

Abstract
Oxytocin (OT) modulates the expression of social and emotional behaviors and consequently has been proposed as a pharmacologic treatment of psychiatric diseases, including autism spectrum disorders and schizophrenia; however, endogenous OT has a short half-life in plasma and poor permeability across the blood-brain barrier. Recent efforts have focused on the development of novel drug delivery methods to enhance brain penetration, but few efforts have aimed at improving its half-life. To explore the behavioral efficacy of an OT analog with enhanced plasma stability, we developed PF-06655075 (PF1), a novel non–brain-penetrant OT receptor agonist with increased selectivity for the OT receptor and significantly increased pharmacokinetic stability. PF-06478939 was generated with only increased stability to disambiguate changes to selectivity versus stability. The efficacy of these compounds in evoking behavioral effects was tested in a conditioned fear paradigm. Both central and peripheral administration of PF1 inhibited freezing in response to a conditioned fear stimulus. Peripheral administration of PF1 resulted in a sustained level of plasma concentrations for greater than 20 hours but no detectable accumulation in brain tissue, suggesting that plasma or cerebrospinal fluid exposure was sufficient to evoke behavioral effects. Behavioral efficacy of peripherally administered OT receptor agonists on conditioned fear response opens the door to potential peripheral mechanisms in other behavioral paradigms, whether they are mediated by direct peripheral activation or feed-forward responses. Compound PF1 is freely available as a tool compound to further explore the role of peripheral OT in behavioral response.
Introduction
Oxytocin (OT) has been proposed as a treatment of a number of psychiatric conditions, including autism spectrum disorder, schizophrenia, depression, post-traumatic stress disorder, and social anxiety. Consequently the OT receptor has been a target of interest throughout the pharmaceutical industry (Striepens et al., 2011; Bakermans-Kranenburg and van I Jzendoorn, 2013); however, the therapeutic potential of OT as a neuroactive compound is limited by its pharmacokinetic properties.
Peripherally administered OT has an extremely short half-life (2–4 minutes in humans) and has poor penetration of the blood-brain barrier (brain-to-plasma ratio of ∼1%) (Fabian et al., 1969; Landgraf, 1981; Ermisch et al., 1985; McEwen, 2004). Nonetheless, early clinical studies have demonstrated behavioral effects of peripheral administration of large quantities of OT in patients with autism. Prolonged i.v. infusion both enhanced the retention of social information and reduced repetitive behaviors (Hollander et al., 2003, 2007). The long infusion duration was hypothesized to circumvent the limitation of OT’s short half-life to facilitate behavioral effects. Intranasal OT administration, a method thought to preferentially introduce the drug to the central nervous system, has also been shown to have social behavioral effects in several psychiatric disorders. In addition to central targeting, intranasal OT administration also results in prolonged elevations of peripheral OT levels (Andari et al., 2010; Carson et al., 2015; Modi et al., 2014). It has been speculated that the elevation in plasma OT may also contribute to the behavioral effects of intranasal OT either through direct activation of peripheral receptors or a feed-forward activation of central receptors (Quintana et al., 2015). To investigate the role of prolonged peripheral OT receptor activation on centrally mediated behavioral effects, we tested the effects of novel long-acting OT receptor agonists on conditioned fear responses.
The inhibitory effect of OT on the production of fear and anxiety behaviors through both peripheral and central mechanisms is well established (Neumann and Landgraf, 2012). Intracerebroventricular (ICV) administration of OT or stimulation of endogenous OT release diminishes the expression of freezing and startle in a conditioned fear paradigm (Knobloch et al., 2012; Toth et al., 2012). Centrally administered OT acts on OT receptors in the central amygdala to inhibit the output of the nuclei and the subsequent behavioral fear response (Viviani et al., 2011; Knobloch et al., 2012). Peripheral administration of OT has been shown to reduce anxiety but not to specifically diminish the expression of fear behavior (de Wied et al., 1987; Uvnas-Moberg et al., 1994; Klenerova et al., 2008; Ayers et al., 2011). The effect of peripheral administration of a lipidated OT analog on anxiety can be seen 24 hours after administration (Mizuno et al., 2015). The difference in efficacy between central and peripheral OT administration on fear behavior could be due to the difference in pharmacokinetics of the peptide in each compartment. The development of OT agonists with enhanced peripheral stability would enable a valid assessment of the behavioral effects of peripherally administered OT.
The roles of: 1) duration of peripheral exposure, 2) peripheral versus central injection, and 3) OT receptor selectivity in reducing fear-potentiated freezing in a fear-conditioning paradigm were investigated through the use of the novel tool compounds, PF-06655075 (PF1) and PF-06478939 (PF2) (Fig. 1). Compound PF1 is a non–brain-penetrant peptide OT receptor agonist with a lipid tail that confers resistance to clearance. The molecule is structurally similar to OT but differs at two amino acid positions, the Pro7 is modified to Gly to enhance selectivity for agonist effects at the OT receptor (OTR) over agonist effects at the V1a receptor, and the Leu8 is converted to a Lys modified with a palmitoyl group to extend the half-life of the peptide. Compound PF2 is similar to PF1 in structure but with only the Leu8 amino acid change from OT, so it maintains the agonist activity at both the OT receptor and the vasopressin receptors, similar to endogenous OT. Compound PF2 allows for the assessment of enhanced stability independent of enhanced selectivity. Comparison of the effects of these tool compounds with those of OT itself demonstrated that peripheral administration of a long-acting OT receptor agonist is sufficient to inhibit a fear response to a similar degree as central OT administration.
Fig. 1.

Structures of novel long-acting oxytocin analogs. Oxytocin (a) was modified with a substitution of the Leu8 to a Lys appended with a polyethylene glycol space and a palmitoyl group in both PF-06655075 (PF1) (b) and PF-06748939 (PF2) (c) to increase the stability of the molecule. PF1 was further modified with a substitution of Gly for the Pro7 to enhance selectivity for the OT receptor.
Material and Methods
Chemistry and Depot Formulation
Peptide Synthesis.
We purchased OT from Sigma-Aldrich (St. Louis, MO). Compounds PF1 and PF2 were synthesized at Chinese Peptide Company (Hangzhou, China) using standard 9-fluorenylmethyloxycarbonyl (FMOC) FMOC solid-phase peptide synthesis methods. Standard amino acids were purchased from Novabiochem (Darmstadt, Germany). The Lys(PegPalm) group was added though Fmoc-Lys(Dde)-OH incorporation into the peptide and subsequent deprotection with hydrazine. Fmoc-NH-PEG8-COOH was added via solid-phase peptide synthesis and deprotected, and then palmitoyl-COOH was added. Once the linear peptide was complete, it was cleaved from the resin, and the formation of the disulfinde bond was achieved in solution with acetic acid and iodine in MeOH. The peptide was purified and analyzed using reverse-phase high-pressure liquid chromatography.
