

ABSTRACT
F-specific RNA phages (FRNAPHs) are considered potential viral indicators of water pollution due to their occurrence and stability in water environments. However, their suitability as viral indicators is not fully elucidated because the characteristics of FRNAPHs are variable depending on the genotype. In this study, for the characterization of infectious FRNAPH genotypes, integrated culture reverse transcription-PCR coupled with the most probable number approach was applied to surface water samples. Further, to recover low concentrations of FRNAPH genotypes, an FRNAPH recovery method was developed. The novel FRNAPH recovery method using a noncharged microfiltration membrane could effectively recover FRNAPH strains without inactivation, while a method using an electronegative microfiltration membrane resulted in the inactivation of some strains. Infectious FRNAPH genotypes in surface water samples were successfully quantified with an efficiency comparable to that of the conventional plaque assay. Genotype I (GI) and GII FRNAPHs tended to be predominant at locations impacted by treated and untreated municipal wastewater, respectively. The numbers and proportions of infectious FRNAPHs tended to be higher during the winter season when water temperature decreased.
IMPORTANCE Properties of FRNAPHs are highly variable depending on their genotypes. Previous typing methods for FRNAPHs are not quantitative and/or are based on molecular assays, which cannot differentiate infective strains from inactive strains. Due to the reasons mentioned above, the utility of FRNAPHs as viral indicators of water pollution has not been fully validated. In this study, a quantitative genotyping method for infectious FRNAPHs was developed and applied to surface water samples. The method enabled characterization of infectious FRNAPH genotypes in terms of their occurrence and seasonality. Moreover, comparison of the method to a conventional molecular assay (reverse transcription-quantitative PCR) enabled characterization of their stability. Our approach can provide novel findings for further validation of FRNAPHs as viral indicators of water pollution.
INTRODUCTION
F-specific coliphages (F+ coliphages) are bacteriophages that infect Escherichia coli cells possessing F pili. F+ coliphages are classified into FDNA phages (FDNAPHs) or FRNA phages (FRNAPHs), depending on whether their genomes consist of single-stranded DNA or single-stranded RNA, respectively (1). FDNAPHs possess a rod-shaped capsid, while FRNAPHs possess an icosahedral capsid with a diameter of 20 to 30 nm. The morphology of FRNAPHs is similar to that of typical human enteric viruses such as caliciviruses and enteroviruses. Due to the similarity of their origin and morphology to those of enteric viruses, FRNAPHs are regarded as potential viral indicators of water contamination and viral fate in water environments and water treatment plants (1, 2). FRNAPHs are further classified into four different genotypes/serotypes, GI to GIV, which are represented by MS2, GA, Qβ, and SP phages, respectively (3). Features such as occurrence in a water environment and stability against water treatment are known to be highly variable among FRNAPH genotypes (4–13). For instance, GII FRNAPHs tend to be the predominant type in raw municipal wastewater, while GI FRNAPHs tend to be predominant after full-scale municipal wastewater treatment (4, 6, 7). GIV FRNAPHs are rarely found in water samples (5, 6, 11, 13). GI FRNAPHs are highly stable in natural environments (8, 10). GII and GIII are the dominant genotypes of FRNAPHs in human feces, while GI and GIV are the dominant genotypes of FRNAPHs in the feces of other animals, such as cow, swine, gull, and goose (2, 14).
