

Abstract
Preference for the Northern (N) ring conformation of the ribose moiety of nucleotide 5′-triphosphate agonists at P2Y1, P2Y2, P2Y4, and P2Y11 receptors, but not P2Y6 receptors, was established using a ring-constrained methanocarba (a 3.1.0-bicyclohexane) ring as a ribose substitute (Kim et al. J. Med. Chem. 2002, 45, 208–218.). We have now combined the ring-constrained (N)-methanocarba modification of adenine nucleotides with other functionalities known to enhance potency at P2 receptors. The potency of the newly synthesized analogues was determined in the stimulation of phospholipase C through activation of turkey erythrocyte P2Y1 or human P2Y1 and P2Y2 receptors stably expressed in astrocytoma cells. An (N)-methanocarba-2-methylthio-ADP analogue displayed an EC50 at the hP2Y1 receptor of 0.40 nM and was 55-fold more potent than the corresponding triphosphate and 16-fold more potent than the riboside 5′-diphosphate. 2-Cl–(N)-methanocarba-ATP and its N6-Me analogue were also highly selective, full agonists at P2Y1 receptors. The (N)-methanocarba-2-methylthio and 2-chloromonophosphate analogues were full agonists exhibiting micromolar potency at P2Y1 receptors, while the corresponding ribosides were inactive. Although β,γ-methylene-ATP was inactive at P2Y receptors, β,γ-methylene-(N)-methanocarba-ATP was a potent hP2Y1 receptor agonist with an EC50 of 160 nM and was selective versus hP2Y2 and hP2Y4 receptors. The rates of hydrolysis of Northern (N) and Southern (S) methanocarba analogues of AMP by rat 5′-ectonucleotidase were negligible. The rates of hydrolysis of the corresponding triphosphates by recombinant rat NTPDase1 and 2 were studied. Both isomers were hydrolyzed by NTPDase 1 at about half the rate of ATP hydrolysis. The (N) isomer was hardly hydrolyzed by NTPDase 2, while the (S) isomer was hydrolyzed at one-third of the rate of ATP hydrolysis. This suggests that new, more stable and selective nucleotide agonists may be designed on the basis of the (N)-conformation, which greatly enhanced potency at P2Y1 receptors.
Introduction
The P2 nucleotide receptors consist of two families: G-protein-coupled receptors (GPCR) termed P2Y, of which seven mammalian subtypes (P2Y1,2,4,6,11,12,13) have been cloned, and ligand-gated cation channels termed P2X, of which seven mammalian subtypes (P2X1–7) have been cloned.1–3 Adenine nucleotides are required for activation of P2Y1, P2Y11, P2Y12, and P2Y13 subtypes, while uracil nucleotides activate P2Y2, P2Y4, and P2Y6 subtypes. P2Y receptors may couple to multiple second messengers, but all except the recently cloned3–5 P2Y12 and P2Y13 receptors lead to activation of phospholipase C (PLC) via Gq, resulting in a rise in intracellular calcium. The Gi-coupled P2Y12 and P2Y13 receptors occur on a more distant branch of the sequence dendrogram than the other members of the P2Y family6 and are closer in sequence to a newly characterized GPCR that is selective for sugar nucleotides (e.g. UDP-glucose).7
The medicinal chemistry of P2 receptors is underdeveloped compared to many other GPCRs.8–10 Reasons for the lack of progress in this field include nonselectivity, chemical instability, heterogeneity, and nonbioavailability of known ligands. Other difficulties in characterizing the receptors include the enzymatic instability of the phosphate groups of nucleotides to form biologically active metabolites. Indeed, most of the known P2 antagonists also inhibit ectonucleotidase activity, thus augmenting the effects of the endogenous agonist(s). Receptor heterogeneity,11 oligomerization,12 and desensitization13 are further complications.
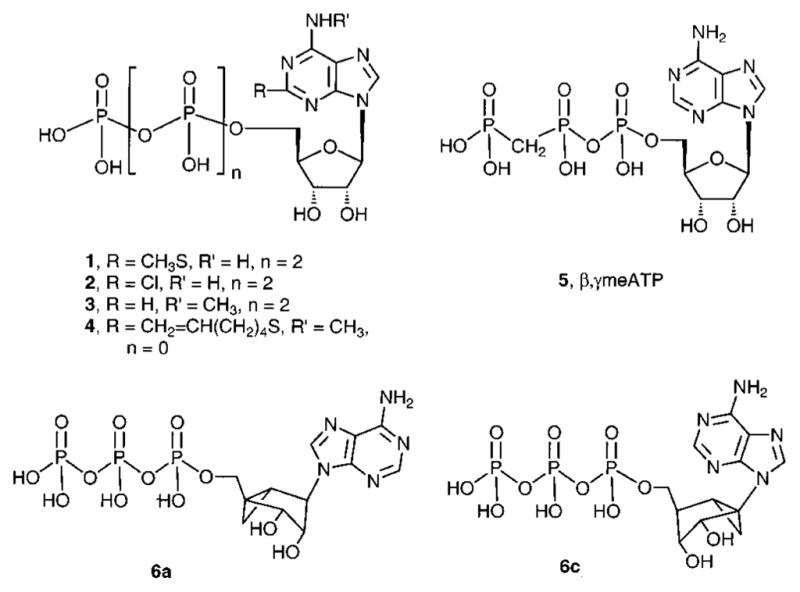
Despite the above-mentioned difficulties, the SAR of ATP analogues (e.g., 1–5; Chart 1) at P2 receptors has been probed.8–10 Selective agonists (P2Y1, P2Y6)14,15 and antagonists (P2Y1, P2Y12, P2X1, P2X7)14,16–19 have been reported for only a few of the subtypes. 2-Methylthio-ATP (1, 2-MeSATP) is one of the most potent agonists at a number of P2Y and P2X receptors.10 It was formerly regarded as selective for P2Y receptors; however, by adjustment for its lability in classical smooth muscle assays in which P2X receptors have been assayed, it is a highly potent agonist within the P2X superfamily.20 At the P2Y1 receptor, which is present in the heart, skeletal and various smooth muscles, prostate, ovary, and brain,21 the potency order for activation is 2-methylthioadenosine 5′-diphosphate (2-MeSADP) > 2-Me-SATP > ADP > ATP, and AMP and UTP are inactive. 2-Chloro-ATP and N6-methyl-ATP (2 and 3, respectively) are moderately potent agonists at P2Y1 receptors.22 MRS 2055 4 (N6-methyl-2-(5-hexenyl)thioadenosine 5′-triphosphate) is an agonist with micromolar potency at a glial adenylate cyclase coupled P2Y receptor (now identified as identical to P2Y12)23,24 but is inactive at P2Y1 receptors. β,γ-Methylene ATP 5 activates P2X1, but not P2Y1, receptors.21,22
Chart 1.
P2Y receptor ligands are being explored for therapeutic applications in the cardiovascular, endocrine, and other systems. A selective P2Y1 receptor agonist may have potential as an antihypertensive or antidiabetic agent.25–27 A selective antagonist of the P2Y1 receptor may be potentially useful in antithrombotic therapy,28,29 since activation of this subtype is associated with the initial shape change during platelet aggregation.29 ATP analogues have been shown to be effective insulin secretagogues.30 A P2Y1 receptor in osteoclasts has been linked to bone disintegration.31 First cloned from chick brain,32 the distribution of the P2Y1 receptor in the human brain has been described33 and abnormalities in P2Y1 receptor levels have been detected in the Alzheimer’s brain.34 A linkage between P2Y1 receptors and the MAP kinase/apoptosis pathway has been proposed.35 Selective agonists and antagonists at the P2Y1 receptor have been reported.10,14
3′,5′-Bisphosphate nucleotides have been explored as selective antagonists of the P2Y1 receptor, and nearly nanomolar affinities have been achieved.14 The ribose moiety of P2Y1 receptor antagonists may be replaced with carbocylics, smaller and larger rings, acyclics, and conformationally constrained rings,14 resulting in retention of affinity for the receptor. The ribose rings of nucleosides and nucleotides can adopt a range of conformations, although their target receptors likely prefer specific conformations. For P2Y1 antagonists we have replaced the ribose moiety with a carbocyclic ring locked in a preferred conformation, a fusion of cyclopropane and cyclopentane rings known as the methanocarba modification.36–38 Two structural variations, depending on the position of the cyclopropane ring, restrict the ring pucker, i.e., hold the ribose-like ring (pseudosugar) in either a Northern (N, 2′-exo) 6a or Southern (S, 2′-endo) 6c envelope conformation (Chart 1), as defined in the pseudorotational cycle. Preference for the Northern (N) ring conformation of the ribose moiety of nucleoside 5′-triphosphate agonists at P2Y1, P2Y2, P2Y4, and P2Y11 receptors, but not P2Y6 receptors, was established using a constrained methanocarba (bicyclic) ring.39
In the present study we combined the ring-constrained (N)-methanocarba modification of adenine nucleotides with other functionalities known to enhance potency at P2 receptors. In several cases, the constrained ring conferred dramatic increases in agonist potencies at P2Y1 receptors, and generally the modification preserved the potencies at P2Y2 receptors.
Results
Chemical Synthesis
We prepared methanocarbocyclic analogues of various synthetic adenine nucleotides (Table 1) in which fused cyclopropane and cyclopentane rings fixed the pseudoribose moiety in a rigid (N)-envelope conformation. The unsubstituted ATP 6a and AMP 7a analogues were reported in a previous study.39 The new nucleotide analogues 8a–15a and 17a were prepared in the ammonium salt form according to the methods shown in Schemes 1 and 2 and were tested biologically as agonists at various P2Y receptors (Table 1). Identity was confirmed using NMR (1H and 31P) and high-resolution mass spectrometry, and purity was demonstrated using high-pressure liquid chromatography (HPLC) in two different solvent systems (Table 2).
Table 1.
