

ABSTRACT
Diversity-generating metabolism leads to the evolution of many different chemicals in living organisms. Here, by examining a marine symbiosis, we provide a precise evolutionary model of how nature generates a family of novel chemicals, the cyanobactins. We show that tunicates and their symbiotic Prochloron cyanobacteria share congruent phylogenies, indicating that Prochloron phylogeny is related to host phylogeny and not to external habitat or geography. We observe that Prochloron exchanges discrete functional genetic modules for cyanobactin secondary metabolite biosynthesis in an otherwise conserved genetic background. The module exchange leads to gain or loss of discrete chemical functional groups. Because the underlying enzymes exhibit broad substrate tolerance, discrete exchange of substrates and enzymes between Prochloron strains leads to the rapid generation of chemical novelty. These results have implications in choosing biochemical pathways and enzymes for engineered or combinatorial biosynthesis.
IMPORTANCE While most biosynthetic pathways lead to one or a few products, a subset of pathways are diversity generating and are capable of producing thousands to millions of derivatives. This property is highly useful in biotechnology since it enables biochemical or synthetic biological methods to create desired chemicals. A fundamental question has been how nature itself creates this chemical diversity. Here, by examining the symbiosis between coral reef animals and bacteria, we describe the genetic basis of chemical variation with unprecedented precision. New compounds from the cyanobactin family are created by either varying the substrate or importing needed enzymatic functions from other organisms or via both mechanisms. This natural process matches successful laboratory strategies to engineer the biosynthesis of new chemicals and teaches a new strategy to direct biosynthesis.
INTRODUCTION
Secondary metabolites are specialized molecules that are often directed outward at other organisms (1, 2). For example, under the sea, soft-bodied animals defend themselves using a diverse array of specialized chemicals, which are required for their survival (3, 4). Symbiotic bacteria, and not the host animal, synthesize many secondary metabolites (5), providing a link between symbiosis, chemistry, the survival of the animal and bacterial associates, and effects on predators and other organisms on coral reefs (6–8). The chemicals underlying these interactions are structurally diverse, comprising families of related compounds that are useful in drug discovery. Slight changes to chemical structure can drastically alter function, yet many marine natural products families are extremely diverse. Compounds such as marine animal defensive chemicals are critical to interactions between organisms (3) so that structural changes have consequences that potentially ripple through the environment. An open question has been, given these many constraints, how can chemical diversity arise?
The evolution of novel chemistry likely relies in part on diversity-generating (DG) metabolism, which features pathways comprised of exceptionally broad-substrate enzymes (1, 9–11). These metabolic pathways natively accept many different substrates, enabling generation of chemical diversity. In turn, this provides organisms with rapidly evolving chemistry to face new challenges.
The tunicate-Prochloron symbiosis provides a model system to understand diversity-generating metabolism. Prochloron cyanobacteria are abundant obligate symbionts found in many tunicates of the family Didemnidae (12). Among the ubiquitous Prochloron compounds are the cyanobactins, a group of ribosomally synthesized and postranslationally modified peptide (RiPP) secondary metabolites (13–18). Cyanobactins in Prochloron are hypervariable in sequence and structure, and their biosynthetic pathways provide a canonical example of diversity generation (9). Cyanobactins originate from simple ribosomally transcribed precursor peptides, exemplified by PatE and TruE, which are then modified by a series of enzymes. Cyanobactins are also widely found in free-living cyanobacteria (13, 15, 19, 20).
Despite previous observations of related cyanobactin pathways in Prochloron (13, 15), the genetic basis underlying chemical diversity was not well understood. Two major hurdles stood in the way: only two basic cyanobactin pathway sequences were known in Prochloron, making it difficult to construct evolutionary hypotheses, and the evolution of the host-symbiont symbiosis was not well characterized. Previously, based upon small subunit (SSU) rRNA gene sequences, the relationships between Prochloron and tunicates appeared stochastic (21–23). However, at least in the tunicate Lissoclinum patella, Prochloron chemistry correlated with host phylogeny (24, 25), indicating that there might be a genetic relationship between symbiont and host. Indeed, a recent phylogeny constructed from 120 available cyanobacterial genomes showed that Prochloron from samples L2 and L3 shared a branch, while those from L1 and L4 shared another (26). This phylogenetic relationship reflected a phylogeny of the host tunicates obtained from 18S rRNA and cytochrome oxidase I genes (24). In many didemnid species, Prochloron is transmitted from parent to offspring through generations (27), which further suggested the possibility of a more specific relationship between symbiont and host phylogeny.
Here, we analyzed sufficient further samples of the Prochloron-tunicate symbiosis to show that, in contrast to previous results, Prochloron and hosts have congruent phylogenies, indicating a specific relationship between symbiont and host. Two new cyanobactin pathways were discovered, which in combination with phylogenetic studies enabled us to finely define the evolutionary trajectory of cyanobactin pathways in tunicates. As a result, we provide a real-world example of how diversity-generating metabolism solves the problem of adapting to new environmental conditions. The evolutionary trajectory closely resembles idealized strategies for combinatorial biosynthesis in the laboratory, setting the stage for the rational engineering of desired chemicals.
MATERIALS AND METHODS
General.
All PCRs were carried using either Platinum Taq High-Fidelity (Invitrogen) or Phusion High-Fidelity DNA Polymerase (NEB), containing 0.1 μl of DNA polymerase (for 10-μl-scale reactions), the supplied buffer (1×), 2 μM each primer, and 0.2 mM each deoxynucleoside triphosphate (dNTP; Invitrogen). Platinum Taq reaction conditions consisted of a hot start (95°C for 5 min), followed by 35 cycles of 95°C for 30 s, variable annealing temperatures for 30 s, and 68°C for variable extension times, with a final extension step of 68°C for 10 min. Phusion reaction conditions consisted of a hot start (95°C for 5 min), followed by 35 cycles of 95°C for 30 s, variable annealing temperatures for 30 s, and 68°C for variable extension times, with a final extension step of 72°C for 10 min. All cloning was carried out by using a TOPO TA cloning kit (with TOP10 chemically competent cells; Invitrogen) after gel extraction using a QIAquick gel extraction kit (Qiagen) or by direct PCR purification using a QIAquick PCR purification kit (Qiagen). Transformed TOP10 cells were grown on LB agar plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal; 40 μg/ml) and ampicillin or kanamycin (both, 50 μg/ml) at 30°C, and individual colonies were grown overnight in LB medium containing the same antibiotics at 30°C, with shaking. Plasmids were isolated using a QIAprep Spin Miniprep kit (Qiagen). Liquid chromatography-mass spectrometry (LC-MS) analysis was carried out using a Micromass Q-TOF Micro and 2795 HT high-performance liquid chromatography (HPLC) system (both, Waters).