The reduction of virus levels in water can occur through removal and inactivation. Removal, e.g., by the filtration process, results in a decrease in the number of viral particles. In the broad sense, the dilution or diffusion of viruses may also be considered the removal of viruses. When the removal occurs, a consistent viral reduction rate should be obtained regardless of virus quantification methods. Inactivation, e.g., by chlorination or other chemical disinfection processes can result in viral particles still remaining, but it causes the loss of viral infectivity. When inactivation occurs, virus quantifications by infectivity assays show decreases, while those by molecular assays such as quantitative PCR show almost negligible changes (15). Inactivation of viruses by UV irradiation, which causes genome damage, also slightly affects quantifications by PCR assays compared to those by infectivity assays, because only a small fraction of a genome is amplified by PCR assays (16). If the removal and inactivation of FRNAPH genotypes could be separately evaluated, the fate of the virus in the water environment or during the water treatment process would be better understood. Currently, the quantitative detection of F+ coliphages is done by plaque (culture) or reverse transcription-quantitative PCR (RT-qPCR) assays (17–22). The plaque assay can detect infectious phages but requires an immense effort for quantitative typing (23). Quantification by the plaque assay is affected by both virus removal and inactivation. In contrast, RT-qPCR-based assays enable rapid quantitative genotyping by applying type-specific primers and TaqMan probes. However, RT-qPCR-based assays cannot differentiate between infective and inactive F+ coliphages. A method coupling plaque isolation with RT-PCR- or other molecular assay-based genotyping was also applied (4, 5, 6, 13, 17, 24). However, it is difficult to isolate and analyze sufficient numbers of each genotype for quantitative typing. Therefore, the presence and fate of a nonpredominant type may be overlooked. The application of RT-PCR-based genotyping after FRNAPH propagation in liquid medium (integrated culture [IC]–RT-PCR) may be effective for the simple genotyping of infectious FRNAPHs. Even though the method is qualitative, quantification can be done using the most probable number (MPN) approach.
To recover FRNAPHs or FRNAPH genotypes present in low numbers, the concentration of FRNAPHs is required. In this regard, a method that can effectively recover FRNAPHs without inactivation is desirable. An adsorption-elution method using an electronegative microfilter (MF) is a rapid and simple virus concentration method (25). However, an acid solution used during the purification process is suggested to inactivate some FRNAPH strains such as Qβ (26). Moreover, in the case of membrane clogging, virus recovery from MF becomes difficult (27). Because our objective was to cultivate FRNAPHs in liquid medium, FRNAPHs recovered on a MF may be directly subjected to cultivation. In this case, the purification or elution of viruses, which may decrease FRNAPH recovery efficiency, can be skipped. Noncharged MFs may also be useful for recovering FRNAPHs. Previous studies have shown that in the presence of a coagulant such as AlCl3 or polyaluminum chloride, virus titers could be reduced by >3 log10 when using a noncharged MF (28, 29). These data suggest that most viruses are retained on a MF and can be propagated by liquid cultivation.
This study aimed to quantitatively investigate the occurrence of infectious FRNAPH genotypes in surface water. The performances of two FRNAPH recovery methods were compared in view of recovery efficiencies for infectious FRNAPH genotypes. The quantitative genotyping of FRNAPHs in surface water samples was conducted by applying the better recovery method coupled with the MPN approach.
MATERIALS AND METHODS
FRNAPH strains.
As representative strains of GI and GIII FRNAPHs, pure culture stocks of MS2 and Qβ phages, respectively, were used. As a representative of GII FRNAPHs, a GA-like phage was isolated from the Katsura River, Kyoto Prefecture, Japan, by a plaque isolation method. Briefly, after application of the plaque assay described below, some plaques were isolated and subjected to propagation in tryptone-glucose broth (TGB) using Salmonella enterica serovar Typhimurium WG49 as the host strain (shown below). Each isolate was subjected to RT-qPCR targeting GI to GIV FRNAPHs (shown below), and an isolate determined to be positive for GII FRNAPHs only was selected as the representative strain. No GIV FRNAPH strain was used in this study because the genotype was not found among the isolates.
FRNAPH strains were propagated and purified before use. Briefly, each FRNAPH strain and S. Typhimurium WG49 in the exponential growth phase were spiked into TGB, which consisted of 10 g/liter of tryptone, 1.0 g/liter of glucose, 8.0 g/liter of NaCl, 0.3 g/liter of CaCl2, 0.15 g/liter of MgSO4, 20 mg/liter of kanamycin, and 100 mg/liter of nalidixic acid. TGB was then incubated overnight at 37°C and was subjected to centrifugation (8,000 rpm, 3 min). The supernatant was filtered through a mixed cellulose ester filter (0.20-μm pore, 25 mm in diameter; Advantec, Tokyo, Japan), and the eluate was recovered as a purified FRNAPH strain (up to 1011 PFU/ml). The purified strains were diluted in phosphate buffer (pH 7) and used for spike tests.
