

ABSTRACT
Strain ST-14, characterized as a member of the genus Cupriavidus, was capable of utilizing 2- and 4-nitrobenzoates individually as sole sources of carbon and energy. Biochemical studies revealed the assimilation of 2- and 4-nitrobenzoates via 3-hydroxyanthranilate and protocatechuate, respectively. Screening of a genomic fosmid library of strain ST-14 constructed in Escherichia coli identified two gene clusters, onb and pob-pca, to be responsible for the complete degradation of 2-nitrobenzoate and protocatechuate, respectively. Additionally, a gene segment (pnb) harboring the genes for the conversion of 4-nitrobenzoate to protocatechuate was unveiled by transposome mutagenesis. Reverse transcription-PCR analysis showed the polycistronic nature of the gene clusters, and their importance in the degradation of 2- and 4-nitrobenzoates was ascertained by gene knockout analysis. Cloning and expression of the relevant pathway genes revealed the transformation of 2-nitrobenzoate to 3-hydroxyanthranilate and of 4-nitrobenzoate to protocatechuate. Finally, incorporation of functional 3-nitrobenzoate dioxygenase into strain ST-14 allowed the recombinant strain to utilize 3-nitrobenzoate via the existing protocatechuate metabolic pathway, thereby allowing the degradation of all three isomers of mononitrobenzoate by a single bacterial strain.
IMPORTANCE Mononitrobenzoates are toxic chemicals largely used for the production of various value-added products and enter the ecosystem through industrial wastes. Bacteria capable of degrading mononitrobenzoates are relatively limited. Unlike other contaminants, these man-made chemicals have entered the environment since the last century, and it is believed that bacteria in nature evolved not quite efficiently to assimilate these compounds; as a consequence, to date, there are only a few reports on the bacterial degradation of one or more isomers of mononitrobenzoate. In the present study, fortunately, we have been able to isolate a Cupriavidus sp. strain capable of assimilating both 2- and 4-nitrobenzoates as the sole carbon source. Results of the biochemical and molecular characterization of catabolic genes responsible for the degradation of mononitrobenzoates led us to manipulate a single enzymatic step, allowing the recombinant host organism to expand its catabolic potential to assimilate 3-nitrobenzoate.
INTRODUCTION
Nitroaromatic compounds are one of the most important groups of industrial chemicals and are currently considered to be priority environmental contaminants (1, 2). Among the nitroaromatics, mononitrobenzoates (MNBAs) are widely employed in the production of dyes, plastics, explosives, pharmaceuticals, polyurethane foams, elastomers, and pesticides (3, 4). At the same time, they are also known to exert toxic effects on living organisms due to their genotoxic and mutagenic properties (5).
In the degradation of MNBAs, 2-nitrobenzoate (2NBA) and 4-nitrobenzoate (4NBA) metabolism occurs mostly via a reductive pathway, with the reduction of a nitro group to an amino group (6–9). In contrast, 3-nitrobenzoate (3NBA) metabolism involves the dioxygenase-dependent removal of the nitro group as nitrite, with the formation of protocatechuate (PCA) (10, 11). Interestingly, in a preliminary study in the late 1950s, oxidative degradation pathways of all three isomers of MNBA were reported for strains of Nocardia, but their complete degradation pathways were not characterized (12). A recent study with Arthrobacter sp. strain SPG revealed 2NBA degradation via a novel oxidative route involving salicylate and catechol as metabolic intermediates (13). 2NBA degradation in Pseudomonas fluorescens KU-7 was extensively studied (6, 14, 15), where 2NBA was metabolized by a nitroreductase (NbaA) and a mutase (NbaB) to furnish 3-hydroxyanthranilate (3HAA). 3HAA was subsequently metabolized by the actions of 3-hydroxyanthranilate dioxygenase (NbaC), decarboxylase (NbaD), dehydrogenase (NbaE), and deaminase (NbaF), ultimately leading to tricarboxylic acid (TCA) cycle intermediates. On the other hand, 2NBA was degraded via anthranilate (AA) by Ralstonia sp. strain SJ98 (16). However, both Arthrobacter protophormiae RKJ100 and Burkholderia terrae KU-15 degraded 2NBA via both AA and 3HAA (7, 17). 4NBA degradation via reductive pathways was also studied for a number of bacterial species, viz., Comamonas acidovorans NBA-10 (18, 19), Ralstonia sp. SJ98 (16), Pseudomonas pickettii YH105 (8), Pseudomonas pickettii YH102 (20), Pseudomonas putida TW3 (9), and Pseudomonas sp. strain 4NT (21). In strain TW3, the genes encoding NAD(P)H-dependent nitroreductase (PnbA) and 4-hydroxylaminobenzoate lyase (PnbB) were reported to catalyze the reduction of 4NBA to 4-hydroxylaminobenzoate and the transformation of the latter to PCA, respectively (9). Genetic information on the reductive metabolism of 4NBA in strains YH105 and YH102 was also reported (8). On the other hand, the only genetic information in regard to 3-nitrobenzoate dioxygenase (MnbAB), which is involved in the oxidative metabolism of 3NBA, was described for Comamonas sp. strain JS46 (10). Figure 1 illustrates the metabolic pathways involved in the bacterial degradation of all three MNBAs.
FIG 1.

Catabolic pathways for the degradation of mononitrobenzoates (6–10, 13, 17). I, 2-nitrobenzoate; II, 2-hydroxylaminobenzoate; III, 3-hydroxyanthranilate; IV, 2-amino-3-carboxymuconic-6-semialdyhyde; V, 2-aminomuconic 6-semialdyhyde; VI, 2-aminomuconate; VII, 4-oxalocrotonate; VIII, acetyl coenzyme A; IX, salicylate; X, anthranilate; XI, catechol; XII, 4-nitrobenzoate; XIII, p-hydroxylaminobenzoate; XIV, 3-nitrobenzoate; XV, 3,4-dihydroxy-3-nitro-cyclohexa-1,5-dienecarboxylate; XVI, protocatechuate; NbaA, 2-nitrobenzoate nitroreductase; NbaB, 2-hydroxylaminobenzoate mutase; NbaC, 3-hydroxyanthranilate dioxygenase; NbaD, 2-amino-3-carboxymuconic-6-semialdyhyde decarboxylase; NbaE, 2-aminomuconatesemialdehyde dehydrogenase; NbaF, 2-aminomuconate deaminase; NbaG, 4-oxalocrotonate decarboxylase; NbaH, 2-oxopent-4-dienoate hydratase; NbaI, 4-hydroxy-2-oxovalerate aldolase; NbaJ, acylating aldehyde dehydrogenase; PnbA, 4-nitrobenzoate nitroreductase; PnbB, 4-hydroxylaminobenzoate lyase; MnbA, 3-nitrobenzoate dioxygenase; MnbB, oxidoreductase; SCoA, deprotonated form of coenzyme A.