Sample collection and processing.
Samples were collected by scuba diving or snorkeling from Papua New Guinea (PNG), the Solomon Islands, Fiji, and Palau, with appropriate legal agreements in place as previously described (25). Freshly collected samples were rinsed with sterile artificial seawater, diced, and placed in RNAlater for later processing in the lab. Frozen samples of the same colonies were also kept for chemical analysis. Diplosoma sp. from Okinawa was kindly provided by Katsuhiro Ueda. Table S1 in the supplemental material describes sample numbers and the location of collection for each sample.
RNAlater-preserved tissue was subjected to an established method for DNA extraction from tunicates, followed by phenol-chloroform extraction and isopropanol precipitation (28). The resulting DNA samples were purified a second time with a Genomic DNA Clean & Concentrator kit (Zymo Research) and further subjected to Illumina sequencing or PCR analysis. Table S2 in the supplemental material describes which samples were used for metagenome sequencing and distinguishes between which samples were sequenced in this study and which were sequenced in previous studies.
We used 19 Prochloron-containing tunicates from different branches of the Didemnidae. Metagenome sequences were analyzed from 11 different tunicate-Prochloron associations, including the didemnid tunicates Lissoclinum patella, Lissoclinum bistratum, Didemnum molle, and Diplosoma sp., from widely different geographic locations. In this study, we specifically extracted and analyzed Prochloron whole-genome sequences and host 18S rRNA gene sequences from the metagenomic sequence. Metagenomes were previously reported for L. patella samples L1 to L4 (25), obtained from Palau, the Solomon Islands, Fiji, and the north coast of PNG. Here, we obtained a further seven Prochloron-tunicate metagenome sequences and partially assembled the Prochloron genomes: L. patella (samples L5 and L6 from southern PNG) L. bistratum (LV5 from eastern PNG), D. molle (samples E11-036 and E11-037 from adjacent locations in PNG), and Diplosoma sp. (one from Okinawa; sample E11-016 from PNG).
Illumina sequencing, assembly, binning, and annotation.
The Illumina HiSeq 2000 data sets of L5 and L6 were previously reported (29). Illumina libraries were prepared for Okinawa, E11-037, E11-036, E11-016, and LV5. Libraries were sequenced using an Illumina HiSeq 2000 sequencer in 101-bp/125-bp paired-end runs (see Table S3 in the supplemental material). The library for E11-036 was sequenced using an Illumina MiSeq sequencer in 251-bp paired-end runs. Raw fasta files were assembled using IDBA_ud (30) (parameters: mink = 20, maxk = 100, step = 20, inner_mink = 10, inner_step = 5, prefix = 3, min_count = 2, min_support = 1, num_threads = 0, seed_kmer = 30, min_contig = 200, similar = 0.95, max_mismatch = 3, min_pairs = 3) in Futuregrid (http://futuresystems.org). The Prochloron genome was recovered from the metagenome by MetaAnnotator (31) in DIAG (http://diagcomputing.org/). Thirty-one bacterial housekeeping genes were used to verify the completeness of each recovered genome. Prochloron genomes were annotated using the RAST server (32–34). Prochloron genomes P1 to P4 (from previously reported L. patella samples L1 to L4 [25]) were obtained from GenBank (accession numbers AGRF00000000, AFSJ00000000, AFSK00000000, and AGGA00000000).
Prochloron genomes were very similar between both D. molle samples and between L5 and L6 although only L6 and one D. molle sample were used in the analysis because of higher quality. Thus, out of 11 metagenomes, 9 were used for full analysis.
Selection of genes for phylogenetic analysis.
Proteins encoded in Prochloron genomes were obtained using the RAST annotation output. Widespread genes present in a single copy in all genomes were identified by performing a blastp search using genome P1 against the eight other Prochloron genomes. Hits were defined as identity of >80%, query coverage of >0.8, and subject coverage of >0.8. Genes that met these criteria and were present in all nine samples were selected by the “regular expression” command (awk '!b[$1,ARGIND]++{if(++a[$1] = = 2)print}'). The orthologous group for each marker was retrieved by a PerlScript (fasta_extract_mult.pl) (see the supplemental material) and used for phylogenetic analysis.
Prochloron 16S rRNA gene sequences were extracted from raw Illumina reads from newly sequenced animals. Prochloron 16S rRNA gene sequences for P1 to P4 were extracted from the GenBank database (accession numbers AGRF00000000, AFSJ00000000, AFSK00000000, and AGGA00000000).
Tunicate 18S sequences were extracted from raw Illumina reads and or amplified from genomic DNA extracted from ascidian samples. Primers AscF2new (5′-CAAGGAAGGCAGCAGGCGCGCAAAT) and AscR5new (5′-GCGGTGTGTACAAAGGGCAGGGA) (28) were used to amplify 18S rRNA genes (see Table S4 in the supplemental material). The PCR product was cloned into a TOPO TA Zero blunt vector (Invitrogen). Individual clones bearing the correct insert sizes were sequenced. Because of deep branching in the 18S sequences, species-specific primers were used in some cases: Diplosoma spp. (DiplosomaF, 5′-GCGTTCGAAGCAGTCTTG; DiplosomaR, 5′-GATCGCCTTTTCGTCGGA); Lissoclinum spp. (LissoclinumF, 5′-TAACGACACTGCGAAAGGC; LissoclinumR, 5′-GCTCGATCCCCGAAGGAC); and Didemnum spp. (DidemnumF, 5′-GTCTACGTGGTCGTGCGGCGACG; DidemnumR, 5′-TTCACGAGCCTCTCGGCCCGC). The sequences of all primers used for 18S rRNA analysis are listed in Table S4 in the supplemental material.
A widespread Prochloron gene, photosystem II complex extrinsic protein precursor U (psbU), was extracted from all nine Prochloron genomes. The sequence was used to design specific primers (201_Fwd, TGAAAAGATTGGTGAGCGTA; 201_Rev, TTAGTAAACGCCAATATTAAAGC). Using these primers, psbU was amplified from 10 additional tunicate samples.
Construction of phylogenetic trees.
Orthologous genes were aligned using t-Coffee (mode mcoffee; output = msf, fasta_aln). To remove poorly aligned regions, the resulting alignment was subsequently trimmed with trimAl, version 1.4 (35), with a gap threshold of 0.4. Maximum-likelihood (ML) trees were constructed using MEGA, version 7.0 (36), using the Jukes-Cantor model and 500 bootstrap replicates to assess node support. We estimated tree topology by Bayesian inference using MrBayes, version 3.2 (37), with the general time-reversible (GTR) substitution model of evolution and default priors. The analysis consisted of 105 generations, with sampling every 102 steps.