Recovery and propagation of infectious FRNAPHs from water.
To determine an appropriate method for recovery and quantitative genotyping of infectious FRNAPHs in the water environment, two types of FRNAPH recovery methods, an adsorption method using an electronegative MF and an entrapment method using a noncharged MF, were compared. For their validation, Qβ was preferentially used because it was presumed that Qβ is easily inactivated during the processes (26). The entrapment method, which showed better recovery, was further validated and was applied to surface water samples.
(i) Adsorption method using an electronegative MF.
For the recovery of FRNAPHs by use of an electronegative MF, a modification of a virus concentration method described by Katayama et al. (25) was applied. Using this method, FRNAPHs were adsorbed on an electronegative MF in the presence of a polyvalent cation and then propagated. For the electronegative MF, a mixed cellulose membrane (type HA, 47 mm in diameter, 0.45-μm pore size: Merck Millipore, Billerica, MA, USA) or a glass fiber membrane (GF/B, 47 mm in diameter, 1.0-μm pore size: GE Healthcare, Buckinghamshire, United Kingdom) was used. For a polyvalent cation solution, 2.5 M MgCl2 or 250 mM AlCl3 was added to the sample at a final concentration of 25 or 0.5 mM, respectively. First, a polyvalent cation solution and S. Typhimurium WG49 at the exponential growth phase were added to the sample. The sample was then passed through an electronegative MF. The MF was then put into a petri dish (90 mm in diameter) containing 10 ml of TGB and incubated overnight at 37°C. After incubation, the culture was subjected to RT-PCR-based detection of the FRNAPH genotypes (IC–RT-PCR) as described below.
(ii) Entrapment method using a noncharged MF.
For the recovery of FRNAPHs using a noncharged MF, FRNAPHs were entrapped on the MF together with S. Typhimurium WG49 in the presence of a coagulant and then propagated. For the noncharged MF, a polyvinylidene fluoride membrane (type HV, 47 mm in diameter, 0.45-μm pore size, 29-ml/min/cm2 flow rate; Merck Millipore) or a polytetrafluoroethylene membrane (type JG, 47 mm in diameter, 0.2-μm pore size, 6.9-ml/min/cm2 flow rate: Merck Millipore) was used. First, the sample pH was adjusted to 5.0 ± 0.3 using H2SO4, and the sample was mixed with 250 mM AlCl3 (final concentration of 0.5 mM) and S. Typhimurium WG49 at the exponential growth phase. The sample was then passed through a noncharged MF. The MF was then put into a petri dish (90 mm in diameter) containing 10 ml of TGB and incubated overnight at 37°C. After incubation, the culture was subjected to RT-PCR-based detection of the FRNAPH genotypes (IC–RT-PCR) as described below.
Detection of FRNAPH genotypes by IC–RT-PCR.
Infectious FRNAPH genotypes were detected by RT-PCR after the overnight incubation. Briefly, 2 μl of the culture was incubated at 95°C for 5 min for RNA extraction, according to a method described previously, with slight modifications (30). Then, the RNA extract was subjected to one-step RT-PCR using the QuantiTect Probe RT-PCR kit (Qiagen, Hilden, Germany). A 2-μl volume of the extract was mixed with 18 μl of a reaction mixture containing 10 μl of 2× QuantiTect Probe RT-PCR master mix (Qiagen), 400 nM (each) genotype-specific forward and reverse primers (31), 150 nM genotype-specific TaqMan probe labeled with 6-carboxyfluorescein (FAM) (31), and 0.4 μl of QuantiTect RT mix (Qiagen). RT-PCR was conducted using a thermal cycler dice real-time system (TaKaRa, Shiga, Japan) under the following thermal cycling conditions: 50°C for 30 min, 95°C for 15 min, and 40 cycles of 95°C for 15 s and 58°C for 60 s. The results were considered possible for an infectious FRNAPH genotype if the resultant cycle threshold (CT) value was no more than 30. If the CT value was higher than 30, the amplification curve was considered to be due to inactive FRNAPHs, which could not multiply during the liquid cultivation process.