Biological Potencies of Methanocarba Derivatives of Adenine Nucleotides and Their Corresponding Riboside Analogues (Stimulation of Phospholipase C through Activation of P2Y Receptors)
| compd | analogue | subtype | a, methanacarba EC50 (nM) of (N)-methanocarba analoguea | b, riboside EC50 (nM) of corresponding ribose analoguea | ref |
|---|---|---|---|---|---|
| 6 | ATP | tP2Y1 | 14 ± 3 | 2800 ± 700 | |
| hP2Y1 | 52 ± 22 | 1500 ± 200 | |||
| hP2Y2 | 91 ± 5 | 85 ± 12 | |||
| hP2Y4 | 32% at 10 μM | c | |||
| hP2Y11 | 35 ± 5% at 10 μM | 17300 ± 2800 | |||
| 7 | AMP | tP2Y1 | d | d | |
| 8 | 2-MeSATP | tP2Y1 | 3.7 ± 1.7 | 8 ± 2 | 40 |
| hP2Y1 | 23 ± 11 | 34 ± 11 | |||
| 9 | 2-MeSADP | tP2Y1 | 0.88 ± 0.37 | 6 ± 3 | 40 |
| hP2Y1 | 0.40 ± 0.23 | 6.4 ± 3.5 | |||
| hP2Y2 | 54 ± 8% at 10 μM | d | |||
| hP2Y4 | d | d | |||
| hP2Y11 | d | d | |||
| 10 | 2-MeSAMP | tP2Y1 | 1190 ± 500 | 15% at 100 μM | |
| hP2Y1 | 590 ± 20 | d | |||
| hP2Y11 | d | d | |||
| 11 | 2-MeS(O)AMP | tP2Y1 | 36.6 ± 12.5 | d | |
| hP2Y1 | d | ||||
| 12 | 2-C1-ATP | tP2Y1 | 3 ± 1 | 720 ± 20 | 22 |
| hP2Y1 | 7 ± 1 | 770 ± 120 | 46 | ||
| hP2Y2 | 1900b | 2300 ± 300 | |||
| 13 | 2-C1-AMP | tP2Y1 | 3900 ± 2900 | d | |
| hP2Y1 | 1890 ± 770 | d | |||
| 14 | N6-Me-ATP | tP2Y1 | 92 ± 32 | 19000 ± 6000 | 22 |
| hP2Y1 | 33b | ||||
| hP2Y2 | 197 ± 2 | ||||
| 15 | 2-C1-N6-MeATP | tP2Y1 | 6 ± 3 | ||
| hP2Y1 | 9 ± 1 | ||||
| hP2Y2 | 8400 ± 3600 | ||||
| 16 | 2-C1-N6-MeAdo-5′-CONH-Me | tP2Y1 | d | ||
| 17 | β,γ-Me-ATP | tP2Y1 | 11,300 ± 1700 | d | 22 |
| hP2Y1 | 158b | d | |||
| hP2Y2 | d | d | |||
| hP2Y4 | d |
Average ± SEM data for 6a and 7a reported in ref 39. A percentage refers to percent of maximal effect.
Average of two determinations.
ATP is an antagonist at hP2Y4 receptors.50
No effect as either agonist or antagonist at 10 μM.
Scheme 1.

Synthesis of (N)-Methanocarbaadenosine 5′-Phosphate Derivativesa
a Reagents: (i) DEAD, Ph3P, THF; (ii) NH3 or CH3NH2, THF; (iii) palladium black, HCO2H, methanol; (iv) NaSCH3, DMF, 90–100 °C; (v) di-tert-butyl N,N-diethylphosphoramidite, tetrazole, THF, room temp, 20 min and then m-CPBA, −78 °C to room temp; (vi) DOWEX 50×8–200, methanol, 60–70 °C, ~80%; (vii) carbonyldiimidazole, DMF, room temp, 6 h; (viii) 5% triethylamine/H2O/MeOH, 2 h; (ix) tributylammonium pyrophosphate or tributylammonium phosphate, DMF, 3 days.
Scheme 2.
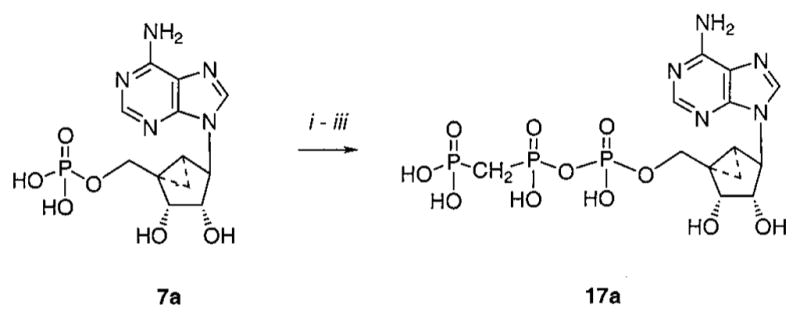
Synthesis of the (N)-Methanocarba Analogue of β,γ-Me-ATPa
a Reagents: (i) carbonyldiimidazole, DMF, room temp, 6 h; (ii) triethylamine/H2O/MeOH (1:10:10), 2 h; (iii) methylenediphosphonic acid, triethylamine, DMF, 3 days.
Table 2.
Synthetic Data for Nucleotide Derivatives, Including Structural Verification Using High-Resolution Mass Spectrometry and Purity Verification Using HPLC
| compd | formula | FAB (M – H+) m/z
|
HPLC retention time in min (purity in %a)
|
yieldb(%) | ||
|---|---|---|---|---|---|---|
| calcd | found | system A | system B unless otherwise noted | |||
| 8a | C13H20N5O12P3S | 561.9964 | 561.9966 | 11.0 (99) | 19.7 (99) | 35 |
| 9a | C13H19N5O9P2S | 482.0301 | 482.0291 | 10.5 (98) | 19.4 (98) | 40 |
| 10a | C13H18N5O6PS | 402.0637 | 402.0650 | 9.0 (98) | 15.6 (99) | 60 |
| 10b | C11H16N5O7PS | 394.0586 | 394.0591 | 14.7 (98) | 16.2 (99) | 16 |
| 11a | C13H18N5O7PS | 418.0586 | 418.0607 | 6.3 (97) | 12.7 (97) | 65 |
| 11b | C11H16N5O8PS | 410.0535 | 410.0545 | 11.2 (97) | 17.1 (97) | 16 |
| 12a | C12H17N5O12P3Cl | 551.9667 | 551.9673 | 7.4 (97) | 19.1 (98) | 30 |
| 13a | C12H15N5O6PCl | 390.0370 | 390.0384 | 7.7 (99) | 11.1(99) | 66 |
| 13b | C10H13N5O7PS | 382.0319 | 382.0322 | 9.2 (99) | 17.5 (99) | 42 |
| 14a | C13H20N5O12P3 | 530.0243 | 530.0247 | 6.0 (98) | 8.8 (98) | 25 |
| 15a | C13H19N5O12P3Cl | 563.9853 | 563.9866 | 11.2 (98) | 6.6 (98) | 32 |
| 16a | C14H17N6O3Cl | 353.1129 | 353.1139 | 10.4 (98) | 10.3c (99) | d |
| 17a | C13H20N5O11P3 | 514.0294 | 514.0306 | 3.2 (98) | 15.6 (97) | 32 |
Purity of each derivative was ≥97%, as determined using HPLC with two different mobile phases. System A: gradient of 0.1 M TEAA/CH3CN from 95/5 to 40/60. System B: gradient of 5 mM TBAP/CH3CN from 80/20 to 40/60. System C: gradient of 0.1 M TEAB/ CH3CN from 100/0 to 90/10 in 20 min.
The percent yields refer to the overall yield for each phosphorylation sequence.
System C.
Synthesis described in ref 45.
As in the previous study,39 since the phosphorus oxychloride method40,41 was unsatisfactory for phosphorylation of (N)-methanocarba nucleosides at the 5′-hydroxyl group, an alternative multistep process was used. This consisted of the formation of a monophosphate using the phosphoramidite method followed by condensation with additional phosphate or pyrophosphate groups using carbonyldiimidazole. By this method, the monophosphate analogues were cleanly converted to either di- or triphosphate via a phosphorimidazolidate intermediate.41 During the sequence, it was necessary to hydrolyze the cyclic carbonates42 38–41, which were formed at the 2′,3′-dihydroxy groups, using a 5% triethylamine solution.
The (N)-methanocarba analogues of various ATP derivatives substituted at the 2- and/or 6-positions of the purine base moiety or at the β,γ position of the triphosphate moiety were prepared by this approach. To construct the pseudoribose ring leading to the (N)-methanocarbaadenosine precursors, a 3.1.0-bicyclohex-ane structure was prepared by the general approach of Lee et al.,43 which utilized ring closure metathesis. An adenine precursor 18 or 19 was condensed with the protected bicycloheptane derivative 2043 using Mitsunobu conditions to give 2139 or 22 (Scheme 1). To introduce an amino or methylamino group at the 6-position, it was necessary to substitute 6-Cl with the corresponding amine following the Mitsunobu reaction to yield 23–26. Deprotection of the 5′-benzyl group of 23–26 was carried out smoothly using Pd black/formic acid, even in the presence of 2-chloro substitution, to give 27, 29, and 30. This was followed by 5′-phosphorylation using a phosphoramidite method44 to give the di-tert-butyl protected monophosphates 31, 34, and 35. Alternatively, the 2-chloro group of 27 was replaced with methylthio to give the nucleoside 28, leading to the protected monophosphate 32. In the process of synthesis of the 2-methylthio-AMP derivative 32, the corresponding sulfoxide 33 was isolated as a byproduct. The monophosphates 31–35 were deprotected and either tested biologically or carried further to the di- and triphosphates by the method shown in Scheme 1. The (N)-methanocarba β,γ-methylene triphosphate analogue 17a was prepared from the unsubstituted AMP equivalent 7a39 using methylene diphosphonate instead of pyrophosphate (Scheme 2).
Three riboside 5′-monophosphates 10b, 11b, and 13b were prepared using methods shown in Scheme 3 for comparison to the corresponding (N)-methanocarba derivatives. The isopropylidene group was used as protection for the 2′- and 3′-hydroxyl groups, and phosphorylation was carried out using the phosphoramidite method, as above.
Scheme 3.