Additionally, a concatenation of alignment of all conserved genes or individual functional categories of genes was done by PerlScript catfasta2phyml.pl (https://github.com/nylander/catfasta2phyml). Here, the 18S rRNA phylogenetic tree was used as a reference to rank each Prochloron gene tree by comparison to the reference. Each individual or concatenated alignment was scored according to its ability to reconstruct the reference phylogeny using Ktreedist (38) by measuring the minimum branch length distance (k-score) and symmetric difference between phylogenetic trees (see Tables S5 and S6 in the supplemental material).
Tree reconciliation analysis.
The Prochloron and host phylogenetic trees were reconciled using Jane, version 4.0 (39). Event costs for cospeciation, duplication, duplication and host switch, losses, and failure to diverge were set to 0, 1, 2, 1, and 1, respectively. The number of generations was set to 1,000, and the population size was set to 1,300 (see Fig. S2 in the supplemental material).
Proteome comparison.
To compare proteomes and construct Venn diagrams, proteins encoded in each Prochloron genome were compared using blastp (see the shell script, proteome_comparison.sh, in the supplemental material). First, a BLAST database that contains all of the unique protein sequences from nine Prochloron genomes was generated. Then, proteins from each Prochloron genome were searched using BLAST against this unique protein database. Identical proteins between each Prochloron genome and the unique protein database were defined as follows: identity of >80%, query coverage of >0.8, and subject coverage of >0.8. These identical proteins were given identical identification numbers and analyzed using an online Venn diagram program (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Annotation and comparison of cyanobactin gene clusters.
PatA and PatG sequences were used as queries for a tblastn search against the Prochloron genomes. Hit contigs with a ≥80% identity to the queries were extracted and annotated online using the RAST server (32–34). The annotated protein sequences were further examined by comparative BLAST analysis for further confirmation. Figure 4 was constructed using Easyfig, version 2.2.2 (40), with blastn output files (penalty, −1; gapextend, 100; gapopen, 100; word_size, 4; outfmt, 6). In Easyfig, imaging options were set as follows: minimum identity, 65; minimum length, 100; maximum E value, 10.
FIG 4.

Similarity of Prochloron cyanobactin pathways. Pathways obtained in this study were compared using blastn. Colors indicate percent identity between the DNA sequences, from red (65% identical) to green (100% identical), as indicated on the figure.
Nucleotide sequence accession numbers.
Prochloron 16S rRNA gene sequences were deposited in the GenBank under the following accession numbers: L6, KU511263; E11-036, KU511264; Okinawa, KU511265; LV5, KU511266; and E11-016, KU511267. Sequences of the psbU genes were deposited in GenBank under the following accession numbers: E11-037, KU514012; E11-021, KU514013; E11-009, KU514014; E11-081, KU514015; E11-046, KU514016; E11-042, KU514017; E11-033, KU514018; E11-027, KU514019; E11-011, KU514020; E11-056, KU514021; Okinawa, KU514022; E11-016, KU514023; E11-036, KU514024; L6, KU514025; and LV5, KU514026.
RESULTS
Prochloron and host tunicates share congruent phylogenies.
Host SSU 18S rRNA gene sequences provided a phylogenetic tree that reflected the known taxonomic relationship between didemnids (Fig. 1B) (23). In contrast, Prochloron 16S rRNA genes from the same samples led to a tree that substantially deviated from the 18S tree topology (see Fig. S1 in the supplemental material). Previously, this lack of congruence was interpreted as a lack of phylogenetic relationship between host and symbiont (23). However, while we were annotating genomes, we noticed trends suggesting a possible evolutionary relationship between host and symbiont at the whole-genome level. We proposed that the apparent stochastic distribution found in 16S genes could be an artifact resulting from the extremely close relationship between Prochloron genomes and consequent lack of variation in the 16S rRNA data set. We compared the sequenced metagenomes to test the hypothesis that Prochloron genomes reflect host phylogeny. Alternatively, the genomes could reflect geography, or there may be no relationship.
FIG 1.

Congruent maximum-likelihood (ML) tree of Prochloron bacteria and host tunicates from the tropical Pacific Ocean. The trees were generated by MEGA, version 7.0. Shapes indicate collection location. Maximum-likelihood bootstrap values and Bayesian clade credibility values are indicated at the nodes (bootstrap values/clade credibility values). (A) An ML tree is shown derived from 1,851 conserved Prochloron genes. (B) ML tree of 18S rRNA gene sequences from the samples identified in panel in A, consistent with a congruent relationship. (C) Location of sampling along a >7,000-km transect in the western Pacific Ocean, centered on Papua New Guinea (map from Mapbox [design copyrighted by Mapbox, data licensed under ODbL, copyrighted by OpenStreetMap contributors]). The same shapes and collection locations are used in Fig. 3.
We concatenated 1,851 Prochloron genes that could be found in all nine samples and performed phylogenetic analysis. The resulting tree was congruent with the host 18S tree, providing support for our hypothesis (Fig. 1; see also Table S5 in the supplemental material). Trees were reconciled using Jane, version 4, providing strong support for the observed congruency (see Fig. S2). Jane, version 4, also provided evidence for a coevolution or codivergence model, but the available evidence is also consistent with a host-switching model (see Discussion).
Statistical analyses of trees derived from individual functional categories of genes also supported phylogenetic congruency in each category (see Fig. S3 and Table S6 in the supplemental material). Most individual genes showed the same trend, but because of the extremely close relationships between Prochloron strains, insufficient variation was observed in most genes to draw firm conclusions. A limitation is the presence of certain classes of repetitive genes in Prochloron genomes (25). These genes were identified as described in Materials and Methods and eliminated from the analysis.
Gene content further reinforced the congruent phylogeny. Samples L1 and L4 are from clade A of L. patella; they share 5,175 genes (Fig. 2A). In contrast, L1 and L2 from different L. patella clades share only 3,393 genes. When representatives of the L. patella clades, D. molle, L. bistratum, and Diplosoma sp. were compared, 1,912 genes were shared in common (Fig. 2B).
FIG 2.

Venn diagrams showing shared gene content between Prochloron samples. (A) Analysis of Prochloron from L. patella clades I (L1 and L4) and II (L2 and L3). (B) Analysis of representatives from L. patella clades (L1), D. molle (E11-036), L. bistratum (LV5), and Diplosoma sp. (E11-016). Values represent the numbers of genes.