Many bacterial species from diverse genera have been reported to degrade MNBAs; however, there are no reports that document the complete degradation of all three isomers by a single strain. In the present study, we report the metabolic pathways for the degradation of MNBAs in Cupriavidus sp. strain ST-14 isolated from municipal-waste-contaminated soil. In addition to biochemical evaluation, we describe the cloning and sequencing of complete sets of catabolic genes and functional analysis of key enzymes involved in the 2NBA and 4NBA degradation pathways. Furthermore, plasmid-mediated transformation of 3NBA dioxygenase from Comamonas sp. JS46 (10) into wild-type Cupriavidus sp. strain ST-14 enabled the latter to utilize 3NBA via PCA, thereby broadening its ability to degrade all three isomers of MNBA.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Strain ST-14, a 2NBA-degrading bacterium, was isolated from a municipal-waste-contaminated soil sample (Dhapa, Kolkata, West Bengal, India) by enrichment culture techniques (see Text S1 in the supplemental material), with 2NBA as the sole source of carbon and energy. The culture was routinely grown in liquid mineral salt medium (MSM) (22) supplemented with either 1.0 g of 2NBA, 0.5 g of 4NBA, 0.5 to 1.0 g of possible pathway intermediates, or 1.0 g of succinate per liter, each individually as the sole carbon source, at 28°C on a rotary shaker (180 rpm). Growth rates were calculated from log-phase cultures based on time course measurements of cell abundances, as described previously (23). Comamonas sp. JS46 (10), a generous gift from Jim C. Spain (Georgia Institute of Technology, USA), was grown in 1/4-diluted lysogeny broth (LB). Escherichia coli strains were routinely grown in LB medium. Where appropriate, ampicillin (100 μg/ml), kanamycin (50 μg/ml), streptomycin (25 μg/ml), tetracycline (12.5 μg/ml), chloramphenicol (12.5 μg/ml), isopropyl-β-d-thiogalactopyranoside (IPTG) (0.5 to 1 mM), or 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) (20 μg/ml) was added. For expression cloning, pET-28a(+) and pCDF-1b (Novagen, Madison, WI) served as expression vectors. Broad-host-range shuttle vectors pBBR1MCS2_START and pBBR1MCS3_START (24) were generously provided by Gordana Maravic-Vlahovicek (University of Zagreb, Croatia). All of the strains and plasmid constructs used in this study are listed in Table 1. Growth was monitored by measuring the optical densities of the cultures at a wavelength of 600 nm in a Cary 100 Bio UV-visible spectrophotometer (Varian, Australia), while CFU were calculated directly based on plate count methods.
TABLE 1.
Bacterial strains and plasmid used in this study
| Bacterial strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Bacterial strains | ||
| Cupriavidus sp. | ||
| ST-14 | 2NBA and 4NBA degrader | This study |
| ST-14Δonb | Non-2NBA utilizing, Kanr; site-directed deletion mutant of the ST-14 onb operon where partial onbA and onbR genes were deleted | This study |
| ST-14Δpnb | Non-4NBA-utilizing, Kanr; site-directed deletion mutant of the ST-14 pnb operon where partial pnbR and pnbA genes and the complete pnbB gene were deleted | This study |
| ST-14-tn103 | Kanr transposon mutant capable of utilizing PCA but not 4NBA | This study |
| ST-14::3NBA | 3NBA utilizing, Kanr; hybrid strain harboring plasmid pBBR1MCS2_START-3NBA | This study |
| ST-14ΔonbpCOMP | 2NBA utilizing, Kanr Tetr; hybrid strain harboring plasmid pBBR1MCS3_START-onbAR | This study |
| ST-14ΔpnbpCOMP | 4NBA utilizing, Kanr Tetr; hybrid strain harboring plasmid pBBR1MCS3_START-pnbAB | This study |
| E. coli | ||
| EPI300-T1R phage T1-resistant plating strain | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ− rpsL (Strr) nupG trfA DHFR | Epicentre |
| EC100D | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ− rpsL (Strr) nupG pir+(DHFR) | Epicentre |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (Tetr)] | Stratagene |
| BL21(DE3) | F− ompT hsdSB(rB− mB−) dcm gal lon λ(DE3) | Stratagene |
| fos45.10 | Fosmid clone showing positive 2NBA growth and a positive PCR-amplified product for the 3HAA dioxygenase gene | This study |
| fos30.12 | Fosmid clone showing positive 4HBA growth and a positive PCR-amplified product for the PCA dioxygenase gene | This study |
| BL21OnbAB | E. coli BL21 cotransformed with plasmids pET-onbA and pCDF-onbB | This study |
| BL21PnbAB | E. coli BL21 transformed with pCDF-pnbAB | This study |
| Comamonas sp. | ||
| JS46 | 3NBA degrader | 10 |
| Plasmids | ||
| CopyControlpCC2fos | Chloramphenicol resistant, linearized at the unique Eco72I site, P1 loxP, dephosphorylated, T7 RNA polymerase promoter flanking the cloning site, E. coli F-factor-based partitioning and copy no. regulation system | Epicentre |
| pET28a(+) | Expression vector; Kanr | Novagen |
| pET-onbA | Kanr; pET28a(+) harboring the onbA gene | This study |
| pET-onbC | Kanr; pET28a(+) harboring the onbC gene | This study |
| pET-pnbB | Kanr; pET28a(+) harboring the pnbB gene | This study |
| pCDF-1b | Expression vector; Strr | Novagen |
| pCDF-onbB | Strr; pCDF-1b harboring the onbB gene | This study |
| pCDF-pnbAB | Strr; pCDF-1b harboring the pnbAB gene | This study |
| pCDF-pnbA | Strr; pCDF-1b harboring the pnbA gene | This study |
| pBBR1MCS2_START | Kanr; Gram-negative broad-host-range shuttle vector | 24 |
| pBBR1MCS3_START | Tetr, Gram-negative broad-host-range shuttle vector | 24 |
| pBBR1MCS2_START-3NBA | pBBR1MCS2_START harboring mnbA, mnbR, and mnbB | This study |
| pBBR1MCS3_START-onbAR | pBBR1MCS3_START harboring onbA and onbR | This study |
| pBBR1MCS3_START-pnbAB | pBBR1MCS3_START harboring pnbA and pnbB | This study |
| pCM184 | Ampr Kanr; Cre/Lox | Addgene |
| pCM184-onb1-2 | pCM184 harboring onb upstream fragment in MCS1 and onb downstream fragment in MCS2 | This study |
| pCM184-pnb1-2 | pCM184 harboring pnb upstream fragment in MCS1 and pnb downstream fragment in MCS2 | This study |
DHFR, dihydrofolate reductase.
For resting-cell transformations, wild-type strain ST-14 or recombinant E. coli BL21(DE3) cells were harvested in late exponential phase by centrifugation at 8,000 × g for 10 min, washed with 50 mM potassium phosphate buffer (pH 7.0), and resuspended in the same buffer to reach an optical density at 600 nm (OD600) of 1.0. 2NBA, 3NBA, 4NBA, or possible pathway intermediates were individually added at a concentration of 0.1 to 0.5 g/liter to the washed-cell suspensions and incubated at 28°C for different periods of time up to 48 h.
Isolation of metabolites and chemical analysis.
After incubation, spent cultures and resting-cell cultures were centrifuged at 8,000 × g for 10 min, and the supernatants were extracted three times with equal volumes of ethyl acetate under acidic conditions. Organic extracts were dried over anhydrous sodium sulfate and evaporated under reduced pressure. Unless stated otherwise, all experiments were performed in triplicate.
To determine substrate consumption and accumulation of metabolites, organic extracts of culture supernatants were analyzed by high-performance liquid chromatography (HPLC) using a Shimadzu model LC20-AT pump system equipped with a diode array model SIL-M20A detector and a C18 reversed-phase column attached to a model SIL-20A autosampler. Biotransformed products were eluted by using an isocratic solvent system at a flow rate of 1.0 ml/min and detected at 254 nm. The mobile phase used for the detection of 2NBA and its pathway metabolites was comprised of aqueous methanol (50:50) supplemented with 0.1% trifluoroacetate, whereas for 4NBA, 3NBA, and their respective metabolites, a mobile phase of acetonitrile and glycine-HCl buffer (40:60, pH 3.5) was used. Metabolites were identified based on comparisons of retention times and UV-visible spectra to those of the authentic compounds analyzed under identical conditions. Quantitative estimation of individual components was made from the standard curve of the respective components created by HPLC under the respective set of analytical conditions. The accumulation of ammonia and nitrite was determined from the culture supernatants of resting-cell suspensions employed for the transformations of MNBAs by using methods described previously (25, 26). O2 uptake by whole bacterial cells was measured by using Clark-type polarographic oxygen electrodes (see Text S1 in the supplemental material for details).