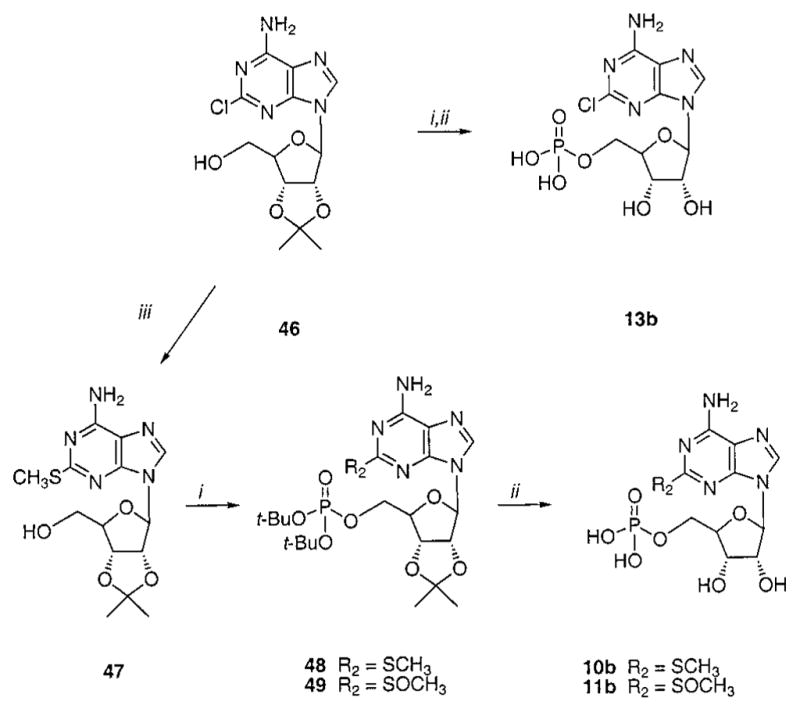
Synthesis of Adenosine 5′-Monophosphate Analoguesa
a Reagents: (i) tetrazole, di-tert-butyl N,N-diethylphosphoramidite, THF, then m-CPBA, CH2Cl2; (ii) DOWEX 50W×8–200, H2O/ MeOH; (iii) NaSCH3, DMF, 90 °C.
Biological Activity
(N)-Methanocarba analogues (denoted “a”, Table 1) were tested for agonist activity by measuring P2Y1-receptor-promoted phospholipase C activity in turkey erythrocyte membranes and by measuring inositol phosphate accumulation in 1321N1 human astrocytoma cells stably expressing the human P2Y1, P2Y2, P2Y4, or P2Y11 receptors. In each case, full concentration effect curves were generated and EC50 values from at least three separate experiments were determined. The agonist potencies of the corresponding ribose-containing nucleotides (denoted “b”) were simultaneously determined for comparison purposes.
Given the high potencies observed with (N)-methanocarba analogues of ATP and UTP and the much greater potency of the (N)-methanocarba as opposed to (S)-methanocarba conformation, we extended our studies to (N)-methanocarba derivatives synthesized containing various functionalities that we and others have shown to produce potent or receptor-selective adenine nucleotide analogues.8–10 As has been observed for adenine nucleotides, substitution at the 2-position, such as 2-methylthio, in the (N)-methanocarba-nucleotide series enhanced P2Y1 receptor potency. Thus, the high P2Y1 potency of 2-MeSATP 8b (Figure 1) was preserved in the corresponding (N)-methanocarba analogue 8a. The most potent P2Y1 agonist in this series was the 2-methylthio diphosphate analogue 9a (Figure 2), which displayed an EC50 of 0.88 nM at the turkey P2Y1 receptor and was 7-fold more potent than the corresponding ribose analogue 9b and 26-fold more potent than 8a. At other receptors 9a was inactive (P2Y4, P2Y11) or weakly active (P2Y2). The (N)-methanocarba-2-methylthio monophosphate analogue 10a and the (N)-methanocarba-2-chloro monophosphate analogue 13a were full agonists exhibiting micromolar potency at the turkey and human P2Y1 receptors. Oxidation of the thioether to a sulfoxide, in 11a, reduced tP2Y1 receptor potency by 30-fold. The corresponding 5′-monophosphate ribosides 10b, 11b, and 13b were inactive at P2Y1 receptors. The (N)-methanocarba-2-chloro 5′-triphosphate analogue 12a was 240-fold more potent at the turkey P2Y1 receptor than the corresponding riboside 12b.
Figure 1.
Effects of the (N)-methanocarba modification of 2-MeS-ATP on phospholipase C activity in 1321N1 astrocytoma cells expressing the hP2Y1 receptor. Concentration-dependent stimulation of inositol phosphate formation by compounds 8a (□) and 8b (■) is shown. The data shown are typical curves for at least three experiments.
Figure 2.
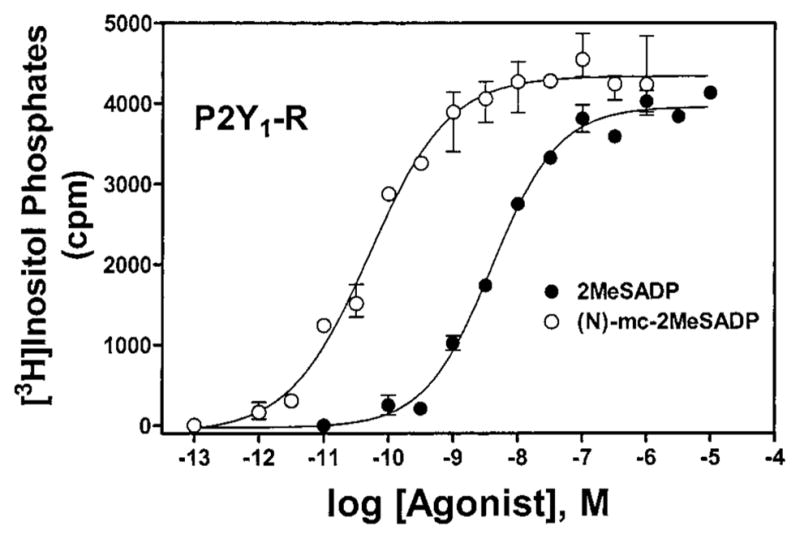
Effects of the (N)-methanocarba modification of 2-MeS-ADP on phospholipase C activity in 1321N1 astrocytoma cells expressing the hP2Y1 receptor. Concentration-dependent stimulation of inositol phosphate formation by compounds 9a (○) and 9b (●) is shown. The data shown are typical curves for at least three experiments.
As for the riboside series, substitution at the exocyclic amine of adenine was tolerated for (N)-methanocarba analogues acting as P2Y receptor agonists. The N6-methyl 5′-triphosphate analogue 14a was a potent agonist at the turkey and human P2Y1 receptors and was 5-fold more potent as a full agonist at the hP2Y1 than at the hP2Y2 receptor. Combination of N6-methyl and 2-chloro modifications in 15a resulted in a very potent agonist for the turkey and human P2Y1 receptors and selectivity for the hP2Y1 receptor versus hP2Y2 receptor of 930-fold. None of the (N)-methanocarba analogues of adenine nucleotide derivatives studied here were agonists at the human P2Y4 receptor (data not shown).
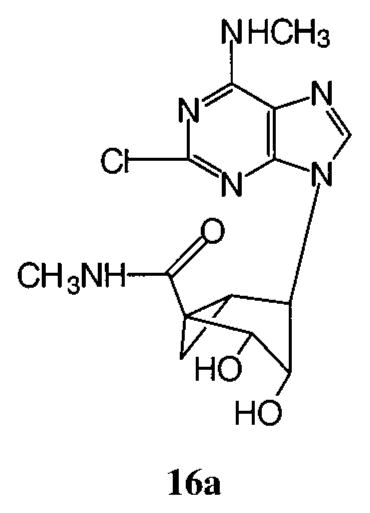
Since the 2-chloro-N6-methyl 5′-triphosphate analogue 15a was a potent P2Y1 receptor agonist, compound 16a45 was designed to evaluate the feasibility of replacing the triphosphate moiety with an uncharged moiety. However, it was inactive either as an agonist or as an antagonist at the turkey P2Y1 receptor. β;,γ-Me-ATP 17b was inactive as an agonist at the P2Y1 receptor.46,47 In contrast, and consistent with the potency enhancement observed for other nucleotide analogues, the (N)-methanocarba-β,γ-methylene analogue 17a was a relatively potent (EC50 = 11 μM) full agonist at the turkey P2Y1 receptor and a very potent (EC50 = 160 nM) agonist at the hP2Y1 receptor (Figure 3). In contrast, this analogue was inactive at the P2Y2 and P2Y4 receptors.
Figure 3.
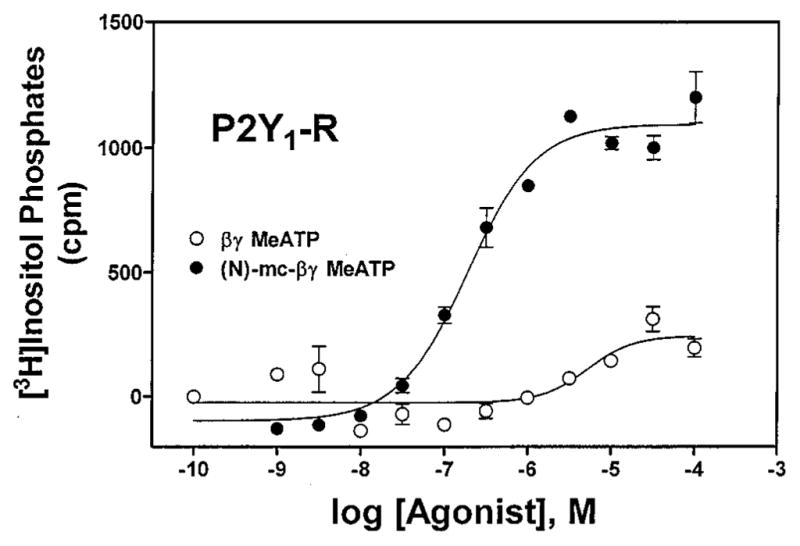
Effects of the (N)-methanocarba modification in triphosphate-modified adenine nucleotides on activation of phospholipase C in 1321N1 astrocytoma cells expressing hP2Y1 receptors, showing concentration-dependent stimulation of inositol phosphate formation by compounds 14a (●) and 14b (○). The data shown are typical curves for at least three experiments.
