

Abstract
Transporter-mediated alterations in bile acid disposition may have significant toxicological implications. Current methods to predict interactions are limited by the interplay of multiple transporters, absence of protein in the experimental system, and inaccurate estimates of inhibitor concentrations. An integrated approach was developed to predict altered bile acid disposition due to inhibition of multiple transporters using the model bile acid taurocholate (TCA). TCA pharmacokinetic parameters were estimated by mechanistic modeling using sandwich-cultured human hepatocyte data with protein in the medium. Uptake, basolateral efflux, and biliary clearance estimates were 0.63, 0.034, and 0.074 mL/min/g liver, respectively. Cellular total TCA concentrations (Ct,Cells) were selected as the model output based on sensitivity analysis. Monte Carlo simulations of TCA Ct,Cells in the presence of model inhibitors (telmisartan and bosentan) were performed using inhibition constants for TCA transporters and inhibitor concentrations, including cellular total inhibitor concentrations ([I]t,cell) or unbound concentrations, and cytosolic total or unbound concentrations. For telmisartan, the model prediction was accurate with an average fold error (AFE) of 0.99–1.0 when unbound inhibitor concentration ([I]u) was used; accuracy dropped when total inhibitor concentration ([I]t) was used. For bosentan, AFE was 1.2–1.3 using either [I]u or [I]t. This difference was evaluated by sensitivity analysis of the cellular unbound fraction of inhibitor (fu,cell,inhibitor), which revealed higher sensitivity of fu,cell,inhibitor for predicting TCA Ct,Cells when inhibitors exhibited larger ([I]t,cell/IC50) values. In conclusion, this study demonstrated the applicability of a framework to predict hepatocellular bile acid concentrations due to drug-mediated inhibition of transporters using mechanistic modeling and cytosolic or cellular unbound concentrations.
Introduction
Transporters play a critical role in the absorption, distribution, and elimination of many drugs and endogenous compounds, such as bile acids. Transporter-mediated drug–bile acid interactions may have significant toxicological implications, such as troglitazone- and bosentan-induced hepatotoxicity due to inhibition of the bile salt export pump (BSEP) (Woodhead et al., 2014, Yang et al., 2014). Transporter inhibition assays have been adopted by the pharmaceutical industry or included in the recent regulatory guidelines to predict drug-drug interactions (DDIs) (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf). However, the static method, based on the ratio of total plasma maximum concentration and IC50 or inhibition constant (Ki) of the inhibitor, may not accurately predict the hepatic disposition of victim substrates. Limitations associated with the static method may explain the lack of cholestatic liability of some multidrug resistance-associated protein (MRP)2 and BSEP inhibitors (Dawson et al., 2012; Pfeifer et al., 2013a). To accurately translate transporter inhibition data (i.e., IC50 or Ki) to the prediction of hepatocellular exposure of victim substrates, a number of factors should be considered.
First, hepatic bile acid exposure is regulated by hepatic uptake transporters [e.g., sodium taurocholate-cotransporting polypeptide (NTCP) and organic anion-transporting polypeptides (OATPs)], as well as canalicular (e.g., BSEP) and basolateral efflux transporters (e.g., MRP3 and MRP4). Often, inhibitors of efflux transporters also inhibit uptake transporters, which may exert protective effects (Leslie et al., 2007). However, the static model based on inhibition data from overexpression systems considers uptake and efflux as isolated processes. To overcome this limitation, mechanistic pharmacokinetic modeling coupled with data from sandwich-cultured hepatocytes has been used to deconvolute the relative contribution of various clearance (CL) pathways to the disposition of rosuvastatin, mycophenolic acid, and 3H-taurocholic acid (TCA) (Pfeifer et al., 2013c; Matsunaga et al., 2014; Yang et al., 2015). Transporters are expressed and properly localized in the sandwich-cultured hepatocyte system, which can be used to assess the function of multiple transporters (Yang et al., 2016). Thus, this cellular model is uniquely suited to evaluate the interplay of multiple transport pathways and predict the net effect due to inhibition of multiple transporters on the hepatic disposition of victim substrates.
Secondly, the presence of protein in plasma is an important physiologic factor. However, albumin at physiologic concentrations [e.g., 4% bovine serum albumin (BSA)] (Doherty et al., 2006; Wolf et al., 2008) has not been added routinely into in vitro experimental systems, such as membrane vesicles, to study transporter-based interactions and assess IC50 or Ki values. In addition, according to the free drug hypothesis, the inhibitory effect is driven by the local unbound concentration of inhibitor, which is the cytosolic unbound inhibitor concentration ([I]u,cyt) for efflux transporters, and the medium unbound inhibitor concentration ([I]u,med) for uptake transporters (Smith et al., 2010). Some high-throughput methods have been used to measure cellular total and unbound inhibitor concentrations ([I]t,cell and [I]u,cell, respectively) (Mateus et al., 2013). However, the isolation of cytosol and measurement of cytosolic total and unbound inhibitor concentrations ([I]t,cyt and [I]u,cyt, respectively) add complexity (Pfeifer et al., 2013b). Thus, [I]t,cyt or [I]u,cyt has not been adopted routinely into the prediction of efflux transporter-based drug interactions. The necessity of measuring the cellular unbound fraction of inhibitor (fu,cell,inhibitor) and/or the cytosolic unbound fraction of inhibitor (fu,cyt,inhibitor) needs to be assessed.
The purpose of this study was to develop an integrated approach to predict altered bile acid disposition mediated by inhibition of multiple transporters in sandwich-cultured human hepatocytes (SCHH), with a focus on TCA, a prototypical bile acid. TCA is generally not metabolized and is commonly used in BSEP and NTCP assays because its transport mechanism is well characterized. First, the hepatobiliary disposition of deuterium-labeled TCA (d8-TCA) was characterized in the presence of 4% BSA, and pharmacokinetic parameters were estimated using mechanistic pharmacokinetic modeling. Total hepatocellular concentrations (Ct,Cells) were identified as the most sensitive model output according to sensitivity analysis. The effect of two model inhibitors, telmisartan and bosentan, on TCA Ct,Cells was predicted based on medium and intracellular inhibitor concentrations (i.e., [I]t,cell, [I]u,cell, [I]t,cyt, and [I]u,cyt, separately) and bile acid transporter inhibition data. The predictive performance of the model was evaluated by comparing the simulation results with experimental data and calculating the average fold error (AFE). To determine the necessity of measuring fu,cell,inhibitor for future studies, sensitivity analyses of fu,cell,inhibitor values for the model inhibitors and a set of theoretical inhibitors were performed. Based on the simulation results, a framework was proposed to help guide future study design.
Materials and Methods
Materials.
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. BioCoat cell culture plates and Matrigel were obtained from BD Biosciences (San Jose, CA). QualGro seeding medium and QualGro hepatocyte culture medium were obtained from Qualyst Transporter Solutions (Durham, NC). d8-TCA, d4-TCA (internal standard for d8-TCA), telmisartan, d3-telmisartan (internal standard for telmisartan), bosentan, and ambrisentan (internal standard for bosentan) were obtained from Toronto Research Chemicals (Toronto, Canada). OmniPur BSA (Fraction V, Heat Shock Isolation) was purchased from Thomas Scientific (Swedesboro, NJ). Pierce bicinchoninic acid protein assay was obtained from Thermo Fisher Scientific (Rockford, IL), and the LDH Cytotoxicity Detection Kit was purchased from Roche Diagnostics (Indianapolis, IN).
Sandwich-Cultured Human Hepatocytes.
B-CLEAR-HU Transporter Certified cryopreserved human hepatocytes (lot numbers HUM4045, HUM4061B, and HUM4059 purchased from Triangle Research Laboratories, Durham, NC) were seeded in QualGro seeding medium at a density of 0.4 × 106 cells/well in 24-well BioCoat plates and 1.75 × 106 cells/well in 6-well BioCoat plates, and cultured in a sandwich configuration (overlaid with Matrigel) in QualGro hepatocyte culture medium, as previously reported (Swift et al., 2010). Donors included one Caucasian male, one Caucasian female, and one Hispanic female ranging in age from 2 to 44 years old with a body mass index ranging from 18.3 to 30.
Uptake and Efflux Studies of d8-TCA in SCHH.
