DNA/RNA helicases are enzymes for eliminating hydrogen bonds between bases of DNA/DNA, DNA/RNA, and RNA/RNA from the unpaired 3′ or 5′ end using the energy of ATP hydrolysis. Due to their unwinding activity, helicases were expected to unwind the structured template and partially annealed primer/template duplexes during PCR. RNA and DNA helicases are classified into several superfamilies (SFs) based on their amino acid sequences (15). An SF1 helicase, UvrD, was previously used to amplify DNA at low temperatures, and this approach was called helicase-dependent amplification (16–18). Because UvrD (a DNA helicase) unwinds blunt-end substrates as well as nicked circular DNA (19), nucleic acids are amplified with a mesophilic DNA polymerase without complicated temperature control. A trial to reduce misamplification during PCR using a helicase has not been reported. We focused on a thermostable DNA/RNA helicase of SF2 from the hyperthermophilic archaeon T. kodakarensis, which grows optimally at 85°C (20). SF2 is the largest superfamily with members that are involved in DNA and RNA metabolism. SF2 includes nine families: Rec-G-like family, RecQ-like family, XPD/Rad3/DinG family, Ski-2-like family, type 1 restriction enzyme helicase subunit (T1R) family, Swi2/Snf family, XPF/Hef/ERCC4/RIG-I nuclease helicase family, DEAD box family, and DEAH/RHA family. Recently, a novel SF2 helicase named Archaea-specific helicase (ASH) was reported, which is specific to Euryarchaeota, with the exception of Thermoproteales and Archaeoglobales (21). SF2 helicases, which unwind DNA duplexes from unpaired 3′ or 5′ ends, contain highly conserved nucleoside triphosphate (NTP) substrate- or metal-binding motifs, such as Walker A and Walker B motifs (22–25). T. kodakarensis possesses several SF2 genes in its genome (26). A DEAD box RNA helicase (Tk-DeaD) belonging to SF2 is dominantly expressed under cold stress conditions (60°C) in T. kodakarensis (27). A recombinant form of Tk-DeaD is stable in a high-ionic-strength buffer containing 500 mM NaCl but is denatured in lower-ionic-strength buffers containing less than 200 mM NaCl, especially at temperatures higher than 60°C (27). Because it is inactivated at temperatures higher than 60°C, Tk-DeaD was not suitable for our purpose. Therefore, we screened for helicases from T. kodakarensis that unwind misannealed primer/template duplexes at high temperatures. In the present study, the effect of an SF2 helicase on PCR was investigated.
MATERIALS AND METHODS
Microorganisms and media.
The strains used in this study are summarized in Table 1. Escherichia coli strains were routinely cultivated at 30°C or 37°C in lysogeny broth (LB) medium. Ampicillin (50 μg · ml−1), kanamycin (20 μg · ml−1), and/or chloramphenicol (25 μg · ml−1) was added to the medium when needed.
TABLE 1.
Strains and primers used in this study
| Strain or primer | Relevant characteristic(s) or sequence (5′ to 3′)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ− thi-1 gyrA96 relA1 | Stratagene |
| BL21-CodonPlus(DE3)-RIL | E. coli B F− ompT hsdS(rB− mB−) dcm+ Tetr gal λ(DE3) endA Hte [argU ileY leuW Camr] | Stratagene |
| Primers | ||
| tk0566-Fw | AAAAAACATATGCTCTTCGTCGTGAGACCGGGA | This study |
| tk0566-Rv | TTTGAATTCTTAGCGTATCATCCTCCCTTCTTCTA | This study |
| tk0928-Fw | AAAAAAACATATGATGGTTGTCCTGAGAATCCC | This study |
| tk0928-Rv | AAAAGTCGACTTAACTAGAGCGGCGCTTTTTGG | This study |
| TK0566-D344A-E345A Fw | GGAACCATAGTGATAGCCGCCATACACACGCTCGAC | This study |
| TK0566-D344A-E345A Rv | GTCGAGCGTGTGTATGGCGGCTATCACTATGGTTCC | This study |
| 16S rRNA gene Fw | ATTCCGGTTGATCCTGCCGGAGGCCACTGC | This study |
| 16S rRNA gene Rv | TCCGGCGATAGGAGGTGATCGAGCCGTAGG | This study |
| toxA Fw | ATGCACCTGACACCCCATTG | This study |
| toxA Rv | TTACTTCAGGTCCTCGCGCG | This study |
| 16 S miss 5′ Rv | ATGGAGGCGAGCTCGAAGCTCGCCCGACAC | This study |
| 16 S miss 3′ Rv | GGCGAGCTCGAAGCTCGCCCGACACAGCTA | This study |
Mismatched nucleotides within the sequences of the 16S rRNA, Tk0566, or Tk0928 gene are underlined.
Expression and purification of helicase candidates.
The genes examined in this study are located at the following sites on the T. kodakarensis genome: Tk0566, bp 486488 to 488986 (negative strand); and Tk0928, bp 806025 to 807410 (negative strand). The Tk0566 and Tk0928 genes were amplified using the primer pairs tk0566-Fw/tk0566-Rv and tk0928-Fw/tk0928-Rv, respectively (Table 1). PCR was performed in a mixture (50 μl) containing 120 mM Tris HCl (pH 8.0), 10 mM KCl, 6 mM (NH4)2SO4, 0.1% Triton X-100, 10 μg · ml−1 bovine serum albumin (BSA), 0.2 mM deoxynucleoside triphosphates (dNTPs), 1 mM MgSO4, 0.2 μM (each) primers, 50 ng of template DNA, and 1 U of KOD-Plus DNA polymerase (Toyobo, Osaka, Japan) in a thermal cycler as follows: 2 min at 98°C, followed by 17 cycles of 15 s at 98°C, 30 s at 55°C, and 8.5 min at 68°C. These amplified fragments of the Tk0566 and Tk0928 genes were separately cloned into the NdeI/EcoRI sites of pET28a and in the NdeI/SalI sites of pET28a, yielding the plasmids pHisTK0566 and pHisTK0928, respectively. E. coli BL21-CodonPlus(DE3)-RIL cells were transformed with these plasmids. TK0566 (the Euryarchaeota-specific helicase EshA from T. kodakarensis [Tk-EshA]) and TK0928 were purified as a recombinant proteins with an N-terminal His tag. The recombinant E. coli cells [BL21-CodonPlus(DE3)/pHisTK0566 and BL21-CodonPlus(DE3)/pHisTK0928] were grown in LB medium containing 20 μg · ml−1 of kanamycin and 25 μg · ml−1 of chloramphenicol at 37°C. Expression of TK0566 (Tk-EshA) and TK0928 was induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside. After a further incubation for 4 h at 37°C, the cells were harvested by centrifugation, resuspended in buffer A (50 mM imidazole, 20 mM Tris HCl [pH 8.0], 500 mM NaCl, and 0.1% Triton X-100), and disrupted by sonication. Cell debris was removed by centrifugation, and each supernatant was incubated at 70°C for 30 min and then centrifuged again. Each resultant supernatant was applied to a column packed with 4 ml of Ni-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen, Tokyo, Japan) and eluted with buffer B (200 mM imidazole, 20 mM Tris HCl [pH 8.0], 500 mM NaCl, and 0.1% Triton X-100). Each purified protein was finally dialyzed against buffer C (20 mM Tris HCl [pH 8.0], 200 mM NaCl, 10% glycerol, and 1 mM dithiothreitol [DTT]). The protein concentration was determined by the Bradford dye-binding assay, using BSA as a standard (28). The A280/A260 ratios of the purified TK0566 (Tk-EshA) and TK0928 were 1.52 and 1.85, respectively.