Preparation of Depot Formulation.
Glycerol dioleate and ethanol (>99%) were purchased from Sigma-Aldrich. Phosphatidylcholine was purchased from Avanti Polar Lipids (Alabaster, AL). Depot formulations of the OT analogs were prepared by mixing equal weight fractions of glycerol dioleate and phosphatidylcholine and 15% ethanol in a glass vial at room temperature until a clear liquid was obtained, followed by the addition of a sufficient volume of a 48-mg/ml solution of the analog to result in 15% water content and 0.7% loading of the analog in the final formulation. The blank formulation was prepared using the same protocol without the analog (Thuresson, 2012).
Pharmacology of PF1, PF2, and Oxytocin
In Vitro Receptor Binding Assay.
Binding assays were performed on membranes prepared from the Chinese hamster ovary hOTR NFAT βLac cell line. Incubation was initiated by the addition of cell membrane solution to 96-well plates containing test drugs and [3H] 8-arginine-vasopressin (0.7 nM final concentration). Nonspecific binding was determined by the presence of a saturating concentration (10 µM) of Relcovaptan (Tocris Bioscience, United Kingdom), a potent OTR antagonist. Membrane-bound [3H] AVP levels were determined by liquid scintillation counting of the filter plates in Ecolume scintillation fluid (MP Biomedicals, Santa Ana, CA). The IC50 value (concentration at which 50% inhibition of specific binding occurs) was calculated by curve fitting. Ki values were calculated according to the Cheng-Prusoff equation: Ki = IC50/(1 + (L/Kd)), where L is the concentration of the radioligand used in the experiment and the Kd value is the dissociation constant for the radioligand (determined previously by saturation analysis).
In Vitro Functional Assay.
Functional activity of compounds at the OT receptor was assessed using a calcium flux fluorometric imaging plate reader assay. Stably transfected Chinese hamster ovary hOTR NFAT βLac cells were plated at a density of 10,000 cells/well in a 384-well plate in growth medium (Dulbecco’s modified Eagle’s medium + Glutamax (Thermo Fisher Scientific, Waltham, MA), 10% dialyzed fetal bovine serum). The next day calcium-sensitive, fluo-4 dye (Invitrogen, Carlsbad, CA) and probenecid (Invitrogen; 2.5 mM) was dissolved in 20 ml warm dye loading buffer (Hanks’ balanced salt solution with calcium and magnesium, 20 mM Hepes, pH 7.4) and added to each well. Plates were incubated for 60–120 minutes in a 37°C cell incubator to allow cells to uptake the dye. The control peptide, OT, and other test compounds were diluted in Hanks’ balanced salt solution with calcium and magnesium, 20 mM Hepes, and 0.1% bovine serum albumin. After dye-loading, the cell plates were placed on the fluorometric imaging plate reader. A total of 15 µl of test compound was added to the 30 µl of dye on the cells, and intracellular calcium was monitored for 90 seconds. If testing for an antagonist response, a concentration of OT that produces an 80% agonist effect was added to the plate (15 µl), and intracellular calcium was monitored for another 90 seconds. Intracellular calcium concentrations were calculated as the maximum increase over basal levels or, in the case of the antagonist assay, as the maximal decrease of the agonist challenge effect. Responses were normalized to control responses. Data were analyzed by curve fitting. For antagonist studies, IC50 was converted to a functional Ki according to the Cheng-Prusoff equation.
Pharmacokinetics of PF1 and Oxytocin
Chemicals, Reagents, and Animals.
OT was purchased from Phoenix Pharmaceuticals (Burlingame, CA). Analytical-grade chemicals and solvents were supplied by the Sigma-Aldrich, Fisher Scientific (Waltham, MA), and Malliinckrodt Baker, Inc. (Phillipsburg, NJ). Control matrices for nonspecific binding studies and concentration analysis were purchased from Bioreclamation, Inc. (Hicksville, NY) or supplied from internal sources. All animal studies were conducted in accordance with Guide for the Care and Use of Laboratory Animals using protocols reviewed and approved by the Pfizer Worldwide Research and Development (WRD) Institutional Animal Care and Use Committee. Male C57BL/6J mice (25–30g; Jackson Laboratories, Bar Harbor, ME) and male jugular vein cannulated Wistar Hannover rats (250–300 g; Charles River Laboratories, Wilmington, MA; and Vital River Laboratories, Bejing, China) were maintained on a 12-hour light/dark cycle for a minimum of 3 days in a temperature- and humidity-controlled environment with free access to food and water. All blood samples were collected into tubes containing EDTA and placed on ice. After centrifugation, the samples were transferred to polypropylene tubes and stored frozen at −20°C to −80°C until analysis.
Nonspecific Binding Studies.
Equilibrium dialysis using standard procedures (Di et al., 2011) were used for determination of the unbound fraction of OT and PF1 in rat plasma (fu,p) (Di et al., 2012) and rat brain homogenates (fu,b) (Di et al., 2011). The duration of the dialysis incubation was extended to 17 hours for PF1 to ensure sufficient time for equilibration. Nonspecific binding of PF1 in cerebrospinal fluid (CSF) was calculated through the use of the measured fraction unbound in plasma correcting for the abundance of albumin in the plasma in comparison with the CSF using eq. 1–4, where [P] is the protein concentration associated with protein binding assuming binding is associated primarily with albumin, and Kd is the dissociation constant:
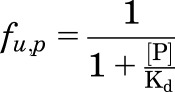 |
(1) |
Rearranging, one obtains:
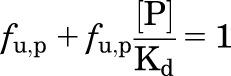 |
(2) |
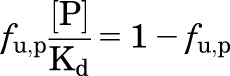 |
(3) |
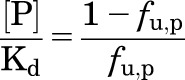 |
(4) |
Simplifying the equation for binding at two protein concentrations, where [P]CSF is the albumin concentration in CSF and [P]p is the albumin concentration in plasma, the fraction unbound is CSF (fu,CSF) was calculated through use of eq. 5:
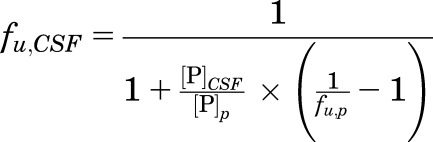 |
(5) |
1. Intravenous administration.
In-life and bioanalytical procedures were conducted at BioDuro, Pharmaceutical Product Development, Inc. (Beijing, China) and at WRD (Groton, CT). Jugular vein-cannulated male Wistar Hannover rats were administered an i.v. bolus of either OT or PF1. The OT i.v. dose (0.1 mg/kg) was formulated in 10% propylene glycol: 90% 20 mM sodium phosphate buffer, pH 7.4. PF1 was formulated in 10% SEDDs: 90% 20 mM, pH 5, phosphate buffer (v/v) (SEDDs = 3:4:3 Miglyol 812: Cremophor RH40: Capmul, v/v/v). Individual animals (n = 3 per dose/compound) were administered a single i.v. bolus dose of compound based on body weight at a volume of 1 ml/kg. Serial blood samples were collected from each rat via a jugular vein cannula before dose administration and at the following time points postdose: 0.033, 0.083, 0.25, 0.5, 1, 2, 4, 7, and 24 hours.