A weakness of the metagenomic analysis was that we had insufficient sampling from the genera Diplosoma and Didemnum. DNA was obtained from an additional eight samples of Diplosoma and D. molle. We selected a Prochloron marker gene that was reliably found in the metagenomes, phylogenetically informative, and essential: psbU (coordinates 31698 to 32078 in contig 2414, GenBank accession number HQ407369.1) encoding an extrinsic protein of the photosystem II complex. The phylogenetic congruency hypothesis predicted that the phylogeny of psbU would be congruent with that of the tunicate 18S rRNA genes. Indeed, these trees were very similar, lending further support to the idea that Prochloron and host phylogenies are congruent (Fig. 3). In contrast, the observed phylogenies did not reflect differences in collection location or external environment. Taken together, phylogenetic evidence shows that Prochloron divergence is related to host divergence and not to other factors.
FIG 3.
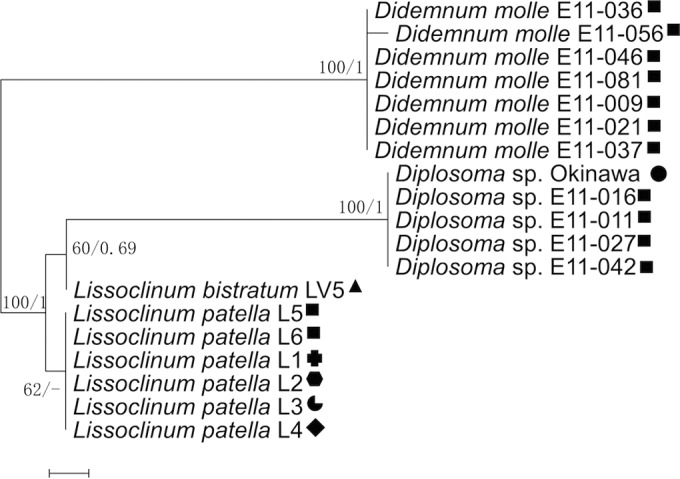
Maximum-likelihood (ML) tree from analysis of the Prochloron psbU gene. The trees were generated by MEGA, version 7.0. Shapes indicate collection location as defined in Fig. 1C. Maximum-likelihood bootstrap values and Bayesian clade credibility values are indicated at the nodes (bootstrap values/clade credibility values).
Discovery of two new cyanobactin pathways.
All seven samples of Lissoclinum spp. examined in this study contained cyanobactin clusters. As previously described, L1 and L4 contained a total of at least three described pat pathways (with two in different locations in the L1 chromosome), while L2 and L3 contained a total of two tru pathways (25). We also found two new cyanobactin clusters, described below.
Using LC-MS, we previously showed that tunicates L5 and L6 contained patellins 3 and 5, while L. bistratum contained bistratamides A and E (28). We therefore predicted that these animals should contain specific types of cyanobactin pathways. The pathways should encode all of the enzymes needed to perform the posttranslational modifications present in the natural products, and the precursor peptide sequences should exactly match the amino acid sequences of the products. Indeed, each animal contained only a single cyanobactin biosynthetic pathway encoding the correct combination of enzymes, and the precursor peptides contained only amino acid sequences that encoded patellins 3 and 5 and bistratamides A and E. Therefore, L5 and L6 contained a novel tru-like cluster, trf (tru from the eastern fields of PNG; GenBank accession number KX100577), encoding patellins 3 and 5 (41), while L. bistratum contained a pat-like cluster, bis (bistratamides cluster; GenBank accession number KX100576), encoding bistratamides A and E (42, 43).
The bis genes and predicted proteins were highly similar to those from the pat pathway (44), with the exception of several with more similarity to homologs from the ten pathway (Table 1). ten was previously found in free-living cyanobacteria from a different genus (21), so that the ten section could conceivably originate in a ten-like pathway in other Prochloron strains or from free-living cyanobacterial relatives. Similarity between bis and ten was observed at the predicted protein level, but significant similarity was not detected by BLAST searches at the nucleotide level.
TABLE 1.
bis cluster
| Locus | Protein homolog(s) or organism (% identity) | Gene homolog(s) (% identity) |
|---|---|---|
| BisA | TruA (97), PatA (97) | truAa (98) |
| BisB | TruB (96), PatB (96) | truB (98), patB (98) |
| BisC | TruC (96), PatC (96) | truC (98), patC (98) |
| BisD | PatD (96), TruD (88) | patD (96), truD (87) |
| BisE | TenE (76) | NAb |
| Leader onlyc | TruE (100), PatE (97) | truE (96), patE (96) |
| BisF1 | TenF (73) | NA |
| BisF2 | PatFe (58) | NA |
| BisGd | TenG (68) | NA/rearrangements |
| Oxidase | Microcystis aeruginosa (78), TenG (73), PatG (73) | |
| Protease | PatG (89), TruG (83) | patGa (85), truG (83) |
| DUF | PatG (96), TruG (94) | patG (96), truG (96) |
| Preoxidase region | PatG (85) | patG (85) |
Gene identity less than expected because of length differences.
NA, not available; nucleotide sequences were not significantly identical in BLAST searches.
Thirty-five amino acids.
Domain boundaries as called by the Conserved Domains Database (CDD)/NCBI.
Pseudogene.
The bis cluster encoded a single precursor peptide, BisE, that contained core peptides corresponding to bistratamides A and E. Interestingly, three core peptides were present, including two copies of the bistratamide A core and one copy of the bistratamide E core, while most tunicate cyanobactin precursors contain two core peptides. The L5/L6 trf cluster encoded a precursor peptide for patellins 3 and 5 and was otherwise nearly identical to tru, with one exception (Table 2).
TABLE 2.
trf cluster
| Locus | Protein homolog(s) (% identity) | Gene homolog (% identity) |
|---|---|---|
| TrfA | TruA (99) | truA (99) |
| TrfB | TruB (100) | truB (100) |
| TrfC | TruC (100) | truC (100) |
| TrfD | TruD (99) | truD (99) |
| TrfE leadera | TruE (97) | truE (99) |
| TrfF1 | TruF1 (100) | truF1 (100) |
| TrfF2 | PatFc (49) | NAd |
| TrfGb | TruG (90) | truGe (94) |
| DUF | TruG (100) | truG (100) |
Consisting of 35 amino acids.
Domain boundaries as called by the Conserved Domains Database (CDD)/NCBI.
Pseudogene.
NA, not available; nucleotide sequences were not significantly identical in BLAST searches.
Except for the first 194 nucleotides.
Comparative analysis of tunicate cyanobactin clusters.
All Prochloron cyanobactin pathways are 96 to 100% identical at the DNA and protein sequence levels in the 5′ end of the pathway, encoding PatA, -B, -C, the substrate-binding domain of PatD, and intergenic sequences (Fig. 4; Tables 1 and 2). The L. patella pathways are about 99 to 100% identical in this region, even when samples are obtained from widely different regions of the Pacific Ocean, while the L. bistratum sample is 96% identical to the others. Similarly, in the extreme 3′ end of the pathway, in the DUF domain of patG, pathways are 96 to 100% identical.