Detection of F+ coliphages by plaque assay.
A plaque assay for the quantification of F+ coliphages was done according to a previous study, which employed S. Typhimurium WG49 as the host strain (32). The liquid cultivation of strain WG49 and F+ coliphages was conducted using TGB.
Collection of surface water samples.
A lake water sample was collected from Lake Biwa, located in Shiga Prefecture, Japan, in June 2014 for validation of the entrapment method using a natural freshwater sample. The water sample was collected in a sterilized polyethylene container and transferred to the laboratory on ice. After transport, the sample was subjected to heating in an autoclave at 121°C for 15 min. The autoclaved lake water was spiked with FRNAPH strains and subjected to the entrapment method, followed by IC–RT-PCR.
Surface water samples were collected monthly from June 2014 to June 2015 at three points along the Katsura River in Kyoto Prefecture, Japan (K1 to K3), and a point on the Furu River, a tributary of the Uji River in Kyoto Prefecture, Japan (F1). At K2 and approximately 20 km upstream of K1, effluents from a municipal wastewater treatment plant are discharged into the Katsura River. K1 and K3 are located 1.9 km upstream and 2.4 km downstream, respectively, of K2. Because the sewer coverage rate is low in the catchment area of the Furu River, the river water at F1 is affected by not fully treated wastewater, e.g., effluents from domestic septic tanks, untreated gray water, etc. At each point, 2-liter samples were collected into a sterilized polyethylene container and transferred to the laboratory on ice. Water quality parameters such as temperature, pH, oxidation-reduction potential, electrical conductivity, turbidity, and dissolved oxygen were measured on-site using a multiparameter water quality meter (U-52G: Horiba, Kyoto, Japan). The samples were subjected to the plate counting assay for F+ coliphages, liquid cultivation of F+ coliphages, and virus concentration within 24 h after collection.
Quantification of F+ coliphages in surface water samples.
F+ coliphages in the surface water samples were quantified by three different assays: IC–RT-PCR coupled with the MPN approach, conventional RT-qPCR, and a conventional plaque assay (Fig. 1).
FIG 1.

Schematic of the F+ coliphage quantification methods.
(i) IC–RT-PCR coupled with the MPN approach.
For the quantitative genotyping of infectious FRNAPHs, the IC–RT-PCR-based genotyping of FRNAPHs was coupled with the MPN approach using 100-, 10-, 1-, and 0.1-ml sample volumes (n = 3 for each volume). The 100-ml sample was subjected to the entrapment method using a noncharged MF (type HV) for the recovery of FRNAPHs. The samples of smaller volumes (0.1 to 10 ml) were directly mixed with TGB containing S. Typhimurium WG49 at the exponential growth phase and incubated at 37°C overnight. After application of the IC–RT-PCR described above, the quantification of infectious FRNAPH genotypes was performed using the MPN approach, which can express quantitative data from qualitative data obtained by serial dilution tests. The concentration of infectious FRNAPH genotypes was determined by referring to an MPN table for three 10-fold dilutions with three tubes at each dilution, provided by Blodgett (33).
(ii) Conventional RT-qPCR.