2. Subcutaneous nondepot administration.
In-life and bioanalytical procedures were conducted at WRD. Male C57BL/6J mice were administered an s.c. injection of either OT or PF-06655075 in a nondepot formulation. The OT doses (1 and 10 mg/kg) were formulated in 20 mM phosphate buffered saline; PF1 (1 mg/kg) was formulated in 10% SEDDs: 90% 50 mM pH 7.4 phosphate buffer (v/v) (SEDDs = 3:4:3 Miglyol 812: Cremophor RH40: Capmul MCM, v/v/v). Individual animals (n = 3 per dose/compound) were administered a single s.c. injection based on body weight. The dose volume for administration of OT and PF1 were 10 ml/kg and 4 ml/kg, respectively. Serial blood samples were collected from each mouse via the retro-orbital sinus before dose administration and at the following time points postdose: 0.25 (OT only), 0.5, 1, 2, 4, 6, and 24 hours (6- and 24-hour sampling limited to PF1 only).
3. Subcutaneous depot administration.
In-life and bioanalytical procedures were conducted at WRD. Male C57BL/6J mice were administered s.c. injection of either OT or PF1 (20 mg/kg) in a depot formulation (prepared as described already). Individual animals (n = 2 per dose/compound) were administered a single s.c. injection. The dose volume for administration was 100 µl per animal for both OT and PF1 formulations. Serial blood samples were collected from each mouse via the retro-orbital sinus at the following time points postdose: 0.5, 1, 2, 4, 6, and 24 hours.
4. Neuropharmacokinetics of s.c. depot administration of PF1.
In-life and bioanalytical procedures were conducted at WRD. Individual jugular vein-cannulated male Wistar Hannover rats (n = 3 per time point) were administered a single s.c. injection of PF1 (10 mg/kg) based on body weight at a dose volume of 4 ml/kg. Rats are used for neuropharmacokinetics for ease of CSF collection. The dose was formulated in the depot formulation. Blood, brain, and CSF samples were obtained at 0.5, 2, 5, 12, 24, and 48 hours postdose. At each time point, blood samples were collected via a jugular vein cannula. Rats were then placed under isoflurane anesthesia, and CSF was collected via puncture of the cisterna magna, placed into polypropylene tubes, and immediately stored on dry ice. The rats were then exsanguinated by a transcardial whole body saline perfusion (heparinized, 10 U/ml, Dulbecco’s phosphate buffered saline containing Mg and Ca; approximately 100 ml perfusion volume), and whole brain was rapidly extracted, rinsed with saline and snap-frozen in liquid nitrogen. All samples were stored at stored frozen at −20°C to −80°C until analysis.
Quantitation of Compound in Biologic Matrices.
Rat and mouse plasma and rat brain tissue and CSF were processed for quantitation of PF1 using protein precipitation followed by analysis using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Mouse plasma was quantitated for OT using protein precipitation followed by LC-MS/MS. Low-bind laboratory ware (tips, plates, tubes) was used throughout sample preparation and analysis to minimize nonspecific binding of PF1 and OT. For quantitation of PF1 and OT in control C57BL/6J mouse plasma, the standard curve was made via serial dilution at concentration ranges of 0.5–1000 ng/ml and 0.5–500 ng/ml, respectively. For quantitation of PF1 in control Wistar Hannover rat plasma, the standard curve was made via serial dilution at a concentration range of 0.1–250 ng/ml. Standard regressions in plasma were performed using linear 1/x2 regression with r2 > 0.98.
For generation of brain tissue homogenate for analysis, brain samples were kept frozen to minimize degradation of PF1 during sample processing. Quantitation of PF1 in brain and CSF samples required a lower limit of quantitation than plasma, so the sample preparation differed. A standard curve was serially diluted in acetonitrile at 20× the desired concentration, and then 5 µl was spiked into 95 µl of blank plasma for a final standard curve concentration range of 10–1000 pg/g for brain homogenate and 10–250 pg/ml for CSF. Linear and quadratic 1/x2 regression was used for brain and CSF, respectively, with r2 > 0.98 for both. Mixed-matrix sample preparation was used for both brain homogenate and CSF. For brain homogenate analysis, 50 µl of blank plasma was added to 50 µl of brain homogenate sample and 50 µl of blank brain homogenate was added to 50 µl of plasma standard and blank. The CSF samples were also extracted using a mixed matrix approach where 25 µl of blank plasma was added to 25 µl of each CSF sample and 25 µl of blank aCSF was added to 25 µl of plasma standard or blank. Samples were then processed as described for plasma.
Samples were analyzed by an LC-MS/MS comprising a AB Sciex API 5500 tandem quadrupole mass spectrometer (AB Sciex Inc., Concord, ON, Canada) with a TurboIon Spray probe (AB Sciex Inc.), and Acquity UPLC H-Class system (Waters, Milford, MA). Instrument settings and potentials were adjusted to provide optimal data. All raw data were processed using Analyst Software version 1.5.1 or 1.5.2 (AB Sciex Inc., Ontario, Canada
Pharmacokinetic and Neuropharmacokinetic Calculations.
The pharmacokinetic parameters were computed by noncompartmental analysis using Watson Bioanalytical LIMS version 7.4 (Thermo Fischer Scientific, Fremont, CA) and WinNonlin version 5.2 (Pharsight, Mountain View, CA). The area under the curve values were calculated using the linear trapezoidal method. Measured total plasma (Cp), brain (Cb; assuming brain tissue density of 1 g/ml), and CSF (CCSF) concentrations were converted to unbound (free) values using the matrix-specific binding factor (fu,p, fu,b, or fu,CSF) to determine free plasma (Cp,u), free brain (Cb,u), and free CSF (CCSF,u) values. All neurocompartmental ratios (Cb/Cp, Cb,u/Cp,u, and CCSF,u/Cp,u) were calculated through use of area under the concentration time curve from time zero to the last measurable concentration values.