Construction of ATPase deficient mutant of Tk-EshA.
The Tk-EshA mutant (Tk-EshA-D344A-E345A) used in this study was generated by a quick-change PCR using plasmid pHisTK0566 as a template. For the construction of mutant, the primer set TK0566-D344A-E345A Fw and TK0566-D344A-E345A Rv (Table 1) was utilized. The PCR product was treated with DpnI (TaKaRa, Tokyo, Japan) to digest the template DNA. E. coli DH5α was transformed by the nicked vector DNA containing the desired mutations to yield plasmid pHisTK0566-D344A-E345A. E. coli BL21-CodonPlus(DE3)-RIL cells were transformed with pHisTK0566-D344A-E345A. The Tk-EshA mutant enzyme (Tk-EshA-D344A-E345A) was purified as a recombinant protein with an N-terminal His tag in the same way as a used for Tk-EshA.
ATPase assay of helicases.
ATPase activity was measured by monitoring phosphate released by ATP hydrolysis. The ATPase activity of helicases was determined using a single-stranded RNA (ssRNA), ssRNA63, or single-stranded DNA (ssDNA), N-DNA70 (Table 2). The reaction mixture (10 μl) composed of 5 nM ssRNA or ssDNA, purified helicase (80 nM TK0566 [Tk-EshA], 200 nM TK0928, and 80 nM Tk-EshA-D344A-E345A), 1 mM ATP, 2 mM MgCl2, and 2 mM DTT, prepared in 50 mM HEPES buffer (pH 7.6), was incubated at 30°C to 110°C for 20 min or 30 min. ATPase activity of Tk-EshA-D344A-E345A was measured using ssDNA as a substrate. After incubation, the reaction mixture was kept on ice to stop the enzymatic reaction. The concentration of free phosphate in the reaction mixture was measured using a Biomol green kit (Enzo Life Sciences, Plymouth Meeting, PA) (30, 31). Thereafter, 10 μl of the reaction mixture, 40 μl of assay buffer (50 mM Tris HCl [pH 7.4] and 200 mM NaCl), and 100 μl of Biomol green reagent (Enzo Life Sciences) were applied to a 96-well plate and left at room temperature for 20 min. Absorbance at 260 nm was measured using an EnVision multilabel reader (PerkinElmer Japan, Yokohama, Japan) to monitor the concentration of free phosphate.
TABLE 2.
Oligonucleotides for helicase assay in this study
| Oligonucleotide | Sequence (5′ to 3′) | Reference |
|---|---|---|
| ssRNA63 | UGGGCUGCAGGUCGACUCUAGAGGAUCCCCGGGCGAGCUCGAAGUCGGGUCUCCCUAUAGUGA | 27 |
| L-HJ-3-54mer | IRDye 700/TCACTCCGCATCTGCCGATTCTGGCTGTGGCGTGTTTCTGGTGGTTCCTAGGTC | 29 |
| N-HJ-3-54mer | TCACTCCGCATCTGCCGATTCTGGCTGTGGCGTGTTTCTGGTGGTTCCTAGGTC | 29 |
| L-5′DNA34 | IRDye 700/GACCTAGGAACCACCAGAAACACGCCACAGCCAG | 29 |
| N-5′DNA34 | GACCTAGGAACCACCAGAAACACGCCACAGCCAG | 29 |
| L-3′DNA54 | IRDye 700/CTGGCTGTGGCGTGTTTCTGGTGGTTCCTAGGTCTTAGCCGTCTACGCCTCACT | This study |
| N-HJ-4 | GACCTAGGAACCACCAGAAACACGCCACAGCCAGGAAGCCGATTGCGAGGCCGTCCTACCATCCTGCAGG | 29 |
| N-B-DNA54 | GACCTAGGAACCACCAGAAACACGCCACAGCCAGAATCGGCAGATGCGGAGTGA | This study |
| L-RNA54 | Cy5.5/CUGGCUGUGGCGUGUUUCUGGUGGUUCCUAGGUCUUAGCCGUCUACGCCUCACU | This study |
| N-DNA70 | GGACGTCCTACCATCCTGCCGGAGCGTTAGCCGAAGGACCTAGGAACCACCAGAAACACGCCACAGCCAG | This study |
| N-DNA34 | CTGGCTGTGGCGTGTTTCTGGTGGTTCCTAGGTC | This study |
Effects of helicases on 16S rRNA gene amplification.
To examine the effect of SF2 helicases on PCR, 16S rRNA genes from T. kodakarensis (the region from bp 2022864 to 2024361 in the genome) was targeted, and genomic DNA from T. kodakarensis was used as a template. PCR was performed in a mixture (20 μl) containing PCR buffer [120 mM Tris HCl (pH 8.0), 10 mM KCl, 6 mM (NH4)2SO4, 0.1% Triton X-100, 10 μg ml−1 BSA, 0.2 mM dNTPs, 2 mM MgSO4], 0.6 μM (each) primers 16S rRNA gene Fw and 16S rRNA gene Rv (Table 1), 20 ng of template DNA, 0.4 U of KOD-Plus DNA polymerase, and 2 to 100 nM helicase (Tk-DeaD, Tk-EshA, TK0928, Tk-EshA-D344A-E345A). PCR was performed in a thermal cycler as follows: 2 min at 94°C, followed by 28 or 45 cycles of 15 s at 94°C, 30 s at 60°C, and 2 min at 68°C. The resultant DNA (1,498 bp) was separated by 1% (wt/vol) agarose gel electrophoresis and visualized by ethidium bromide (EtBr) staining. To examine the PCR specificity, band intensities on agarose gels were measured using ImageJ software (National Institutes of Health, Bethesda, MD). The relative amounts of PCR products were normalized by the intensities of the different size bands (bands a [target], b [noise], and c [noise]; see Fig. 3A and C). Each band (a, b, or c) was in the absence of Tk-EshA, which was set to 100%.
FIG 3.

Effect of thermostable helicases on PCR specificity. (A) A 1,498-bp region of 16S rRNA genes from T. kodakarensis was amplified by 28 or 45 repeating cycles in the presence of Tk-DeaD (left panel, 28 cycles), Tk-EshA (center panel, 28 or 45 repeating cycles), or TK0928 (right panel, 28 repeating cycles). The concentration of the helicases was 0, 2, 10, 50, or 100 nM. DNA size markers are shown in lane M. (B) A 1,498-bp region of 16S rRNA genes from T. kodakarensis was amplified by 28 repeating cycles in the presence of 0, 2, 10, 50, or 100 nM Tk-EshA-D344A-E345A. The 100-bp DNA ladder is shown in lane M. (C) Relative amounts of PCR products amplified by 28 repeating cycles upon addition of Tk-EshA. In the gels shown in the center of panel A, band a is the target product and bands b and c are nonspecific products. The relative amounts of desired and nonspecific amplification products were measured using ImageJ software. In the graph shown on the right, the circles, squares, and triangles denote the relative amounts of products a, b, and c, respectively. The relative amounts of PCR products were normalized based on the intensities of the bands. Each band (a, b, or c) was in the absence of Tk-EshA, which was set to 100%.
Effect of Tk-EshA on PCR specificity using competitive misannealing primers.