Construction of a fosmid library, phenotypic screening, subcloning, and sequencing of gene clusters.
Genomic DNA from ST-14 was isolated by using a QIAamp DNA minikit (Qiagen Inc., USA). A genomic library was prepared with the pCC2FOS fosmid vector provided in the CopyControl HTP fosmid library production kit (Epicentre, Madison, WI). The genomic DNA was randomly sheared by mechanical methods using a needle, and fragments of ∼40 kb were gel purified. Fragments were converted into blunt-end DNA by using End-Repair enzyme mix, followed by ligation into the pCC2FOS vector. Ligated DNA was packaged into a phage head with MaxPlax Lambda packaging extracts and transduced into the EPI300-T1R phage T1-resistant E. coli plating strain. Selection of positive transductants was done on LB agar plates containing 12.5 μg/ml chloramphenicol. The resulting library was replica plated onto MSM agar supplemented individually with 2NBA (0.8 g/liter), 4NBA (0.5 g/liter), or PCA (1.0 g/liter) as the substrate to screen for the respective substrate-utilizing clones. Subsequently, positive clones were selected, and the presence of pathway-specific catabolic genes was confirmed by PCR using primers listed in Table 2 (see Text S1 in the supplemental material). DNA isolated by using the FosmidMAX DNA purification kit (Epicentre, Madison, WI) from the fosmid clones was then digested individually with BamHI, PstI, XhoI, SmaI, EcoRI, NotI, and SacII, and the DNA fragments (1 to 8 kb) were subcloned into pBlueScript-SK(+) and transformed into E. coli XL1-Blue. The recombinant plasmids were isolated and subjected to sequencing with an ABI Prism 377 automated sequencer (Perkin-Elmer, Applied Biosystems) using M13 forward and reverse primers. Identity matches for the sequenced genes were performed by BLAST analysis (version 2.2.12; National Center for Biotechnology Information). Gaps between genes were bridged by the conventional primer-walking method (27).
TABLE 2.
Primers used in this study
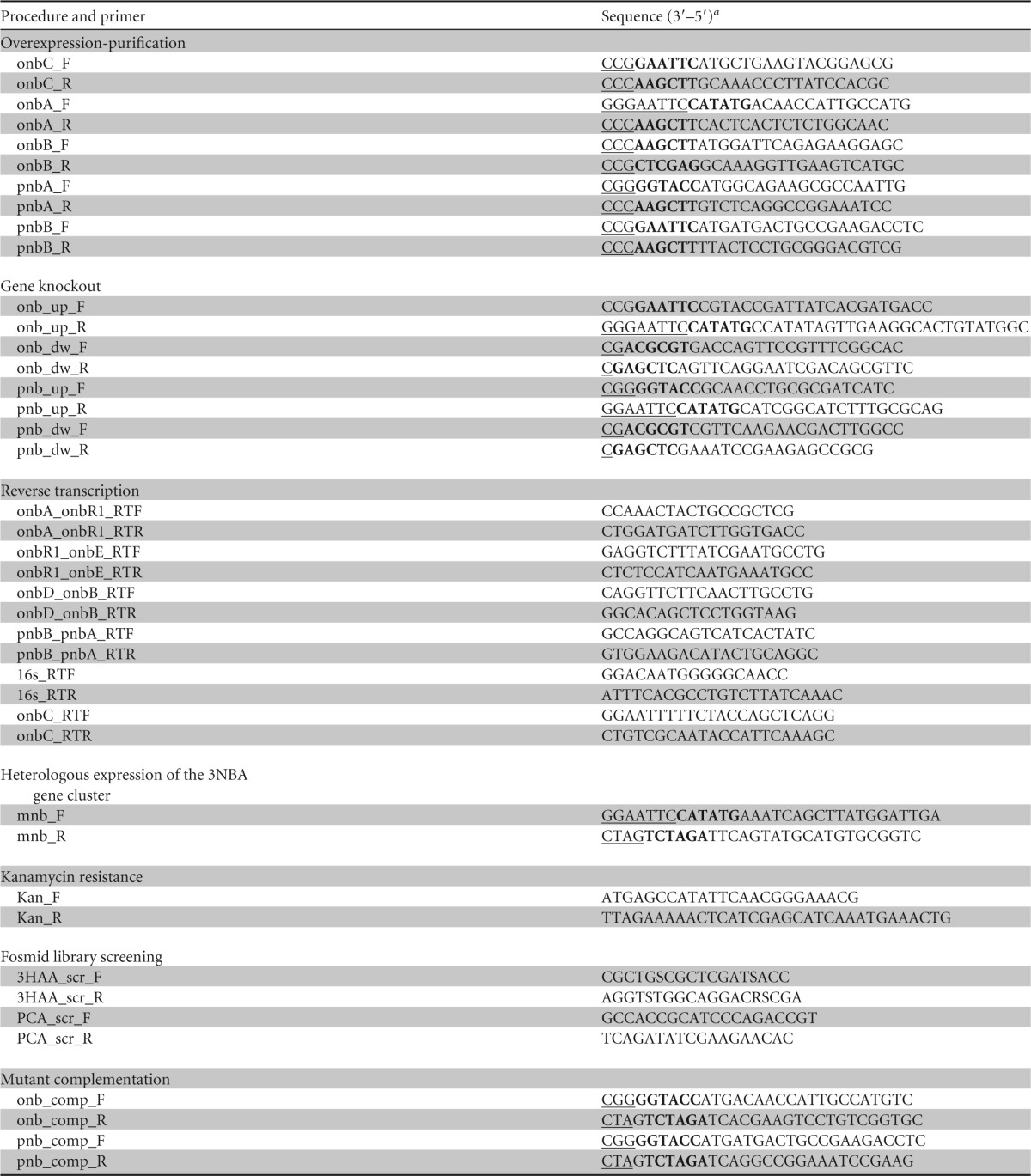
a Restriction recognition sequences are in boldface type, and enhancer sequences are underlined.
Cloning and expression of recombinant proteins.
The DNA fragments encoding 2NBA reductase (onbA), 2-hydroxylaminobenzoate mutase (onbB), 3HAA ring cleavage dioxygenase (onbC), 4NBA reductase (pnbA), p-hydroxylaminobenzoate lyase (pnbB), and 4NBA reductase-lyase (pnbAB) were amplified from the ST-14 genome by using the primer pairs onbA_F and onbA_R, onbB_F and onbB_R, onbC_F and onbC_R, pnbA_F and pnbA_R, pnbB_F and pnbB_R, and pnbB_F and pnbA_R, respectively, each possessing the desired restriction sites (Table 2). The amplified DNA fragments were gel purified, digested with the appropriate restriction enzymes, and cloned into the linearized pET-28a(+) plasmid for the expression of OnbA, OnbC, and PnbB. For the expression of OnbB, PnbA, and PnbAB, the pCDF1b plasmid served as the expression vector. The respective recombinant plasmids, designated pET-onbA, pET-onbC, pET-pnbB, pCDF-onbB, pCDF-pnbA, and pCDF-pnbAB, were transformed/cotransformed into E. coli BL21(DE3) cells, and the transformants were selected on LB agar plates supplemented with kanamycin, streptomycin, or kanamycin plus streptomycin as required. The positive clones were identified by colony-screening PCR, and DNA sequencing of the recombinant plasmid was performed to exclude the possibility of mutations of the amplified genes. For overexpression of recombinant proteins, positive clones were grown in LB at 37°C with the appropriate antibiotic marker(s) to achieve an OD600 of 0.5, followed by induction with IPTG at a final concentration of 0.5 to 1.0 mM. The clones were incubated at 28°C for 3 h or at 16°C for 16 h as required.