Because of the well-established lability of nucleotide analogues in pharmacological studies, the enzymatic stability of the simple (N)-methanocarba and (S)-methanocarba analogues of AMP and ATP was investigated (Tables 3 and 4). Neither the (N)-methanocarba nor (S)-methanocarba 5′-monophosphate derivatives 7a and 7c, respectively, were substrates for recombinant rat ecto- 5′-nucleotidase (Table 3).48 Hydrolysis of the (N)-methanocarba and (S)-methanocarba derivatives of ATP 6a and 6c, respectively, by recombinant rat ectoapyrase (NTPDase1) and ecto-ATPase (NTPDase2)49 stably expressed in CHO cells was also examined (Table 4). The (N)-methanocarba-ATP analogue was resistant to hydrolysis by NTPDase2 (4% as effective as ATP as a substrate), whereas the corresponding (S)-methanocarba-ATP was hydrolyzed by NTPDase2 at one-third (34%) the rate of ATP hydrolysis. In contrast, both molecules were good substrates for NTPDase1, at about half the rate of ATP hydrolysis (52% and 57%, respectively).
Table 3.
Hydrolysis Rates of Monophosphate Derivatives as Substrates of Purified Recombinant Rat Ecto-5′-nucleotidase
| substrate | activitya (%) |
|---|---|
| AMP | 100 |
| 7a, (N)-methanocarba-AMP39 | 0.14 |
| 7c, (S)-(±)-methanocarba-AMP39 | 0.0 |
Values are means of two experiments with duplicate determinations in each. The catalytic activity in the presence of AMP (100%) corresponded to 5.6 nmol/(min μg) protein.
Table 4.
Hydrolysis of the Triphosphate Derivatives as Substrates of Rat NTPDase1 (Ectoapyrase) and NTPDase2 (Ecto-ATPase) Stably Expressed in CHO Cells
| substrate | activitya (%)
|
|
|---|---|---|
| NTPDase1 | NTPDase2 | |
| ATP | 100 | 100 |
| 6a, (N)-methanocarba-ATP39 | 52 | 4 |
| 6c, (S)-(±)-methanocarba-ATP39 | 57 | 34 |
The hydrolysis rate with the two derivatives as substrate is compared to that of ATP (100%) for each enzyme. Values are means of two experiments with duplicate determinations in each. Nontransfected CHO cells were analyzed as control. They contained no significant activity for the extracellular hydrolysis of ATP. 100% values correspond to the following: NTPDase1, 42.1 nmol/(min 106 cells); NTPDase2, 13.5 nmol/(min 106 cells).
Discussion
In the previous study,39 we concluded that substitution of the ribose moiety with fused cyclopentane and cyclopropane rings in a pseudorotational (N)-conformation produced agonists at certain adenine and uracil nucleotide activated P2Y receptors. At P2Y1, P2Y2, P2Y4, and P2Y11 receptors, but not P2Y6 receptors, an (N)-methanocarba analogue was much more potent than the corresponding molecule constrained in the Southern conformation. Here, we have extended the synthesis to multiple substituted analogues of ring-constrained (N)-methanocarba-ATP, ADP, and AMP and illustrated that these derivatives exhibit as high or even higher potency for activation than the corresponding riboside, depending on the P2Y subtype involved. In some cases, alreadypotent ATP analogues were increased in potency upon (N)-methanocarba modification. In other cases, an inactive compound became biologically active upon such substitution.
A key finding in this study is that remarkably potent agonists, especially at the P2Y1 receptor, can be produced by introducing the (N)-methanocarba modification into molecules having other substitutions known to increase P2Y receptor potency. This was very notable in the (N)-methanocarba analogues of 2-Cl-ATP 13a and N6-Me-ATP 14a, which exhibited increases in potency in the (N)-methanocarba form versus ribosides of >200-fold. However, perhaps the most remarkable enhancement of activity occurred with β,γ-Me-ATP, although a full agonist at P2X receptors is not an agonist (or antagonist) at the P2Y1 and other P2Y receptors. However, the (N)-methanocarba analogue of β,γ-Me-ATP was a full agonist at the turkey and human P2Y1 receptors, exhibiting an EC50 for the human receptor of 160 nM. We have not yet extended this analysis to a direct comparison of relative activities of (N)- versus (S)-conformers to molecules that also possess other functional groups, e.g., 2-MeS or N6-CH3 substitution, known to increase binding affinity and selectivity for P2Y receptor agonists.
As we have observed with many different molecules that were synthesized and tested previously, results in human and turkey P2Y1 receptors were similar. The potencies determined in assays of phospholipase C in turkey erythrocyte membranes corresponded closely to the EC50 values determined for the same molecules at the recombinant human P2Y1 receptor stably expressed in 1321N1 human astrocytoma cells. This correlation was closest with a series of analogues containing the (N)-methanocarba substitution with various functional groups, e.g., 2-Cl, 2-MeS, N6-CH3, that we and others had previously demonstrated to increase P2Y receptor agonist potency in the riboside series. An explanation for the relatively large difference in apparent potency of the (N)-methanocarba analogue of β,γ-Me-ATP at the turkey versus human P2Y1 receptor is not readily apparent.
Although not studied in detail here, the (N)-methanocarba analogues exhibit resistance to hydrolysis relative to naturally occurring nucleotides. This observation may be particularly significant for 5′-monophosphate derivatives, since we have shown that they potently and selectively activate P2Y1 receptors but may be some-what stable to dephosphorylation by 5′-nucleotidase. However, it should be pointed out that this resistance to hydrolysis would at most only make a minor contribution to the very large difference in potencies seen between certain (N)-methanocarba compounds and their natural ribose counterpart. That is, the Km/Vmax values of ecto-nucleotidases on 1321N1 cells do not result in sufficient nucleotide hydrolysis to result in large shifts in the activation curves of, for example, ATP or UTP. More work will be required to obtain a clearer view on the capacity of the (N)-methanocarba molecules to serve as substrates for nucleotide-metabolizing ectoenzymes.
Thus, we established that constraining the ribose-like ring as methanocarba analogues was an important principle for producing P2Y receptor-subtype selectivity, similar to our previous demonstration of this phenomenon for P2Y1 receptor antagonists. The (N)-methanocarba analogues generally displayed increased potency over the corresponding nucleotide agonists at the P2Y1 and P2Y2 receptors. The high potency of several of the derivatives (e.g., the selective P2Y1 receptor agonist 9a) suggested the possibility of preparation of high-affinity radioligands in the (N)-methanocarba series. The potent and selective P2Y1 agonists (EC50 in nM at the tP2Y1 receptor unless otherwise noted) found in the present study were the following: 9a (0.88), 12a (3), 15a (9), 8a (23), 17a (160 at human), and 10a (590). Compound 14a was roughly equipotent at P2Y1 and P2Y2 receptors.
In conclusion, remarkable P2Y1 potency was generated in derivatives that combined the (N)-methanocarba modification with other functional groups known to enhance potency and/or selectivity in P2 receptor agonists. These molecules provide a new armamentarium for the study of P2Y receptors. Moreover, they provide an attractive new chemical backbone on which to build highly potent and selective P2Y receptor agonists and antagonists that, in lacking a ribose moiety, are not nucleotides, and therefore, they are less likely to bind to the myriad of nucleotide binding proteins that are not P2Y receptors. Furthermore, enhanced stability to some nucleotidase enzymes is indicated in the methanocarba series. These findings promise to be useful for defining the microscopic determinants of the binding sites on these receptors and for designing novel pharmacological probes and/or therapeutic agents.
Experimental Section
1. Chemical Synthesis
Nucleosides and synthetic reagents were purchased from Sigma Chemical Co. (St. Louis, MO) and Aldrich (Milwaukee, WI). 2,6-Dichloropurine was obtained from Sigma. The protected intermediate 21 was synthesized in our laboratory as described.43,45 (N)-Methanocarba analogues of ATP 6a and AMP 7a and the corresponding (±)-(S)-methanocarba analogues 6c and 7c, respectively, were prepared as described.39 Compound 16a was prepared as described.45
1H NMR spectra were obtained with a Varian Gemini-300 spectrometer (300 MHz) using D2O/CDCl3 and CD3OD as a solvent. 31P NMR spectra were recorded at room temperature by use of a Varian XL-300 spectrometer (121.42 MHz); orthophosphoric acid (85%) was used as an external standard.
Purity of compounds was checked using a Hewlett-Packard 1090 HPLC apparatus equipped with an SMT OD-5-60 RP-C18 analytical column (250 mm × 4.6 mm; Separation Methods Technologies, Inc., Newark, DE) in two solvent systems.
System A was a linear gradient solvent system consisting of 0.1 M TEAA/CH3CN from 95/5 to 40/60 in 20 min, and the flow rate was 1 mL/min. System B was a linear gradient solvent system consisting of 5 mM TBAP/CH3CN from 80/20 to 40/60 in 20 min, and the flow rate was 1 mL/min. System C was a linear gradient solvent system consisting of 0.1 M TEAB/CH3CN from 100/0 to 90/10 in 20 min, and the flow rate was 1 mL/min.
Peaks were detected by UV absorption using a diode array detector. All derivatives tested for biological activity showed ≥97% purity in the HPLC systems.
Low-resolution CI(NH3) (chemical ionization) mass spectra were carried out with Finnigan 4600 mass spectrometer, and high-resolution EI (electron impact) mass spectrometry was carried out with a VG7070F mass spectrometer at 6 kV. High-resolution FAB (fast-atom bombardment) mass spectrometry was performed with a JEOL SX102 spectrometer using 6 kV Xe atoms following desorption from a glycerol matrix.
Purification of the nucleotide analogues for biological testing was carried out on DEAE-A25 Sephadex columns as described above.
General Phosphorylation Procedure: Synthesis of (N)-Methanocarbaadenosine Derivatives (23–26). (1′S,2′ R,3′S,4′R,5′S)-4-(6-Amino-9H-purin-9-yl)-1-[(phenylmethoxy)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (23)
To a solution of 21 (0.2 g, 0.47 mmol) in i-PrOH (3 mL) was added NH3 (2 M solution in i-PrOH, 5 mL, 10 mmol), and the reaction mixture was heated at 90 °C in a closed tube for 15 h for complete reaction. The resulting mixture was concentrated under reduced pressure, and the residue obtained was purified by flash chromatography using 9/1 CHCl3/MeOH to furnish 0.182 g of 23 (95%). 1H NMR (CDCl3) δ 8.35 (s, 1H), 8.30 (s, 1H), 7.37 (s, 5H), 5.87 (bs, 2H), 5.31 (d, 1H, J = 6.8 Hz), 5.13 (s, 1H), 4.64 (qAB, 2H, J = 11.7, 20.5 Hz), 4.51 (d, 1H, J = 6.8 Hz), 3.97 (d, 1H, J = 10.7 Hz), 3.35 (d, 1H, J = 10.7 Hz), 1.72–1.62 (m, 1H), 1.55 (s, 3H), 1.32–1.26 (m, 1H), 1.23 (s, 3H), 1.00–0.93 (m, 1H).