Between these extreme ends, significant recombination has occurred, generating many different functional classes of cyanobactin natural products (Fig. 5). (Here and elsewhere, the concept of recombination, and not a biochemical mechanism, is implied.) The second half of PatD, the YcaO domain, is either tru-like or pat-like; these two groups are about 74% identical to each other in the YcaO domain. The difference in the YcaO domains dictates whether cysteine and serine/threonine are heterocyclized (pat and bis) or whether only cysteine is heterocyclized (tru and trf) (45).
FIG 5.

Recombination in the pathway generates many different functional classes of cyanobactin natural products. (A) Precursor peptide sequence alignments show that the leader peptide portion is nearly constant. At bottom, consensus (Con) and alternative substitutions (Alt) are shown. Important residues for recognition sequences I, II, and III are shown in yellow, blue, and gray, respectively. Hypervariable core peptide sequences are underlined in red. Note that each precursor encodes 2 or 3 core peptides. Total consensus and mutations for each position in the core peptide are shown in the box at right. (B) Pathway comparison in variable regions using tblastx. This represents an informative subset of alignments from the sequences shown in Fig. 4. (C) Cyanobactin structures synthesized by various pathways. Functional groups are indicated by the same colors as found in gene regions shown in panel B. Genetic changes shown in panels A and B are responsible for the resulting chemical differences.
The leader peptides encoded by E genes are nearly identical among all pathways (Fig. 5A). The major differences between E genes are found in the core peptides, which are hypervariable, leading to many different compounds (21). Multiple E peptides containing different core sequences can exist within single animals. These different E peptides apparently exist as single copies within a set of otherwise complete, identical cyanobactin gene clusters. For example, the genome of L1 contains two identical cyanobactin biosynthetic pathways in separate regions of the chromosome, but with different core peptides. Because the E genes are sometimes present in differing quantities, it is clear that animals sometimes contain more than one strain of Prochloron. This provides fertile ground for generation of E variants.
Downstream of the E genes, the comparison becomes more complicated. pat from L1/L4 and tru from L2/L3 contain one and two different F genes, respectively. bis and trf each contain genes for two F proteins, one of which is a pseudogene in each case (Fig. 5). Only a single F protein is required for high yield in recombinant expression experiments, even when two such genes are present, so that a pseudogene second copy is consistent with the known functions of the pathways (9).
The F proteins are all different from each other and exhibit a complex pattern of substitution. In bis, the potentially functional F protein is more closely related to TenG from the tenuecyclamide pathway (free-living cyanobacteria) than to any Prochloron gene. In trf, TrfF1 is identical to TruF1, the prenylating protein in the tru pathway (46). Indeed, the trf products are prenylated, according to MS analysis (28). The F proteins have functional consequences in that TruF1 from L2/L3 and TrfF1 prenylate cyclic peptides, while the other F proteins have unknown (nonenzymatic) functions (9, 21). Potentially, they are required to bind or chaperone the cyclic peptides.
Finally, the G proteins contain macrocyclase domains (47). In pat and bis pathways in which the thiazoline moiety and potentially the oxazoline moiety are oxidized to thiazole/oxazole moieties, the G proteins also contain an oxidase domain. The G proteins have undergone a complex series of rearrangements where the oxidase domain has crossed in several times. In particular, the bis oxidase protein is more similar to that of ten; in ten and bis products, oxazole moieties are prevalent, whereas they may not exist in the pat family. Thus, perhaps a special oxidase is required to efficiently modify the oxazoline moiety. In contrast, the latter halves of the G protein domains are very similar between all pathways described here. The macrocyclase domain of bis is relatively highly divergent from the macrocyclase domains of pat and tru, suggesting that different functionality may be required to efficiently macrocyclize these precursors. All known pat pathway products are hepta- or octapeptides. The bis products are hexapeptides, which are more similar to the products of the ten pathway, the hexapeptide tenuecyclamides (48). Although PatG and TruG are able to macrocyclize hexapeptides in vitro, perhaps the efficiency is too low for in vivo biosynthesis, and a ten-like protein is therefore required.
In phylogenetic trees, the A, B, and C proteins and the heterocyclase domain of D and DUF domain of G proteins are congruent with those of the tunicate hosts (Fig. 6). However, the swapped regions (YcaO domain of D, F proteins, and oxidase/macrocyclase domain of G) (Fig. 4) do not follow a phylogenetic pattern consistent with their horizontal origin but, instead, reflect chemical differences found in products.
FIG 6.

Comparison of the maximum-likelihood (ML) tree of the core cyanobactin genes with Lissoclinum spp. hosts. The trees were generated by MEGA, version 7.0. Maximum likelihood bootstrap values and Bayesian clade credibility values are indicated at the nodes (bootstrap values/clade credibility values). (A) ML tree of concatenated nucleotide sequences of the A, B, and C genes and heterocyclase domain of D and DUF domain of G genes. (B) ML tree of 18S rRNA nucleotide sequences from the same samples.
DISCUSSION
Diversity-generating metabolism enables organisms to rapidly generate highly diverse chemical repertoires, which then undergo selection in response to challenge. Here, we obtained additional cyanobactin biosynthetic pathways from marine animals and demonstrated a phylogenetic congruence between symbiont and host. Using these data, we propose the evolutionary trajectory of cyanobactin biosynthesis. Two types of changes are readily apparent. First, substrates change by modifications to the core peptides encoded by the E genes. Second, enzymes are exchanged between otherwise identical pathways that expand the available posttranslational chemistry. Taken together, these changes lead to natural libraries of small molecules.
Here, using whole-genome data and a larger sample size, we show that there is a clear relationship between symbiont and host phylogeny. In contrast, there is no relationship between Prochloron and environmental habitat since adjacent animals found in the same habitats contain quite different Prochloron strains. In fact, several of the samples collected in this study were found in adjacent positions on the reef, yet Prochloron had more in common with samples from the same species collected thousands of kilometers away than with samples from the neighboring different animal species. This clearly indicates strong host specificity rather than specificity resulting from abiotic factors.
As a caveat, here we studied a subset of tunicates where the relationship with Prochloron is stable and probably obligate; there are other, more complex cases where the relationships may be different (27, 49). Exceptions to the rule may be found in further sampling, and species other than those studied here may exhibit different patterns.