Contextual and Cue Fear Conditioning
Animals.
All animal studies were conducted in accordance with Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) using protocols reviewed and approved by the Pfizer WRD Institutional Animal Care and Use Committee. Male C57BL/6J mice (25–30 g; Jackson Laboratories) were maintained on a 12-hour light/dark cycle for a minimum of 3 days in a temperature- and humidity-controlled environment with free access to food and water before behavioral testing.
Fear Conditioning.
Freezing in response to a shock-associated novel environment and tone was assessed in male mice. The fear-conditioning protocol took place over 2 days (LeDoux, 2003). Coulbourn Instruments (Whitehall, PA) instrument chambers combined with Freeze Frame software (Coulbourn Instruments) were used to digitally assess freezing behavior via a camera mounted on the ceiling of the chamber. Before conditioning, each chamber was calibrated to ensure consistent amperage across the shock grids. On day 1, mice were placed in the chamber with the stimulus, and house lights illuminated; the mice were allowed 2 minutes of free exploration. After the first 2 minutes, a 30-second 2-Hz clicking sound was presented as the conditioned stimulus (CS). The last 2 seconds of the CS was paired with a 2-second 1.5-mA foot shock (unconditioned stimulus, US). After 90 seconds, the CS and US were presented a second time. After another 30 seconds, the animals were returned to their home cages for 24 hours. On day 2, the animals were experimentally manipulated and returned to the chambers. Freezing to context was measured for the initial 150 seconds. The CS was then presented for 30 seconds, and freezing was measured for another 150 seconds (freezing to cue). Freezing behavior was analyzed with FreezeView (Coulbourn Instruments) in 1-second bouts, sensitivity setting at 9.68, and aggregated into 30-second bins. Data are expressed as percentage of time freezing and analyzed with GraphPad Prism (GraphPad Software, San Diego,CA) using a one-way analysis of variance (ANOVA), followed by a Holm-Sidak post-hoc test or unpaired t test based on experimental design.
1. Subcutaneous PF1 depot administration.
To test the effect of peripheral PF1 on the expression of fear-conditioned freezing, PF1 (16 mg/kg in depot formulation) or the depot vehicle was administered s.c. 5 hours before testing in the chamber on day 2. Pretreatment time was based on the time of maximal concentration in the pharmacokinetic study. A positive control group given propranolol s.c. (10 mg/kg in 10% hydroxypropylbetacyclodextrin) 30 minutes before testing on day 2 and a negative control group conditioned in the absence of the US on day 1 were also tested. Freezing to both the context and cue were assessed after the pharmacologic manipulation.
2. Multidosing of OT.
To test the effect of peripheral OT administration, OT was administered in a multidose fashion to correct for the short t½ of OT. Either OT (10 mg/kg in saline; Sigma-Aldrich) or vehicle was dosed s.c. at 5 hours, 3 hours, and 1 hour before testing in the chamber on day 2. The dosing regimen was designed to mimic the sustained activation of peripheral OT receptors achieved with PF1. Freezing to both the context and cue were assessed after the pharmacologic manipulation.
3. Intracerebroventricular PF1 and PF2 administration.
To assess central effects of PF1 on the expression of fear conditioned freezing, animals were dosed ICV using a freehand technique with either 5 μg of PF1 (in 2 µl of 5:5:90 PEG400, DMSO, aCSF), 5 μg of PF2 (in 2 µl of 5:5:90 PEG400, DMSO, aCSF), 1 µg of OT (in 2 µl aCSF; Sigma no. 081M8715V, Sigma), or vehicle (2 µl 5:5:90 PEG400, DMSO, aCSF) 15 minutes before testing in the chamber on day 2. Compound PF2 was included to disambiguate the effect of enhanced stability versus enhanced selectivity achieved with PF1, as it has similar stability to PF1 but similar selectivity to OT. Freezing to both the context and cue were assessed after the pharmacologic manipulation.
Results
Pharmacology of PF1, PF2, and OT
The tool compounds PF1 and PF2 are analogs of OT specifically designed with an appended lipid tail to enhance binding to plasma proteins, which protects the active peptide portion of the molecule from enzymatic degradation (Fig. 1). To impart this property, both compounds have a single amino acid substitution changing the Leu8 of OT to a Lys that is further modified with a short polyethylene glycol spacer to a palmitoyl group. The half-time of these compounds is extended by dosing in a depot formulation that complexes with the lipid tail and slowly releases the compounds. Additionally, PF1 has a Pro7 to Gly substitution, which changes the pharmacology of the compound with respect to functional activity at the vasopressin (V1a) receptor. Compound PF1 is an antagonist at V1a receptor with a binding Ki of 4 nM, whereas OT and PF2 are potent agonists at the V1a receptor. V1a receptor antagonism also inhibits the expression of fear-conditioned freezing (Stoehr et al., 1993) and from a pharmacologic perspective is preferable to V1a agonism in a drug because of the cardiovascular effects associated with V1a agonists (Favory et al., 2009).
Table 1 shows the pharmacologic profiles of the lipidated analogs compared with OT. OT and PF2 are full agonists of the OT and V1a receptors, whereas PF1 is a full agonist at OTR but an antagonist at V1aR, presumably owing to the Pro7 amino acid change.
TABLE 1.
| Compound Name and Description | Oxytocin (OT) | PF-06655075 (PF1) | PF-06478939 (PF2) |
|---|---|---|---|
| nM | |||
| OTR agonist EC50 | 0.039 (n = 70) | 0.025 (n = 5) | 0.01 (n = 6) |
| OTR % agonist | 100 (n = 70) | 93 (n = 5) | 92 (n = 6) |
| OTR binding Ki | 0.48 (n = 7) | 0.037 (n = 1) | 0.076 (n = 2) |
| V1a agonist EC50 | 7.66 (n = 70) | >10,000 (n = 5) | 0.078 (n = 3) |
| V1a % agonist | 94 (n = 70) | N/A | 97 (n = 3) |
| V1a Ki (nM) | 16.1 (n = 8) | 4.37 (n = 7) | 0.327 (n = 2) |
OTR, oxytocin receptor.
Pharmacokinetics and Neuropharmacokinetics
Nonspecific Binding Studies.
The fu,p of OT in rat is 0.89, indicating that OT is not highly bound to plasma proteins. In contrast, PF1 exhibited high protein binding, both to plasma proteins and nonspecific binding interactions in brain tissue homogenate. The fu,p and fu,b were 0.0019 and 0.0006, respectively, for PF1. Because of the high extent of binding to plasma proteins, the fu,CSF was calculated to enable the understanding of the free concentration relationships across the plasma, brain, and CSF compartments. The calculated fu,CSF was 0.16.