Preparation of cell extracts.
Wild-type strain ST-14 grown individually on MSM supplemented with 2NBA, 4NBA, or succinate and recombinant E. coli strains grown on LB were centrifuged at 8,000 × g for 10 min, and the cell pellets were washed twice with 50 mM potassium phosphate buffer (pH 7.0). Prior to lysis, the pellet was resuspended in the same buffer (OD600 = 1.0), loaded into a precooled French press (One Shot model 182 constant cell disruption system with an 8.0-ml lysis chamber; Constant System Ltd., United Kingdom), and lysed at 30,000 lb/in2 (207 MPa) for two cycles. Cell lysates were centrifuged at 20,000 × g for 30 min at 4°C, and the supernatants were used as cell-free enzymes for further studies. The protein concentration was measured by the Bradford method, and bovine serum albumin (BSA) was used as the standard (28).
Enzyme assays.
2NBA and 4NBA reductase activities were monitored spectrophotometrically (Cary 100 Bio UV-visible spectrophotometer; Varian, Australia) by measuring the 2NBA- and 4NBA-dependent decrease in the absorbance at 340 nm due to the oxidation of NADH or NADPH using the cell extracts of strain ST-14 grown in the presence of 2NBA and 4NBA, respectively. The same assay method was used to monitor the activity of the overexpressed reductases OnbA and PnbA. PCA and 3HAA ring cleavage dioxygenase activities were monitored according to protocols described previously by Stanier and Ingraham (29) and Hasegawa et al. (14), respectively. One unit of enzyme activity is defined as the amount of enzyme required for the production of 1 μmol of product per min. Specific activity is expressed as units per milligram of protein.
Mutation.
onb and pnb deletion mutants were generated by using the allelic-exchange vector pCM184 (30). DNA fragments flanking each of the onb and pnb target sequences were amplified by using primers (Table 2) upstream of onb (onb_up_F and onb_up_R), downstream of onb (onb_down_F and onb_down_R), upstream of pnb (pnb_up_F and pnb_up_R), and downstream of pnb (pnb_down_F and pnb_down_R). Amplified fragments that corresponded to the upstream and downstream flanking regions of each of the onb and pnb target sequences were introduced into multiple cloning site 1 (MCS1) and MCS2 of pCM184, and the resulting allelic-exchange vectors were electroporated individually into electrocompetent ST-14 cells according to methods described previously by Taghavi et al. (31). Mutation was established by the deletion of onb and pnb target sequences, confirmed by diagnostic PCR, and based on the inability of the mutant strains to utilize 2NBA or 4NBA as a carbon source. For complementation experiments, deleted genes were PCR amplified, cloned into suitable vectors, and electroporated into the respective mutant strains (ST-14Δonb and ST-14Δpnb), and the transformants were checked for their ability to degrade individual nitrobenzoates (see Text S1 in the supplemental material for details).
Transposome mutagenesis.
Transposome mutagenesis assays were performed by using the EZ-Tn5⟨R6Kγori/KAN-2⟩Tnp transposome kit (Epicentre, Madison, WI). Electrocompetent ST-14 cells were electroporated with the EZ-Tn5⟨R6Kγori/KAN-2⟩ transposon and plated onto LB agar medium containing kanamycin (LB-kanamycin agar). Clones that did not exhibit growth on 4NBA-MSM plates were selected and rescreened on PCA-MSM plates. Transposon-containing, Kanr mutants capable of utilizing PCA but not 4NBA were selected, and their genomic DNA was isolated, sheared, and digested with MluI. Following end repair, digested fragments were ligated and transformed into E. coli EC100D pir+ cells (Epicentre, Madison, WI). Transformants capable of growing on LB-kanamycin agar plates were selected, followed by plasmid isolation and sequencing of the rescued genomic fragments using the KAN-2 FP-1 forward and R6KAN-2 RP-1 reverse primers supplied with the kit.
RNA isolation, cDNA preparation, and reverse transcription (RT) analysis.
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used to isolate total RNA from mid-exponential-phase cultures of strain ST-14 that were grown on 2NBA, 4NBA, 4-hydroxybenzoate (4HBA), PCA, or succinate as the sole carbon source, according to the manufacturer's instructions. DNA contaminants present in the RNA samples were removed by treatment with RNase-free DNase I (Fermentas, Lithuania) for 1 h at 37°C, followed by inactivation of the enzyme with EDTA for 10 min at 70°C. Following isolation, total RNA was quantified spectrophotometrically at 260 nm, and purity was checked by calculating the 260/280 absorbance ratio. RNA integrity was checked by agarose (2.0%) gel electrophoresis, and 1 μg of DNA-free RNA was used for cDNA synthesis. In the process, secondary RNA structures were heat denatured at 70°C for 5 min, snap-chilled, and incubated at 42°C for 1 h with the addition of RevertAid reverse transcriptase (Fermentas, Lithuania) and Ribolock RNase inhibitor (Fermentas, Lithuania). Reverse primers used for gene-specific cDNA preparation are listed in Table 2. PCR amplification was performed by using 1 μl of the above-described reaction mixture (cDNA) as the template. 16S rRNA was used as an endogenous control.
Construction of a 3NBA-degrading recombinant Cupriavidus strain.
In Comamonas sp. JS46, the oxygenase component (MnbA) and the oxidoreductase component (MnbB) were identified as the enzymes responsible for ring dioxygenation of 3NBA (10). A 2.8-kb DNA fragment that carried the mnbB, mnbR (a regulator), and mnbA genes was PCR amplified from the genomic DNA of strain JS46 by using primers mnbF and mnbR (Table 2). The amplified DNA fragment was cloned into the broad-host-range shuttle vector pBBR1MCS2_START (24), followed by electroporation into wild-type Cupriavidus sp. ST-14 cells. The transformants were selected on LB-kanamycin agar plates. Expression of 3NBA ring-hydroxylating dioxygenase was confirmed based on the growth of the selected clones (recombinant strain ST-14::3NBA) on 3NBA (0.5 g/liter) as the sole carbon source in MSM supplemented with kanamycin. Resting-cell incubations for the transformation of 3NBA and preparation of cell extracts to analyze in vitro enzyme activity using ST-14::3NBA cells that were grown on 3NBA were conducted as described above for the wild-type strain.
Nucleotide sequence accession numbers.
The nucleotide sequences reported in this work were deposited in the DDBJ/EMBL/GenBank database under accession numbers KT000585 for the 16S rRNA gene sequence, KT033704 for the pob-pca locus, KT033703 for the onb locus, and KT004672 for the pnb locus.
RESULTS
Isolation and identification of MNBA-degrading bacteria.
By using enrichment culture techniques, a Gram-negative, aerobic, nonsporulating, peritrichously flagellated bacterial strain, designated ST-14, was isolated from municipal-waste-contaminated soil (Dhapa, Kolkata, India) through the utilization of 2NBA as a sole carbon source. Physiological and biochemical characterization revealed that it is oxidase positive, urease positive, and catalase positive; β-glucosidase and β-galactosidase activities were absent. Phylogenetic analysis of strain ST-14 using the partial 16S rRNA gene sequence (1,393 bp) positioned it within the genus Cupriavidus, displaying 98.42%, 98.35%, and 98.19% identities to Cupriavidus alkaliphilus ACS-732T, Cupriavidus taiwanensis LMG-19424T, and Cupriavidus oxalatica LMG-2235T, respectively (see Text S1 and Fig. S1 in the supplemental material).