PCR was performed in a mixture (20 μl) containing PCR buffer as described above, 0.6 μM (each) primers 16S rRNA gene Fw and 16S rRNA gene Rv, 0.6 μM misannealing primer 16 S miss 5′ Rv or 16 S miss 3′ Rv (Table 1), 20 ng of template DNA, 0.4 U of KOD-Plus DNA polymerase, and 2 to 100 nM Tk-EshA. Five bases at the 5′ end of primer 16 S miss 5′ Rv and five bases at the 3′ end of primer 16 S miss 3′ Rv were not complementary to the template DNA. PCR was performed in a thermal cycler as follows: 2 min at 94°C, followed by 30 cycles of 15 s at 94°C, 30 s at 60°C, and 2 min at 68°C. The target DNA was amplified as 1,498-bp DNA with primers 16S rRNA gene Fw and 16S rRNA gene Rv. In contrast, an 805-bp DNA was synthesized by primers 16 S miss 5′ Rv and 16S rRNA gene Fw. Another misamplified product (800 bp) was synthesized by primers 16 S miss 3′ Rv and 16S rRNA gene Fw. PCR was performed with specific primers, 16S rRNA gene Fw, 16S rRNA gene Rv, and 16 S miss 5′ Rv or 16 S miss 3′ Rv, in the presence or absence of Tk-EshA. The products were separated by 2% (wt/vol) agarose gel electrophoresis and visualized by EtBr staining.
Effect of Tk-EshA on toxA amplification.
As a high-GC-content target, the toxA region of Pseudomonas aeruginosa chromosomal DNA (bp 1240584 to 1242500; GC content, 69%) was used. PCR was performed in a mixture (20 μl) containing PCR buffer as described above, 0.6 μM (each) primers toxA Fw and toxA Rv (Table 1), 20 ng of template DNA, 0.4 U of KOD-Plus DNA polymerase, and 50 or 100 nM Tk-EshA. PCR was performed in a thermal cycler as follows: 2 min at 96°C followed by 28 cycles of 15 s at 96°C, 30 s at 62°C, 65°C, or 68°C, and 2.5 min at 68°C. The sample was separated by 2% (wt/vol) agarose gel electrophoresis and visualized by EtBr staining. The PCR efficiency of the resultant toxA DNA (1,917 bp) was evaluated.
Effect of Tk-EshA on PCR by family A DNA polymerases.
Enzymes from T. aquaticus and Thermus thermophilus were used as family A DNA polymerases (PolI type). Genomic DNA from T. kodakarensis was used as a template, and the region of 16S rRNA genes (bp 2022864 to 2024361) was targeted. PCR was performed in a mixture (20 μl) containing 20 mM Tris HCl (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 4 mM MgSO4, 0.1% Triton X-100, 0.2 mM dNTPs, 0.6 μM (each) primers 16S rRNA gene Fw and 16S rRNA gene Rv (Table 1), 20 ng of template DNA, 0.5 U of Taq DNA polymerase (New England BioLabs Japan, Tokyo) or 2.5 U of Tth DNA polymerase (Roche Diagnostics Japan, Tokyo), and 2 to 100 nM Tk-EshA. PCR was performed in a thermal cycler as follows: 2 min at 94°C, followed by 30 cycles of 15 s at 94°C, 30 s at 60°C, and 2 min at 68°C. The sample was separated by 2% (wt/vol) agarose gel electrophoresis and visualized by EtBr staining. The resultant DNA (1,498 bp) was evaluated.
Preparation of substrate DNAs for helicase activity.
To examine unwinding activity for helicase evaluation, various double-stranded DNA (dsDNA) substrates (forked, 5′ overhung, 3′ overhung, and blunt end) were prepared. The oligonucleotide sequences used in this study were modified based on a previous report (29). Oligonucleotides L-HJ-3-54mer, L-5′DNA34, and L-3′DNA54 (Table 2) were fluorescently labeled at the 5′ terminus with IRDye 700 (Integrated DNA Technologies, Coralville, IA). To prepare forked, 5′ overhung, 3′ overhung, and blunt-end DNAs, the labeled oligonucleotides L-HJ-3-54mer, L-5′DNA34, L-3′DNA54, and L-HJ-3-54mer were annealed with nonlabeled N-HJ-4, N-HJ-3-54mer, N-5′DNA34, and N-B-DNA54, respectively, in a solution containing 20 mM Tris HCl (pH 8.0) and 50 mM NaCl, heated at 95°C for 3 min, and cooled to 25°C for 1 h. To prepare the RNA/DNA duplex substrate, oligonucleotide L-RNA54 labeled at the 5′ terminus with Cy5.5 (Integrated DNA Technologies) was annealed with nonlabeled N-DNA70 (Table 2) under the same conditions as for dsDNA substrates.
Monitoring the unwinding activity of helicases.
The unwinding assay of helicases was performed using a trap DNA according to a previous report, with slight modifications (29). Trap DNA was included to capture produced single-stranded DNA at the time of double-strand separation. The reaction was carried out at 50°C for 40 min in a mixture (20 μl) containing 20 mM Tris HCl (pH 8.0), 10 mM MgSO4, 2 mM DTT, 0.01% BSA, 0.01% Triton X-100, 3.3 mM ATP, 2 pmol of the DNA substrate, 4 or 10 pmol of the trap DNA, and a helicase (Tk-DeaD, Tk-EshA, TK0928, or Tk-EshA-D344A-E345A). When blunt-end dsDNA was used, 10 pmol of the trap DNA was added to the reaction mixture. The sequence of the trap DNA (N-DNA34) for 5′ overhung (L-5′DNA34 and N-HJ-3-54mer) and 3′ overhung (L-3′DNA54 and N-5′DNA34) DNAs is shown in Table 2. The N-5′DNA34 oligonucleotide was used to trap single-stranded DNA from forked (L-HJ-3-54mer and N-HJ-4) and RNA/DNA hybrid (L-RNA54 and N-DNA70) substrate DNAs, and N-HJ-3-54mer was used for blunt-end substrate DNA (L-HJ-3-54mer and N-B-DNA54) (Table 2). The unwinding reaction was terminated by immediately transferring the tube to ice. Thereafter, 1 μl of loading buffer (0.5 mg · ml−1 bromophenol blue, 50% glycerol, 100 mM EDTA, and 1% SDS) and 7 μl of water were added to 2 μl of the reaction sample. These samples (10 μl) were resolved on a 13% native polyacrylamide gel by electrophoresis at 15 mA for 50 min. Fluorescence on the gel was detected by an Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE). The unwinding activity of helicases was quantified by measuring band intensities using the application software (Li-Cor).
RESULTS
Temperature dependency of ATPase activity.
Three candidate SF2 helicases were selected based on differences in their molecular masses (Tk-DeaD, 46 kDa; TK0566, 96 kDa; TK0928, 53 kDa). TK0566 and TK0928 were expressed in E. coli cells, and the recombinant forms were purified to near homogeneity (Fig. 1A). Helicases unwind folded DNA and/or RNA, generally coupled with NTP hydrolysis. The ATPase activity of the DEAD box RNA helicase (Tk-DeaD) was described previously (27), and the optimum temperature for Tk-DeaD activity, which is dependent on the presence of ssRNA, is 50°C. To examine the enzymatic properties of TK0566 and TK0928, ATPase activity for ssRNA was first measured at 30°C to 110°C by monitoring phosphate released from ATP. TK0566 and TK0928 showed the highest ATPase activity at 80°C and 90°C, respectively (Fig. 1B). The highest ATPase activity of TK0566 (158 nmol/min/mg) was almost 5.3-fold higher than that of TK0928 (30 nmol/min/mg). To examine the nucleic acid dependency of helicase, ATPase activity of TK0566 and TK0928 for ssDNA was also measured at 80°C and 90°C, respectively. ATPase activity of TK0566 showed no significant difference in the presence of ssDNA or of ssRNA (Fig. 1B). ATPase activity of TK0928 in the presence of ssDNA was almost 3-fold lower than in the presence of ssRNA (Fig. 1B). The ATPase activity in the absence of RNA or DNA was slightly detected by adding TK0566 (13.7 nmol/min/mg) and TK0928 (0.19 nmol/min/mg). They would be caused by prebound RNA derived from E. coli, as E. coli cells were used for recombinant helicase expression.