The literature is replete with examples in which phylogenetic congruency and other biological data are used to infer coevolution or cospeciation (50). Some evidence obtained in this study suggests that Prochloron and hosts are coevolving or codiverging. For example, analysis with Jane, version 4, strongly supported the coevolution model. However, we do not believe that our data support coevolution for the following reasons. First, Prochloron secondary metabolite pathways are known to occur sporadically in a manner that is consistent with exchange between populations, rather than gene loss (51). Second, the host exhibits more sequence divergence than the symbionts. Normally, in coevolution one would expect the reverse to be true due to the faster doubling time of bacteria (52). Finally, Prochloron is absent from many branches of Didemnidae that fall inside the 18S rRNA tree shown in Fig. 1, as well as from most ascidian species in Didemnidae in general. Common examples include Didemnum vexillum, Lissoclinum perforatum, and many others. (This may indicate symbiont loss as well.) An additional problem is that there is no fossil record for tunicates, making timing of potential cospeciation events highly problematic and limiting the methods that can be employed.
Thus, it is unlikely that the phylogenetic congruency between symbiont and host reflects coevolution. Instead, we propose that the phylogenetic congruency reflects a host-switching model (53), in which specific Prochloron genotypes prefer specific host types, or vice versa. This preference could be mediated by cell surface proteins, which are extremely varied in Prochloron (25), by Prochloron secondary metabolism, which is associated with host phylogeny (24), or by a combination of these or other factors.
Despite this complexity, the observed congruence is useful for understanding the genetic origin of diversity generation in the cyanobactins. With the existence of multiple, closely related clusters, an evolutionary model can be proposed (Fig. 7). The overall gene content of Prochloron cyanobactin pathways is relatively stable. Large portions of the 5′ and 3′ ends of the gene cluster are nearly gene sequence-identical between different animals and pathway types and follow the phylogeny of producing bacteria and host tunicates. This near-identity at the ends of the pathways provides fertile ground for recombination in central regions involved in product diversification. A second region in which recombination is observed centers around the F proteins. The dynamic nature of this region is highlighted by the presence of pseudogenes in several pathways. Finally, in cyanobactin E genes, the sequences flanking core peptides are repetitive, facilitating recombination.
FIG 7.

Evolutionary model of cyanobactin biosynthesis. Because cyanobactin enzymes exhibit DG properties of broad-substrate tolerance, genetic swapping (likely recombination) leads to generation of new variants. Our model is as follows. (A) Capture of new functional enzymes. The pat pathway captured a ten-like cluster, possibly from free-living cyanobacteria, leading to the ability to produce hexapeptides (only heptapeptides and octapeptides are known from pat-like clusters). Subsequently, a second copy of the F gene was degraded into a pseudogene via point mutation. In panels A and B, colors are meant to make it easier to see the origin of genes but have no functional meaning. (B) Evolution of new precursor peptides. Precursor peptides likely evolve based upon duplication, divergence, and recombination. One way that this could work is shown here, with the duplication of precursors within a pathway based upon what is known from other cyanobactin pathways (18). This enables ready generation of sequence variants. (C) Example of how duplicated sequences might enable generation of precise variants for DG metabolism. Protein sequence is shown in place of gene sequence for ease of comprehension. Sequences L1a and L1b are real sequences from L. patella L1, while Hyp is a hypothetical, diverged intermediate. Important residues for recognition sequences I, II, and III are shown in yellow, blue, and gray, respectively.
Thus, the genetic model is that different Prochloron strains harbor cyanobactin clusters with conserved DNA segments that facilitate swapping of DNA sequences. The observed genetic pattern is most consistent with a recombinational mechanism. Two factors further help swapping: (i) the population structure of Prochloron, where cells are closely related and multiple strains (rarely) co-occur within single animals (21), and (ii) the presence of two or more nearly identical cyanobactin pathways on a single chromosome (25). The existence of these multiple pathways is a direct example of duplication and divergence. However, recombination then acts to smooth over divergence between pathways, leaving functional differences behind. This is facilitated by diversity-generating metabolism since each enzyme is natively broadly substrate tolerant.
The recombinational hypothesis is most obvious in substrate evolution in cyanobactin pathways. In substrate evolution, the core peptides diversify, while the enzymes remain constant. In several cyanobactin pathways of free-living organisms, multiple E genes exist in a tandem array, including several that are obviously nonfunctional (lacking core sequences) (14). Only a small subset of these is actually converted into products. This enables an accordion-like strategy of evolution, wherein E genes are copied and then recombined to produce diversified core peptides within a single precursor. There is significant evidence of this in that Prochloron E genes have been identified encoding one, two, and three (bis) core peptides, indicating facile recombination.
A computational comparison of biosynthetic gene clusters across sequenced genomes has revealed that different families of gene clusters take different evolutionary trajectories (54). Among these, there are many examples of concerted evolution, in which genes are more similar to their orthologs within a strain than to the paralogs between strains. It was suggested that clusters or portions of clusters using concerted evolution may be a good starting point for biosynthetic engineering. The cyanobactin pat and tru pathways similarly have several features that might be explained by concerted evolution, such as the recombination of precursor peptides and high similarities between specific portions of the biosynthetic clusters. These cyanobactin pathways are highly plastic and are well known for their ability to be engineered, providing some validation for the idea that pathways subject to concerted evolution are good for engineering. A special feature of the tunicate-derived pathways is their extremely close genetic identities, which allow the evolutionary pathways to be precisely defined (13, 21).
As a relevant contrast, several elegant studies show how closely related pathways diverge by swapping functional motifs (55–59). In these studies, the proteins are not as closely related to each other as those described here so that multiple mutations may be involved in creating functional swaps. Future work will determine whether these differences complicate engineering efforts or whether concerted evolution is a general indication of a diversity-generating metabolic pathway that is immediately useful for engineering.
In summary, here we provide evidence of the existence of diversity-generating metabolic pathways and propose their mode of evolution. Absolutely essential to this evolution, all enzymes in the cyanobactin pathways so far characterized are exceptionally broad-substrate tolerant (9). By swapping core peptides and enzymes, cyanobactin pathways rapidly (on an evolutionary time scale) access compounds with different functions. In turn, the resulting compounds are bioactive and are present as a major proportion of the extractable metabolites of the animals. This mechanism circumvents a presumed and widely cited limitation in the evolution of secondary metabolites, wherein structural changes usually lead to nonfunctional compounds and are thus proposed not to be evolutionarily favored. The presence of broad-substrate biosynthetic pathways enables populations to access many different types of compounds without sacrificing the original, active metabolites. New compounds that confer a selective advantage may then become fixed, and in some circumstances the pathway may lose its diversity-generating character.
Supplementary Material
ACKNOWLEDGMENTS
Computer time was obtained on FutureGrid (developed and supported by NSF 0910812 and NSF DBI-0959894), National Science Foundation-funded MRI-R2 project DBI-0959894 (DIAG), and the University of Utah Center for High Performance Computing.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00860-16.