Growth characteristics.
Strain ST-14 was found to be capable of utilizing both 2NBA and 4NBA as sole sources of carbon and energy, and the optimum temperature and pH for growth were found to be 28°C and 7.0, respectively. Growth rates of strain ST-14 in the presence of 2NBA (1.0 g/liter) and 4NBA (0.5 g/liter) were 0.387/h and 0.227/h, respectively, under optimal growth conditions. Growth profiles of strain ST-14 based on the utilization of these compounds correlated well with substrate degradation, indicating no major accumulation of a metabolite(s) (Fig. 2). AA and PCA, the possible metabolic intermediates in the degradation of 2NBA and 3NBA/4NBA, respectively, were utilized individually as the sole source of carbon and energy by strain ST-14 with specific growth rates of 0.353/h and 0.462/h, respectively, whereas 3NBA did not serve as a carbon and energy source.
FIG 2.

Growth of Cupriavidus sp. strain ST-14 in MSM in the presence of MNBAs as sole carbon sources. (A) Utilization of 2NBA (1 g/liter). (B) Utilization of 4NBA (0.5 g/liter). ●, growth; △, remaining substrate concentration; ■, remaining substrate concentration in the uninoculated control. Vertical bars represent means ± standard deviations from triplicate measurements.
Metabolism of nitrobenzoates.
Cell-fee extracts of ST-14 grown with 2NBA catalyzed the time-, protein-, and 2NBA-dependent oxidation of NADPH (Table 3). Apart from 2NBA nitroreductase activity, the release of ammonia (0.85 to 0.9 mol/mol of 2NBA) during degradation of 2NBA by resting-cell cultures and the utilization of AA as the sole carbon source indicated a possible pathway of degradation of 2NBA via AA in strain ST-14. However, the above-mentioned proposition was invalidated due to a low level of oxygen consumption in the presence of AA by cells of strain ST-14 grown with 2NBA (Table 4). Although 3HAA (at a concentration of 0.1 to 0.5 g/liter) could not be utilized, a high rate of oxygen uptake on 3HAA was observed for cells of strain ST-14 grown with 2NBA (Table 4) and consequently indicates that the degradation of 2NBA proceeds via 3HAA. These results were confirmed by the detection of 3HAA as a metabolic intermediate in the supernatants of 2NBA-degrading log-phase cultures, which was identified by HPLC analysis (data not shown). In addition, characteristic ring cleavage spectra were also revealed during the transformation of 3HAA by cell extracts of cultures of strain ST-14 grown with 2NBA, indicating the presence of 3HAA dioxygenase (Table 3).
TABLE 3.
Specific activities of enzymes involved in 2NBA and 4NBA degradation by Cupriavidus sp. strain ST-14
| Substrate | Activity (U/mg protein)a |
||||||
|---|---|---|---|---|---|---|---|
| 2NBA nitroreductase in the presence of: |
4NBA nitroreductase in the presence of: |
3HAA dioxygenase | PCA dioxygenase | Ammonia-releasing enzyme | |||
| NADPH | NADH | NADPH | NADH | ||||
| 2NBA | 0.128 | 0.012 | <0.001 | <0.001 | 0.046 | <0.001 | 0.26 |
| 4NBA | <0.001 | <0.001 | 0.244 | 0.021 | <0.001 | 0.284 | 0.22 |
| Succinate | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
For determination of specific activities, cell extracts were used as the sources of enzymes except for ammonia-releasing activity, where whole cells were used, and specific activities were measured from the results of transformation of MNBAs in resting-cell suspensions.
TABLE 4.
Rate of oxygen uptake with various compounds by resting-cell suspensions of strain ST-14 grown on different substrates
| Compound | Oxygen uptake rate (nmol O2 consumed min−1 mg−1 of protein) of cells grown ona: |
|||
|---|---|---|---|---|
| 2NBA | 4NBA | 4HBA | PCA | |
| 2NBA | 0.0 | 0.0 | 0.0 | 0.0 |
| 3HAA | 35 | 0.0 | 0.0 | 0.0 |
| AA | 2.7 | 0.0 | 0.0 | 0.0 |
| PCA | 0.0 | 50.1 | 48.5 | 57.5 |
| 4NBA | 0.0 | 0.0 | 0.0 | 0.0 |
| 4HBA | 0.0 | 5.7 | 27.4 | 4.3 |
All values are corrected for endogenous O2 uptake.
Similarly, an initial reductase-mediated metabolism of 4NBA was confirmed on the basis of NADPH consumption during the metabolism of 4NBA by cell extracts of cultures of strain ST-14 grown with 4NBA (Table 3). This was supported by the identification of liberated ammonia (0.88 to 0.94 mol/mol of 4NBA) in resting-cell cultures of strain ST-14. A high rate of oxygen uptake by cells grown with 4NBA in the presence of PCA (Table 4), the detection of PCA in the supernatants of log-phase cultures grown with 4NBA by HPLC analysis (data not shown), and the presence of protocatechuate-3,4-dioxygenase activity (Table 3) in cell extracts obtained from cells grown with 4NBA confirmed 4NBA metabolism via PCA in strain ST-14.
Note that cell extracts of cultures grown with 4NBA could not transform 2NBA and vice versa (Table 3). Moreover, reductases involved in the metabolism of 2NBA and 4NBA prefer NADPH over NADH as the reducing cofactor, and the turnover ratio is ∼10:1 in favor of NADPH (Table 3), while background NADPH reductase activity in cell extracts of cultures grown with 2NBA and 4NBA are <0.001 U/mg of protein (Table 3). For the degradation of 2NBA and 4NBA, an almost stoichiometric amount of ammonia (0.85 to 0.94 mol/mol of 2NBA/4NBA) was released, with ammonia-releasing specific activities of 0.26 and 0.22 U/mg protein (Table 3), respectively, which nearly fit the respective growth rates. Both 2NBA and 4NBA metabolic pathways were found to be inducible in vivo, since oxygen uptake or spectral changes as described above were not observed for cells grown on succinate. Moreover, ST-14 cells grown on PCA did not exhibit 4NBA reductase activity, which indicated the possible involvement of multiple operons in the degradation of 4NBA.
Cloning and sequence analysis of the gene cluster(s) involved in 2NBA and 4NBA degradation.
To understand the metabolic pathways of degradation of MNBAs, a fosmid library comprising 1,080 clones was constructed by using genomic DNA from strain ST-14. Screening of the library on 2NBA-MSM agar plates supplemented with chloramphenicol yielded four clones. This result indicated the possible recruitment of genes involved in the mineralization of 2NBA within the clones. The presence of 3HAA dioxygenase within the clones was verified by PCR. Isolation of fosmid DNA from one of the fosmid clones (fos45.10) (Table 1) followed by digestion, subcloning, and sequence analysis revealed the presence of a 14.1-kb gene cluster, designated onb, which showed similarities to the nba cluster of Pseudomonas fluorescens strain KU-7 (Fig. 3). BLAST analysis showed that the onb cluster consists of 15 complete open reading frames (ORFs) whose putative functions and identities with similar gene products are described in Table S1 in the supplemental material. The genes of the onb cluster (onbX1X2FCAR1EHJIGDBX3) are organized in the same direction and are contiguous with respect to the complete set of 2NBA catabolic genes, which is in contrast to the nba cluster in KU-7 (6), where the nbaA and nbaY genes are found in the opposite orientation with respect to other nba genes (Fig. 3). In addition, there are insertions of several genes of unknown functions between nbaB and nbaE in the nba gene cluster (6). NbaR in KU-7, which was described as the regulatory protein involved in the expression of the nba gene cluster, showed high levels of sequence similarity with OnbR1. Apart from the results described above, the organization of the onb gene cluster (Fig. 3), and the expression profile as evaluated by reverse transcription analysis (see Fig. S3 in the supplemental material), OnbR1 may be described as the putative regulatory protein of the operon. OnbR2, encoded by the other putative regulatory gene sequence, which was detected at one of the extreme ends of the onb gene cluster and oriented in the reverse direction with respect to 2NBA catabolic genes (Fig. 3), displayed relatively low identity with OnbR1 and NbaR (see Table S1 in the supplemental material). Moreover, no expression of OnbR2 was detected by reverse transcription analysis, suggesting no role of the latter in 2NBA degradation (data not shown).