FIG 1.

ATPase activity of Tk-EshA, TK0928, and Tk-EshA-D344A-E345A. (A) SDS-PAGE with Coomassie brilliant blue staining of purified recombinant proteins. Lane M1, molecular mass markers (β-galactosidase, 120,000 Da; BSA, 95,000 Da; glutamine dehydrogenase, 68,000 Da; ovalbumin, 50,000 Da; carbonic anhydrase, 36,000 Da; myoglobin, 27,000 Da; lysozyme, 20,000 Da; and aprotinin, 10,000 Da); lane M2, molecular mass markers (phosphorylase b, 97,000 Da; albumin, 66,000 Da; ovalbumin 45,000 Da; and trypsin inhibitor, 20,100 Da); lane 1, Tk-EshA (TK0566); lane 2, TK0928; and lane 3, Tk-EshA-D344A-E345A. (B) Temperature-dependent ATPase activity of Tk-EshA (■, left panel), TK0928 (▲, center panel), and Tk-EshA-D344A-E345A (◽, right panel), which depends on the presence of ssRNA. ATPase activities of Tk-EshA (at 80°C) and TK0928 (at 90°C) for ssDNA are shown in the box (Tk-EshA, left panel; TK0928, center panel). The activity for ssDNA is shown relative to that for ssRNA, set to 100%.
Unwinding activity of thermostable helicases.
To assess the enzymatic property, the unwinding activity of thermostable helicases (Tk-DeaD, TK0566, and TK0928) for forked double-stranded DNA was measured. The substrate was fluorescently labeled with 5′ IRDye 700, and a trap DNA was added to prevent reannealing of the unwound DNA during the unwinding reaction. As a result, by adding TK0566, the intensity of forked DNA was decreased, and dsDNA with trap DNA was increased (Fig. 2B). Addition of Tk-DeaD also reduced the intensity of forked DNA, but its effect was less than that of adding TK0566 (Fig. 2A). On the other hand, unwound product for TK0928 was not observed (Fig. 2C). This result showed that Tk-DeaD and TK0566 had an unwound activity for forked dsDNA but that TK0928 did not unwind the forked dsDNA at 50°C. Based on the amino acid sequence, TK0566 orthologs have been classified in the group of Archaea-specific helicases (21), which is specific to Euryarchaeota, with the exception of Thermoproteales and Archaeoglobales (21). The ortholog is not conserved in the Crenarchaeota. In the present study, TK0566 has been designated a Euryarchaeota-specific helicase, EshA (Tk-EshA), and used for further study. The Walker B motif of helicase is known to play an important role for ATPase activity (15). We constructed mutant Tk-EshA by replacing conserved Asp344 and Glu345 with Ala, and the mutant was designated Tk-EshA-D344A-E345A. ATPase activity of Tk-EshA-D344A-E345A was measured at 30°C to 100°C in the presence of ssDNA, and no activity was detected at any of the temperatures examined (Fig. 1B). In addition, unwinding activity of Tk-EshA-D344A-E345A for forked dsDNA was not detected (Fig. 2D).
FIG 2.
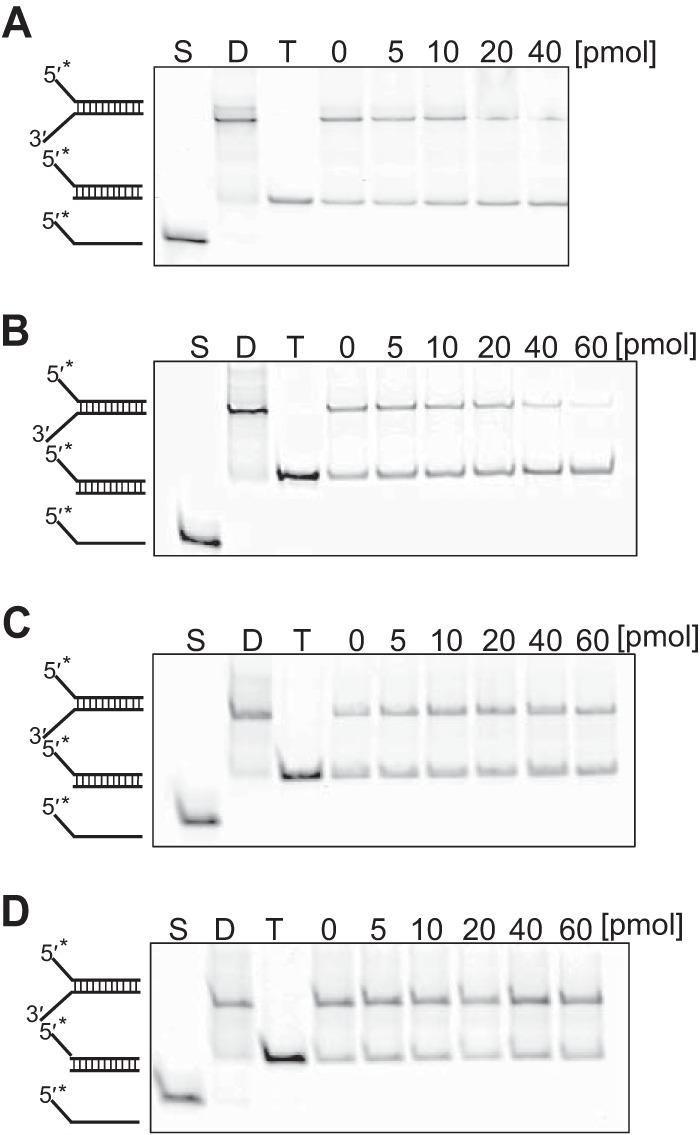
Unwinding activity of thermostable helicase for forked dsDNA. Native PAGE demonstration was used in detecting the helicase activity for forked dsDNA. Tk-DeaD (A), Tk-EshA (B), TK0928 (C), or Tk-EshA-D344A-E345A (D) was added to assay mixtures containing 2 pmol of forked DNA labeled with 5′ IRDye 700. The amount of Tk-EshA was 0, 5, 10, 20, 40, or 60 pmol. *, fluorescent label; S, single-stranded DNA; D, dsDNA of each substrate; T, trapped dsDNA.
Effects of thermostable helicases on PCR specificity.