Uptake and efflux studies of d8-TCA were performed in SCHH, as reported previously, with minor modifications (Pfeifer et al., 2013c). Briefly, on day 6 of culture, SCHH seeded in 24-well plates were preincubated with standard (Ca2+-containing) or Ca2+-free (Ca2+/Mg2+-free buffer containing EGTA) Hanks’ balanced salt solution (HBSS) for 10 minutes. Incubation with standard HBSS maintains the integrity of the tight junctions, whereas incubation with Ca2+-free HBSS disrupts the tight junctions, allowing the contents in the bile canaliculi to be washed into the medium (B-CLEAR technology; Qualyst Transporter Solutions, Durham, NC). Following preincubation, the uptake phase was initiated by treating the SCHH with dosing solution (1 μM d8-TCA in 0.3 mL/well standard HBSS, with 4% BSA) for up to 20 minutes. At the end of the uptake phase, the dosing solution was removed, and the SCHH were washed twice with standard or Ca2+-free HBSS at 37°C for 1 minute, and incubated with the third application of buffer for a 15-minute efflux. Accumulation of d8-TCA in Cells + Bile (standard HBSS) and Cells (Ca2+-free HBSS) during uptake (2, 5, 10, and 20 minutes) and efflux (2, 5, 10, and 15 minutes) phases was determined by terminal sampling of triplicate wells at each time point. During the efflux phase, incubation buffer also was collected at 2, 5, 10, and 15 minutes. At the end of incubation, the hepatocytes were washed with ice-cold standard HBSS three times, and the samples were stored at −80°C for future analysis.
Determination of Kinetic Parameters for d8-TCA Using Mechanistic Modeling.
A model scheme incorporating linear uptake and efflux CL was adopted (Pfeifer et al., 2013c; Yang et al., 2015) and was fit to d8-TCA Cells + Bile, Cells, and incubation medium total mass-time data from three individual SCHH experiments (Fig. 1A). The model fitting was performed with Phoenix WinNonlin, v6.3 (Certara, St. Louis, MO) using the stiff estimation method and a power model to account for residual error. Differential eqs. 1–5 describe the changes in the amount of TCA in the different compartments in this model.
Fig. 1.

(A) Model schemes depicting disposition of d8-TCA in SCHH using standard (Cells + Bile) HBSS (left) and Ca2+-free (Cells) HBSS (right). (B) d8-TCA mass versus time data in SCHH lysate (left) and incubation buffer (right). Closed symbols/solid lines represent d8-TCA in Cells + Bile or standard HBSS, and open symbols/dashed lines represent d8-TCA in Cells or Ca2+-free HBSS. Experimental data (circles) represent the mean ± S.E.M. (n = 3 SCHH preparations in triplicate per group). The simulated profiles (lines) were generated from eqs. 1–5 using the mean of best-fit parameter estimates from three SCHH datasets (Table 2).
Mass in standard HBSS:
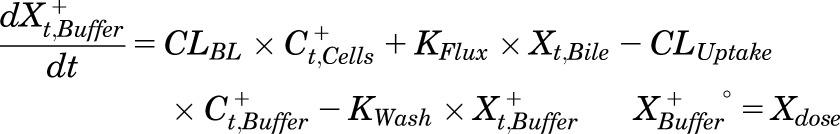 |
(1) |
Mass in Ca2+-free HBSS:
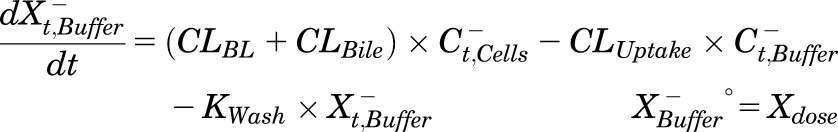 |
(2) |
Mass in Cells:
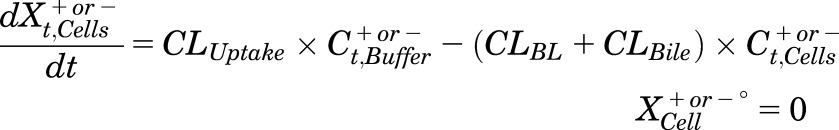 |
(3) |
Mass in Bile (standard HBSS):
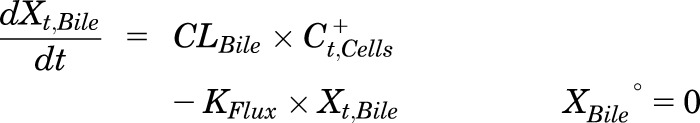 |
(4) |
Mass in Cells + Bile (standard HBSS):
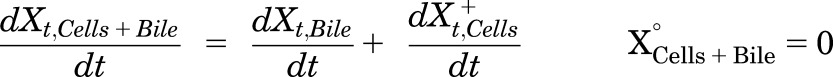 |
(5) |
where Ct,Cells represents the total intracellular concentration, and was calculated as XCells/VCells; VCells was calculated and fixed using the protein content of each preparation and a value of 7.4 μL/mg protein (Pfeifer et al., 2013c; Yang et al., 2015); “+” and “−” refer to Ca2+-containing (standard HBSS) and Ca2+-free HBSS, respectively; Xt,Cells represents the total amount in Cells; Xt,Cells+Bile represents the total amount in Cells + Bile; Xt,Bile represents the total amount in Bile; Ct,Buffer represents the total Buffer concentration; VBuffer was set as a constant (0.3 mL); CLUptake represents total uptake CL; CLBL represents total basolateral efflux CL; CLBile represents total biliary CL; and KFlux represents the first-order rate constant that describes the flux from bile networks into the medium due to periodic contraction of the bile canalicular networks (Oshio and Phillips, 1981; Phillips et al., 1982; Lee et al., 2010; Pfeifer et al., 2013c). CL units (µL/min/mg protein) were converted to mL/min/g liver based on the protein content in liver tissue (90 mg protein/g liver) (Sohlenius-Sternbeck, 2006). To represent the 1-minute wash step, Kwash was activated for 1 minute at the end of the 20-minute uptake phase using an if-then statement. Kwash was fixed at 1 × 104 min−1, which was sufficient to eliminate the d8-TCA from the buffer compartment based on simulations. Initial parameter estimates were obtained from previous reports for 3H-TCA (Yang et al., 2015).
Sensitivity Analyses of Model Output.
Sensitivity analyses were conducted using Berkeley-Madonna v.8.3.11 to identify the most sensitive SCHH model output with respect to changes in CL. Different model outputs measured in the SCHH experiment, including the (A) total concentration of TCA in Cells (Ct,Cells), (B) total amount in Cells + Bile (Xt,Cells+Bile), (C) Xt,Bile, (D) ratio of the total amount of TCA in Cells to the total amount of TCA in Cells + Bile (Xt,Cells/Xt,Cells+Bile), (E) ratio of the total amount of TCA in Bile to the total amount of TCA in Cells (Xt,Bile/Xt,Cells), and (F) ratio of the total amount in Bile to the total amount in Cells + Bile (Xt,Bile/Xt,Cells+Bile), were simulated throughout the time course assuming CLUptake and CLEfflux were inhibited by 0- to 0.99-fold. CLEfflux was defined as CLBile + CLBL, assuming CLBile and CLBL were impaired to the same extent. The simulated fold changes of the model output values at steady state (120 minutes) were plotted against the fraction of inhibition of CLUptake and CLEfflux in a three-dimensional fashion using SigmaPlot v.11, Systat Software Inc. (San Jose, CA). The fraction of inhibition was calculated as (CL − CLinhibitor)/CL, where CL and CLinhibitor represent the CL in the absence and presence of inhibitor, respectively. A higher fraction of inhibition means more potent inhibition.
Determination of Cellular and Cytosolic Total and Unbound Concentrations of Inhibitors ([I]t,cell, [I]t,cyt, [I]u,cell, and [I]u,cyt, respectively).
SCHH seeded in 6-well plates were preincubated with Ca2+-free HBSS for 10 minutes, followed by a 20-minute incubation with dosing solution of model inhibitors (1 and 10 μM telmisartan, or 0.8 and 8 μM bosentan in standard HBSS with 4% BSA). After the incubation, cells were washed with ice-cold HBSS three times and stored at −80°C for future analysis. All the incubations in this study were performed at 37°C.