FIG 3.

Organization of the onb gene cluster of Cupriavidus sp. ST-14 and comparison with the nba gene cluster of Pseudomonas fluorescens KU-7. The designated genes are shown in colored boxes, while the colored arrows represent the respective gene products along with their putative functions. FMN, flavin mononucleotide; MFS, major facilitator superfamily.
To identify the genes involved in 4NBA degradation, the same fosmid library was screened on 4NBA-MSM agar plates supplemented with chloramphenicol. Unfortunately, no colony that could utilize 4NBA as a sole carbon source appeared on the plates. However, biochemical results suggested the metabolism of 4NBA via PCA, and the latter could also be utilized by strain ST-14 as a sole carbon source. Based on the above-described characteristics, screening of the fosmid library on PCA-MSM agar plates resulted in the isolation of five clones, suggesting the recruitment of genes involved in PCA degradation. Primers designed for the protocatechuate-3,4-dioxygenase alpha subunit (Table 2), which supported PCR amplification of the target gene sequence from strain ST-14, also amplified the dioxygenase in all five clones. Isolation of fosmid DNA from one of the five clones (fos30.12) (Table 1) followed by digestion, subcloning, and sequence analysis revealed the presence of 11 complete ORFs in a 13.1-kb gene cluster. Sequence analysis revealed that the gene cluster, termed pob-pca, harbored all the genes necessary for the degradation of 4HBA via PCA (Fig. 4). The putative functions of the individual ORFs and their identities with the similar gene products are described in Table S1 in the supplemental material, showing significant similarities with similar genes present in Cupriavidus necator JMP134 (DSM4058, later reclassified as Cupriavidus pinatubonensis JMP134) (32).
FIG 4.

Organization of the pnb and pob-pca gene clusters of Cupriavidus sp. ST-14. The designated genes are shown in colored boxes, while the colored arrows represent the respective gene products along with their putative functions.
To detect nitroreductase and/or lyase genes responsible for the conversion of 4NBA to PCA, transposome mutagenesis targeting the genomic DNA of strain ST-14 was performed. Transposome mutagenesis yielded a mutant (ST-14-tn103) (Table 1) showing positive growth on PCA-MSM agar but no growth on 4NBA-MSM agar, suggesting the impairment of the metabolic pathway responsible for the transformation of 4NBA to PCA by this mutant. Subcloning and sequence analysis of ST-14-tn103 identified a gene cluster, termed pnb, comprised of the pnbR, pnbB, and pnbA genes (Fig. 4). The putative functions of the individual ORFs and their identities with similar gene products are described in Table S1 in the supplemental material, showing significant similarities with similar genes present in strain YH105. Thus, it appeared that 4NBA assimilation in strain ST-14 was mediated by the mutual cooperation of the pnb and pob-pca gene clusters (Fig. 4).
Expression of genes and activity of their products.
To confirm the functional roles of the onbA, onbB, onbC, pnbA, and pnbB gene products, the gene products were recombinantly expressed as His-tagged proteins in E. coli BL21(DE3), and their activities were evaluated by spectral and HPLC analyses. Figure S2 in the supplemental material shows the SDS-PAGE profiles of OnbA, OnbB, OnbC, PnbA, and PnbB as His-tagged proteins having molecular masses in concurrence with the theoretical subunit molecular masses of 26.4, 23.8, 23.9, 27.2, and 23.8 kDa, respectively. The specific activities of purified OnbA, OnbC, and PnbA, determined spectrophotometrically, were found to be 12.78, 4.55, and 63.91 U/mg of protein, respectively. In addition, HPLC analyses revealed time-dependent transformations of 2NBA (3.19 μg/min/mg protein) to 3HAA and of 4NBA (21.09 μg/min/mg protein) to PCA in resting-cell cultures of clones BL21OnbAB and BL21PnbAB (Table 1), respectively, justifying the functional roles of the recombinant proteins.
Reverse transcription analysis.
To understand the transcription profiles of the onb and pnb gene clusters, RT-PCR analyses were performed, which revealed the upregulation of intergenic regions, viz., onbA_onbR1, onbR1_onbE, and onbD_onbB, and the structural gene onbC when ST-14 cells were grown in the presence of 2NBA (see Fig. S3 in the supplemental material). Similarly, ST-14 cells grown with 4NBA showed the upregulation of the pnbB_pnbA intergenic fragment. In contrast, ST-14 cells grown in the presence of succinate did not exhibit such upregulation, confirming the fact that the onb and pnb gene clusters are polycistronic and strictly inducible in nature (see Fig. S3 in the supplemental material).
Mutational analyses.
To validate the in vivo involvement of the onb and pnb gene clusters in 2NBA and 4NBA metabolism in ST-14, deletion mutant strains ST-14Δonb and ST-14Δpnb (Table 1) were constructed. Consequently, strains ST-14Δonb and ST-14Δpnb lost their ability to utilize 2NBA and 4NBA, respectively, as sole sources of carbon and energy. However, ST-14Δonb was able to grow individually in the presence of PCA and 4NBA, and ST-14Δpnb could utilize PCA and 2NBA independently. Moreover, the 2NBA and 4NBA degradation properties of the mutant strains were successfully complemented by the introduction of the deleted genes into strains ST-14Δonb and ST-14Δpnb (see Text S1 and Fig. S4 in the supplemental material). This result confirmed the functional role of the deleted genes in the 2NBA and 4NBA degradation pathways.
Construction of strain ST-14::3NBA and 3NBA metabolism.
Hybrid strain ST-14::3NBA, harboring the 3NBA ring-hydroxylating dioxygenase system (Fig. 5A), was found to utilize 3NBA as a sole source of carbon (0.5 g/liter) with a specific growth rate of 0.31/h, which indicated plasmid-mediated constitutive expression of the two-component 3NBA dioxygenase system in this strain (Fig. 5B). The stability of the plasmid was ascertained from 20 consecutive batch culture experiments with the hybrid strain. However, under nonselective conditions, after five consecutive batch cultures, curing of the plasmid was revealed based on PCR amplification profiles of 3NBA dioxygenase and kanamycin resistance genes using primers as described above (Table 2).
FIG 5.

(A) Organization of the mnb gene cluster of Comamonas sp. JS46 and functional roles of MnbAB and PcaGHBL in the degradation of 3NBA in strain ST-14::3NBA. MnbA, 3NBA dioxygenase; MnbB, oxidoreductase; MnbR, transcriptional regulator; PcaGHBL, protocatechuate-degrading gene cluster, as shown in Fig. 4. (B) Growth of ST-14::3NBA upon utilization of 3NBA as the sole carbon and energy source. ●, growth; △, remaining substrate concentration; ■, remaining substrate concentration in the uninoculated control. Vertical bars represent means ± standard deviations from triplicate measurements.