To examine the effects of Tk-DeaD, Tk-EshA, and TK0928 on PCR amplification, the helicases were added to PCR mixtures, and the amplified patterns of DNA were compared. The genomic DNA of T. kodakarensis was used as a template, and full-length 16S rRNA genes (1,498 bp) were amplified by a DNA polymerase (KOD polymerase). Misamplified products (noise DNAs) were detected in the absence of helicases (Fig. 3A). In contrast, when 50 nM Tk-EshA was added to the PCR mixture, the signals of noise DNAs were decreased (Fig. 3A). The addition of an excess amount of Tk-EshA (100 nM) almost eliminated noise DNAs and partially decreased the target product. However, by increasing PCR cycles from 28 to 45, the target band was detected in the presence of 100 nM Tk-EshA. In the presence of Tk-DeaD or TK0928, no significant difference was observed. Noise DNAs were still detected in the presence of 100 nM Tk-DeaD or TK0928. Because TK0928 showed a lower ATPase activity and did not show unwound activity, an even larger concentration of it (1 μM) was added to the PCR mixture; however, no DNA amplification was observed (data not shown). We concluded that only Tk-EshA shows a unique noise reduction effect in PCR among the tested SF2 enzymes. To determine the effect of Tk-EshA on PCR, the amounts of amplified DNAs were compared by measuring the intensity of each band on the agarose gel. In the absence of Tk-EshA, three bands (band a, target; band b, noise; band c, noise) were detected (Fig. 3A and C). By increasing the amount of Tk-EshA, the signals of noise DNAs (bands b and c) were gradually decreased. The signal of the targeted DNA (band a) was also gradually decreased by increasing the amount of Tk-EshA (Fig. 3C); however, the rate of this decrease was lower than those of the other two bands. This result suggested that Tk-EshA dominantly inhibits the misannealing of primers and decreases misamplification during PCR, resulting in specific DNA amplification of the region targeted by primers. An excess amount of Tk-EshA inhibited specific DNA amplification. Tk-EshA-D344A-E345A was also added to the PCR mixture, and the effect was examined (Fig. 3B). In the presence of Tk-EshA-D344A-E345A, noise DNAs still remained, indicating that Tk-EshA-D344A-E345A does not show specific noise reduction. In the presence of a high concentration (100 nM) of Tk-EshA-D344A-E345A, DNA amplification was inhibited. An excess amount of Tk-EshA-D344A-E345A inhibited DNA amplification, probably due to unspecific Tk-EshA-D344A-E345A binding to DNA, resulting in prevention of DNA polymerase access.
Substrate specificity of Tk-EshA.
We measured the unwinding activity of Tk-EshA for various DNA substrates (5′ overhung, 3′ overhung, and blunt end) to understand how Tk-EshA unwinds misannealed primer/template duplexes. These substrates were fluorescently labeled with 5′ IRDye 700, and a trap DNA was added to prevent reannealing of the unwound DNA during the unwinding reaction. The intensity of 3′ overhung DNAs was decreased and the intensity of duplex DNA with trap DNA was increased by increasing the amount of Tk-EshA (Fig. 4A, upper panel). In contrast, unwound molecules were not produced by Tk-EshA from 5′ overhung or blunt-end DNAs (Fig. 4A, middle and lower panels). The relative unwinding efficiency of Tk-EshA for each substrate is compared with that for forked DNA set to 100% in Fig. 4B. The unwinding activity for 3′ overhung DNA was the highest among the substrates (Fig. 4B), indicating that Tk-EshA unwound misannealed primers from the 3′ terminus. In addition, we measured the unwinding activity of Tk-EshA for RNA/DNA hybrid duplexes. The intensity of RNA/DNA hybrid duplexes was decreased and that of DNA duplexes with trap DNA was increased by increasing the amount of Tk-EshA (Fig. 5). This result showed that Tk-EshA also unwound RNA/DNA hybrid duplexes.
FIG 4.
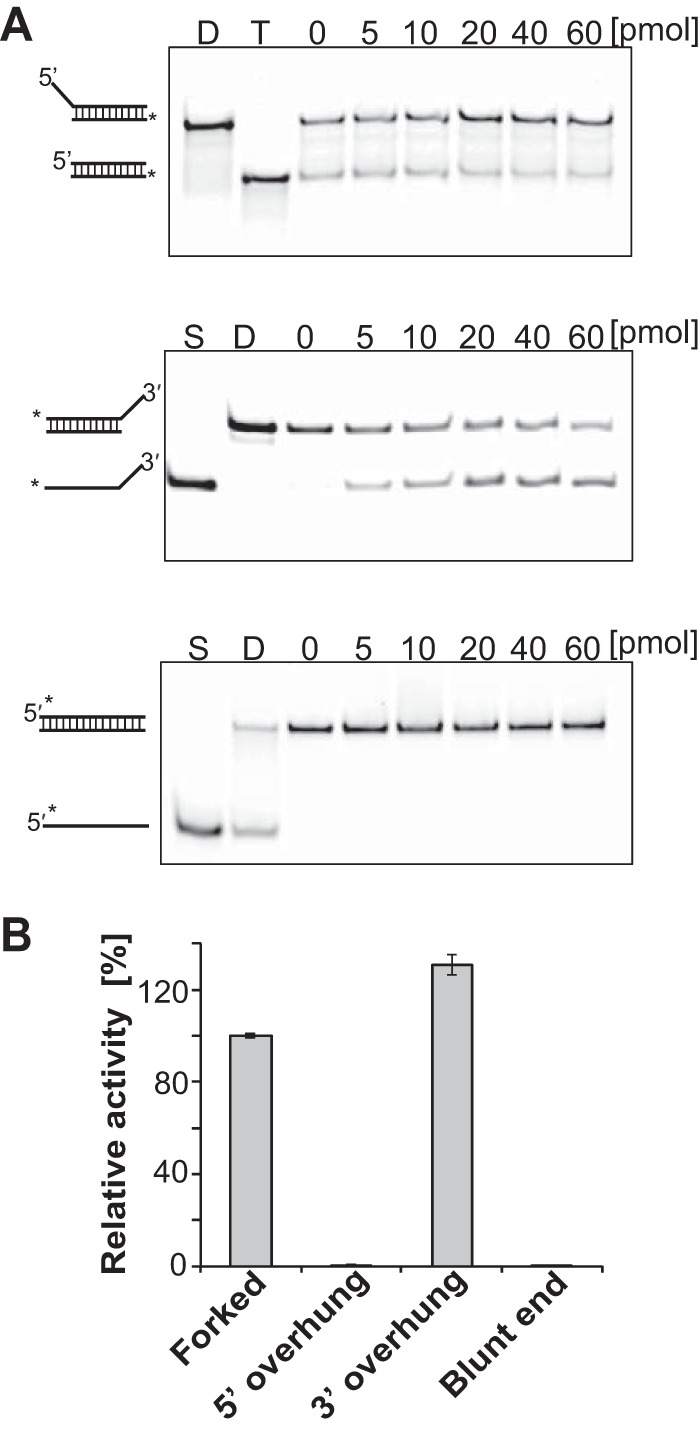
Substrate specificity of Tk-EshA. (A) Native PAGE demonstration used in the quantitative helicase assay. Tk-EshA was added to assay mixtures containing 2 pmol 5′ overhung (upper panel), 3′ overhung (middle panel), or blunt-end (lower panel) DNA labeled with 5′ IRDye 700. The amount of Tk-EshA was 0, 5, 10, 20, 40, or 60 pmol. *, fluorescent label; S, single-stranded DNA; D, dsDNA of each substrate; T, trapped dsDNA. Because the substrates were trapped by the trap DNA when unwound by Tk-EshA, the position of the substrate band changed. (B) Activities of Tk-EshA for various substrates (forked, 5′ overhung, 3′ overhung, and blunt-end DNAs) relative to that for forked DNA, set to 100%.
FIG 5.