For the quantitative genotyping of all FRNAPHs, FRNAPHs were quantified by a conventional RT-qPCR assay after the concentration (25) and RNA extraction steps. Briefly, 1,000 ml of a sample was mixed with 2.5 M MgCl2 (to a final concentration of 25 mM) and passed through an electronegative MF (type HA, 90 mm in diameter, 0.45-μm pore size: Merck Millipore). Then, 200 ml of 0.5 mM H2SO4 (pH 3.0) and 10 ml of 1.0 mM NaOH (pH 10.8) were successively passed through the MF. The final filtrate was recovered as a primary concentrate in a tube containing 50 μl of 100 mM H2SO4 (pH 1.0) and 100 μl of 100× Tris-EDTA buffer (pH 8.0). The primary concentrate was further subjected to centrifugal ultrafiltration-based concentration using a Centriprep YM-50 (Merck Millipore) to obtain a final volume of 650 μl. The secondary concentrate was subjected to a conventional two-step RT-qPCR-based quantification of FRNAPHs. Murine norovirus (MNV), propagated in the RAW 264.7 cell line (34), was spiked with 140 μl of the secondary concentrate as a process control to evaluate the reliability of the RNA extraction/RT-qPCR procedures. Viral RNA was extracted from 140 μl of the secondary concentrate using a QIAamp viral RNA minikit (Qiagen) to obtain 60 μl of RNA extract according to the manufacturer's instructions. The extracted viral RNA was subjected to RT using a high-capacity cDNA reverse transcription kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. The obtained cDNA was subjected to TaqMan-based qPCR assays for the quantitative detection of GI to GIV FRNAPH genes using a thermal cycler dice real-time system (TaKaRa). A reaction mixture (25 μl) was prepared by mixing 5.0 μl of cDNA, 12.5 μl of TaqMan gene expression master mix (Life Technologies), 1.0 μl each of 10 μM forward and reverse primers, and 0.5 μl of 5.0 μM TaqMan probe. Sequences of primers and TaqMan probes for the detection of MNV and FRNAPH were derived from previous studies by Kitajima et al. (35) and Wolf et al. (31), respectively. Thermal cycling for FRNAPHs and the spiked MNV was conducted using the following conditions: 95°C for 15 min and then, for FRNAPHs, 50 cycles of 95°C for 15 s, 58°C for 60 s, and 95°C for 15 min or, for spiked MNV, 50 cycles of 95°C for 15 s and 56°C for 60 s (31, 35). Gene copy numbers were calculated from a standard curve generated using 10-fold serial dilutions (concentrations from 104 to 100 copies/reaction) of plasmid DNA containing the target sequences.
(iii) Conventional plaque assay.
For the quantification of the total number of infectious F+ coliphages, a conventional plaque assay, described above, was performed. A sample (50 ml) dispensed into petri dishes in volumes of 5 ml was subjected to the assay.
Statistical analysis.
Statistical analyses were performed with Microsoft Excel 2010 (Microsoft). Student's t test was performed to determine whether infectivity indices, which were defined as differences between the log10-transformed number of infectious FRNAPHs (MPN by IC–RT-PCR–MPN assay) and the total number (copies by RT-qPCR assay) for each FRNAPH genotype, were significantly different during the summer season and the winter season. Tukey's multiple-comparison test was performed to determine whether infectivity indices were significantly different over the sampling sites. In both analyses, if the resultant P value was 0.05 or lower, the difference was determined to be significant.
RESULTS
Comparison of FRNAPH recovery methods.
The FRNAPH strains of each genotype were independently spiked into 50 ml of Milli-Q water and recovered by the adsorption method using an electronegative MF and by the entrapment method using a noncharged MF. The recovered FRNAPHs were qualitatively detected by IC–RT-PCR. The adsorption method using the type HA membrane as the MF and MgCl2 as the cation solution produced positive results for MS2, even if its spiked amount was approximately 100 PFU (Table 1). In contrast, the method generated positive results for GA-like phage or Qβ only when their spiked amounts were on the order of 102 PFU or higher. The FRNAPH numbers in the MF filtrates were less than 28% of those in the feed water (determined by plaque assay, n = 3 for each FRNAPH strain; data not shown), indicating that >72% of the FRNAPHs were retained on the MF. The use of the GF/B membrane as the electronegative MF or AlCl3 as the cation solution also resulted in poor recovery efficiencies for infectious Qβ. The entrapment method using the type HV membrane gave positive results for all tested FRNAPH strains even if their spiked amounts were 100 PFU or lower (Table 2). The FRNAPH numbers in the type HV membrane filtrates were less than 1.9% of those in the feed water (plaque assay, n = 3 for each FRNAPH strain; data not shown). These data indicate that most (>98%) of the FRNAPHs were trapped on the type HV MF without losing infectivity. The use of the type JG membrane as the noncharged MF also showed a reasonable recovery of Qβ; however, based on the flow rate, the use of the type HV membrane seems to be preferable.