In Vivo Studies.
The pharmacokinetic profiles of OT and PF1 were evaluated through four methods of administration: 1) i.v. administration with plasma collection to assess inherent pharmacokinetic properties (Fig. 2a), 2) s.c. administration in the nondepot formulation with plasma collection to determine the concentration-time profiles (Fig. 2b), 3) s.c. administration in the depot formulation with plasma collection to parallel the conditions in the behavioral studies to establish the exposure-response relationship (Figs. 2, c and d), and 4) s.c. administration of the depot formulation with brain, plasma, and CSF collection to compare the peripheral and central levels of exposure (Fig. 2e, f).
Fig. 2.

Pharmacokinetics of OT and PF-06655075 (PF1). (a) PF1 has an extended t½ in comparison with OT after i.v. administration to rats, (b) s.c. non–depot administration of PF1 and OT to mice, (c) incorporation of OT or PF1 into the depot formulation resulting in sustained total concentrations after s.c. administration versus the non–depot vehicle and simulation of s.c. multidosing OT in a non–depot vehicle, (d) corresponding free concentration time profiles in the depot formulation and after OT multidosing presented on the time scale of the behavioral experiments (red dashed line indicates time of testing), (e) total concentration-time profiles (brain, plasma, and CSF) of PF1 in rats, and (f) corresponding free concentration-time profiles (brain, plasma, CSF) of PF1 in rats.
After i.v. administration to rats, OT was rapidly cleared from systemic circulation with a plasma clearance and steady-state volume of distribution of 17.1 ml/min/kg and 0.085 liters/kg, respectively, resulting in a short half-life of 0.12 hour. Concentrations of OT were detectable out to 1 hour postdose, after which all values were below the limit of quantitation. In contrast, PF1 exhibited reduced plasma clearance (1.1 ml/mg/kg) and an increased steady-state volume of distribution (0.61 liters/kg), resulting in a half-life of 8.5 hours, significantly prolonged in comparison with OT. Concentrations of PF1 were quantifiable for the duration of the study. The pharmacokinetic profiles of OT (dose normalized) and PF1 are illustrated in Fig. 2a.
After s.c. administration in a nondepot formulation to mice, PF1 exhibited an extended half-life in comparison with OT, 3.2 hours versus 0.5 hour, respectively (Fig. 2b). Administration of PF1 at 1 mg/kg in the nondepot formulation resulted in a total maximal plasma concentration (Cmax) of 331 nM (free Cmax = 0.6 nM) at 2 hours postdose. The total Cmax of OT at 1 mg/kg was 216 nM (free Cmax = 191 nM) and occurred at 0.25 hour postdose. The plasma concentrations of OT increased in a dose-proportional manner, with the increase in dose from 1 to 10 mg/kg. The concentration-time profile of the single 10 mg/kg s.c. administration of OT was modeled to obtain pharmacokinetic parameters; subsequently, simulations were conducted to predict the pharmacokinetic profile of OT after the multidose paradigm in the behavioral study (Fig. 2, c and d).
Incorporation of either PF1 or OT in the depot formulation further increased the duration of exposure of the molecules in vivo, as the depot formulation facilitates a sustained release of the compounds from the s.c. space into circulation (Fig. 2, c and d). In mice, s.c. administration of OT at 20 mg/kg in the depot formulation resulted in a total Cmax of 65.4 nM (free Cmax = 58.2 nM) and a half-life of 7.3 hours. In contrast, s.c. administration of PF1 resulted in a total Cmax of 552 nM (free Cmax = 1.1 nM) and a half-life of 309 hours. The plasma concentrations of PF1 were maintained for the 24-hour study duration. In alignment with the free drug hypothesis (Tillement et al., 1988), as PF1 exhibits a high level of binding to proteins, it is presumed to be nonfunctional while bound, and only the free concentrations of the compound should be considered pharmacologically active. OT differs from PF1 as it is not highly bound to plasma proteins; consequently, the total and free concentrations of OT are similar.
The neuropharmacokinetics of PF1 were assessed through the use of s.c. depot administration. The s.c. administration of PF1 in the depot formulation resulted in prolonged exposure in the plasma, with minimal concentrations detectable in the central compartments (Fig. 2, e and f). The Cb/Cp of PF1 was 0.004, correcting for the nonspecific binding in the matrices resulted in a Cp,u/Cb,u of 0.001. Brain concentrations were measurable throughout the time course, although these concentrations are consistent with an incomplete perfusion of the cerebral vasculature. Calculations were completed using the measured plasma concentrations, cerebral blood volume, and a perfusion efficiency of 93% (Chugh et al., 2009), which resulted in calculated brain values in alignment with the measured values, suggesting that there is no accumulation in the brain parenchyma (Fig. 2e). The CCSF,u/Cp,u was 0.02, indicating passive diffusion across the blood-CSF barrier in the expected range for a peptide in this molecular weight range. The maximal free concentration of PF1 detected in the CSF was 0.009 nM, which would result in occupancy of approximately 20% of exposed OT receptors. It is possible, as a result of the proximity of a large density of OT receptors to the ventricles (primarily those in the paraventricular nucleus of the hypothalamus surrounding the third ventricle), that this level of CSF concentration could result in the activation of central OT receptors.
Effect on Fear-Conditioned Freezing
Prolonged peripheral exposure of PF1 in the depot formulation is sufficient to induce centrally mediated behavioral effects. There was a significant main effect of treatment on the expression of freezing in response to the conditioned context [one-way ANOVA; F(3,62) = 24.43, P < 0.0001] and the context + cue [one-way ANOVA; F(3,62) = 9.766, P < 0.0001] (Fig. 3a,b). The s.c. dosing of PF1 resulted in significantly reduced freezing to both context (post hoc Holm-Sidak P < 0.0001) and context + cue (post hoc Holm-Sidak P < 0.0001) on the day after CFC training (Fig. 3, a and b) to a similar degree as propranolol (context: Holm-Sidak P < 0.0001; context + cue Holm-Sidak P < 0.05), a known modulator of the centrally mediated conditioned fear response (Rodriguez-Romaguera et al., 2009).
Fig. 3.

Effect of PF-06655075 (PF1) on fear-induced freezing. Peripheral administration of PF1 inhibits freezing to a similar degree as propranolol in response to both context (a) and context + cue (b). However, multidose administration of OT does not significantly inhibit freezing in either context (b) or context + cue (c). Central administration of PF1, PF2, and OT inhibits the expression of freezing to both context (e) and context + cue (f). *P < 0.05; **P < 0.01; ***P < 0.001.