Cells were fractionated, as reported previously, with minor modification (Pfeifer et al., 2013b). Briefly, hepatocytes from the same treatment group were pooled and homogenized by passing the cells in fractionation buffer through a 27-g needle 20 times to disrupt the cell membranes. The resultant cell lysate was subject to 10,000g centrifugation for 10 minutes at 4°C to isolate cytosol (supernatant) from other cell debris. The protein content of cell lysate was determined by Pierce bicinchoninic acid protein assay. The lactate dehydrogenase (LDH) activity of each fraction (i.e., cell lysate, cytosol, and suspended pellet) was measured using a LDH cytotoxicity detection kit to reflect LDH recovery from cytosol. Glucose-6-phosphatase, succinate dehydrogenase, and acid phosphatase activity of each fraction were measured to assess microsomal, mitochondrial, and lysosomal contamination, respectively. The percentage of the organelle-specific enzyme activity measured in each fraction was calculated in comparison with the whole lysate to assess recovery. The unbound fraction (fu) was determined by equilibrium dialysis, as previously reported (Pfeifer et al., 2013b). Briefly, triplicate aliquots of samples (dosing solution, cell lysate, and cytosol) were loaded into a 96-well equilibrium dialysis apparatus (HTDialysis, Gales Ferry, CT) and incubated at 37°C for 8 hours with shaking, which was sufficient to achieve equilibrium for most compounds (Banker et al., 2003). The fu was back-calculated based on eq. 6 to account for dilution during the homogenization and fractionation process, as described previously (Kalvass et al., 2007).
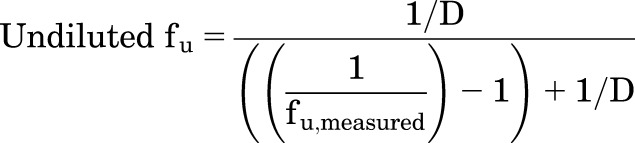 |
(6) |
where D represents the dilution factor.
The total mass of inhibitors in cell lysate and cytosol samples was measured. Cellular concentrations were calculated by dividing the mass by the estimated cellular volume of 7.4 μL/mg protein (Pfeifer et al., 2013c; Yang et al., 2015); cytosolic concentrations were calculated by dividing the mass by the estimated cytosolic volume, assuming cytosolic volume represents 70% of cellular volume (Grunberg et al., 2009). Unbound inhibitor concentrations ([I]u) were calculated as the product of total inhibitor concentration ([I]t) and fu.
Simulation of Inhibitor Effects on TCA Disposition and Comparison with Experimental Results.
d8-TCA Ct,Cells were measured after the following treatments: a 10-minute preincubation of SCHH with telmisartan (1 or 10 μM) or bosentan (0.8 or 8 μM) in Ca2+-free HBSS with 4% BSA, followed by a 10-minute coincubation with d8-TCA (1 μM) and telmisartan or bosentan in standard HBSS with 4% BSA. TCA Ct,Cells after 10-minute uptake were simulated using eqs. 1–5 and CLinhibitor values, which were calculated using eqs. 7–10.
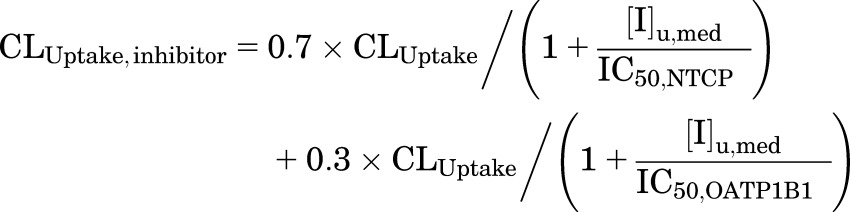 |
(7) |
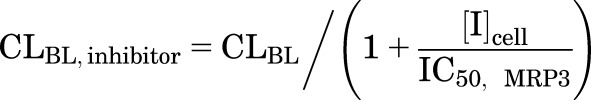 |
(8) |
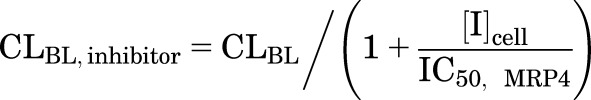 |
(9) |
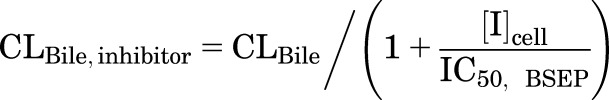 |
(10) |
where CLUptake,inhibitor, CLBL,inhibitor, and CLBile,inhibitor represent the CLUptake, CLBL, and CLBile of TCA in the presence of inhibitors, respectively; [I]u,med represents the unbound concentration of inhibitor in the medium; [I]cell represents the cellular inhibitor concentration; and different types of cellular concentration were used in the simulation, including [I]t,cell, [I]u,cell, [I]t,cyt, [I]u,cyt (obtained as described in the previous section; values are shown in Table 3). The mean IC50 values for each transporter in Table 1 were used with the assumptions that NTCP and OATPs contribute 70% and 30%, respectively, to CLUptake (Shitara et al., 2003; De Bruyn et al., 2014), BSEP mediates CLBile (Noe et al., 2002; Chandra and Brouwer, 2004; Hayashi et al., 2005), and CLBL is governed by MRP3 (Zhang et al., 2003) or MRP4 (Rius et al., 2006). Because the relative contribution of MRP3 and MRP4 is unknown, two extremes were simulated assuming MRP3 (eq. 8) or MRP4 (eq. 9) contributes 100% to CLBL. Monte Carlo simulations of 40 individuals were performed 10 times using parameter estimates and the associated variance (Table 2); CL was assumed to be normally distributed. The fold changes in the TCA Ct,Cells in the presence versus the absence of inhibitors were calculated and compared between predicted and observed results. Arithmetic mean and 95% confidence intervals of the fold changes were reported. The precision of the prediction was evaluated using the AFE (eq. 11) (Vildhede et al., 2016).
TABLE 3.
Measured total and unbound concentrations of inhibitors ([I]t and [I]u, µM) in the medium ([I]med), whole-cell lysate ([I]cell), and cytosol ([I]cyt)
SCHH were treated with telmisartan (1 or 10 µM) and bosentan (0.8 or 8 µM) for 20 min in the presence of 4% BSA. Data were generated from n = 1 SCHH preparation. Unbound concentration and fu data were expressed as mean values obtained from triplicate measurements, and values in parentheses represent ranges. Total concentrations were from single measurements.
| Inhibitor | Medium | Cell Lysate | Cytosol | ||||||
|---|---|---|---|---|---|---|---|---|---|
| [I]t,med | [I]u,med | fu,med | [I]t,cell | [I]u,cell | fu,cell,inhibitor | [I]t,cyt | [I]u,cyt | fu,cyt,inhibitor | |
| Telmisartan | 1 | 0.012 (0.0098–0.014) | 0.012 (0.0098–0.0134) | 16 | 2.1 (1.6–2.5) | 0.13 (0.099–0.16) | 16 | 0.85 (0.8–0.9) | 0.053 (0.050–0.056) |
| 10 | 0.20 (0.18–0.22) | 0.02 (0.018–0.022) | 40 | 3.7 (2.7–4.8) | 0.094 (0.068–0.12) | 35 | 2.8 (1.8–3.4) | 0.080 (0.052–0.098) | |
| Bosentan | 0.8 | 0.031 (0.023–0.039) | 0.039 (0.029–0.048) | 1.9 | 0.79 (0.66–0.93) | 0.41 (0.34–0.48) | 1.7 | 0.21 (0.21–0.21) | 0.12 (0.12–0.12) |
| 8 | 0.45 (0.44–0.47) | 0.057 (0.055–0.058) | 17 | 3.8 (3.1–4.5) | 0.22 (0.18–0.26) | 14 | N/A | N/A | |
N/A, not available.
TABLE 1.
Inhibition constants (µM) of telmisartan and bosentan against transporters involved in the hepatic uptake and efflux of TCA
| Clearance | Transporter | Telmisartan | Reference | Bosentan | Reference |
|---|---|---|---|---|---|
| CLUptake | NTCP | 60 (Ki) | (Dong et al., 2014) | 18 (Ki) | (Leslie et al., 2007; Lepist et al., 2014) |
| 36 (IC50) | |||||
| OATP1B1 | 0.44 (Ki) | (Hirano et al., 2006) | 18a | ||
| CLBile | BSEP | 16–16.2 (IC50) | (Morgan et al., 2013; Lai, 2014) | 23–42 (IC50) | (Morgan et al., 2013) (Lepist et al., 2014) |
| CLBL | MRP4 | 11–36 (IC50) | (Sato et al., 2008) | 22 (IC50) | (Morgan et al., 2013) |
| (Morgan et al., 2013) | |||||
| MRP3 | 60 (IC50) | (Morgan et al., 2013) | 42 (IC50) | (Morgan et al., 2013) |
Not available and therefore assumed to be the same as NTCP.
TABLE 2.