TABLE 1.
Recovery of FRNAPHs spiked into 50 ml of Milli-Q water by the adsorption method using electronegative MFs
| Spiked FRNAPH (PFU) | No. of runs with recovered FRNAPHs/total no.a |
||||
|---|---|---|---|---|---|
| Type HA membrane (n = 3) |
GF/B membrane (n = 1), AlCl3, Qβ (GIII) | ||||
| MgCl2 |
AlCl3, Qβ (GIII) | ||||
| MS2 (GI) | GA-like (GII) | Qβ (GIII) | |||
| 0.5 × 105–5.0 × 105 | 3/3 | ||||
| 0.5 × 104–5.0 × 104 | 3/3 | 3/3 | 0/1 | ||
| 0.5 × 103–5.0 × 103 | 3/3 | 1/3 | 0/1 | ||
| 0.5 × 102–5.0 × 102 | 3/3 | 1/3 | 0/3 | 0/3 | 0/1 |
| 0.5 × 101–5.0 × 101 | 3/3 | 0/3 | 0/3 | 0/3 | 0/1 |
| 0.5 × 100–5.0 × 100 | 2/3 | 0/3 | 0/3 | 0/3 | 0/1 |
| 0.5 × 10−1–5.0 × 10−1 | 0/3 | 0/3 | 0/3 | 0/1 | |
Recovery of FRNAPHs was done by the absorption method using type HA or GF/B MFs and 2.5 mM MgCl2 or 0.5 mM AlCl3 as the cation solution for MS2, GA, and Qβ phages.
TABLE 2.
Recovery of FRNAPHs spiked into 50 ml of Milli-Q water by the entrapment method using noncharged MFs
| Spiked FRNAPH (PFU) | No. of runs with recovered FRNAPHs/total no.a |
|||
|---|---|---|---|---|
| Type HV membrane (n = 3) |
Type JG membrane (n = 1), Qβ (GIII) | |||
| MS2 (GI) | GA-like (GII) | Qβ (GIII) | ||
| 0.5 × 103–5.0 × 103 | 1/1 | |||
| 0.5 × 102–5.0 × 102 | 1/1 | |||
| 0.5 × 101–5.0 × 101 | 3/3 | 3/3 | 3/3 | 1/1 |
| 0.5 × 100–5.0 × 100 | 2/3 | 3/3 | 2/3 | 1/1 |
| 0.5 × 10−1–5.0 × 10−1 | 2/3 | 0/3 | 0/3 | 0/1 |
| 0.5 × 10−2–5.0 × 10−2 | 0/3 | 0/3 | 0/3 | 0/1 |
Recovery of FRNAPHs was done by the entrapment method using type HV or JG noncharged MFs for MS2, GA-like, and Qβ phages.
Recovery of simultaneously spiked FRNAPH genotypes from lake water samples.
The effectiveness of the entrapment method was further validated using 50 ml of autoclaved lake water samples. In this assay, to evaluate the effect of competition on the propagation of FRNAPH genotypes, the FRNAPH strains of three genotypes (GI to GIII) were simultaneously spiked into a lake water sample. Three runs (1 to 3), in which a genotype was spiked to achieve a relatively low concentration (3.9 × 100 to 1.3 × 101 PFU/sample) in comparison to the concentrations of other genotypes (7.7 × 102 to 2.7 × 103), were performed in triplicate tests (Table 3). Except for Qβ in one of the triplicate tests in run 3, in which it was spiked to be nonpredominant, all spiked FRNAPH genotypes were detected by the entrapment method, followed by IC–RT-PCR (Table 3). These results suggest that the entrapment method coupled with IC–RT-PCR is effective in the detection of even nonpredominant genotypes in environmental freshwater samples.
TABLE 3.