To determine whether prolonged peripheral stimulation of the OT receptor accounted for the behavioral effect of PF1 when given peripherally, we tested whether maintaining a constant plasma level of OT would have a similar effect on the fear response (Fig. 2, c and d). Compared with vehicle, multidose OT failed to alter the fear response to context (unpaired two-tailed Student’s t test, P = 0.3323) or context + cue (P = 0.8616) when given in a multidose fashion before testing on day 2.
To compare the effect of peripheral versus central PF1 administration on fear-conditioned freezing, OT, PF1 or PF2 was administered ICV. There was a significant main effect of treatment condition on both context [one-way ANOVA; F(3,47) = 13.48, P < 0.0001] and context + cue [one-way ANOVA; F(3,47) = 8.104, P < 0.001] induced freezing. Compared with vehicle, OT, PF1, and PF2 all reduced fear conditioned freezing in both paradigms (Fig. 3, e and f). Strikingly, OT eliminated freezing after ICV administration (context: Holm-Sidak P < 0.0001; context + cue Holm-Sidak P < 0.0001). Compound PF1, in contrast, significantly reduced freezing but did not eliminate the behavior (context: Holm-Sidak P < 0.01; context + cue Holm-Sidak P < 0.05). To test whether the difference in receptor selectivity between OT and PF1 accounted for the difference in the magnitude of response, compound PF2 (which has the stability of PF1 but the binding profile of OT) was also tested. Compound PF2 reduced freezing to both context and context + cue to a greater extent than PF1 but did not completely eliminate freezing (context: Holm-Sidak P < 0.01; context + cue Holm-Sidak P < 0.05).
Discussion
Peripheral administration of the long-acting, non–brain-penetrant OTR agonist PF1 inhibits the expression of fear-potentiated freezing. Compound PF1 evokes this behavioral effect despite not being detectable at levels that indicate permeability into brain tissue, suggesting a peripheral mechanism of action. Given that similar inhibition of freezing occurs with central administration of the agonist, however, it is hypothesized that the peripheral efficacy of PF1 may be mediated by feed-forward effects. The development of this novel molecule presents a unique opportunity to study the peripheral effects of OT without the limitation of rapid proteolytic degradation.
The central activity of both compounds PF1 and PF2 is comparable to that of OT in the fear-conditioning assay, indicating that it is the OTR agonist activity responsible for this effect rather than the V1a agonist activity intrinsic to both OT and PF2. From a safety perspective, compound PF1 is preferable to compound PF2 because of the cardiovascular effects associated with V1a agonism. Therefore, compound PF1 was fully profiled pharmacokinetically.
Functional Properties of Compound PF1.
The compound PF1 was designed to be resistant to degradation through the addition of a lipid tail. The tail facilitates binding to circulating proteins, primarily albumin, which protects the molecule from proteolysis and decreases overall systemic clearance (Meijer and Van der Sluijs, 1987; Yang et al., 2014). The dramatic stability of the molecule after i.v. administration demonstrates the resistance to clearance conferred on the molecule relative to OT with an increase in half-life from 0.12 hour to 8.5 hours. Most of the PF1 maintained in circulation is bound to plasma proteins, as indicated by the low fraction unbound in plasma. The resistance to clearance translates to prolonged functionality through the maintenance of steady-state free concentrations of the molecule based on dissociation from the carrier proteins (Yang et al., 2014). A similar long-lasting functional effect can be seen in a previously described mechanistically similar but structurally distinct lipidated OT agonist; however, the pharmacokinetics of this molecule are unknown (Mizuno et al., 2015). The sustained exposure of PF1 is further extended by the formulation of the compound in the depot vehicle by impacting the rate of absorption (Chitkara et al., 2006). Whereas the depot formulation extended the duration of exposure of both OT and PF1 through the slow release of the lipophilic molecules from the lipid matrix, the interaction of the lipid tail of PF1 with the lipid components of the depot formulation made the effect much more dramatic. The depot formulation was designed to allow for sustained release of the payload from a lipid-based gel at a fixed rate over at least 48 hours. The combined increased stability of PF1 and slow-release kinetics from the depot formulation stabilized the free drug concentrations in mice at 1 nM in plasma and 0.02 nM in CSF for at least 24 hours. Thus, ample opportunity is afforded to activate peripheral receptors and to diffuse into the CSF to evoke behavioral effects without the limitation of short peripheral half-time of OT.
Mechanism of Action.
As both peripheral and central administration of compound PF1 reduce freezing in a fear-associated environment, it is clear that this effect can be mediated by a central mechanism. In fact, the central circuitry underlying the expression-freezing response has been well characterized and shown to be modulated by OT. Neurons expressing OT receptors in the lateral central amygdala regulate the expression of freezing behavior through their projections to the brainstem, particularly the periaqueductal gray, via the medial nucleus of the central amygdala (Viviani and Stoop, 2008; Knobloch et al., 2012). The extent of the control of freezing behavior by OT was evident as ICV administration of OT eliminated fear-conditioned freezing on our study, although the site of action was not directly assessed. Unlike OT, however, PF1 also was equally efficacious in reducing freezing behavior after peripheral administration. There are two potential mechanisms through which peripheral PF1 could inhibit the expression of freezing behavior: 1) activation of peripheral OT receptors that feed back to the central nervous system, and 2) activation of central OT receptors that feed-forward to stimulate endogenous OT release.
Compound PF1 could act peripherally through OT receptors located throughout the body in both male and female reproductive tissue, kidney, heart, and vascular tissue (Gimpl et al., 2008). The endogenous peptide is additionally found in the adrenals, vagus nerve, thymus, and pancreas, suggesting multifarious peripheral functions (Gimpl et al., 2008; Llewellyn-Smith et al., 2012). Activity in these tissues could exert behavioral effects through the autonomic nervous system, the hypothalamic-pituitary axis, or the peripheral sensory nervous system. Each of these systems plays a role in the modulation of the expression of stress-associated behaviors and in the modulation of fear-associated behaviors observed in this study could be due to changes in baseline anxiety states. In fact, it has been recently demonstrated that peripheral, but not central, administration of OT reduces anxiety in a fear conditioned startle assay (Ayers et al., 2011); however, direct peripheral activation of OT receptors through prolonged or high dosing of OT could alternatively result in receptor downregulation, limiting behavioral efficacy, as may have happened with the multidosing regimen in this study (Gimpl et al., 2008).