Compounds 24 and 25 were synthesized from 21, and 26 was synthesized from 22. All were produced in 90–95% yields.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-9H-purin-9-yl)-1-[(phenylmethoxy)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (24)
1H NMR (CDCl3) δ 8.41 (s, 1H), 8.22 (s, 1H), 7.40–7.30 (m, 5H), 6.01 (bs, 1H), 5.30 (d, 1H, J = 7.2 Hz), 5.12 (s, 1H), 4.62 (qAB 2H, J = 12.1, 20.9 Hz), 4.51 (d, 1H, J = 7.2 Hz), 3.94 (d, 1H, J = 9.9 Hz), 3.35 (d, 1H, J = 9.9 Hz), 3.21 (s, 3H), 1.70–1.62 (m, 1H), 1.55 (s, 3H), 1.29–1.25 (m, 1H), 1.22 (s, 3H), 0.98–0.92 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[(phenylmethoxy)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (25)
1H NMR (CDCl3) δ 8.22 (s, 1H), 7.40–7.33 (m, 5H), 6.48 (bs, 2H), 5.32 (d, 1H, J = 7.15 Hz), 5.07 (s, 1H), 4.61 (qAB, 2H, J = 12.1, 19.2 Hz), 4.51 (d, 1H, J = 7.2 Hz), 3.94 (d, 1H, J = 9.9 Hz), 3.42 (d, 1H, J = 9.9 Hz), 1.64–1.56 (m, 1H), 1.55 (s, 3H), 1.30–1.26 (m, 1H), 1.24 (s, 3H), 0.98–0.933 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-2-chloro-9H-pu-rin-9-yl)-1-[(phenylmethoxy)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (26)
1H NMR (CDCl3) δ 8.15 (s, 1H), 7.40–7.28 (m, 5H), 6.29 (bs, 1H), 5.32 (d, 1H, J = 7.1 Hz), 5.05 (s, 1H), 4.61 (qAB, 2H, J = 12.4, 17.6 Hz), 3.90 (d, 1H, J = 10.2 Hz), 3.42 (d, 1H, J = 10.2 Hz), 3.17 (bs, 3H), 1.60–1.56 (m, 1H), 1.54 (s, 3H), 1.30–1.26 (m, 1H), 1.24 (s, 3H), 0.96–0.92 (m, 1H).
Procedure for Debenzylation of 24–26
See ref 39.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[hydroxymethyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (27)
1H NMR (CDCl3) δ 7.90 (s, 1H), 5.56 (d, 1H, J = 7.4 Hz), 4.79 (s, 1H), 4.66 (d, 1H, J = 7.4 Hz), 4.26 (d, 1H, J = 11.5 Hz), 3.37 (d, 1H, J = 11.5 Hz), 1.78–1.70 (m, 1H), 1.55 (s, 3H), 1.26 (s, 3H), 1.20–1.14 (m, 1H), 1.20–0.96 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-9H-purin-9-yl)-1-[hydroxymethyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (29)
1H NMR (CDCl3) δ 8.36 (s, 1H), 7.77 (s, 1H), 5.61 (d, 1H, J = 7.4 Hz), 4.78 (s, 1H), 4.65 (d, 1H, J = 7.4 Hz), 4.33 (d, 1H, J = 11.5 Hz), 3.25 (d, 1H, J = 11.5 Hz), 3.20 (bs, 3H), 1.79–1.74 (m, 1H), 1.55 (s, 3H), 1.25 (s, 3H), 1.17–1.13 (m, 1H), 1.01–0.96 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-2-chloro-9H-pu-rin-9-yl)-1-[hydroxymethyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (30)
1H NMR (CD3OD) δ 8.14 (s, 1H), 5.30 (d, 1H, J = 7.1 Hz), 4.87 (s, 1H), 4.61 (d, 1H, J = 7.1 Hz), 3.91 (d, 1H, J = 11.5 Hz), 3.52 (d, 1H, J = 11.5 Hz), 3.00 (bs, 3H), 1.64–1.60 (m, 1H), 1.44 (s, 3H), 1.18 (s, 3H), 1.08–1.04 (m, 1H), 0.92–0.87 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-1-[hydroxymethyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (28)
Compound 27 (30 mg, 0.086 mmol) was dissolved in DMF (1.5 mL) and treated with sodium thiomethoxide (90 mg, 1.3 mmol). The reaction mixture was heated in a sealed tube at 90 °C for 1.5 h. The solvent was removed under vacuum, and the product was purified by preparative thin-layer chromatography (9/1 CHCl3/MeOH) to furnish 25 mg of 28 (80%). 1H NMR (CDCl3) δ 7.76 (s, 1H), 5.82 (bs, 2H), 5.56 (d, 1H, J = 7.1 Hz), 4.78 (s, 1H), 4.71 (d, 1H, J = 7.1 Hz), 4.25 (d, 1H, J = 11.5 Hz), 3.35 (d, 1H, J = 11.5 Hz), 2.59 (s, 3H), 1.71–1.66 (m, 1H), 1.55 (s, 3H), 1.26 (s, 3H), 1.18–1.14 (m, 1H), 0.99–0.93 (m, 1H).
Protected (N)-Methanocarbaadenosine 5′-Monophosphate Derivatives (31–35)
These monophosphate derivatives were synthesized by procedures similar to those used to prepare the unsubstituted adenine analogue.39
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[(di-tert-butyl phosphate)methyl]bicyclo[3.1.0]hex-ane-2,3-(O-isopropylidene) (31)
1H NMR (CDCl3) δ 8.23 (s, 1H), 6.91 (bs, 2H), 5.41 (d, 1H, J = 7.1 Hz), 5.16 (s, 1H), 4.64 (d, 1H, J = 7.1 Hz), 4.59 (dd, 1H, J = 5.5, 11.0 Hz), 3.98 (dd, 1H, J = 6.3, 11.0 Hz), 1.84–1.76 (m, 1H), 1.63 (s, 3H), 1.61 (s, 9H), 1.59 (s, 9H), 1.42–1.37 (m, 1H), 1.33 (s, 3H), 1.18–1.13 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-1-[(di-tert-butyl phosphate)methyl]bicyclo[3.1.0]-hexane-2,3-(O-isopropylidene)(32)and(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-methylsulfoxy-9H-purin-9-yl)-1-[(di-tert-butyl phosphate)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (33)
To a mixture of 28 (20 mg, 0.055 mmol) and tetrazole (0.012 g, 0.165 mmol) in anhydrous THF (2 mL) was added di-tert-butyl-N,N-diethylphosphoramidite (0.023 mL, 0.083 mmol), and the mixture was stirred at room temperature overnight. The reaction mixture was cooled to −78 °C, and a solution of 70% mCPBA (0.012 g, 0.066 mmol) in 1 mL of CH2Cl2 was warmed to room temperature. A total of 1 mL MeOH was added, and the mixture was concentrated and purified by preparative TLC using 10% MeOH in CHCl3 to furnish 32 (12 mg) and 33 (12 mg).
32: 1H NMR (CDCl3) δ 7.98 (s, 1H), 5.87 (bs, 2H), 5.31 (d, 1H, J = 7.2 Hz), 5.07 (s, 1H), 4.59 (d, 1H, J = 7.2 Hz), 4.49 (dd, 1H, J = 5.8, 11.0 Hz), 3.83 (dd, 1H, J = 6.3, 11.0 Hz), 2.59 (s, 3H), 1.75–1.70 (m, 1H), 1.55 (s, 3H), 1.49 (s, 18H), 1.30–1.27 (m, 1H), 1.23 (s, 3H), 1.05–1.00 (m, 1H).
33: 1H NMR (CDCl3) δ 8.26 (s, 1H), 6.66 (bs, 2H), 5.33 (d, J = 7.14 Hz, 1H), 5.22 (s, 1H), 4.60–4.50 (m, 2H), 3.81–3.71 (m, 2H), 2.93 (s, 3H), 1.76–1.70 (m, 1H), 1.54 (s, 3H), 1.50 (s, 9H), 1.34–1.30 (m, 1H), 1.25 (s, 3H), 1.23 (s, 9H), 1.07–1.02 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-9H-purin-9-yl)-1-[(di-tert-butyl phosphate)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (34)
1H NMR (CDCl3) δ 8.45 (s, 1H), 8.05 (s, 1H), 6.04 (bs, 1H), 5.30 (d, 1H, J = 7.2 Hz), 5.08 (s, 1H), 4.57 (d, 1H, J = 7.2 Hz), 4.47 (dd, 1H, J = 5.8, 11.0 Hz), 3.90 (dd, 1H, J = 6.3, 11.0 Hz), 3.20 (bs, 3H), 1.79–1.71 (m, 1H), 1.54 (s, 3H), 1.51 (s, 9H), 1.50 (s, 9H), 1.29–1.26 (m, 1H), 1.22 (s, 3H), 1.08–1.03 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-2-chloro-9H-pu-rin-9-yl)-1-[(di-tert-butyl phosphate)methyl]bicyclo[3.1.0]-hexane-2,3-(O-isopropylidene) (35)
1H NMR (CDCl3) δ 8.04 (s, 1H), 5.38 (d, 1H, J = 7.1 Hz, 1H), 5.04 (s, 1H), 4.58 (d, 1H, J = 7.1 Hz), 4.42–4.52 (m, 1H), 3.90–4.08 (m, 1H), 3.20 (bs 3H), 1.79–1.71 (m, 1H), 1.54 (s, 3H), 1.51 (s, 9H), 1.50 (s, 9H), 1.29–1.26 (m, 1H), 1.22 (s, 3H), 1.08–1.03 (m, 1H).