Growth profiles (Fig. 5B) of ST-14::3NBA indicated no major accumulation of the metabolite(s) in the medium. Nonetheless, as the nitro group is released spontaneously from the dioxygenated product of 3NBA (10), the release of nitrite (0.92 to 0.97 mol/mol of 3NBA) was detected by resting-cell transformation studies during 3NBA metabolism by ST-14::3NBA. Apart from the results described above, by using a culture of ST-14::3NBA grown with 3NBA, determination of oxygen uptake individually in the presence of 3NBA (25.78 nmol O2/min/mg protein) and PCA (52.7 nmol O2/min/mg protein), the presence of protocatechuate-3,4-dioxygenase (0.292 U/mg protein) in the cell extract, and detection of trace amounts of PCA as the metabolic intermediate in the supernatants of 3NBA-degrading log-phase cultures supported 3NBA catabolism. Moreover, the activity of the 3NBA ring-hydroxylating dioxygenase was evaluated by HPLC analysis based on the time-dependent transformation of 3NBA (2.9 μg/min/mg protein) in a resting-cell culture of E. coli XL1-Blue harboring pBBR1MCS2_START-3NBA.
DISCUSSION
NBAs are one of the largest groups of chemicals that enter the ecosystem as a part of industrial wastes, and they are significantly toxic (5, 33). Strain ST-14, isolated from municipal-waste-contaminated soil and identified as a member of the genus Cupriavidus, could utilize both 2- and 4-NBAs individually as sole sources of carbon and energy. Moreover, cloning of 3NBA dioxygenase from Comamonas sp. JS46 (10) followed by its expression in wild-type strain ST-14 allowed the recombinant strain of Cupriavidus to utilize 3NBA.
Although there are reports of a few biochemical studies on the bacterial degradation of MNBAs, in most cases, a single isomer of MNBAs was targeted. In the reductive metabolism of nitroaromatics, a variety of bacterial strains were reported to express nitroreductases that either act on specific substrates or have broad substrate specificities (34–37). The specific activity of purified 2NBA reductase obtained from ST-14 is fairly comparable to that of KU-7 (14). On the other hand, 4NBA reductase of ST-14 displayed much higher activity than that of NBA-10 (18, 19). Again, in comparison to biochemical observations, gene information is limited for the evaluation of the pathways of degradation of MNBAs (6, 8, 10, 20). In the present study, the gene cluster (onbFCAR1EHJIGDB) identified for 2NBA degradation is compact, unidirectional, and polycistronic in nature, residing in a single operonic structure, and is induced in the presence of 2NBA but not succinate. A similar nba operon was reported for strain KU-7 (6); however, it differed in gene organization: the upper pathway genes were located at a distant locus separated by six unidentified ORFs, and the 2NBA reductase (nbaA) was in the reverse orientation. These data point toward a possible complex regulation of 2NBA degradation in strain KU-7. On the other hand, the pnb gene cluster was identified for the degradation of 4NBA via PCA, where the latter compound was metabolized by genes residing in a multioperonic pob-pca gene cluster, which harbors the genes for 4HBA degradation. Moreover, it was observed that either the intergenic spaces are short or there are overlaps of stop and potential start codons in the onb and pnb gene clusters, which were indicative of translational coupling and operonic structures (38). Nevertheless, gene knockout experiments showed a disruption of 2NBA and 4NBA degradation in mutant strains ST-14Δonb and ST-14Δpnb, which also established that these genes appear to be present individually as a single functional copy in the genome of strain ST-14.
Based on biochemical and molecular analyses, strain ST-14 possesses the genetic architecture to degrade all three MNBAs, excluding the genes for 3NBA dioxygenase, which is responsible for the transformation of 3NBA to PCA, with the spontaneous release of a nitro group as nitrite. In the present study, to compensate for said catabolic deficiency, functional 3NBA dioxygenase was incorporated into strain ST-14, which resulted in the acquirement of additional catabolic potential to assimilate 3NBA.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Jim C. Spain and Gordana Maravic-Vlahovicek for generous gifts of strain JS46 and the shuttle vectors pBBR1MCS2_START and pBBR1MCS3_START used in this study. We acknowledge Robert A. Kanaly for carefully editing the manuscript.
Financial support for this work was provided by the Bose Institute, Kolkata, India. P.P.C. and S.D. were supported by fellowships from the Council of Scientific and Industrial Research and the Department of Atomic Energy, Government of India, while S.B. was supported by a fellowship from the Bose Institute.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00739-16.
REFERENCES
- 1.Kovacic P, Somanathan R. 2014. Nitroaromatic compounds: environmental toxicity, carcinogenicity, mutagenicity, therapy and mechanism. J Appl Toxicol 34:810–824. doi: 10.1002/jat.2980. [DOI] [PubMed] [Google Scholar]
- 2.Spain JC. 1995. Biodegradation of nitroaromatic compounds. Annu Rev Microbiol 49:523–555. doi: 10.1146/annurev.mi.49.100195.002515. [DOI] [PubMed] [Google Scholar]
- 3.Peres CM, Agathos SN. 2000. Biodegradation of nitroaromatic pollutants: from pathways to remediation. Biotechnol Annu Rev 6:197–220. doi: 10.1016/S1387-2656(00)06023-3. [DOI] [PubMed] [Google Scholar]
- 4.Ju KS, Parales RE. 2010. Nitroaromatic compounds, from synthesis to biodegradation. Microbiol Mol Biol Rev 74:250–272. doi: 10.1128/MMBR.00006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grummt T, Wunderlich HG, Chakraborty A, Kundi M, Majer B, Ferk F, Nersesyan AK, Parzefall W, Knasmuller S. 2006. Genotoxicity of nitrosulfonic acids, nitrobenzoic acids, and nitrobenzylalcohols, pollutants commonly found in ground water near ammunition facilities. Environ Mol Mutagen 47:95–106. doi: 10.1002/em.20172. [DOI] [PubMed] [Google Scholar]
- 6.Iwaki H, Muraki T, Ishihara S, Hasegawa Y, Rankin KN, Sulea T, Boyd J, Lau PC. 2007. Characterization of a pseudomonad 2-nitrobenzoate nitroreductase and its catabolic pathway-associated 2-hydroxylaminobenzoate mutase and a chemoreceptor involved in 2-nitrobenzoate chemotaxis. J Bacteriol 189:3502–3514. doi: 10.1128/JB.01098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandey G, Paul D, Jain RK. 2003. Branching of o-nitrobenzoate degradation pathway in Arthrobacter protophormiae RKJ100: identification of new intermediates. FEMS Microbiol Lett 229:231–236. doi: 10.1016/S0378-1097(03)00844-9. [DOI] [PubMed] [Google Scholar]