REFERENCES
- 1.Williams DH, Stone MJ, Hauck PR, Rahman SK. 1989. Why are secondary metabolites (natural products) biosynthesized? J Nat Prod 52:1189–1208. doi: 10.1021/np50066a001. [DOI] [PubMed] [Google Scholar]
- 2.Olivera BM. 2006. Conus peptides: biodiversity-based discovery and exogenomics. J Biol Chem 281:31173–31177. doi: 10.1074/jbc.R600020200. [DOI] [PubMed] [Google Scholar]
- 3.Loh TL, Pawlik JR. 2014. Chemical defenses and resource trade-offs structure sponge communities on Caribbean coral reefs. Proc Natl Acad Sci U S A 111:4151–4156. doi: 10.1073/pnas.1321626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawlik JR. 2011. The chemical ecology of sponges on Caribbean reefs: natural products shape natural systems. Bioscience 61:888–898. doi: 10.1525/bio.2011.61.11.8. [DOI] [Google Scholar]
- 5.Flórez LV, Biedermann PHW, Engl Kaltenpoth TM. 2015. Defensive symbioses of animals with prokaryotic and eukaryotic microorganisms. Nat Prod Rep 32:904–936. doi: 10.1039/C5NP00010F. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt EW. 2015. The secret to a successful relationship: lasting chemistry between ascidians and their symbiotic bacteria. Invertebr Biol 134:88–102. doi: 10.1111/ivb.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson MC, Mori T, Rückert C, Uria AR, Helf MJ, Takada K, Gernert C, Steffens UAE, Heycke N, Schmitt S, Rinke C, Helfrich EJN, Brachmann AO, Gurgui C, Wakimoto T, Kracht M, Crüsemann M, Hentschel U, Abe I, Matsunaga S, Kalinowski J, Takeyama H, Piel J. 2014. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506:58–62. doi: 10.1038/nature12959. [DOI] [PubMed] [Google Scholar]
- 8.Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. 2007. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula,” the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J Nat Prod 70:67–74. doi: 10.1021/np060361d. [DOI] [PubMed] [Google Scholar]
- 9.Tianero MD, Pierce E, Sardar D, Raghuraman S, McIntosh JA, Heemstra JR, Schonrock Z, Covington BC, Cox JE, Bachmann BO, Olivera BM, Ruffner DE, Schmidt EW. 2016. Metabolic model for diversity-generating metabolism. Proc Natl Acad Sci U S A 113:1772–1777. doi: 10.1073/pnas.1525438113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischbach MA, Clardy J. 2007. One pathway, many products. Nat Chem Biol 3:353–355. doi: 10.1038/nchembio0707-353. [DOI] [PubMed] [Google Scholar]
- 11.Jones CG, Firn RD, Malcolm SB. 1991. On the evolution of plant secondary chemical diversity. Philos Trans R Soc Lond B Biol Sci 333:273–280. doi: 10.1098/rstb.1991.0077. [DOI] [Google Scholar]
- 12.Hirose E, Neilan BA, Schmidt EW, Murakami A. 2009. Enigmatic life and evolution of Prochloron and related cyanobacteria inhabiting colonial ascidians, p 161–189. In Gault PM, Marler HJ (ed), Handbook on cyanobacteria. Nova Science Publishers, New York, NY. [Google Scholar]
- 13.Donia MS, Ravel J, Schmidt EW. 2008. A global assembly line for cyanobactins. Nat Chem Biol 4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, et al. . 2013. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat Prod Rep 30:108–160. doi: 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leikoski N, Fewer DP, Sivonen K. 2009. Widespread occurrence and lateral transfer of the cyanobactin biosynthesis gene cluster in cyanobacteria. Appl Environ Microbiol 75:853–857. doi: 10.1128/AEM.02134-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leikoski N, Liu L, Jokela J, Wahlsten M, Gugger M, Calteau A, Permi P, Kerfeld CA, Sivonen K, Fewer DP. 2013. Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem Biol 20:1033–1043. doi: 10.1016/j.chembiol.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Sivonen K, Leikoski N, Fewer DP, Jokela J. 2010. Cyanobactins-ribosomal cyclic peptides produced by cyanobacteria. Appl Microbiol Biotechnol 86:1213–1225. doi: 10.1007/s00253-010-2482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donia MS, Schmidt EW. 2011. Linking chemistry and genetics in the growing cyanobactin natural products family. Chem Biol 18:508–519. doi: 10.1016/j.chembiol.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peraud O, Biggs JS, Hughen RW, Light AR, Concepcion GP, Olivera BM, Schmidt EW. 2009. Microhabitats within venomous cone snails contain diverse actinobacteria. Appl Environ Microbiol 75:6820–6826. doi: 10.1128/AEM.01238-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziemert N, Ishida K, Quillardet P, Bouchier C, Hertweck C, de Marsac NT, Dittmann E. 2008. Microcyclamide biosynthesis in two strains of Microcystis aeruginosa: from structure to genes and vice versa. Appl Environ Microbiol 74:1791–1797. doi: 10.1128/AEM.02392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW. 2006. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat Chem Biol 2:729–735. doi: 10.1038/nchembio829. [DOI] [PubMed] [Google Scholar]
- 22.Münchhoff J, Hirose E, Maruyama T, Sunairi M, Burns BP, Neilan BA. 2007. Host specificity and phylogeography of the prochlorophyte Prochloron sp., an obligate symbiont in didemnid ascidians. Environ Microbiol 9:890–899. doi: 10.1111/j.1462-2920.2006.01209.x. [DOI] [PubMed] [Google Scholar]
- 23.Yokobori S, Kurabayashi A, Neilan BA, Maruyama T, Hirose E. 2006. Multiple origins of the ascidian-Prochloron symbiosis: molecular phylogeny of photosymbiotic and non-symbiotic colonial ascidians inferred from 18S rDNA sequences. Mol Phylogenet Evol 40:8–19. doi: 10.1016/j.ympev.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 24.Kwan JC, Tianero MDB, Donia MS, Wyche TP, Bugni TS, Schmidt EW. 2014. Host control of symbiont natural product chemistry in cryptic populations of the tunicate Lissoclinum patella. PLoS One 9:e95850. doi: 10.1371/journal.pone.0095850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donia MS, Fricke WF, Partensky F, Cox J, Elshahawi SI, White JR, Phillippy AM, Schatz MC, Piel J, Haygood MG, Ravel J, Schmidt EW. 2011. Complex microbiome underlying secondary and primary metabolism in the tunicate-Prochloron symbiosis. Proc Natl Acad Sci U S A 108:E1423–E1432. doi: 10.1073/pnas.1111712108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Baracaldo P. 2015. Origin of marine planktonic cyanobacteria. Sci Rep 5:17418. doi: 10.1038/srep17418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirose E. 2015. Ascidian photosymbiosis: diversity of cyanobacterial transmission during embryogenesis. Genesis 53:121–131. doi: 10.1002/dvg.22778. [DOI] [PubMed] [Google Scholar]