Alternatively, PF1 in the CSF could act on OT receptors located within the cell layers immediately surrounding the ventricles within the central nervous system. OT receptors are highly concentrated in the paraventricular nucleus and the supraoptic nucleus of the hypothalamus, and these regions are located adjacent to ventricular space (Yoshimura et al., 1993). OT receptors in these nuclei are autoregulatory and activation can upregulate the axonal release of endogenous OT (Neumann et al., 1996). Consequently, the behavioral effects could be driven by an upregulation of OT release within the amygdalar circuit. Induction of OT release driven by activation of hypothalamic OT receptors is likely to cause a similar effect as induction through optogenetic stimulation: the inhibition of fear potentiated freezing though site-specific axonal release (Knobloch et al., 2012).
From Fear Conditioning to Social Cognition.
Fear conditioning was used as a behavioral readout in this study because the role of OT and the associated circuitry has been extensively characterized; however, there are some limitations to the use of this model as a behavioral readout for drug discovery: 1) Fear conditioning both in our hands and in the literature shows considerable intersubject variability (MacNamara et al., 2015). In our studies, we needed 12–16 subjects owing to variability in the initial levels of freezing. Experience, age, and genetic influence can produce random variation in fear- conditioning experiments, necessitating the use of larger group sizes and thus limiting its utility (Wehner and Radcliffe, 2004). 2) Although the expression of freezing is through a discrete circuit, fear or anxiety can be modulated through both peripheral and central mechanisms. The ambiguity in the compartment through which peripheral effects are evoked makes it difficult to optimize chemical matter. 3) Lastly, whereas the role of OT in fear and anxiety is intriguing, much of the impetus for commercialization of OT is for the treatment of social impairments. Unfortunately, there currently are few well-validated, high-throughput assays of social behavior. As the potential of OT as a therapeutic is dependent on the development and validation of a viable behavioral or physiologic read of efficacy, Pfizer is making compound PF1 freely available to academic collaborators to validate their models and paradigms.
Conclusion
Peripheral administration of PF1 results in sustained concentrations in both plasma and CSF but no concentrations in the brain parenchyma. This distribution is sufficient to inhibit fear-conditioned freezing, but it has yet to be determined whether this effect is through direct peripheral action or through feed-forward release of central OT. On request, PF1 is available from Pfizer for use as a research tool to independent investigators to further explore the role of peripheral OT receptor activation.
Abbreviations
- ANOVA
analysis of variance
- Cb
concentration in brain
- CCSF
concentration in CSF
- Cp
concentration in plasma
- CS
conditioned stimulus
- CSF
cerebrospinal fluid
- fu,b
fraction unbound in brain
- fu,CSF
fraction unbound in CSF
- fu,p
fraction unbound in plasma
- ICV
intracerebroventricular
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- OT
oxytocin
- OTR
oxytocin receptor
- PF1
PF-06655075
- PF2
PF-06478939
- US
unconditioned stimulus
- V1a
vasopressin receptor 1a
- WRD
Worldwide Research and Development
Authorship Contributions
Participated in research design: Modi, Buhl, Fonseca, Kablaoui.
Conducted experiments: Majchrzak, Doran, Osgood, Vanase-Frawley.
Contributed new reagents or analytical tools: McInnes, Darvari, Feyfant.
Performed data analysis: Modi, Buhl, Fonseca, Kablaoui.
Wrote or contributed to the writing of the manuscript: Modi, Buhl, Fonseca, Kablaoui.
Footnotes
This work is entirely funded by the research and development budget of the Pfizer Neuroscience Research Unit.
References
- Andari E, Duhamel JR, Zalla T, Herbrecht E, Leboyer M, Sirigu A. (2010) Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci USA 107:4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers LW, Missig G, Schulkin J, Rosen JB. (2011) Oxytocin reduces background anxiety in a fear-potentiated startle paradigm: peripheral vs central administration. Neuropsychopharmacology 36:2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakermans-Kranenburg MJ, van I Jzendoorn MH. (2013) Sniffing around oxytocin: review and meta-analyses of trials in healthy and clinical groups with implications for pharmacotherapy. Transl Psychiatry 3:e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson DS, Berquist SW, Trujillo TH, Garner JP, Hannah SL, Hyde SA, Sumiyoshi RD, Jackson LP, Moss JK, Strehlow MC, et al. (2015) Cerebrospinal fluid and plasma oxytocin concentrations are positively correlated and negatively predict anxiety in children. Mol Psychiatry 20:1085–1090. [DOI] [PubMed] [Google Scholar]
- Chitkara D, Shikanov A, Kumar N, Domb AJ. (2006) Biodegradable injectable in situ depot-forming drug delivery systems. Macromol Biosci 6:977–990. [DOI] [PubMed] [Google Scholar]
- Chugh BP, Lerch JP, Yu LX, Pienkowski M, Harrison RV, Henkelman RM, Sled JG. (2009) Measurement of cerebral blood volume in mouse brain regions using micro-computed tomography. Neuroimage 47:1312–1318. [DOI] [PubMed] [Google Scholar]
- de Wied D, Gaffori O, Burbach JP, Kovács GL, van Ree JM. (1987) Structure activity relationship studies with C-terminal fragments of vasopressin and oxytocin on avoidance behaviors of rats. J Pharmacol Exp Ther 241:268–274. [PubMed] [Google Scholar]
- Di L, Umland JP, Chang G, Huang Y, Lin Z, Scott DO, Troutman MD, Liston TE. (2011) Species independence in brain tissue binding using brain homogenates. Drug Metab Dispos 39:1270–1277. [DOI] [PubMed] [Google Scholar]
- Di L, Umland JP, Trapa PE, Maurer TS. (2012) Impact of recovery on fraction unbound using equilibrium dialysis. J Pharm Sci 101:1327–1335. [DOI] [PubMed] [Google Scholar]
- Ermisch A, Barth T, Rühle HJ, Skopková J, Hrbas P, Landgraf R. (1985) On the blood-brain barrier to peptides: accumulation of labelled vasopressin, DesGlyNH2-vasopressin and oxytocin by brain regions. Endocrinol Exp 19:29–37. [PubMed] [Google Scholar]