Recovered estimates of d8-TCA total uptake clearance (CLUptake), basolateral efflux clearance (CLBL), biliary clearance (CLBile), and KFlux in the presence of 4% BSA
Estimates were based on the model scheme and time-course data depicted in Fig.1. The model was fit to data generated from n = 3 independent SCHH preparations (triplicate measurements) separately.
| Parameter Estimate | Mean | SD | CV% |
|---|---|---|---|
| CLUptake (mL/min/g liver) | 0.63 | 0.12 | 20 |
| CLBL (mL/min/g liver) | 0.034 | 0.011 | 32 |
| CLBile (mL/min/g liver) | 0.074 | 0.030 | 36 |
| KFlux (min−1) | 0.018 | 0.0015 | 8 |
 |
(11) |
Sensitivity Analyses of Model Input.
The sensitivity of fu,cell,inhibitor, a compound-specific parameter for telmisartan and bosentan, on model output (TCA Ct,Cells) was evaluated. Monte Carlo simulations were performed to predict the fold changes in the TCA Ct,Cells at steady state (120 minutes) using parameters and the associated variance in Table 2, and eqs. 1–5 and 7, 8, and 10, where [I]cell = [I]t,cell × fu,cell,inhibitor. Different fu,cell,inhibitor values (0.02–1) were used in the simulations. [I]t,cell and [I]u,med in SCHH incubated with telmisartan (1 or 10 μM) and bosentan (0.8 or 8 μM) were obtained from Table 3, as described above.
Furthermore, to generalize the sensitivity analysis of fu,cell,inhibitor to a broader range of inhibitors, TCA Ct,Cells at steady state (120 minutes) in the presence of theoretical inhibitors with different ([I]t,cell/IC50) values (ranging from 0.5 to 60) was simulated assuming fu,cell,inhibitor = 1 or 0.02, respectively. In these simulations, IC50 represented the inhibitory potency against efflux transporters, and CLBL and CLBile were assumed to be inhibited to the same extent. All simulations were performed with and without 50% inhibition of CLUptake.
Bioanalysis.
Lysis solution [500 µL 70% methanol/30% water containing internal standard (25 nM d4-TCA, d3-telmisartan, or ambrisentan)] was added to each well of previously frozen 24-well or 6-well plates containing study samples. Plates were shaken for ∼15 minutes, and the cell lysate solution was filtered, evaporated to dryness, and reconstituted. Medium samples (100 μL) were extracted with 300 µL 100% methanol containing internal standard, filtered, evaporated, and reconstituted. Standards and quality control samples were prepared by adding a known amount of standards into a blank cell plate or medium, followed by the same sample processing methods with test samples. The d8-TCA samples were reconstituted in 60% methanol/40% water containing 10 mM ammonium acetate and analyzed by liquid chromatography with triple quadrupole mass spectrometry (LC-MS/MS) using a Shimadzu binary high performance liquid chromatography (HPLC) system (Columbia, MD) and Thermo Electron TSQ Quantum Discovery MAX (Waltham, MA) with an Ion Max ESI source using negative electrospray ionization mode. Samples (10 μL) were injected onto a 100 × 1.0 mm Hypersil Gold C18 column (Thermo Scientific, Bellefonte, PA). The mobile phase was methanol/water with 0.5 mM ammonium acetate at a flow rate of 50 μL/min. The transitions monitored (parent m/z > product m/z) were 522 > 128 and 518 > 124 for d8-TCA and d4-TCA. The calibration curve range was 0.5–100 pmol/well. Telmisartan samples were reconstituted in 70% methanol/30% water with 0.1% formic acid and analyzed by the same LC-MS/MS system using positive electrospray ionization mode. The mobile phase was methanol/water with 0.1% formic acid at a flow rate of 50 μL/min. The transitions monitored were 515.2 > 276.2 and 518.2 > 279.2 for telmisartan and d3-telmisartan, respectively. The calibration curve range was 0.01–10 pmol/well. Bosentan samples were reconstituted in 60% methanol/30% water with 0.1% formic acid and analyzed by LC-MS/MS using a Shimadzu binary HPLC system (Columbia, MD) and Applied Biosystems (Foster City, CA) API-3000 mass spectrometer operated in positive electrospray ionization mode. Samples (10 μL) were loaded onto a 100 × 1.0-mm Hypersil Gold C18 column (Thermo Scientific, Bellefonte, PA). The mobile phase was acetonitrile/water with 0.2% formic acid. The transitions monitored were 522.3 > 202.2 for bosentan and 379.1 > 303.1 for ambrisentan. The calibration curve range was 0.05–50 pmol/well. Acceptance criteria for percentage of accuracy of back-calculated values was 15–20%. TCA accumulation in cell lysate was corrected for nonspecific binding to the BioCoat plate without cells.
Results
Hepatobiliary Disposition of d8-TCA in SCHH.
The model scheme depicted in Fig. 1A and eqs. 1–5 was used to describe SCHH data (TCA in Cells + Bile, Cells, and incubation medium) from three human livers. Data were analyzed as three independent data sets and were well described by the mechanistic model (individual fits are not shown). The mean (±S.E.M.) data and simulated mass-time profiles generated using the mean of best-fit parameter estimates from the three SCHH data sets (Table 2) are presented in Fig. 1B. After 20-minute uptake, TCA Ct,Cells was 5.6 μM. The mean kinetic parameters and the associated variance estimated by fitting the differential eqs. 1–5 to TCA mass-time data from three independent SCHH preparations are presented in Table 2. The estimated total CLUptake of TCA was approximately one order of magnitude greater than total CLBile and total CLBL estimates; TCA CLBile was approximately twofold greater than CLBL. These parameter estimates were used in subsequent simulations.
Sensitivity Analyses of Model Output.
To identify a model output that was sensitive to impairment in both CLUptake and CLEfflux, the simulated fold changes in different endpoints of the SCHH assay (at steady state) were plotted against the fraction of inhibition of CLUptake and CLEfflux in Fig. 2, where CLEfflux = CLBL + CLBile. The most sensitive model output to both CLUptake and CLEfflux of TCA was Ct,Cells. Ct,Cells decreased to 0.01-fold of baseline when CLUptake was inhibited by 99% and CLEfflux was not inhibited, and increased to approximately 15-fold of baseline when CLEfflux was inhibited by 99% and CLUptake was not inhibited. Other endpoints were only sensitive to either CLUptake (e.g., Xt,Cells+Bile) or CLEfflux (e.g., Xt,Bile, Xt,Bile/Xt,Cells, and Xt,Bile/Xt,Cells+Bile), and the fold changes were less pronounced. Therefore, the TCA Ct,Cells was chosen as the model output in subsequent simulations to reflect the altered hepatobiliary disposition of TCA in the presence of inhibitors.
Fig. 2.

Impact of impaired CLUptake and CLEfflux (CLEfflux = CLBL + CLBile) of TCA on different model outputs in SCHH: (A) TCA total concentration in Cells (Ct,Cells), (B) TCA total amount in Cells + Bile (Xt,Cells+Bile), (C) TCA total amount in Bile (Xt,Bile), (D) ratio of the total amount of TCA in Cells to the total amount of TCA in Cells + Bile (Xt,Cells/Xt,Cells+Bile), (E) ratio of the total amount of TCA in Bile to the total amount of TCA in Cells (Xt,Bile/Xt,Cells), and (F) ratio of the total amount of TCA in Bile to the total amount of TCA in Cells + Bile (Xt,Bile/Xt,Cells+Bile). The Z-axis represents the fold change compared with baseline (shown in the color map on the right), based on simulations of TCA accumulation at steady state. (C, E, and F) have been rotated to improve visibility of the three-dimensional surface.
Determination of Cellular and Cytosolic Total and Unbound Concentrations of Inhibitors.
After 20-minute incubation with SCHH, the total and unbound concentrations of telmisartan and bosentan in medium, whole-cell lysates, and cytosol were measured; the results are reported in Table 3. The cytosol was isolated with ∼100% recovery (based on the LDH assay; data not shown) and low contamination of subcellular organelles (3% recovery of the enzyme marker for microsomal contamination; 5% recovery of the enzyme marker for mitochondrial contamination). As shown in Table 3, telmisartan was highly bound in the whole-cell lysate and cytosol (fu,cell,inhibitor = 0.09–0.13 and fu,cyt,inhibitor = 0.05–0.08); the cytosolic unbound telmisartan concentrations were only 5.3–7% of the total concentrations in the cell. The unbound fraction of bosentan was higher than telmisartan (fu,cell,inhibitor = 0.22–0.41 and fu,cyt,inhibitor = 0.12); the cytosolic unbound bosentan concentration was 11% of the total concentration in the whole cell. More than one-half of the amount of telmisartan (62–70%) and bosentan (58–63%) in the whole-cell lysate was recovered in the cytosol. Considering that cytosolic volume represents ∼70% of the cellular volume (Grunberg et al., 2009), the cytosolic and cellular total concentrations were similar.