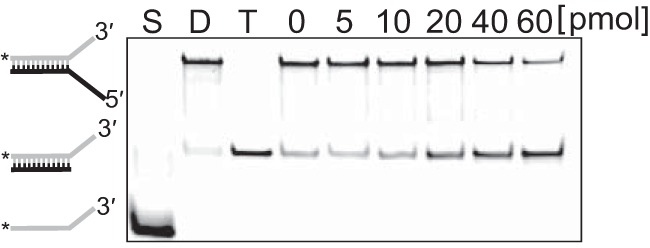
Unwinding activity of Tk-EshA for DNA/RNA hybrid duplexes. Native PAGE demonstration used in the detection of helicase activity. Tk-EshA was added to assay mixtures containing 2 pmol of the RNA/DNA hybrid composed of oligonucleotides, RNA54 labeled at the 5′ terminus with Cy5.5, and DNA70. The amount of Tk-EshA was 0, 5, 10, 20, 40, or 60 pmol. Gray line, RNA; black line, DNA; *, fluorescent label; S, ssRNA; D, RNA/DNA hybrid substrate; T, trapped RNA/DNA hybrid.
Effect of Tk-EshA on PCR containing competitive misannealed primers.
To evaluate the action of Tk-EshA on partially annealed primers, oligonucleotides that partially anneal to the target region were designed. Five bases at the 5′ end of primer 16 S miss 5′ Rv and five bases at the 3′ end of primer 16 S miss 3′ Rv were designed to be partially complementary to the target 16S rRNA genes. Full-length 16S rRNA genes (1,498 bp) were amplified by primers 16S rRNA gene Fw and 16S rRNA gene Rv (Fig. 6A, lane FR). When primers 16 S miss 5′ Rv and 16S rRNA gene Fw were used for DNA amplification, an 805-bp DNA was produced (Fig. 6A, lane FR5′R). Furthermore, when primers 16 S miss 3′ Rv and 16S rRNA gene Fw were used, an 800-bp DNA was produced (Fig. 6A, lane FR3′R). Amplification of 16S rRNA genes was performed with primers 16S rRNA gene Fw and 16S rRNA gene Rv in the presence of primer 16 S miss 3′ Rv or 16 S miss 5′ Rv. In the absence of Tk-EshA, the target band (1,498 bp) and noise bands (805 bp and 800 bp) were detected in PCR conditions with a 5′ overhung primer (16 S miss 5′ Rv) and a 3′ overhung primer (16 S miss 3′ Rv). In the presence of 50 nM Tk-EshA, noise DNAs (800 bp and 805 bp) produced by misannealed primers disappeared, while target DNA (1,498 bp) was detected (Fig. 6B and C).
FIG 6.

Effect of Tk-EshA addition on PCR in the presence of competitive misannealing primers. (A) The amplification pattern in the absence of Tk-EshA is shown. Lane F, in the presence of only forward primer, 16S rRNA gene Fw; lane R, in the presence of only reverse primer, 16S rRNA gene Rv; lane FR, in the presence of 16S rRNA gene Fw and 16S rRNA gene Rv; lane FR5′R, in the presence of 16S rRNA gene Fw, 16S rRNA gene Rv, and competitive primer (16 S miss 5′ Rv); and lane FR3′R, in the presence of 16S rRNA gene Fw, 16S rRNA gene Rv, and competitive primer (16 S miss 3′ Rv). 16S rRNA genes (1,498 bp) of T. kodakarensis each were targeted in PCR. (B and C) The amplification patterns with competitive primers in the presence of Tk-EshA are shown. PCR was performed with primers 16S rRNA gene Fw and 16S rRNA gene Rv in the presence of the misannealing primer 16 S miss 5′ Rv (5′ overhung) (B) or 16 S miss 3′ Rv (3′ overhung) (C). DNA (1,498 bp) amplified by primers 16S rRNA gene Fw and 16S rRNA gene Rv is indicated by a black arrowhead. DNAs (800 bp and 805 bp) amplified by primer 16S rRNA gene Fw and primer 16 S miss 5′ Rv or primer 16 S miss 3′ Rv are indicated by white arrowheads. The concentration of Tk-EshA was 0, 2, 10, 50, or 100 nM. Lane M shows the 100-bp DNA ladder.
Effect of Tk-EshA addition on PCR efficiency with a high-GC-content DNA template.
Generally, PCR from high-GC-content templates is not successfully completed due to unfavorable secondary structures formed in the DNA template and/or misannealing of primers. Tk-EshA was examined for the amplification of high-GC-content template DNAs. Genomic DNA from Pseudomonas aeruginosa was used as the template, and full-length toxA (length, 1,917 bp; GC content, 69%) was amplified by a DNA polymerase (KOD-Plus polymerase) from T. kodakarensis. Some noise DNAs were detected in addition to toxA DNA in the absence of Tk-EshA (Fig. 7). These noise DNAs were not eliminated by increasing the annealing temperature from 62°C to 68°C. In contrast, noise DNAs were eliminated by adding 50 nM Tk-EshA. When 100 nM Tk-EshA was added, toxA DNA was also eliminated. This result means that Tk-EshA reduced the level of misamplified DNAs when added at an appropriate concentration but also reduced target DNA amplification when an excess amount was added.
FIG 7.
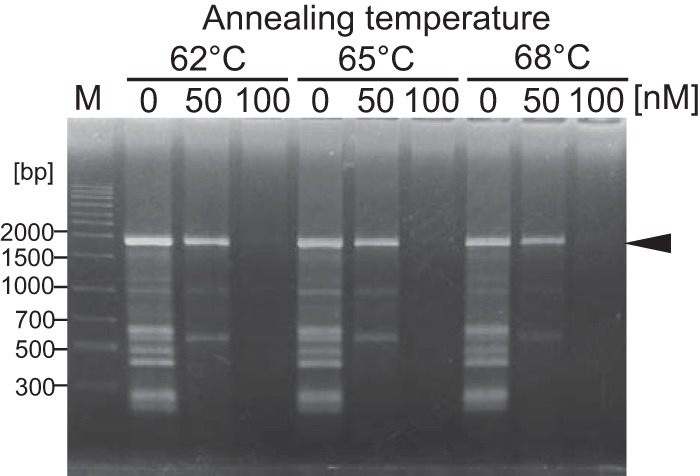
Effect of Tk-EshA addition on PCR with a high-GC-content template. The toxA region (1,917 bp) with a high GC content (69%) from the Pseudomonas aeruginosa genome was amplified in the presence or absence of Tk-EshA. The annealing temperature in the PCR was 62°C, 65°C, or 68°C. The concentration of Tk-EshA was 0, 50, or 100 nM. DNA size markers are shown in lane M. The target DNA (1,917 bp) amplified by primers toxA Fw and toxA Rv is indicated by a black arrowhead.
Effect of Tk-EshA on PCR by family A DNA polymerases from T. aquaticus and T. thermophilus.
The above-mentioned experiments were carried out using a family B DNA polymerase (KOD polymerase from T. kodakarensis). Another type of DNA polymerase (family A) from T. aquaticus (Taq polymerase) and T. thermophilus (Tth polymerase) is also commonly used for PCR. Taq and Tth polymerases belong to family A and do not possess proofreading activity. We examined the effect of Tk-EshA on PCR using Taq and Tth polymerases. Genomic DNA from T. kodakarensis was used as the template, and full-length 16S rRNA genes (1,498 bp) were amplified. Noise DNAs amplified by Taq and Tth polymerases were also eliminated by Tk-EshA (Fig. 8A and B), as in the case of the family B DNA polymerase (KOD polymerase from T. kodakarensis).
FIG 8.