- 28.Tianero MD, Kwan JC, Wyche TP, Presson AP, Koch M, Barrows LR, Bugni TS, Schmidt EW. 2015. Species specificity of symbiosis and secondary metabolism in ascidians. ISME J 9:615–628. doi: 10.1038/ismej.2014.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwan JC, Donia MS, Han AW, Hirose E, Haygood MG, Schmidt EW. 2012. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci U S A 109:20655–20660. doi: 10.1073/pnas.1213820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng Y, Leung HC, Yiu SM, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Leung HC, Yui SM, Chin FYL. 2014. MetaCluster-TA: taxonomic annotation for metagenomic data based on assembly-assisted binning. BMC Genomics 15(Suppl 1):S12. doi: 10.1186/1471-2164-15-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomason JA III, Stevens R, Vonstein V, Wattam AR, Xia F. 2015. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S, Stecher G, Tamura K. 22 March 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soria-Carrasco V, Talavera G, Igea J, Castresana J. 2007. The K tree score: quantification of differences in the relative branch length and topology of phylogenetic trees. Bioinformatics 23:2954–2956. doi: 10.1093/bioinformatics/btm466. [DOI] [PubMed] [Google Scholar]
- 39.Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R. 2010. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol Biol 5:16. doi: 10.1186/1748-7188-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carroll AR, Coll JC, Bourne DJ, MacLeod JK, Zabriskie TM, Ireland CM, Bowden BF. 1996. Patellins 1–6 and trunkamide A: novel cyclic hexa-, hepta- and octa-peptides from colonial ascidians, Lissoclinum sp. Aust J Chem 49:659–667. [Google Scholar]
- 42.Degnan BM, Hawkins CJ, Lavin MF, McCaffrey EJ, Parry DL, Watters DJ. 1989. Novel cytotoxic compounds from the ascidian Lissoclinum bistratum. J Med Chem 32:1354–1359. doi: 10.1021/jm00126a035. [DOI] [PubMed] [Google Scholar]
- 43.Perez LJ, Faulkner DJ, Fenical W. 2003. Bistratamides E–J, modified cyclic hexapeptides from the Philippines ascidian Lissoclinum bistratum. J Nat Prod 66:247–250. doi: 10.1021/np0204601. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. 2005. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc Natl Acad Sci U S A 102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McIntosh JA, Donia MS, Schmidt EW. 2010. Insights into heterocyclization from two highly similar enzymes. J Am Chem Soc 132:4089–4091. doi: 10.1021/ja9107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sardar D, Lin Z, Schmidt EW. 2015. Modularity of RiPP enzymes enables designed synthesis of decorated peptides. Chem Biol 22:907–916. doi: 10.1016/j.chembiol.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee J, McIntosh J, Hathaway BJ, Schmidt EW. 2009. Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J Am Chem Soc 131:2122–2124. doi: 10.1021/ja8092168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banker R, Carmeli S. 1998. Tenuecyclamides A–D, cyclic hexapeptides from the cyanobacterium Nostoc spongiaeforme var. tenue. J Nat Prod 61:1248–1251. doi: 10.1021/np980138j. [DOI] [PubMed] [Google Scholar]
- 49.Nielsen DA, Pernice M, Schliep M, Sablok G, Jeffries TC, Kühl M, Wangpraseurt D, Ralph PJ, Larkum AW. 2015. Microenvironment and phylogenetic diversity of Prochloron inhabiting the surface of crustose didemnid ascidians. Environ Microbiol 17:4121–4132. doi: 10.1111/1462-2920.12983. [DOI] [PubMed] [Google Scholar]
- 50.de Vienne DM, Refrégier G, López-Villavicencio M, Tellier A, Hood ME, Giraud T. 2013. Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol 198:347–385. doi: 10.1111/nph.12150. [DOI] [PubMed] [Google Scholar]
- 51.Donia MS, Fricke WF, Ravel J, Schmidt EW. 2011. Variation in tropical reef symbiont metagenomes defined by secondary metabolism. PLoS One 6:e17897. doi: 10.1371/journal.pone.0017897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiers ET, West SA. 2015. Evolving new organisms via symbiosis: when, how do symbiotic partnerships become new, integrated organisms? Science 348:392–394. doi: 10.1126/science.aaa9605. [DOI] [PubMed] [Google Scholar]
- 53.Charleston MA, Robertson DL. 2002. Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst Biol 51:528–535. doi: 10.1080/10635150290069940. [DOI] [PubMed] [Google Scholar]
- 54.Medema MH, Cimermancic P, Sali A, Takano E, Fischbach MA. 2014. A systematic computational analysis of biosynthetic gene cluster evolution: lessons for engineering biosynthesis. PLoS Comput Biol 10:e1004016. doi: 10.1371/journal.pcbi.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ziemert N, Lechner A, Wietz M, Millan-Aguinaga N, Chavarria KL, Jensen PR. 2014. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc Natl Acad Sci U S A 111:E1130–E1139. doi: 10.1073/pnas.1324161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ueoka R, Uria AR, Reiter S, Mori T, Karbaum P, Peters EE, Helfrich EJ, Morinaka BI, Gugger M, Takeyama H, Matsunaga S, Piel J. 2015. Metabolic and evolutionary origin of actin-binding polyketides from diverse organisms. Nat Chem Biol 11:705–712. doi: 10.1038/nchembio.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gallagher KA, Jensen PR. 2015. Genomic insights into the evolution of hybrid isoprenoid biosynthetic gene clusters in the MAR4 marine streptomycete clade. BMC Genomics 16:960. doi: 10.1186/s12864-015-2110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulze CJ, Donia MS, Siqueira-Neto JL, Ray D, Raskatov JA, Green RE, McKerrow JH, Fischbach MA, Linington RG. 2015. Genome-directed lead discovery: biosynthesis, structure elucidation, and biological evaluation of two families of polyene macrolactams against Trypanosoma brucei. ACS Chem Biol 10:2373–2381. doi: 10.1021/acschembio.5b00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fisch KM, Gurgui C, Heycke N, van der Sar SA, Anderson SA, Webb VL, Taudien S, Platzer M, Rubio BK, Robinson SJ, Crews P, Piel J. 2009. Polyketide assembly lines of uncultivated sponge symbionts from structure-based gene targeting. Nat Chem Biol 5:494–501. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.