Comparison of Simulated and Observed TCA Disposition in the Presence of Inhibitors.
The fold changes in the TCA Ct,Cells in the presence of inhibitors (telmisartan and bosentan) versus in the absence of inhibitors were simulated and compared with experimental results (Table 4). Monte Carlo simulations were performed for 40 individuals and repeated 10 times using either eq. 8 or eq. 9 assuming either MRP3 or MRP4 mediated the basolateral efflux of TCA. The simulation results were similar, and therefore only the simulations based on MRP3 inhibition were presented because MRP3 expression was reported to be higher than MRP4 expression in human liver and hepatocytes (Vildhede et al., 2015; Wisniewski et al., 2016). In the prediction of telmisartan’s effect on TCA Ct,Cells, the AFE of simulations using [I]u,cell and [I]u,cyt was 1.0 and 0.99, respectively. The 95% confidence interval of the simulation results overlapped with the range of observed data. When [I]t,cell and [I]t,cyt were used in the simulation, TCA Ct,Cells was overpredicted and the AFE was 1.4 and 1.3, respectively. In the prediction of bosentan’s effect, the mechanistic model slightly overpredicted the fold change for TCA Ct,Cells, with an AFE of 1.2–1.3, no matter which inhibitor concentration was used. According to the simulations (Supplemental Fig. 1), telmisartan-induced changes in TCA Ct,Cells increased as the uptake phase was extended. After a 30-minute uptake phase, the simulated TCA Ct,Cells for telemisartan based on [I]t,cell was threefold of the simulation based on [I]u,cyt.
TABLE 4.
Experimentally observed and simulated alteration of TCA total concentration in Cells (Ct,Cells) due to telmisartan and bosentan
Observed data are presented as fold change in the presence compared with the absence of inhibitors. SCHH were pretreated with telmisartan (1 and 10 µM) or bosentan (0.8 and 8 µM) for 10 min, followed by coincubation with d8-TCA and telmisartan or bosentan for 10 min. Observed data represented arithmetic mean (range) measured in n = 1 SCHH preparation in duplicate. Monte Carlo simulations for 40 individuals were performed 10 times using parameter estimates and associated variance (Table 2), different inhibitor concentrations (Table 3), and IC50 data (Table 1) assuming CLUptake was mediated by NTCP (70%) and OATPs (30%), CLBile was mediated by BSEP, and CLBL was governed by MRP3. Simulation data are presented as arithmetic mean of 10 simulations (95% confidence interval). Average fold errors were calculated based on eq. 11.
| Inhibitor | Dosing Concentration | Fold Change in TCA Ct,Cells | ||||
|---|---|---|---|---|---|---|
| Observation | Simulation | |||||
| [I]t,cell | [I]u,cell | [I]t,cyt | [I]u,cyt | |||
| Telmisartan | 1 µM | 0.91 (0.87–0.95) | 1.3 (1.3–1.4) | 1.0 (0.99–1.1) | 1.3 (1.3–1.3) | 1.0 (0.94–1.1) |
| 10 µM | 1.1 (1.0–1.1) | 1.4 (1.3–1.4) | 1.0 (0.98–1.0) | 1.3 (1.3–1.3) | 0.96 (0.95–0.98) | |
| Average fold error | 1.4 | 1.0 | 1.3 | 0.99 | ||
| Bosentan | 0.8 µM | 0.88 (0.83–0.92) | 1.0 (0.99–1.0) | 0.99 (0.96–1.0) | 1.0 (0.99–1.0) | 0.99 (0.97–1.0) |
| 8 µM | 0.81 (0.80–0.82) | 1.2 (1.2–1.3) | 1.0 (1.0–1.1) | 1.1 (1.1–1.2) | N/A | |
| Average fold error | 1.3 | 1.2 | 1.3 | N/A | ||
N/A, not available.
Sensitivity Analyses of Model Inputs.
Because the use of [I]u or [I]t affected the simulation of TCA Ct,Cells differently for telmisartan and bosentan, sensitivity analysis of fu,cell,inhibitor for telmisartan and bosentan was performed by simulating TCA Ct,Cells using [I]t,cell of telmisartan and bosentan and various fu,cell,inhibitor values (0.02–1) (Table 3). Simulated TCA Ct,Cells at steady state were expressed as the mean and S.D. of fold changes over baseline (without inhibitors) (shown in Fig. 3). The TCA Ct,Cells was sensitive to changes in fu,cell,inhibitor for telmisartan but not for bosentan at both the low and high dosing concentrations. At the low dosing concentration, the mean fold change in the TCA Ct,Cells increased from 0.8 to 2.5 when the fu,cell,inhibitor for telmisartan changed from 0.02 to 1; the mean fold change in the TCA Ct,Cells increased from 0.8 to 1 when the fu,cell,inhibitor for bosentan changed from 0.02 to 1. At the high dosing concentration, the mean fold change in the TCA Ct,Cells ranged from 0.9 to 4 when the fu,cell,inhibitor for telmisartan changed from 0.02 to 1; the mean fold change ranged from 0.8 to 1.5 when the fu,cell,inhibitor for bosentan changed from 0.02 to 1 (Fig. 3).
Fig. 3.

Sensitivity analysis of cellular unbound fraction of inhibitor (fu,cell,inhibitor) for telmisartan and bosentan. Fold changes in the TCA Ct,Cells at steady state compared with baseline (without inhibitors), in the presence of telmisartan and bosentan, were simulated based on the average IC50 values (Table 1), cellular total inhibitor concentration (Table 3), and different fu,cell,inhibitor values using a Monte Carlo simulation approach. Data were expressed as mean and S.D. of 40 simulated individuals.
To explore the differential sensitivity of TCA Ct,Cells to fu,cell,inhibitor for different inhibitors, the TCA Ct,Cells in the presence of a set of theoretical inhibitors with various ([I]t,cell/IC50) values were simulated using fu,cell,inhibitor = 1 and 0.02 (Fig. 4). For inhibitors with the same ([I]t,cell/IC50) value, if the inhibitor exhibited no intracellular binding (i.e., fu,cell,inhibitor = 1), the simulated fold change in the TCA Ct,Cells was greater than when the inhibitor exhibited extensive intracellular binding (i.e., fu,cell,inhibitor = 0.02). As the ([I]t,cell/IC50) value increased, the difference in simulated TCA Ct,Cells between fu,cell,inhibitor = 1 and 0.02 increased. For inhibitors with ([I]t,cell/IC50) > 1, the predicted TCA Ct,Cells when fu,cell,inhibitor = 1 was more than twice of the predicted TCA Ct,Cells when fu,cell,inhibitor = 0.02. These relationships were the same with or without 50% inhibition of CLUptake (data not shown).
Fig. 4.
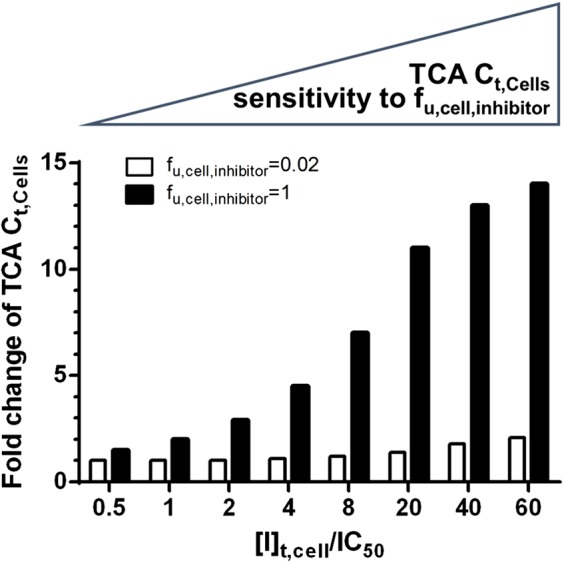
The sensitivity of the predicted TCA Ct,Cells to changes in fu,cell,inhibitor as a function of ([I]t,cell/IC50) values for a set of theoretical inhibitors. TCA Ct,Cells in the presence of theoretical inhibitors with different ([I]t,cell/IC50) values were simulated assuming fu,cell,inhibitor = 1 (black bar) and fu,cell,inhibitor = 0.02 (white bar). The fold changes of TCA Ct,Cells compared to baseline (without inhibitors) are plotted on the y-axis.
Discussion
In this study, an integrated approach was developed to predict the net effect of inhibition of multiple transporters on the hepatocellular disposition of the model bile acid TCA based on inhibition constants and SCHH data using mechanistic modeling. Importantly, the intracellular binding of inhibitors was considered in the simulations, and a strategy was proposed to determine whether it is necessary to measure the intracellular binding a priori.