General Procedure for Deprotection of (N)-Methanocarbaadenosine 5′-Monophosphate Derivatives (Synthesis of 13a, 10a, 11a, 36, and 37)
Deprotection was carried out by procedures similar to those used to prepare the unsubstituted adenine analogue.39
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-1-[phosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (10a)
1H NMR (D2O) δ 8.53 (s, 1H), 4.91 (s, 1H), 4.48 (dd, 1H, J = 4.7, 11.0 Hz), 4.05 (d, 1H, J = 6.6 Hz), 3.69 (dd, 1H, J = 4.7, 11.0 Hz), 2.63 (s, 3H), 1.92–1.84 (m, 1H), 1.58–1.40 (m, 1H), 1.18–0.98 (m, 1H). 31P NMR (D2O) δ 0.613.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-methylthiooxy-9H-purin-9-yl)-1-[phosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (11a)
1H NMR (D2O) δ 8.83 (s, 1H), 5.46 (d, J = 6.04 Hz, 1H), 4.52 (dd, J = 4.94, 10.71 Hz, 1H), 4.06 (d, J = 6.04 Hz, 1H), 3.72 (dd, J = 4.94, 10.71 Hz, 1H), 2.99 (s, 3H), 2.04–1.92 (m, 1H), 1.26–1.22 (m, 1H), 1.04–0.98 (m, 1H). 31P NMR (D2O) δ 0.623.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[phosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-di-ol (13a)
1H NMR (D2O) δ 8.60 (s, 1H), 4.72 (s, 1H), 4.48–4.40 (m, 1H), 3.99 (d, 1H, J = 6.6 Hz), 3.62–3.76 (m, 1H), 1.94–1.82 (m, 1H), 1.60–1.52 (m, 1H), 1.08–0.95 (m, 1H).
The imidazolate 42, derived from 13a, displayed a single 31P NMR resonance in D2O at δ −11.824.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-9H-purin-9-yl)-1-[phosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (36)
1H NMR (D2O) δ 8.47 (s, 1H), 8.26 (s, 1H), 4.87 (s, 1H), 4.76 (d, 1H, J = 6.6 Hz), 4.46 (dd, 1H, J = 5.0, 11.0 Hz), 3.99 (d, 1H, J = 6.6 Hz), 3.66 (dd, 1H, J = 5.0, 11.0 Hz), 3.12 (bs, 3H), 1.88–1.82 (m, 1H), 1.54–1.48 (m, 1H), 1.00–0.92 (m, 1H).
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-2-chloro-9H-pu-rin-9-yl)-1-[phosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (37)
1H NMR (D2O) δ 8.42 (s, 1H), 4.71 (s, 1H), 4.52 (m, 1H), 3.97 (d, 1H, J = 6.3 Hz), 3.72–3.62 (m, 1H), 3.01 (bs, 3H), 1.72–1.80 (m, 1H), 1.62–1.48 (m, 1H), 1.04–0.94 (m, 1H).
General Method of Synthesis of (N)-Methanocarba-ATP Analogues (8a, 12a, 14a, and 15a)
These triphosphate derivatives were synthesized by procedures similar to the procedure used to prepare the unsubstituted adenine analogue39 and the procedure for 5′-diphosphate analogue 9a.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-1-[diphosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (9a)
A mixture of 10a (14 mg, 0.035 mmol) and 1,1′-carbonyldiimidazole (28 mg, 0.175 mmol) in 1 mL of anhydrous DMF was stirred at room temperature for 6 h. Et3N/H2O/ MeOH (1:10:10 by volume, 2 mL) was added, and the mixture was stirred at room temperature for an additional 2 h. The mixture was concentrated to dryness under vacuum. To the residue, phosphoric acid (20 mg, 0.21 mmol) was added and dried under high vacuum. This was suspended in anhydrous DMF (2 mL), and Et3N (0.1 mL) was added. The mixture was stirred at room temperature for 3 days. Triethylammonium bicarbonate (2 mL) was added, all of the solvent was removed under vacuum, and the residue was purified using Sephadex column chromatography, eluting with 150 mL of water and 150 mL of ammonium bicarbonate to furnish 9a (5.0 mg). 1H NMR (D2O) δ 8.34 (s, 1H), 4.87 (s, 1H), 4.71 (d, J = 6.2 Hz, 1H), 4.54–4.48 (m, 1H), 4.02 (d, J = 6.2 Hz, 1H), 3.85–3.78 (m, 1H), 2.58 (s, 3H), 1.91–1.86 (m, 1H), 1.54–1.46 (m, 1H), 1.02–0.96 (m, 1H). 31P NMR (D2O) δ −9.63, −10.70.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-methylthio-9H-purin-9-yl)-1-[triphosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (8a)
1H NMR (D2O) δ 8.15 (s, 1H), 4.10–3.98 (m, 1H), 3.90–3.74 (m, 1H), 3.67–3.61 (m, 1H), 3.57–3.52 (m, 1H), 2.59 (s, 3H), 2.02–1.84 (m, 1H), 1.62–1.46 (m, 1H), 1.06–0.94 (m, 1H). 31P NMR (D2O) δ 0.02, −9.6, −10.8.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-2-chloro-9H-purin-9-yl)-1-[triphosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (12a)
1H NMR (D2O) δ 8.22 (s, 1H), 4.86 (s, 1H), 4.72 (d, 1H, J = 5.2 Hz), 3.84–3.74 (m, 2H), 2.14–2.02 (m, 1H), 1.62–1.54 (m, 1H), 1.06–0.96 (m, 1H). 31P NMR (D2O) δ −10.55, −10.93, −22.79.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-9H-purin-9-yl)-1-[triphosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-di-ol (14a)
1H NMR (D2O) δ 8.44 (s, 1H), 8.24 (s, 1H), 4.85 (d, 1H, J = 5.2 Hz), 4.75 (s, 1H), 4.57 (dd, 1H, J = 5.2, 11.3 Hz), 4.01 (d, 1H, J = 5.2 Hz), 3.83 (dd, 1H, J = 4.7, 11.3 Hz), 3.09 (bs, 3H), 1.96–1.86 (m, 1H), 1.56–1.48 (m, 1H), 1.04–0.96 (m, 1H). 31P NMR (D2O) δ −10.03, −10.67, −22.54.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Methylamino-2-chloro-9H-pu-rin-9-yl)-1-[triphosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (15a)
1H NMR (D2O) δ 8.42 (s, 1H), 4.86 (s, 1H), 4.74 (s, 1H), 4.64–4.55 (m, 1H), 4.00 (s, 1H), 3.95–3.72 (m, 1H), 3.08 (bs, 3H), 1.96–1.82 (m, 1H), 1.58–1.44 (m, 1H), 1.08–0.96 (m, 1H). 31P NMR (D2O) δ −8.05, −10.73, −22.19.
(1′S,2′R,3′S,4′R,5′S)-4-(6-Amino-9H-purin-9-yl)-1-[b,g-methylenetriphosphoryloxymethyl]bicyclo[3.1.0]hexane-2,3-diol (17a)
A mixture of compound 7a (11 mg, 0.03 mmol)39 and 1,1′-carbonyldiimidazole (24 mg, 0.15 mmol) in DMF (2 mL) was stirred at room temperature for 6 h. Et3N/H2O/MeOH (1:10:10 by volume, 1 mL) was added, and the mixture was stirred for 2 h. All of the solvent was removed under vacuum to dryness. The residue and methylene diphosphonic acid (0.32 g, 0.18 mmol) were dried under high vacuum for 1 h and suspended in anhydrous DMF (2 mL), 0.1 mL Et3N was added, and the mixture was stirred at room temperature for 3 days. Triethylammonium bicarbonate (2 mL) was added, and all of the solvent was removed under vacuum and purified using Sephadex column chromatography, eluting with 100 mL of water and 100 mL of ammonium bicarbonate to furnish 17a (4.0 mg). 1H NMR (D2O) δ 8.70 (s, 1H), 8.41 (s, 1H), 4.98 (s, 1H), 4.59 (dd, J = 4.95, 10.72 Hz, 1H), 4.00 (d, J = 6.04 Hz, 1H), 3.82 (dd, J = 4.95, 10.72 Hz, 1H), 2.36 (bs, 2H), 1.98–1.86 (m, 1H), 1.58–1.52 (m, 1H), 1.06–0.98 (m, 1H). 31P NMR (D2O) δ 15.454, 9.024, −10.341.
Phosphoric Acid Mono[(2R,3R,4S,5R)-5-(6-amino-2-chloro-9H-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-ylmethyl]ester (13b)
This monophosphate was synthesized by procedures similar to those used to prepare the unsubstituted adenine analogue.39 1H NMR (D2O) δ 8.23 (s, 1H), 5.93 (d, J = 6.04 Hz, 1H), 4.39 (s, 1H), 4.27 (s, 1H), 3.92–3.82 (m, 2H).
(2R,3R,4S,5R)-2-(6-Amino-2-methylsulfanyl-9H-purin-9-yl]tetrahydrofuran-3,4-(O-isopropylidene) (47)
Compound 46 (30 mg, 0.088 mmol) was dissolved in DMF (1.5 mL) and treated with sodium thiomethoxide (30 mg, 0.44 mmol). The reaction mixture was heated in a sealed tube at 90 °C for 1.5 h. The solvent was removed under vacuum, and the product was purified using preparative thin-layer chromatography (9/1 CHCl3/MeOH) to furnish 20 mg of 47 (65%). 1H NMR (CDCl3) δ 7.7 (s, 1H), 5.81 (d, J = 4.39 Hz, 1H), 5.62 (s, 2H), 5.34–5.28 (m, 1H), 5.12 (d, J = 4.39 Hz, 1H), 4.47 (s, 1H), 3.98–3.76 (m, 2H), 2.56 (s, 3H), 1.63 (s, 3H), 1.38 (s, 3H).
Phosphoric Acid Mono[(2R,3R,4S,5R)-5-(6-amino-2-methylsulfanyl-9H-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-ylmethyl]ester (10b) and Phosphoric Acid Mono-[(2R,3R,4S,5R)-5-(6-amino-2-methylsulfinyl-9H-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-ylmethyl]ester (11b)
Di-tert-butyl-N,N-diethylphosphoramidite (0.02 mL, 0.075 mmol) was added to a mixture of 47 (18 mg, 0.05 mmol) and tetrazole (17 mg, 0.05 mmol) in anhydrous THF (2 mL), and the mixture was stirred at room temperature overnight. The reaction mixture was cooled to −78 °C, treated with a solution of 70% mCPBA (15 mg, 0.06 mmol) in 1 mL of CH2Cl2, and warmed to room temperature. A total of 1 mL of MeOH was added, and the mixture was concentrated and purified by preparative TLC using 10% MeOH in CHCl3 to furnish 48 (10 mg) and 49 (10 mg). These were treated with 30 mg of Dowex 50×8–200 in 1/1 H2O/MeOH at 80 °C for 1 h. The Dowex resin was filtered, and the filtrate was concentrated to furnish 10b (3 mg, 15%) and 11b (3 mg, 15%).