- Fabian M, Forsling ML, Jones JJ, Pryor JS. (1969) The clearance and antidiuretic potency of neurohypophysial hormones in man, and their plasma binding and stability. J Physiol 204:653–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favory R, Salgado DR, Vincent JL. (2009) Investigational vasopressin receptor modulators in the pipeline. Expert Opin Investig Drugs 18:1119–1131. [DOI] [PubMed] [Google Scholar]
- Gimpl G, Reitz J, Brauer S, Trossen C. (2008) Oxytocin receptors: ligand binding, signalling and cholesterol dependence. Prog Brain Res 170:193–204. [DOI] [PubMed] [Google Scholar]
- Hollander E, Bartz J, Chaplin W, Phillips A, Sumner J, Soorya L, Anagnostou E, Wasserman S. (2007) Oxytocin increases retention of social cognition in autism. Biol Psychiatry 61:498–503. [DOI] [PubMed] [Google Scholar]
- Hollander E, Novotny S, Hanratty M, Yaffe R, DeCaria CM, Aronowitz BR, Mosovich S. (2003) Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology 28:193–198. [DOI] [PubMed] [Google Scholar]
- Klenerova V, Sery O, Hynie S. (2008) Corticotropin-releasing hormone receptor subtypes in the rat anterior pituitary after two types of restraint stress. Ann N Y Acad Sci 1148:415–420. [DOI] [PubMed] [Google Scholar]
- Knobloch HS, Charlet A, Hoffmann LC, Eliava M, Khrulev S, Cetin AH, Osten P, Schwarz MK, Seeburg PH, Stoop R, et al. (2012) Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 73:553–566. [DOI] [PubMed] [Google Scholar]
- Landgraf R. (1981) Simultaneous measurement of arginine vasopressin and oxytocin in plasma and neurohypophyses by radioimmunoassay. Endokrinologie 78:191–204. [PubMed] [Google Scholar]
- LeDoux J. (2003) The emotional brain, fear, and the amygdala. Cell Mol Neurobiol 23:727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn-Smith IJ, Kellett DO, Jordan D, Browning KN, Travagli RA. (2012) Oxytocin-immunoreactive innervation of identified neurons in the rat dorsal vagal complex. Neurogastroenterol Motil 24:e136–e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNamara A, Rabinak CA, Fitzgerald DA, Zhou XJ, Shankman SA, Milad MR, Phan KL. (2015) Neural correlates of individual differences in fear learning. Behav Brain Res 287:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BB. (2004) Brain-fluid barriers: relevance for theoretical controversies regarding vasopressin and oxytocin memory research. Adv Pharmacol 50:531–592, 655–708. [DOI] [PubMed] [Google Scholar]
- Meijer DK, Van der Sluijs P. (1987) The influence of binding to albumin and alpha 1-acid glycoprotein on the clearance of drugs by the liver. Pharm Weekbl Sci 9:65–74. [DOI] [PubMed] [Google Scholar]
- Mizuno A, Cherepanov SM, Kikuchi Y, Fakhrul AA, Akther S, Deguchi K, Yoshihara T, Ishihara K, Shuto S, Higashida H. (2015) Lipo-oxytocin-1, a novel oxytocin analog conjugated with two palmitoyl groups, has long-lasting effects on anxiety-related behavior and social avoidance in CD157 knockout mice. Brain Sci 5:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi ME, Connor-Stroud F, Landgraf R, Young LJ, Parr LA. (2014) Aerosolized oxytocin increases cerebrospinal fluid oxytocin in rhesus macaques. Psychoneuroendocrinology 45:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann I, Douglas AJ, Pittman QJ, Russell JA, Landgraf R. (1996) Oxytocin released within the supraoptic nucleus of the rat brain by positive feedback action is involved in parturition-related events. J Neuroendocrinol 8:227–233. [DOI] [PubMed] [Google Scholar]
- Neumann ID, Landgraf R. (2012) Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci 35:649–659. [DOI] [PubMed] [Google Scholar]
- Quintana DS, Alvares GA, Hickie IB, Guastella AJ. (2015) Do delivery routes of intranasally administered oxytocin account for observed effects on social cognition and behavior? A two-level model. Neurosci Biobehav Rev 49:182–192. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Romaguera J, Sotres-Bayon F, Mueller D, Quirk GJ. (2009) Systemic propranolol acts centrally to reduce conditioned fear in rats without impairing extinction. Biol Psychiatry 65:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoehr JD, Cheng SW, Serlin MC, Cramer CP, North WG. (1993) A physiological role of vasopressin in the expression of conditioned fear. Ann N Y Acad Sci 689:674–676. [DOI] [PubMed] [Google Scholar]
- Striepens N, Kendrick KM, Maier W, Hurlemann R. (2011) Prosocial effects of oxytocin and clinical evidence for its therapeutic potential. Front Neuroendocrinol 32:426–450. [DOI] [PubMed] [Google Scholar]
- Thuresson K, Tiberg F, Johansson M, Harwigsson I, Joabsson F, Johnsson M; Camurus AB, assignee (2005) Liquid Depot Formulations WO/2005/117830.
- Tillement JP, Urien S, Chaumet-Riffaud P, Riant P, Bree F, Morin D, Albengres E, Barre J. (1988) Blood binding and tissue uptake of drugs. Recent advances and perspectives. Fundam Clin Pharmacol 2:223–238. [DOI] [PubMed] [Google Scholar]
- Toth I, Neumann ID, Slattery DA. (2012) Central administration of oxytocin receptor ligands affects cued fear extinction in rats and mice in a timepoint-dependent manner. Psychopharmacology (Berl) 223:149–158. [DOI] [PubMed] [Google Scholar]
- Uvnäs-Moberg K, Ahlenius S, Hillegaart V, Alster P. (1994) High doses of oxytocin cause sedation and low doses cause an anxiolytic-like effect in male rats. Pharmacol Biochem Behav 49:101–106. [DOI] [PubMed] [Google Scholar]
- Viviani D, Charlet A, van den Burg E, Robinet C, Hurni N, Abatis M, Magara F, Stoop R. (2011) Oxytocin selectively gates fear responses through distinct outputs from the central amygdala. Science 333:104–107. [DOI] [PubMed] [Google Scholar]
- Viviani D, Stoop R. (2008) Opposite effects of oxytocin and vasopressin on the emotional expression of the fear response. Prog Brain Res 170:207–218. [DOI] [PubMed] [Google Scholar]
- Wehner JM and Radcliffe RA (2004). Cued and contextual fear conditioning in mice. Curr Protoc Neurosci (Chapter 8, Unit 8 5C). [DOI] [PubMed] [Google Scholar]
- Yang F, Zhang Y, Liang H. (2014) Interactive association of drugs binding to human serum albumin. Int J Mol Sci 15:3580–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura R, Kiyama H, Kimura T, Araki T, Maeno H, Tanizawa O, Tohyama M. (1993) Localization of oxytocin receptor messenger ribonucleic acid in the rat brain. Endocrinology 133:1239–1246. [DOI] [PubMed] [Google Scholar]