The following assumptions were made for the mechanistic modeling. Linear kinetics was assumed because the unbound concentration of TCA in the medium (fu,med × 1 µM, where fu,med refers to the unbound fraction of TCA in the medium equivalent to 0.15 in 4% BSA) (Wolf et al., 2008) was below the Km of TCA for the uptake transporter NTCP (5–20 µM) and OATPs (5.8–71.8 µM) (Shitara et al., 2003; Nozawa et al., 2004; Mita et al., 2006; De Bruyn et al., 2014). In addition, the cellular total concentration of TCA (5.6 µM) after 20-minute uptake was below the Km for the efflux transporters BSEP (6.2 µM) (Hayashi et al., 2005), MRP3 (30 µM), and MRP4 (7.7 µM); if intracellular binding is taken into account, the cellular unbound concentration of TCA would be even lower. Passive diffusion was not included in the model because active uptake plays a major role in the hepatocellular accumulation of TCA (Shitara et al., 2003; Mita et al., 2006).
To simulate the effects of inhibitors on TCA disposition, eqs. 7–10 were used. Due to low TCA concentrations, eqs. 7–10 held true regardless of the mechanisms of inhibition and the IC50 value was substituted for Ki. The inhibitory effects of metabolites of telmisartan and bosentan were assumed to be negligible. There are no literature reports about inhibitory effects of telmisartan metabolites on human bile acid transporters. Although a bosentan metabolite, Ro 47-8634, was reported to be an inhibitor of rat Bsep (Ki = 8.5 µM) (Fattinger et al., 2001), the intracellular concentration of this metabolite in human SCHH is less than 5% of bosentan (Matsunaga et al., 2016). In addition, the concentration of this metabolite in human plasma (Dingemanse et al., 2002) and feces (Weber et al., 1999) is much lower than bosentan. Both MRP3 and MRP4 have been reported to contribute to the basolateral efflux of TCA without consensus on the relative contribution. The expression of MRP3 is higher than MRP4 in human liver and hepatocytes, whereas the affinity of TCA toward MRP4 (Km = 7.7 µM) (Rius et al., 2006) is higher than MRP3 (Km = 30 µM) (Zhang et al., 2003). Akita et al. (2002) reported that TCA was not transported to a significant degree by MRP3. Therefore, two extreme scenarios were simulated assuming 100% contribution of either MRP3 or MRP4; the simulation results were similar, so MRP3 was selected as the main basolateral efflux transporter for subsequent simulations.
In the current study, a physiologic concentration of protein (4% BSA) was added to mimic the in vivo scenario. Using the mechanistic model, the estimated total CLUptake of TCA was 0.63 mL/min/g liver (Table 2) and the unbound CLUptake of TCA was 4.2 mL/min/g liver (calculated as total CLUptake/fu,med). This value is close to the reported unbound CLUptake (2.2 mL/min/g liver) (Yang et al., 2015). CLBL and CLBile were similar to values reported previously (CLBL = 0.042 mL/min/g liver and CLBile = 0.14 mL/min/g liver) (Yang et al., 2015).
In this study, we leveraged SCHH and a mechanistic model to evaluate the net effect of uptake and efflux. The comparison between simulated and experimental results for telmisartan and bosentan provided an example of the applicability of this approach to predict the net effect of inhibition at multiple sites on the disposition of a model bile acid (Table 4). This applicability is important because the interplay of multiple transporters is common. Examples of dual inhibitors of BSEP and NTCP include the nonhepatotoxic drugs pioglitazone, telmisartan, and reserpine (Morgan et al., 2010; Dong et al., 2014), as well as the hepatotoxic compound troglitazone (Morgan et al., 2010; Yang et al., 2014). Some compounds are dual inhibitors of both uptake and basolateral efflux of TCA, such as α-naphthylisothiocyanate (Guo et al., 2014).
The slight differences between model predicted and experimental results observed for bosentan’s effect on TCA Ct,Cells could be attributed to the model assumptions discussed earlier. It should be noted that inhibitor-mediated alterations in TCA Ct,Cells were not extensive due to the short 10-minute uptake phase in this study and simultaneous inhibition of uptake and efflux. A more pronounced alteration in TCA Ct,Cells could be achieved by extending the uptake phase (Supplemental Fig. 1). However, accurate measurement of the TCA Ct,Cells after an uptake phase >30 minutes is technically challenging in sandwich-cultured hepatocytes. When Ca2+ is present during an extended uptake phase, the tight junctions reseal, yielding a measured Xt,Cells+Bile instead of Xt,Cells (Pfeifer et al., 2013c).
This is the first study to evaluate the impact of using different cellular inhibitor concentrations to predict transporter-mediated interactions in SCHH. Use of cytosolic concentrations marginally improved the prediction of telmisartan’s effects; the AFE dropped by ≤0.1 when cytosolic inhibitor concentration instead of [I]cell was used. This difference was minor because telmisartan was recovered primarily in the cytosol (62–70% of the total mass) and the cytosolic concentration approximated the cellular concentration. The impact of using cytosolic inhibitor concentration instead of [I]cell would likely be greater for drugs that are trapped in subcellular organelles, such as furamidine (Pfeifer et al., 2013b). Different enzymatic markers were used to evaluate the purity and recovery of cytosol. However, membrane-anchored proteins (e.g., the endoplasmic reticulum marker glucose-6-phosphatase) would not be able to detect whether content in the endoplasmic reticulum lumen had been released into the cytosol. Lumen protein markers [e.g., ERp57 (Coe et al., 2010) or Glucosidase II (Zuber et al., 2000)] could be measured in future studies to exclude this possibility.
In drug-drug interaction (DDI) evaluations, [I]t,cell is used commonly to avoid false-negative predictions by assessing the worst-case scenario, but this value can lead to false-positive predictions. Pfeifer et al. (2013a) reported that using [I]u,cell of ritonavir correctly predicted no clinical MRP2-mediated DDI between ritonavir and 99mTc-mebrofenin, whereas predictions based on [I]t,cell of ritonavir led to a false-positive prediction of DDI liability. In the case of telmisartan, simulations using [I]t,cell and [I]t,cyt slightly overpredicted TCA Ct,Cells compared with simulations using [I]u,cell and [I]u,cyt. Unlike telmisartan, simulations for bosentan’s effect on TCA Ct,Cells were similar regardless of whether total or unbound, cellular or cytosol, concentrations of bosentan were employed (Table 4). Sensitivity analysis revealed the differential sensitivity of TCA Ct,Cells to fu,cell,inhibitor for telmisartan and bosentan (Fig. 3). This difference suggested that, although it is ideal to use [I]u, it is not necessary to measure fu,cell,inhibitor and use [I]u,cell for every inhibitor. Simulations of a set of theoretical inhibitors showed that inhibitors with high ([I]t,cell/IC50) values were more sensitive to changes in fu,cell,inhibitor (Fig. 4). For example, when ([I]t,cell/IC50) was >1, the simulation assuming no protein binding overpredicted TCA Ct,Cells by two times or more. Inhibitors with large ([I]t,cell/IC50) values either tend to accumulate in the cells or serve as strong inhibitors of efflux transporters. In these cases, ignoring protein binding would greatly impact the prediction, and thus, fu,cell,inhibitor needs to be measured. The ([I]t,cell/IC50) value of telmisartan was 3.6 at the 10 µM dose level, and the ([I]t,cell/IC50) value of bosentan was 0.8 at the 8 µM dose level. This difference could explain the greater sensitivity of predicted TCA Ct,Cells to changes in fu,cell,inhibitor of telmisartan compared with bosentan.
Based on the results of these studies, a framework was proposed to predict the net effect of drug–bile acid interactions mediated by inhibition of multiple transporters (Fig. 5). The kinetic parameters (CLUptake, CLBile, and CLBL) of the victim bile acid (e.g., TCA) are estimated by mechanistic modeling; in the presence of inhibitors, the CL values are affected by [I]u,med or [I]cell and IC50 or Ki values. The choice of which [I]cell value to use (e.g., [I]t,cell, [I]t,cyt, [I]u,cell, [I]u,cyt) depends on the sensitivity of the model output to fu,cell,inhibitor, which is determined by the ([I]t,cell/IC50) value of the inhibitor. If this value is high, the model output, Ct,Cells, is sensitive to changes in fu,cell,inhibitor. In these cases, it is critical to measure fu,cell,inhibitor, as demonstrated in this study. For inhibitors that sequester in subcellular organelles, it may be necessary to isolate cytosol and measure [I]u,cyt. Finally, the altered hepatocellular disposition of the victim bile acid, namely Ct,Cells, can be simulated using CLinhibitor (calculated using eqs. 7–10). This approach could be applied to evaluate transporter-mediated interactions involving other victim substrates (e.g., hepatotoxic bile acids), which would have significant toxicological implications.