10b: 1H NMR (D2O) δ 8.39 (s, 1H), 6.11 (d, J = 5.5 Hz, 1H), 4.82–4.95 (m, 1H), 4.45 (s, 1H), 4.26 (s, 1H), 3.92–3.82 (m, 2H), 2.59 (s, 3H).
11b: 1H NMR (D2O) δ 8.49 (s, 1H), 6.11 (d, J = 5.5 Hz, 1H), 4.81–4.94 (m, 1H), 4.45 (s, 1H), 4.26 (s, 1H), 3.92–3.82 (m, 2H), 2.82 (s, 3H).
2. Enzymatic Analyses
2.1. Hydrolysis of 5′-Triphosphate Derivatives by Recombinant Rat Ecto-5′-nucleotidase
Catalytically active recombinant ecto-5′-nucleotidase was expressed in insect cells using the baculovirus system and purified as described in ref 48. Ecto-5′-nucleotidase (0.5 μg) was incubated in Tris-buffered saline (TPS: 10 mM Tris, 150 mM NaCl, 0.4 mM MgCl2, pH 7.4) with the corresponding substrate (AMP, 7a, or 7c) at a final concentration of 100 μM. After 10 min of incubation at 37 °C, the reaction was terminated by addition of 5% TCA. The sample was centrifuged for 10 min at 12000g (4 °C). Inorganic phosphate was determined in the supernatant fraction according to the method of Lanzetta et al.51
2.2. Hydrolysis of 5′-Triphosphate Derivatives by Rat NTPDase1 (Ectoapyrase) and NTPDase2 (Ecto-ATPase) Stably Expressed in CHO Cells. Cell Transfection
CHO cells stably transfected with a plasmid DNA containing NTPDase1 (ectoapyrase) or NTPDase2 (ecto-ATPase) have previously been described.49 They were cultured in in HAM’s FC-12 medium containing 10% fetal calf serum, 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco BRL, Eggenstein, Germany) as described in ref 52.
2.3. Measurement of Ectonucleotidase Activity
Transfected CHO cells were seeded in multiwell plates (20 000 cells per well, 1.88 cm2). Surface-located enzyme activity of intact cells was determined 24 h later at 37 °C. Cells were washed twice with physiological saline solution (140 mM NaCl, 5 mM KCl, 10 mM glucose, 1 mM MgCl2, 2 mM CaCl2, 20 mM Hepes, pH 7.4) and subsequently incubated in 500 μL of the identical saline solution containing 50 mM of nucleotide substrate (ATP, 6a, or 6c). Aliquots of the culture supernatant were collected at varying time points and subjected to high-performance liquid chromatography (HPLC) analysis as previously described.49 The absorbance at 260 nm was continuously monitored, and the nucleotide and nucleoside concentrations were determined from the area under each absorbance peak.
4. Pharmacological Analyses
P2Y-receptor-promoted stimulation of inositol phosphate formation was measured at human P2Y1, P2Y2, P2Y4, and P2Y11 receptors stably expressed in 1321N1 human astrocytoma cells as previously described.46,50 The K0.5 values were averaged from 3 to 8 independently determined concentration–effect curves for each compound. Turkey erythracytes were incubated in inositol-free medium (DMEM; Gibco, Gaithersburg, MD) with 0.5 mCi of 2-[3H]myo-inositol (20 Ci/mmol; American Radiolabeled Chemicals, Inc., St. Louis, MO) for 18–24 h in a humidified atmosphere of 95%/5% air/CO2 at 37 °C. tP2Y1 receptor-promoted phospholipase C activity was measured in 25 μL of [3H]inositol-labeled ghosts (approximately 175 μg of protein, 200–500000 cpm/assay) in a medium containing 424 μM CaCl2, 0.91 mM MgSO4, 2 mM EGTA, 115 mM KCl, 5 mM KH2PO4, and 10 mM Hepes, pH 7.0. Assays (200 μL final volume) contained 1 μM GTPγS and the indicated concentrations of nucleotide analogues. Membranes were incubated at 30 °C for 5 min, and total [3H]inositol phosphates were quantified by anion exchange chromatography as previously described.47,53
5. Data Analysis
Agonist potencies were calculated using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). EC50 values (mean ± standard error) represent the concentration at which 50% of the maximal effect is achieved. Relative efficacies (%) were determined by comparison with the effect produced by a maximal effective concentration of UTP or UDP in the same experiment.
All concentration–effect curves were repeated in at least three separate experiments, carried out in duplicate or triplicate using different membrane preparations.
Acknowledgments
This work was supported by USPHS Grants GM38213 and HL54889 (to T.K.H.) and by the Deutsche Forschungsgemeinschaft (Grant SFB 269, A4) (to H.Z.). R.G.R. thanks Gilead Sciences (Foster City, CA) for financial support. We thank Kelly Soltysiak for proofreading this manuscript.
References
- 1.Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA, Schwabe U, Williams M. Toward a revised nomenclature for P1 and P2 receptors. Trends Pharmacol Sci. 1997;18:79–82. doi: 10.1016/s0165-6147(96)01038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King BF, Townsend-Nicholson A. Recombinant P2Y receptors: the UCL experience. J Auton Nerv Syst. 2000;81:164–170. doi: 10.1016/s0165-1838(00)00134-x. [DOI] [PubMed] [Google Scholar]
- 3.Communi D, Suarez Gonzalez N, Detheux M, Brézillon S, Lannoy V, Parmentier M, Boeynaems JM. Identification of a novel human ADP receptor coupled to Gi. J Biol Chem. 2001;276:41479–41485. doi: 10.1074/jbc.M105912200. [DOI] [PubMed] [Google Scholar]
- 4.Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 5.Zhang FL, Luo L, Gustafson E, Lachowicz J, Smith M, Qiao X, Liu YH, Chen G, Pramanik B, Laz TM, Palmer K, Bayne M, Monsma FJ., Jr ADP is the cognate ligand for the orphan G protein-coupled receptor SP1999. J Biol Chem. 2001;276:8608–8615. doi: 10.1074/jbc.M009718200. [DOI] [PubMed] [Google Scholar]
- 6.Nicholas RA. Identification of the P2Y12 Receptor: A Novel Member of the P2Y Family of Receptors Activated by Extracellular Nucleotides. Mol Pharmacol. 2001;60:416–420. [PubMed] [Google Scholar]
- 7.Chambers JK, Macdonald LE, Sarau HM, Ames RS, Freeman K, Foley JJ, Zhu Y, McLaughlin MM, Murdock P, McMillan L, Trill J, Swift A, Aiyar N, Taylor P, Vawter L, Naheed S, Szekeres P, Hervieu G, Scott C, Watson JM, Murphy AJ, Duzic E, Klein C, Bergsma DJ, Wilson S, Livi GP. A G protein-coupled receptor for UDP-glucose. J Biol Chem. 2000;275:10767–10771. doi: 10.1074/jbc.275.15.10767. [DOI] [PubMed] [Google Scholar]
- 8.Bhagwhat SS, Williams M. P2 Purine and Pyrimidine Receptors: Emerging superfamilies of G protein and ligand-gated ion channel receptors. Eur J Med Chem. 1997;32:183–193. [Google Scholar]
- 9.Fischer B. Therapeutic applications of ATP-(P2) receptors agonists and antagonists. Expert Opin Ther Pat. 1999;9:385–399. [Google Scholar]
- 10.Jacobson KA, Knutsen LJS. P1 and P2 purine and pyrimidine receptors. In: Abbracchio MP, Williams M, editors. Purinergic and Pyrimidinergic Signalling I; Handbook of Experimental Pharmacology. 151/I. Springer-Verlag; Berlin, Germany: 2001. pp. 129–175. [Google Scholar]
- 11.Torres GE, Haines WR, Egan TM, Voigt MM. Co-expression of P2X1 and P2X5 receptor subunits reveals a novel ATP-gated ion channel. Mol Pharmacol. 1998;54:989–993. doi: 10.1124/mol.54.6.989. [DOI] [PubMed] [Google Scholar]
- 12.Yoshioka K, Saitoh O, Nakata H. Heteromeric association creates a P2Y-like adenosine receptor. Proc Natl Acad Sci US A. 2001;98:7617–7622. doi: 10.1073/pnas.121587098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baurand A, Eckly A, Bari N, Leon C, Hechler B, Cazenave JP, Gachet C. Desensitization of the platelet aggregation response to ADP: differential down-regulation of the P2Y1 and P2cyc receptors. Thromb Haemostasis. 2000;84:484–491. [PubMed] [Google Scholar]
- 14.Nandanan E, Jang SY, Moro S, Kim H, Siddiqi MA, Russ P, Marquez VE, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. Synthesis, biological activity, and molecular modeling of ribose-modified adenosine bisphosphate analogues as P2Y1 receptor ligands. J Med Chem. 2000;43:829–842. doi: 10.1021/jm990249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malmsjö M, Adner M, Harden TK, Pendergast W, Edvinsson L, Erlinge D. The stable pyrimidines UDPβS and UTPγS discriminate between the P2 receptors that mediate vascular contraction and relaxation of the rat mesenteric artery. Br J Pharmacol. 2000;131:51–56. doi: 10.1038/sj.bjp.0703536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Theobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- 17.King BF, Liu M, Pintor J, Gulaix J, Miras-Portugal MT, Burnstock G. Diinosine pentaphosphate (IP5I) is a potent antagonist at recombinant rat P2X1 receptors. Br J Pharmacol. 1999;128:981–988. doi: 10.1038/sj.bjp.0702876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambrecht G. Agonists and antagonists acting at P2X receptors: selectivity profiles and functional implications. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;362:340–350. doi: 10.1007/s002100000312. [DOI] [PubMed] [Google Scholar]
- 19.Humphreys BD, Virginio C, Surprenant A, Rice J, Dubyak GR. Isoquinolines as antagonists of the P2X7 nucleotide receptor: high selectivity for the human versus rat receptor homologues. Mol Pharmacol. 1998;54:22–32. doi: 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- 20.Jacobson KA, King BF, Burnstock G. Pharmacological characterization of P2 (nucleotide) receptors. Celltransmissions. 2000;16:3–16. [Google Scholar]