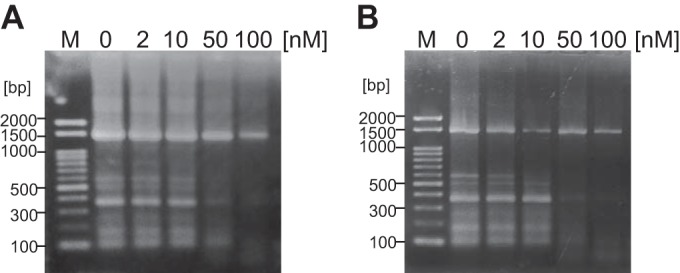
Effect of Tk-EshA addition on PCR by family A DNA polymerases. 16S rRNA genes (1,498 bp) from the T. kodakarensis genome were targeted in PCR. 16S rRNA genes were amplified in the presence or absence of Tk-EshA. The concentration of Tk-EshA was 0, 2, 10, 50, or 100 nM. Lane M shows the 100-bp DNA ladder. PCR results using a DNA polymerase from T. aquaticus (A) or from T. thermophilus (B) are shown.
DISCUSSION
PCR is a technique that is used in various genetic experiments, such as gene cloning, genotyping, and mutation introduction into DNA. It is also applied to investigate genetic diagnosis and detect pathogenic microorganisms. However, accurate DNA amplification is often hampered by the misannealing of primers, especially when long DNAs and high-GC-content DNAs are targeted. DNA/RNA helicases eliminate hydrogen bonds between DNA or RNA bases using energy from ATP hydrolysis. Helicases are expected to unwind the secondary structure of the template and misannealed primer/template duplexes in PCR. To achieve noise reduction in PCR by eliminating primer misannealing, a thermostable helicase was included in PCR amplifications. Three putative SF2 helicases from T. kodakarensis were examined. The enzyme characteristics of Tk-DeaD were examined previously (27). It exhibits maximal ATPase activity and unwinding activity specific for single-strand paired RNA at 50°C, which is lower than the temperature growth limit of T. kodakaraensis. The optimum temperatures for Tk-EshA and TK0928 activity were 80°C and 90°C, respectively (Fig. 1B). Tk-DeaD and Tk-EshA showed unwinding activity for forked DNA, while TK0928 did not show the activity (Fig. 2A to C). Among the tested helicases, only TK0566, designated Tk-EshA (Euryarchaeota-specific helicase from T. kodakarensis), showed a noise reduction effect (Fig. 3A). Tk-DeaD also showed slight unwinding activity for forked DNA. However, the thermolabile property of Tk-DeaD would not be suitable for noise reduction in PCR. In the present study, the amount of desired amplification products relative to the amount of nonspecific products was increased depending on the concentration of Tk-EshA (Fig. 3C). Tk-EshA unwound 3′ overhung DNA (Fig. 4), suggesting that Tk-EshA unwinds misannealed primer/template duplexes from the 3′ terminus during the annealing step of PCR, while primers perfectly bound to the target region are not peeled off, because a 5′ overhang region forms (Fig. 9). This idea is supported by data showing that Tk-EshA did not unwind 5′ overhung or blunt-end DNA duplexes. In addition, Tk-EshA dominantly unwound misannealed 5′ overhung and 3′ overhung primers rather than target-specific primers (Fig. 6). This result suggests that Tk-EshA unwound the 3′ overhung primer (16 S miss 3′ Rv) from template DNA or primer and peeled it off. Tk-EshA also reduced noise DNAs produced by the 5′ overhung primer (16 S miss 5′ Rv) and primer 16S rRNA gene Fw, suggesting that Tk-EshA accesses the 3′ end of the template and unwinds primer/template duplexes. When an excess amount of Tk-EshA was added, the specific target DNA was eliminated. Tk-EshA might attack the primer that anneals to the specific target, and DNA synthesis by DNA polymerases might be inhibited by Tk-EshA, because it competes for Mg2+ for the polymerization reaction. When Tk-EshA is used to reduce PCR noise, a series of Tk-EshA concentrations should be prepared in advance. By adding Tk-EshA, noise bands were efficiently eliminated in the amplification of GC-rich toxA (Fig. 7). Tk-EshA addition was more effective than increasing the annealing temperature. As for the mechanism underlying the effect of Tk-EshA, two possibilities are considered. One is a primer-peeling action, as shown in Fig. 9. Tk-EshA is predicted to peel off partially annealed primers. The second possibility is that Tk-EshA enhances the unwinding of template DNA. Tk-EshA could unwind template DNA, resulting in the easy access of primers to the target. Tk-EshA seems to be functional by one or both of these mechanisms.
FIG 9.

Model for noise reduction in PCR by Tk-EshA. In the absence of Tk-EshA, primers misanneal to various homologous regions, resulting in the amplification of noise DNAs. In the presence of Tk-EshA, misannealed primers are peeled off, and specific primers dominantly anneal to the target region, resulting in the reduced amplification of noise DNAs.
Comparative analysis found Tk-EshA orthologs only in the phylum Euryarchaeota of the domain Archaea, not in the domains Bacteria and Eucarya. Tk-EshA orthologs were identified in methanogens and extreme halophiles but not in thermophilic bacteria. Euryarchaeota-specific helicase (ESH) possesses the distinct consensus sequence rich in cysteine at the N terminus. This region is predicted to be a metal-binding motif (21). Tk-EshA also has a cysteine-rich sequence. Its Euryarchaeota-specific existence led us to hypothesize that Tk-EshA is functional only for family B DNA polymerases and not for family A DNA polymerases, because family A DNA polymerase is not found in the domain Archaea. However, Tk-EshA was effective for family A DNA polymerases in our evaluation. Taq and Tth DNA polymerases are classified into family A and used for PCR. Tk-EshA also inhibited misamplification in the family A polymerase reaction (Fig. 8). Taq and Tth polymerases are applicable for the amplification refractory mutation system (ARMS) to detect SNPs in genetic diagnosis, because they do not possess 3′-to-5′ exonuclease activity. Tk-EshA would be useful in reducing the occurrence of false positives.
The elimination of nonspecific amplification is important for a wide range of PCR-based technologies, including the detection of viral and bacterial infections and identification of SNPs. RecA and MutS are used to enhance the specificity of PCR (11–14). Given that Tk-EshA functions in a different manner than RecA and MutS, a combined effect is expected by using them together. Furthermore, Tk-EshA seems to be applicable not only for PCR but also for other methods to detect nucleic acids, such as transcription-mediated amplification, ligase chain reaction, and nucleic acid sequence-based amplification (32–37), because Tk-EshA also unwinds RNA/DNA hybrids (Fig. 5).