Fig. 5.

Proposed framework to predict altered bile acid disposition in SCHH mediated by inhibition of multiple transporters. Black solid boxes represent experimental observations, and black double lines depict the simulation output, as detailed in Discussion. The kinetic parameters, including uptake clearance (CLUptake), biliary clearance (CLBile), and basolateral efflux clearance (CLBL), of the victim bile acid (e.g., taurocholic acid), are estimated by using mechanistic modeling; in the presence of inhibitors, the clearance values (CLinhibitor) are estimated using eqs. 7–10 based on inhibitor concentrations in the medium or cells ([I]cell) and IC50 or Ki values. The choice of which [I]cell to use depends on the ([I]t,cell/IC50) value of the inhibitor, where [I]t,cell represents cellular total concentration of inhibitor. If this value is high, the cellular unbound fraction of inhibitor (fu,cell,inhibitor) should be measured to estimate [I]u,cell, where [I]u,cell represents the cellular unbound concentration of inhibitor. Otherwise, [I]t,cell can be used. Finally, the altered cellular total concentrations (Ct,Cells) of the victim bile acid are simulated using CLinhibitor.
Supplementary Material
Acknowledgments
Phoenix software was generously provided to the Division of Pharmacotherapy and Experimental Therapeutics, University of North Carolina Eshelman School of Pharmacy, by Certara as a member of the Pharsight Academic Center of Excellence Program. The authors thank Dr. Weslyn Friley for analytical support.
Abbreviations
- AFE
average fold error
- BSA
bovine serum albumin
- BSEP
bile salt export pump
- CL
clearance
- CLBile
biliary CL
- CLBL
basolateral efflux CL
- CLEfflux
sum of CLBL and CLBile
- CLinhibitor
CL in the presence of inhibitor
- CLUptake
uptake CL
- Ct,Cells
total concentration in Cells
- d8-TCA
deuterium-labeled TCA
- DDI
drug-drug interaction
- fu
unbound fraction
- fu,cell,inhibitor
cellular unbound fraction of inhibitor
- fu,cyt,inhibitor
cytosolic unbound fraction of inhibitor
- fu,med
unbound fraction in the medium
- HBSS
Hanks’ balanced salt solution
- [I]cell
cellular inhibitor concentration
- [I]t
total inhibitor concentration
- [I]t,cell
cellular total inhibitor concentration
- [I]t,cyt
cytosolic total inhibitor concentration
- [I]u
unbound inhibitor concentration
- [I]u,cell
cellular unbound inhibitor concentration
- [I]u,cyt
cytosolic unbound inhibitor concentration
- [I]u,med
medium unbound concentration of inhibitor
- Ki
inhibition constant
- LDH
lactate dehydrogenase
- MRP
multidrug resistance-associated protein
- NTCP
sodium taurocholate cotransporting polypeptide
- OATP
organic anion-transporting polypeptide
- SCHH
sandwich-cultured human hepatocytes
- TCA
taurocholic acid
- Xt,Bile
total amount in Bile
- Xt,Cells
total amount in Cells
- Xt,Cells+Bile
total amount in Cells + Bile
Authorship Contributions
Participated in research design: Guo, Yang, K. R. Brouwer, K. L. R. Brouwer.
Conducted experiments: Guo, St. Claire.
Performed data analysis: Guo, Yang, K. R. Brouwer, K. L. R. Brouwer.
Wrote or contributed to the writing of the manuscript: Guo, Yang, K. L. R. Brouwer.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant R01GM041935]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. C.G. is supported, in part, by the University of North Carolina Royster Society of Fellows.
This work was previously presented, in part, as a poster presentation at the following workshops: Guo C, Brouwer KR, Yang K, St. Claire R, and Brouwer, KLR (2014) Prediction of altered bile acid disposition by drugs using an integrated approach: Sandwich-cultured human hepatocytes, mechanistic modeling and simulation, in 19th North American International Society for the Study of Xenobiotics (ISSX) Meeting/29th Japanese Society for the Study of Xenobiotics (JSSX) Meeting; 2014 Oct 19-23; San Francisco, CA. ISSX, Washington, DC; and Guo C, Yang K, and Brouwer KLR (2015) Prediction of hepatic efflux transporter-mediated DDIs: When does variability in IC50 or intracellular unbound fraction of inhibitors matter? 20th North American ISSX Meeting; 2015 Oct 18-22; Orlando, FL. ISSX, Washington, DC.
B-CLEAR is covered by US Pat. No. 6,780,580 and other US and International patents both issued and pending.
Dr. K.L.R. Brouwer is a co-inventor of the sandwich-cultured hepatocyte technology for quantification of biliary excretion (B-CLEAR) and related technologies, which have been licensed exclusively to Qualyst Transporter Solutions, LLC. Dr. K.R. Brouwer and Dr. R.L. St. Claire are employed by Qualyst Transporter Solutions, LLC.
Current affiliation: DILIsym Services Inc., Research Triangle Park, North Carolina
 This article has supplemental material at jpet.aspetjournals.org.
This article has supplemental material at jpet.aspetjournals.org.
References
- Akita H, Suzuki H, Hirohashi T, Takikawa H, Sugiyama Y. (2002) Transport activity of human MRP3 expressed in Sf9 cells: comparative studies with rat MRP3. Pharm Res 19:34–41. [DOI] [PubMed] [Google Scholar]
- Banker MJ, Clark TH, Williams JA. (2003) Development and validation of a 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J Pharm Sci 92:967–974. [DOI] [PubMed] [Google Scholar]
- Chandra P, Brouwer KLR. (2004) The complexities of hepatic drug transport: current knowledge and emerging concepts. Pharm Res 21:719–735. [DOI] [PubMed] [Google Scholar]
- Coe H, Jung J, Groenendyk J, Prins D, Michalak M. (2010) ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J Biol Chem 285:6725–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson S, Stahl S, Paul N, Barber J, Kenna JG. (2012) In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab Dispos 40:130–138. [DOI] [PubMed] [Google Scholar]
- De Bruyn T, Sempels W, Snoeys J, Holmstock N, Chatterjee S, Stieger B, Augustijns P, Hofkens J, Mizuno H, Annaert P. (2014) Confocal imaging with a fluorescent bile acid analogue closely mimicking hepatic taurocholate disposition. J Pharm Sci 103:1872–1881. [DOI] [PubMed] [Google Scholar]
- Dingemanse J, Bodin F, Weidekamm E, Kutz K, van Giersbergen P. (2002) Influence of food intake and formulation on the pharmacokinetics and metabolism of bosentan, a dual endothelin receptor antagonist. J Clin Pharmacol 42:283–289. [DOI] [PubMed] [Google Scholar]
- Doherty MM, Poon K, Tsang C, Pang KS. (2006) Transport is not rate-limiting in morphine glucuronidation in the single-pass perfused rat liver preparation. J Pharmacol Exp Ther 317:890–900. [DOI] [PubMed] [Google Scholar]
- Dong Z, Ekins S, Polli JE. (2014) Quantitative NTCP pharmacophore and lack of association between DILI and NTCP inhibition. Eur J Pharm Sci 66C:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattinger K, Funk C, Pantze M, Weber C, Reichen J, Stieger B, Meier PJ. (2001) The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 69:223–231. [DOI] [PubMed] [Google Scholar]
- Grünberg W, Staufenbiel R, Constable PD, Dann HM, Morin DE, Drackley JK. (2009) Liver phosphorus content in Holstein-Friesian cows during the transition period. J Dairy Sci 92:2106–2117. [DOI] [PubMed] [Google Scholar]
- Guo C, He L, Yao D, A J, Cao B, Ren J, Wang G, Pan G. (2014) Alpha-naphthylisothiocyanate modulates hepatobiliary transporters in sandwich-cultured rat hepatocytes. Toxicol Lett 224:93–100. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Takada T, Suzuki H, Onuki R, Hofmann AF, Sugiyama Y. (2005) Transport by vesicles of glycine- and taurine-conjugated bile salts and taurolithocholate 3-sulfate: a comparison of human BSEP with rat Bsep. Biochim Biophys Acta 1738:54–62. [DOI] [PubMed] [Google Scholar]
- Hirano M, Maeda K, Shitara Y, Sugiyama Y. (2006) Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos 34:1229–1236. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS, Pollack GM. (2007) Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p-glycoprotein efflux ratios. Drug Metab Dispos 35:660–666. [DOI] [PubMed] [Google Scholar]
- Lai Y. (2014) Transporters in Drug Discovery and Development: Detailed Concepts and Best Practice, Woodhead Publishing, Sawston, UK. [Google Scholar]