- 21.Janssens R, Communi D, Pirotton S, Samson M, Parmentier M, Boeynaems JM. Cloning and tissue distribution of the human P2Y1 receptor. Biochem Biophys Res Commun. 1996;221:588–593. doi: 10.1006/bbrc.1996.0640. [DOI] [PubMed] [Google Scholar]
- 22.Burnstock G, Fischer B, Maillard M, Ziganshin A, Ralevic V, Knight G, Brizzolara A, von Isakovics A, Boyer JL, Harden TK, Jacobson KA. Structure activity relationships for derivatives of adenosine-5′-triphosphate as agonists at P2 purinoceptors: heterogeneity within P2X- and P2Y-subtypes. Drug Dev Res. 1994;31:206–219. doi: 10.1002/ddr.430310308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyer JL, Siddiqi S, Fischer B, Romera-Avila T, Jacobson KA, Harden TK. Identification of potent P2Y purinoceptor agonists that are derivatives of adenosine 5′-monophosphate. Br J Pharmacol. 1996;118:1959–1964. doi: 10.1111/j.1476-5381.1996.tb15630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin J, Tomlinson W, Kirk IP, Kim YB, Humphries RG, Kunapuli SP. The C6-2B glioma cell P2YAC receptor is pharmacologically and molecularly identical to the platelet P2Y12 receptor. Br J Pharmacol. 2001;133:521–528. doi: 10.1038/sj.bjp.0704114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley MR. Oxygen-induced pulmonary vasodilation is mediated by adenosine triphosphate in newborn lambs. J Cardiovasc Pharmacol. 1997;30:102–109. doi: 10.1097/00005344-199707000-00015. [DOI] [PubMed] [Google Scholar]
- 26.Loubatières-Mariani M-M, Hillaire-Buys D, Chapal J, Bertrand G, Petit P. P2 purinoceptor agonists: New insulin secretagogues potentially useful in the treatment of non-insulin-dependent diabetes mellitus. In: Jacobson KA, Jarvis MF, editors. Purinergic Approaches in Experimental Therapeutics. Chapter 13. Wiley; New York: 1997. pp. 253–260. [Google Scholar]
- 27.Fernandez-Alvarez J, Hillaire-Buys D, Loubatieres-Mariani MM, Gomis R, Petit P. P2 receptor agonists stimulate insulin release from human pancreatic islets. Pancreas. 2001;22:69–71. doi: 10.1097/00006676-200101000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Boyer JL, Adams M, Ravi RG, Jacobosn KA, Harden TK. 2-Chloro-N6-methyl-(N)-methanocarba-2-deoxyadenosine-3,5-bisphosphate is a selective high affinity P2Y1 receptor antagonist that blocks the ADP-activated P2Y1 receptor of platelets without effect on the adenylyl cyclase linked P2Y receptor. Br J Pharmacol. in press. [Google Scholar]
- 29.Eckly A, Gendrault JL, Hechler B, Cazenave JP, Gachet C. Differential involvement of the P2Y1 and P2YT receptors in the morphological changes of platelet aggregation. Thromb Haemostasis. 2001;85:694–701. [PubMed] [Google Scholar]
- 30.Fischer B, Chulkin A, Boyer JL, Harden KT, Gendron FP, Beaudoin AR, Chapal J, Hillaire-Buys D, Petit P. 2-Thioether 5′-O-(1-thiotriphosphate)adenosine derivatives as new insulin secretagogues acting through P2Y-Receptors. J Med Chem. 1999;42:3636–3646. doi: 10.1021/jm990158y. [DOI] [PubMed] [Google Scholar]
- 31.Hoebertz A, Meghji S, Burnstock G, Arnett TR. Extracellular ADP is a powerful osteolytic agent: evidence for signaling through the P2Y1 receptor on bone cells. FASEB J. 2001;15:1139–1148. doi: 10.1096/fj.00-0395com. [DOI] [PubMed] [Google Scholar]
- 32.Moore D, Iritani S, Chambers J, Emson P. Immunohistochemical localization of the P2Y1 purinergic receptor in Alzheimer’s disease. Neuroreport. 2000;11:3799–3803. doi: 10.1097/00001756-200011270-00041. [DOI] [PubMed] [Google Scholar]
- 33.Webb TE, Simon J, Krishek BJ, Bateson AN, Smart TG, King BF, Burnstock G, Barnard EA. Cloning and functional expression of a brain G-protein-coupled ATP receptor. FEBS Lett. 1993;324:219–225. doi: 10.1016/0014-5793(93)81397-i. [DOI] [PubMed] [Google Scholar]
- 34.Moore D, Chambers J, Waldvogel H, Faull R, Emson PJ. Regional and cellular distribution of the P2Y1 purinergic receptor in the human brain: striking neuronal localisation. J Comp Neurol. 2000;421:374–384. doi: 10.1002/(sici)1096-9861(20000605)421:3<374::aid-cne6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 35.Sellers LA, Simon J, Lundahl TS, Cousens DJ, Humphrey PP, Barnard EA. Adenosine nucleotides acting at the human P2Y1 receptor stimulate mitogen-activated protein kinases and induce apoptosis. J Biol Chem. 2001;276:16379–16390. doi: 10.1074/jbc.M006617200. [DOI] [PubMed] [Google Scholar]
- 36.Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 37.Ezzitouni A, Russ P, Marquez VE. (1S,2R)-[(Benzyloxy)-methyl]cyclopent-3-enol. A versatile synthon for the preparation of 4′,1′a-methano and 1′,1′a-methano carbocyclic nucleosides. J Org Chem. 1997;62:4870–4873. [Google Scholar]
- 38.Siddiqui MA, Ford H, Jr, George C, Marquez VE. Synthesis, conformational analysis and biological activity of a rigid carbocyclic analogue of 2′-deoxyaristeromycin built on a bicyclo[3.1.0]hexane template. Nucleosides Nucleotides. 1996;15:235–250. [Google Scholar]
- 39.Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. Methanocarba modification of uracil and adenine nucleotides: High potency of Northern ring conformation at P2Y1, P2Y2, P2Y4, and P2Y11 but not P2Y6 receptors. J Med Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer B, Boyer JL, Hoyle CHV, Ziganshin AU, Brizzolara AL, Knight GE, Zimmet J, Burnstock G, Harden TK, Jacobson KA. Identification of potent, selective P2Y-purinoceptor agonists. Structure–activity-relationships for 2-thioether derivatives of adenosine 5′-triphosphate. J Med Chem. 1993;36:3937–3946. doi: 10.1021/jm00076a023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burgess K, Cook D. Synthesis of nucleoside triphosphates. Chem Rev. 2000;100:2047–2059. doi: 10.1021/cr990045m. [DOI] [PubMed] [Google Scholar]
- 42.Hampton A, Patel AD, Maeda M, Hai TT, Chang CD, Kang JB, Kappler F, Abo M, Preston RK. Use of adenine nucleotide derivatives to assess the potential of exo-active-site-directed reagents as species- or isozyme-specific enzyme inactivators. 3. Synthesis of adenosine 5′-triphosphate derivatives with N6- or 8-substituents bearing iodoacetyl groups. J Med Chem. 1982;25:373–381. doi: 10.1021/jm00346a009. [DOI] [PubMed] [Google Scholar]
- 43.Lee K, Cass C, Jacobson KA. Synthesis using ring closure metathesis and effect on nucleoside transport of a (N)-methanocarba S-(4-nitrobenzyl)thioinosine derivative. Org Lett. 2001;3:597–599. doi: 10.1021/ol006999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perich JW, Johns RB. Di-tert-butyl N,N-diethylphosphora-midite. A new phosphitylating agent for the efficient phosphorylation of alcohols. Synthesis. 1988;2:142–144. [Google Scholar]
- 45.Lee K, Ravi RG, Ji X-d, Marquez VE, Jacobson KA. Ring-constrained (N)methanocarba-nucleosides as adenosine receptor agonists: Independent 5′-uronamide and 2′-deoxy modifications. Bioorg Med Chem Lett. 2001;11:1333–1337. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schachter JB, Li Q, Boyer JL, Nicholas RA, Harden TK. Second messenger cascade specificity and pharmacological selectivity of the human P2Y1-purinoceptor. Br J Pharmacol. 1996;118:167–173. doi: 10.1111/j.1476-5381.1996.tb15381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyer JL, Downes CP, Harden TK. Kinetics of activation of phospholipase C by P2Y purinergic receptor agonists and guanine nucleotides. J Biol Chem. 1989;264:884–890. [PubMed] [Google Scholar]
- 48.Servos J, Reiländer H, Zimmermann H. Catalytically active soluble ecto-5′-nucleotidase purified after heterologous expression as a tool for drug screening. Drug Dev Res. 1998;45:269–276. [Google Scholar]
- 49.Heine P, Braun N, Zimmermann H. Functional characterization of rat ecto-ATPase and ecto-ATP diphosphohydrolase after heterologous expression in CHO cells. Eur J Biochem. 1999;262:102–107. doi: 10.1046/j.1432-1327.1999.00347.x. [DOI] [PubMed] [Google Scholar]
- 50.Kennedy C, Qi AD, Herold CL, Harden TK, Nicholas RA. ATP, an agonist at the rat P2Y4 receptor, is an antagonist at the human P2Y4 receptor. Mol Pharmacol. 2000;57:926–931. [PubMed] [Google Scholar]
- 51.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 52.Kegel B, Braun N, Heine P, Maliszewski CR, Zimmermann H. An ecto-ATPase and an ecto-ATP diphosphohydrolase are expressed in rat brain. Neuropharmacology. 1997;36:1189–1200. doi: 10.1016/s0028-3908(97)00115-9. [DOI] [PubMed] [Google Scholar]
- 53.Harden TK, Hawkins PT, Stephens L, Boyer JL, Downes P. Phosphoinositide hydrolysis by guanosine 5′-(gamma-thio]triphosphate-activated phospholipase C of turkey erythrocyte membranes. Biochem J. 1988;252:583–593. doi: 10.1042/bj2520583. [DOI] [PMC free article] [PubMed] [Google Scholar]