Recovery of FRNAPHs spiked into 50 ml of autoclaved lake water by the entrapment method using a noncharged MF (n = 3 for each run)a
| Run | Phage (genotype) | Spiked amt (PFU) | No. of positive samples/total no. |
|---|---|---|---|
| 1 | MS2 (GI) | 3.9 × 100 | 3/3 |
| GA-like (GII) | 8.1 × 102 | 3/3 | |
| Qβ (GIII) | 2.7 × 103 | 3/3 | |
| 2 | MS2 (GI) | 7.7 × 102 | 3/3 |
| GA-like (GII) | 4.0 × 100 | 3/3 | |
| Qβ (GIII) | 2.7 × 103 | 3/3 | |
| 3 | MS2 (GI) | 7.7 × 102 | 3/3 |
| GA-like (GII) | 8.1 × 102 | 3/3 | |
| Qβ (GIII) | 1.3 × 101 | 2/3 |
Underlining indicates the phage strains (genotypes) that were spiked to achieve a relatively low concentration in a sample compared to other phage strains (genotypes).
Performance of IC–RT-PCR–MPN for indigenous FRNAPHs in surface water.
Indigenous FRNAPHs were quantitatively detected in the surface water samples by three different methods: the IC–RT-PCR–MPN method, conventional RT-qPCR, and conventional plaque assays (Fig. 1).
To further validate the IC–RT-PCR–MPN assay, the quantities of each infectious FRNAPH genotype (IC–RT-PCR–MPN assay) in surface water samples were summed up and compared to those of total infectious F+ coliphages (plaque assay) (Fig. 2). In total, 41 of 52 samples (88%) were positive in both assays. Among the positive samples, a correlation was observed between the sum for each genotype and the total of F+ coliphages (r = 0.77, P < 0.01). Besides, 36 out of the 41 positive samples showed less than a 1 log10 difference between the assays. In most cases, the difference between upper and lower 95% confidence intervals for an MPN value is around 1.0 log10 (33), indicating that the IC–RT-PCR–MPN assay can effectively quantify FRNAPH genotypes in surface water samples. However, exceptions were seen, particularly if the results of the IC–RT-PCR–MPN assay were negative (Fig. 2). Only samples taken at F1 showed such unusual results (collected from June to September 2014) (Fig. 3).
FIG 2.
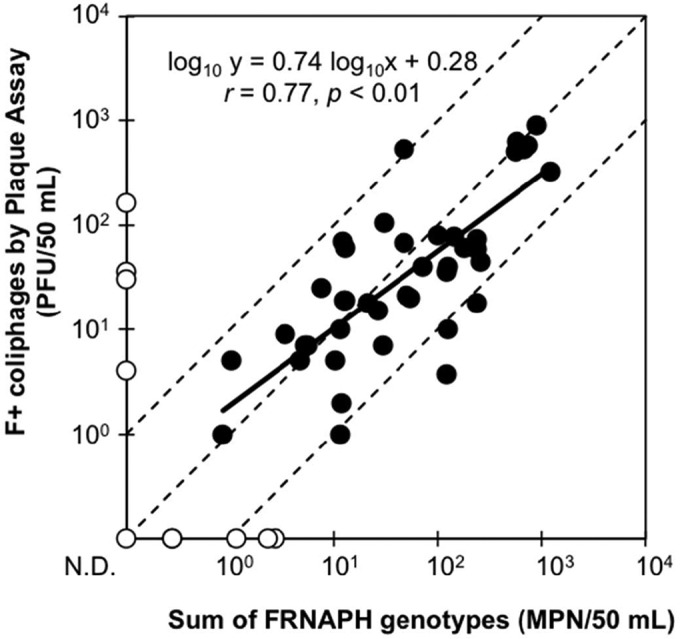
Relationship between the concentrations of all infectious FRNAPH genotypes and infectious F+ coliphages in the surface water samples. The total concentrations of all infectious FRNAPH genotypes were determined by summing up the quantities of each FRNAPH genotype obtained using the IC–RT-PCR–MPN assay. The concentrations of F+ coliphages were determined by the conventional plaque assay. The white circles indicate negative results for all infectious FRNAPH genotypes and/or infectious F+ coliphages. The limits of detection for all infectious FRNAPH genotypes and infectious F+ coliphages were 1.5 × 10−1 MPN/50 ml and 1.0 × 100 PFU/50 ml, respectively. The solid line is a regression line. Slope, intercept, r, and P values of the regression line are shown in the figure. The circles located in the area between the dashed lines indicate that the differences between the quantities determined by the two assays are within 1 log10.