- Lee JK, Marion TL, Abe K, Lim C, Pollock GM, Brouwer KLR. (2010) Hepatobiliary disposition of troglitazone and metabolites in rat and human sandwich-cultured hepatocytes: use of Monte Carlo simulations to assess the impact of changes in biliary excretion on troglitazone sulfate accumulation. J Pharmacol Exp Ther 332:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepist EI, Gillies H, Smith W, Hao J, Hubert C, St Claire RL, 3rd, Brouwer KR, Ray AS. (2014) Evaluation of the endothelin receptor antagonists ambrisentan, bosentan, macitentan, and sitaxsentan as hepatobiliary transporter inhibitors and substrates in sandwich-cultured human hepatocytes. PLoS One 9:e87548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EM, Watkins PB, Kim RB, Brouwer KLR. (2007) Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1) by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther 321:1170–1178. [DOI] [PubMed] [Google Scholar]
- Mateus A, Matsson P, Artursson P. (2013) Rapid measurement of intracellular unbound drug concentrations. Mol Pharm 10:2467–2478. [DOI] [PubMed] [Google Scholar]
- Matsunaga N, Kaneko N, Staub AY, Nakanishi T, Nunoya K, Imawaka H, Tamai I. (2016) Analysis of the metabolic pathway of bosentan and of the cytotoxicity of bosentan metabolites based on a quantitative modeling of metabolism and transport in sandwich-cultured human hepatocytes. Drug Metab Dispos 44:16–27. [DOI] [PubMed] [Google Scholar]
- Matsunaga N, Wada S, Nakanishi T, Ikenaga M, Ogawa M, Tamai I. (2014) Mathematical modeling of the in vitro hepatic disposition of mycophenolic acid and its glucuronide in sandwich-cultured human hepatocytes. Mol Pharm 11:568–579. [DOI] [PubMed] [Google Scholar]
- Mita S, Suzuki H, Akita H, Hayashi H, Onuki R, Hofmann AF, Sugiyama Y. (2006) Vectorial transport of unconjugated and conjugated bile salts by monolayers of LLC-PK1 cells doubly transfected with human NTCP and BSEP or with rat Ntcp and Bsep. Am J Physiol Gastrointest Liver Physiol 290:G550–G556. [DOI] [PubMed] [Google Scholar]
- Morgan RE, Trauner M, van Staden CJ, Lee PH, Ramachandran B, Eschenberg M, Afshari CA, Qualls CW, Jr, Lightfoot-Dunn R, Hamadeh HK. (2010) Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci 118:485–500. [DOI] [PubMed] [Google Scholar]
- Morgan RE, van Staden CJ, Chen Y, Kalyanaraman N, Kalanzi J, Dunn RT, 2nd, Afshari CA, Hamadeh HK. (2013) A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci 136:216–241. [DOI] [PubMed] [Google Scholar]
- Noé J, Stieger B, Meier PJ. (2002) Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology 123:1659–1666. [DOI] [PubMed] [Google Scholar]
- Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I. (2004) Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther 308:438–445. [DOI] [PubMed] [Google Scholar]
- Oshio C, Phillips MJ. (1981) Contractility of bile canaliculi: implications for liver function. Science 212:1041–1042. [DOI] [PubMed] [Google Scholar]
- Pfeifer ND, Goss SL, Swift B, Ghibellini G, Ivanovic M, Heizer WD, Gangarosa LM, Brouwer KLR. (2013a) Effect of ritonavir on (99m)technetium-mebrofenin disposition in humans: a semi-PBPK modeling and in vitro approach to predict transporter-mediated DDIs. CPT Pharmacometrics Syst Pharmacol 2:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer ND, Harris KB, Yan GZ, Brouwer KLR. (2013b) Determination of intracellular unbound concentrations and subcellular localization of drugs in rat sandwich-cultured hepatocytes compared with liver tissue. Drug Metab Dispos 41:1949–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer ND, Yang K, Brouwer KLR. (2013c) Hepatic basolateral efflux contributes significantly to rosuvastatin disposition I: characterization of basolateral versus biliary clearance using a novel protocol in sandwich-cultured hepatocytes. J Pharmacol Exp Ther 347:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, Oshio C, Miyairi M, Katz H, Smith CR. (1982) A study of bile canalicular contractions in isolated hepatocytes. Hepatology 2:763–768. [DOI] [PubMed] [Google Scholar]
- Rius M, Hummel-Eisenbeiss J, Hofmann AF, Keppler D. (2006) Substrate specificity of human ABCC4 (MRP4)-mediated cotransport of bile acids and reduced glutathione. Am J Physiol Gastrointest Liver Physiol 290:G640–G649. [DOI] [PubMed] [Google Scholar]
- Sato M, Iwanaga T, Mamada H, Ogihara T, Yabuuchi H, Maeda T, Tamai I. (2008) Involvement of uric acid transporters in alteration of serum uric acid level by angiotensin II receptor blockers. Pharm Res 25:639–646. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Li AP, Kato Y, Lu C, Ito K, Itoh T, Sugiyama Y. (2003) Function of uptake transporters for taurocholate and estradiol 17beta-D-glucuronide in cryopreserved human hepatocytes. Drug Metab Pharmacokinet 18:33–41. [DOI] [PubMed] [Google Scholar]
- Smith DA, Di L, Kerns EH. (2010) The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 9:929–939. [DOI] [PubMed] [Google Scholar]
- Sohlenius-Sternbeck AK. (2006) Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol In Vitro 20:1582–1586. [DOI] [PubMed] [Google Scholar]
- Swift B, Pfeifer ND, Brouwer KLR. (2010) Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev 42:446–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vildhede A, Mateus A, Khan EK, Lai Y, Karlgren M, Artursson P, Kjellsson MC. (2016) Mechanistic modeling of pitavastatin disposition in sandwich-cultured human hepatocytes: a proteomics-informed bottom-up approach. Drug Metab Dispos 44:505–516. [DOI] [PubMed] [Google Scholar]
- Vildhede A, Wiśniewski JR, Norén A, Karlgren M, Artursson P. (2015) Comparative proteomic analysis of human liver tissue and isolated hepatocytes with a focus on proteins determining drug exposure. J Proteome Res 14:3305–3314. [DOI] [PubMed] [Google Scholar]
- Weber C, Gasser R, Hopfgartner G. (1999) Absorption, excretion, and metabolism of the endothelin receptor antagonist bosentan in healthy male subjects. Drug Metab Dispos 27:810–815. [PubMed] [Google Scholar]
- Wiśniewski JR, Vildhede A, Norén A, Artursson P. (2016) In-depth quantitative analysis and comparison of the human hepatocyte and hepatoma cell line HepG2 proteomes. J Proteomics 136:234–247. [DOI] [PubMed] [Google Scholar]
- Wolf KK, Brouwer KR, Pollack GM, Brouwer KLR. (2008) Effect of albumin on the biliary clearance of compounds in sandwich-cultured rat hepatocytes. Drug Metab Dispos 36:2086–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhead JL, Yang K, Siler SQ, Watkins PB, Brouwer KLR, Barton HA, Howell BA. (2014) Exploring BSEP inhibition-mediated toxicity with a mechanistic model of drug-induced liver injury. Front Pharmacol 5:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Guo C, Woodhead JL, St Claire RL, 3rd, Watkins PB, Siler SQ, Howell BA, Brouwer KLR. (2016) Sandwich-cultured hepatocytes as a tool to study drug disposition and drug-induced liver injury. J Pharm Sci 105:443–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Pfeifer ND, Köck K, Brouwer KLR. (2015) Species differences in hepatobiliary disposition of taurocholic acid in human and rat sandwich-cultured hepatocytes: implications for drug-induced liver injury. J Pharmacol Exp Ther 353:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Woodhead JL, Watkins PB, Howell BA, Brouwer KLR. (2014) Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin Pharmacol Ther 96:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DW, Gu HM, Vasa M, Muredda M, Cole SP, Deeley RG. (2003) Characterization of the role of polar amino acid residues within predicted transmembrane helix 17 in determining the substrate specificity of multidrug resistance protein 3. Biochemistry 42:9989–10000. [DOI] [PubMed] [Google Scholar]
- Zuber C, Spiro MJ, Guhl B, Spiro RG, Roth J. (2000) Golgi apparatus immunolocalization of endomannosidase suggests post-endoplasmic reticulum glucose trimming: implications for quality control. Mol Biol Cell 11:4227–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.