- 8.Yabannavar AV, Zylstra GJ. 1995. Cloning and characterization of the genes for p-nitrobenzoate degradation from Pseudomonas pickettii YH105. Appl Environ Microbiol 61:4284–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes MA, Williams PA. 2001. Cloning and characterization of the pnb genes, encoding enzymes for 4-nitrobenzoate catabolism in Pseudomonas putida TW3. J Bacteriol 183:1225–1232. doi: 10.1128/JB.183.4.1225-1232.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Providenti MA, Shaye RE, Lynes KD, McKenna NT, O'Brien JM, Rosolen S, Wyndham RC, Lambert IB. 2006. The locus coding for the 3-nitrobenzoate dioxygenase of Comamonas sp. strain JS46 is flanked by IS1071 elements and is subject to deletion and inversion events. Appl Environ Microbiol 72:2651–2660. doi: 10.1128/AEM.72.4.2651-2660.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nadeau LJ, Spain JC. 1995. Bacterial degradation of m-nitrobenzoic acid. Appl Environ Microbiol 61:840–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartwright NJ, Cain RB. 1959. Bacterial degradation of the nitrobenzoic acids. Biochem J 71:248–261. doi: 10.1042/bj0710248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arora PK, Sharma A. 2015. New metabolic pathway for degradation of 2-nitrobenzoate by Arthrobacter sp. SPG. Front Microbiol 6:551. doi: 10.3389/fmicb.2015.00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa Y, Muraki T, Tokuyama T, Iwaki H, Tatsuno M, Lau PC. 2000. A novel degradative pathway of 2-nitrobenzoate via 3-hydroxyanthranilate in Pseudomonas fluorescens strain KU-7. FEMS Microbiol Lett 190:185–190. doi: 10.1111/j.1574-6968.2000.tb09284.x. [DOI] [PubMed] [Google Scholar]
- 15.Muraki T, Taki M, Hasegawa Y, Iwaki H, Lau PC. 2003. Prokaryotic homologs of the eukaryotic 3-hydroxyanthranilate 3,4-dioxygenase and 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase in the 2-nitrobenzoate degradation pathway of Pseudomonas fluorescens strain KU-7. Appl Environ Microbiol 69:1564–1572. doi: 10.1128/AEM.69.3.1564-1572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samanta SK, Bhushan B, Chauhan A, Jain RK. 2000. Chemotaxis of a Ralstonia sp. SJ98 toward different nitroaromatic compounds and their degradation. Biochem Biophys Res Commun 269:117–123. doi: 10.1006/bbrc.2000.2204. [DOI] [PubMed] [Google Scholar]
- 17.Iwaki H, Hasegawa Y. 2007. Degradation of 2-nitrobenzoate by Burkholderia terrae strain KU-15. Biosci Biotechnol Biochem 71:145–151. doi: 10.1271/bbb.60419. [DOI] [PubMed] [Google Scholar]
- 18.Groenewegen PE, Breeuwer P, Van Helvoort JM, Langenhoff AA, De Vries FP, De Bont JA. 1992. Novel degradative pathway of 4-nitrobenzoate in Comamonas acidovorans NBA-10. J Gen Microbiol 138:1599–1605. doi: 10.1099/00221287-138-8-1599. [DOI] [PubMed] [Google Scholar]
- 19.Groenewegen PE, De Bont JA. 1992. Degradation of 4-nitrobenzoate via 4-hydroxylaminobenzoate and 3,4-dihydroxybenzoate in Comamonas acidovorans NBA-10. Arch Microbiol 158:381–386. doi: 10.1007/BF00245369. [DOI] [PubMed] [Google Scholar]
- 20.Newman LM, Zylstra GJ, Bang S-W, Perry LL. 2000. Chapter 6. Microbial degradation of mononitrophenols and mononitrobenzoates. In Spain JC, Hughes JB, Knackmuss H-J (ed), Biodegradation of nitroaromatic compounds and explosives. CRC Press, Boca Raton, FL. doi: 10.1201/9781420032673. [DOI] [Google Scholar]
- 21.Haigler BE, Spain JC. 1993. Biodegradation of 4-nitrotoluene by Pseudomonas sp. strain 4NT. Appl Environ Microbiol 59:2239–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mallick S, Chatterjee S, Dutta TK. 2007. A novel degradation pathway in the assimilation of phenanthrene by Staphylococcus sp. strain PN/Y via meta-cleavage of 2-hydroxy-1-naphthoic acid: formation of trans-2,3-dioxo-5-(2′-hydroxyphenyl)-pent-4-enoic acid. Microbiology 153:2104–2115. doi: 10.1099/mic.0.2006/004218-0. [DOI] [PubMed] [Google Scholar]
- 23.Ferrera I, Gasol JM, Sebastian M, Hojerova E, Koblizek M. 2011. Comparison of growth rates of aerobic anoxygenic phototrophic bacteria and other bacterioplankton groups in coastal Mediterranean waters. Appl Environ Microbiol 77:7451–7458. doi: 10.1128/AEM.00208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obranic S, Babic F, Maravic-Vlahovicek G. 2013. Improvement of pBBR1MCS plasmids, a very useful series of broad-host-range cloning vectors. Plasmid 70:263–267. doi: 10.1016/j.plasmid.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Rand M, Greenberg AE, Taras MJ. 1976. Standard methods for the examination of water and wastewater. American Public Health Association, Washington, DC. [Google Scholar]
- 26.Smibert RM, Krieg NR. 1981. General characterization, p 409–443. In Gerhardt P, Murray RGE, Costilow RN, Nester EW, Wood WA, Krieg NR, Phillips GB (ed), Manual of methods for general bacteriology. American Society for Microbiology, Washington, DC. [Google Scholar]
- 27.Parker JD, Rabinovitch PS, Burmer GC. 1991. Targeted gene walking polymerase chain reaction. Nucleic Acids Res 19:3055–3060. doi: 10.1093/nar/19.11.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Stanier RY, Ingraham JL. 1954. Protocatechuic acid oxidase. J Biol Chem 210:799–808. [PubMed] [Google Scholar]
- 30.Marx CJ, Lidstrom ME. 2002. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques 33:1062–1067. [DOI] [PubMed] [Google Scholar]
- 31.Taghavi S, van der Lelie D, Mergeay M. 1994. Electroporation of Alcaligenes eutrophus with (mega)plasmids and genomic DNA fragments. Appl Environ Microbiol 60:3585–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pérez-Pantoja D, De la Iglesia R, Pieper DH, González B. 2008. Metabolic reconstruction of aromatic compounds degradation from the genome of the amazing pollutant-degrading bacterium Cupriavidus necator JMP134. FEMS Microbiol Rev 32:736–794. doi: 10.1111/j.1574-6976.2008.00122.x. [DOI] [PubMed] [Google Scholar]
- 33.Pouretedal HR, Keshavarz MH. 2011. Prediction of toxicity of nitroaromatic compounds through their molecular structures. J Iran Chem Soc 8:78–89. doi: 10.1007/BF03246204. [DOI] [Google Scholar]
- 34.Somerville CC, Nishino SF, Spain JC. 1995. Purification and characterization of nitrobenzene nitroreductase from Pseudomonas pseudoalcaligenes JS45. J Bacteriol 177:3837–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kutty R, Bennett GN. 2005. Biochemical characterization of trinitrotoluene transforming oxygen-insensitive nitroreductases from Clostridium acetobutylicum ATCC 824. Arch Microbiol 184:158–167. doi: 10.1007/s00203-005-0036-x. [DOI] [PubMed] [Google Scholar]
- 36.Blasco R, Castillo F. 1993. Characterization of a nitrophenol reductase from the phototrophic bacterium Rhodobacter capsulatus E1F1. Appl Environ Microbiol 59:1774–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe M, Nishino T, Takio K, Sofuni T, Nohmi T. 1998. Purification and characterization of wild-type and mutant “classical” nitroreductases of Salmonella typhimurium L33R mutation greatly diminishes binding of FMN to the nitroreductase of S. typhimurium. J Biol Chem 273:23922–23928. doi: 10.1074/jbc.273.37.23922. [DOI] [PubMed] [Google Scholar]
- 38.Gold L. 1988. Posttranscriptional regulatory mechanisms in Escherichia coli. Annu Rev Biochem 57:199–233. doi: 10.1146/annurev.bi.57.070188.001215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.