ACKNOWLEDGMENTS
This research was mainly supported by a research grant (AS262Z02420P to S.F.) from JST (Japan Science and Technology Agency, Research for Promoting Technological Seeds) A-STEP. The investigation of helicase activity for RNA/DNA duplexes was supported by a grant from JST-SENTAN. Bioinformatic analysis was supported by a grant from the Japan Society for the Promotion of Science (JSPS) (KAKENHI grant 26292045).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Mullis KB, Faloona FA. 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol 155:335–350. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- 2.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 3.Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. 1985. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 4.Ehling-Schulz M, Fricker M, Scherer S. 2004. Identification of emetic toxin producing Bacillus cereus strains by a novel molecular assay. FEMS Microbiol Lett 232:189–195. doi: 10.1016/S0378-1097(04)00066-7. [DOI] [PubMed] [Google Scholar]
- 5.Duggan DB, Ehrlich GD, Davey FP, Kwok S, Sninsky J, Goldberg J, Baltrucki L, Poiesz BJ. 1988. HTLV-I-induced lymphoma mimicking Hodgkin's disease. Diagnosis by polymerase chain reaction amplification of specific HTLV-I sequences in tumor DNA. Blood 71:1027–1032. [PubMed] [Google Scholar]
- 6.Victor T, du Toit R, van Zyl J, Bester AJ, van Helden PD. 1991. Improved method for the routine identification of toxigenic Escherichia coli by DNA amplification of a conserved region of the heat-labile toxin A subunit. J Clin Microbiol 29:158–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roux KH. 2009. Optimization and troubleshooting in PCR. Cold Spring Harb Protoc 2009:pdb.ip66. [DOI] [PubMed] [Google Scholar]
- 8.Chou Q, Russell M, Birch DE, Raymond J, Bloch W. 1992. Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications. Nucleic Acids Res 20:1717–1723. doi: 10.1093/nar/20.7.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kellogg DE, Rybalkin I, Chen S, Mukhamedova N, Vlasik T, Siebert PD, Chenchik A. 1994. TaqStart antibody: hot start PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase. Biotechniques 16:1134–1137. [PubMed] [Google Scholar]
- 10.Mizuguchi H, Nakatsuji M, Fujiwara S, Takagi M, Imanaka T. 1999. Characterization and application to hot start PCR of neutralizing monoclonal antibodies against KOD DNA polymerase. J Biochem 126:762–768. doi: 10.1093/oxfordjournals.jbchem.a022514. [DOI] [PubMed] [Google Scholar]
- 11.Stefanska A, Gaffke L, Kaczorowska AK, Plotka M, Dabrowski S, Kaczorowski T. 4 September 2015. Highly thermostable RadA protein from the archaeon Pyrococcus woesei enhances specificity of simplex and multiplex PCR assays. J Appl Genet. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12.Shigemori Y, Mikawa T, Shibata T, Oishi M. 2005. Multiplex PCR: use of heat-stable Thermus thermophilus RecA protein to minimize nonspecific PCR products. Nucleic Acids Res 33:e126. doi: 10.1093/nar/gni111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukui K, Bessho Y, Shimada A, Yokoyama S, Kuramitsu S. 2013. Thermostable mismatch-recognizing protein muts suppresses nonspecific amplification during polymerase chain reaction (PCR). Int J Mol Sci 14:6436–6453. doi: 10.3390/ijms14036436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisen JA. 1998. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res 26:4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singleton MR, Dillingham MS, Wigley DB. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 16.An L, Tang W, Ranalli TA, Kim HJ, Wytiaz J, Kong H. 2005. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J Biol Chem 280:28952–28958. doi: 10.1074/jbc.M503096200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong YJ, Park K, Kim DE. 2009. Isothermal DNA amplification in vitro: the helicase-dependent amplification system. Cell Mol Life Sci 66:3325–3336. doi: 10.1007/s00018-009-0094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Artiushin S, Tong Y, Timoney J, Lemieux B, Schlegel A, Kong H. 2011. Thermophilic helicase-dependent DNA amplification using the IsoAmp SE experimental kit for rapid detection of Streptococcus equi subsp. equi in clinical samples. J Vet Diagn Invest 23:909–914. doi: 10.1177/1040638711416968. [DOI] [PubMed] [Google Scholar]
- 19.Runyon GT, Lohman TM. 1989. Escherichia coli helicase II (UvrD) protein can completely unwind fully duplex linear and nicked circular DNA. J Biol Chem 264:17502–17512. [PubMed] [Google Scholar]
- 20.Atomi H, Fukui T, Kanai T, Morikawa M, Imanaka T. 2004. Description of Thermococcus kodakaraensis sp. nov., a well studied hyperthermophilic archaeon previously reported as Pyrococcus sp. KOD1. Archaea 1:263–267. doi: 10.1155/2004/204953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chamieh H, Ibrahim H, Kozah J. 2016. Genome-wide identification of SF1 and SF2 helicases from archaea. Gene 576:214–228. doi: 10.1016/j.gene.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Jankowsky E, Fairman-Williams ME. 2010. An introduction to RNA helicases: superfamilies, families, and major themes, p 1–31. In Jankowsky E (ed), RNA helicases. The Royal Society of Chemistry, London, United Kingdom. [Google Scholar]
- 23.Tanner NK, Linder P. 2001. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell 8:251–262. doi: 10.1016/S1097-2765(01)00329-X. [DOI] [PubMed] [Google Scholar]
- 24.Caruthers JM, McKay DB. 2002. Helicase structure and mechanism. Curr Opin Struct Biol 12:123–133. doi: 10.1016/S0959-440X(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 25.Fairman-Williams ME, Guenther UP, Jankowsky E. 2010. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukui T, Atomi H, Kanai T, Matsumi R, Fujiwara S, Imanaka T. 2005. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res 15:352–363. doi: 10.1101/gr.3003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimada Y, Fukuda W, Akada Y, Ishida M, Nakayama J, Imanaka T, Fujiwara S. 2009. Property of cold inducible DEAD-box RNA helicase in hyperthermophilic archaea. Biochem Biophys Res Commun 389:622–627. doi: 10.1016/j.bbrc.2009.09.038. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Ogino H, Ishino S, Haugland GT, Birkeland NK, Kohda D, Ishino Y. 2014. Activation of the MCM helicase from the thermophilic archaeon, Thermoplasma acidophilum by interactions with GINS and Cdc6-2. Extremophiles 18:915–924. doi: 10.1007/s00792-014-0673-6. [DOI] [PubMed] [Google Scholar]
- 30.Harder KW, Owen P, Wong LK, Aebersold R, Clark-Lewis I, Jirik FR. 1994. Characterization and kinetic analysis of the intracellular domain of human protein tyrosine phosphatase beta (HPTP beta) using synthetic phosphopeptides. Biochem J 298:395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin B, Pallen CJ, Wang JH, Graves DJ. 1985. Use of fluorinated tyrosine phosphates to probe the substrate specificity of the low molecular weight phosphatase activity of calcineurin. J Biol Chem 260:14932–14937. [PubMed] [Google Scholar]
- 32.Stary A, Schuh E, Kerschbaumer M, Gotz B, Lee H. 1998. Performance of transcription-mediated amplification and ligase chain reaction assays for detection of chlamydial infection in urogenital samples obtained by invasive and noninvasive methods. J Clin Microbiol 36:2666–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langabeer SE, Gale RE, Harvey RC, Cook RW, Mackinnon S, Linch DC. 2002. Transcription-mediated amplification and hybridization protection assay to determine BCR-ABL transcript levels in patients with chronic myeloid leukemia. Leukemia 16:393–399. doi: 10.1038/sj.leu.2402392. [DOI] [PubMed] [Google Scholar]
- 34.Fire A, Xu SQ. 1995. Rolling replication of short DNA circles. Proc Natl Acad Sci U S A 92:4641–4645. doi: 10.1073/pnas.92.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demidov VV. 2002. Rolling-circle amplification in DNA diagnostics: the power of simplicity. Expert Rev Mol Diagn 2:542–548. doi: 10.1586/14737159.2.6.542. [DOI] [PubMed] [Google Scholar]
- 36.Compton J. 1991. Nucleic acid sequence-based amplification. Nature 350:91–92. doi: 10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- 37.van der Vliet GM, Schukkink RA, van Gemen B, Schepers P, Klatser PR. 1993. Nucleic acid sequence-based amplification (NASBA) for the identification of mycobacteria. J Gen Microbiol 139:2423–2429. doi: 10.1099/00221287-139-10-2423. [DOI] [PubMed] [Google Scholar]