FIG 3.

Quantification of infectious FRNAPHs in the surface water samples collected at K1 (A), K2 (B), K3 (C), and F1 (D). Quantitative genotyping was performed by the IC–RT-PCR–MPN assay. N.D. on the horizontal axis indicates not detected.
Occurrence of infectious FRNAPH genotypes in surface water.
The quantification data for each genotype in the samples examined by the IC–RT-PCR–MPN assay are shown in Fig. 3. At K1, K2, and K3, which were supposed to be impacted by treated municipal wastewater, GI tended to be the predominant FRNAPH genotype. On the contrary, at F1, which was supposed to be impacted by not fully treated wastewater, GII FRNAPHs tended to be predominant.
At K1, the concentrations of infectious GI FRNAPHs were higher during the winter season (from October to April) than during the summer season, i.e., they were detected at 7.5 × 10−1 to 1.2 × 102 MPN/50 ml during the winter season but only in one sample (7.5 × 100 MPN/50 ml) during the summer season (Fig. 3). Such a seasonality of GI FRNAPHs was more noticeable at F1, where only one of the six samples was positive (1.8 × 10−1 MPN/50 ml) during the summer season while six out of the seven samples were positive (1.8 × 10−1 to 2.3 × 102 MPN/50 ml) during the winter season. However, at K2, GI FRNAPHs were constantly detected at concentrations ranging from 1.2 × 101 to 5.5 × 102 MPN/50 ml, while at K3, their concentrations fluctuated highly (11/13 positives, 5.5 × 10−1 to 5.5 × 102 MPN/50 ml).
At K1, K2, and K3, GII FRNAPHs were detected in all samples at concentrations ranging from 1.8 × 10−1 to 2.2 × 101 MPN/50 ml, from 1.2 × 100 to 7.5 × 101 MPN/50 ml, and from 3.6 × 10−1 to 5.5 × 102 MPN/50 ml, respectively. At the three sampling sites, the highest concentrations of GII FRNAPHs were observed in February 2015; however, the seasonality of the genotype at the sites was not obvious. On the contrary, at F1, GII FRNAPHs were detected only in the samples collected during the winter season (7/7 positives, 4.6 × 10−1 to 5.5 × 102 MPN/50 ml).
The positive rates and concentrations of GIII FRNAPHs tended to be lower than those of GI and GII FRNAPHs. Except for K1, which showed a particularly low positive rate of GIII FRNAPHs (3/13 positives), GIII FRNAPHs tended to be prevalent during the winter season.
GIV FRNAPHs were found at K2 from January to March 2015, and their concentrations were quite low (1.8 × 10−1 to 4.6 × 10−1 MPN/50 ml).
Comparison of direct RT-qPCR and IC–RT-qPCR.
In water environments, viruses lose their infectivity due to several environmental factors. Molecular assays such as direct RT-qPCR can detect such inactivated viruses, but cultural assays cannot. In other words, molecular assays can detect all targets, regardless of their infectivity, while culture-based assays can detect only infective targets. Thus, by comparing the results of a molecular assay with those of a culture-based assay, the degree of virus inactivation can be estimated. The quantification efficiency of RT-qPCR for MNV spiked into a virus concentrate (adsorption-elution method) was sufficiently high (>23% compared to MNV spiked into Milli-Q water; data not shown), indicating that RNA extraction and subsequent RT-qPCR were performed with acceptable efficiencies (7). For GI, GII, and GIII FRNAPHs, which showed relatively high positive rates, the ratios between the direct RT-qPCR assay (molecular assay) and culture-based IC–RT-qPCR assays (infectivity index) are compared in Fig. 4. The infectivity index was calculated only if the sample was positive by both the direct RT-qPCR and IC–RT-qPCR assays.
FIG 4.
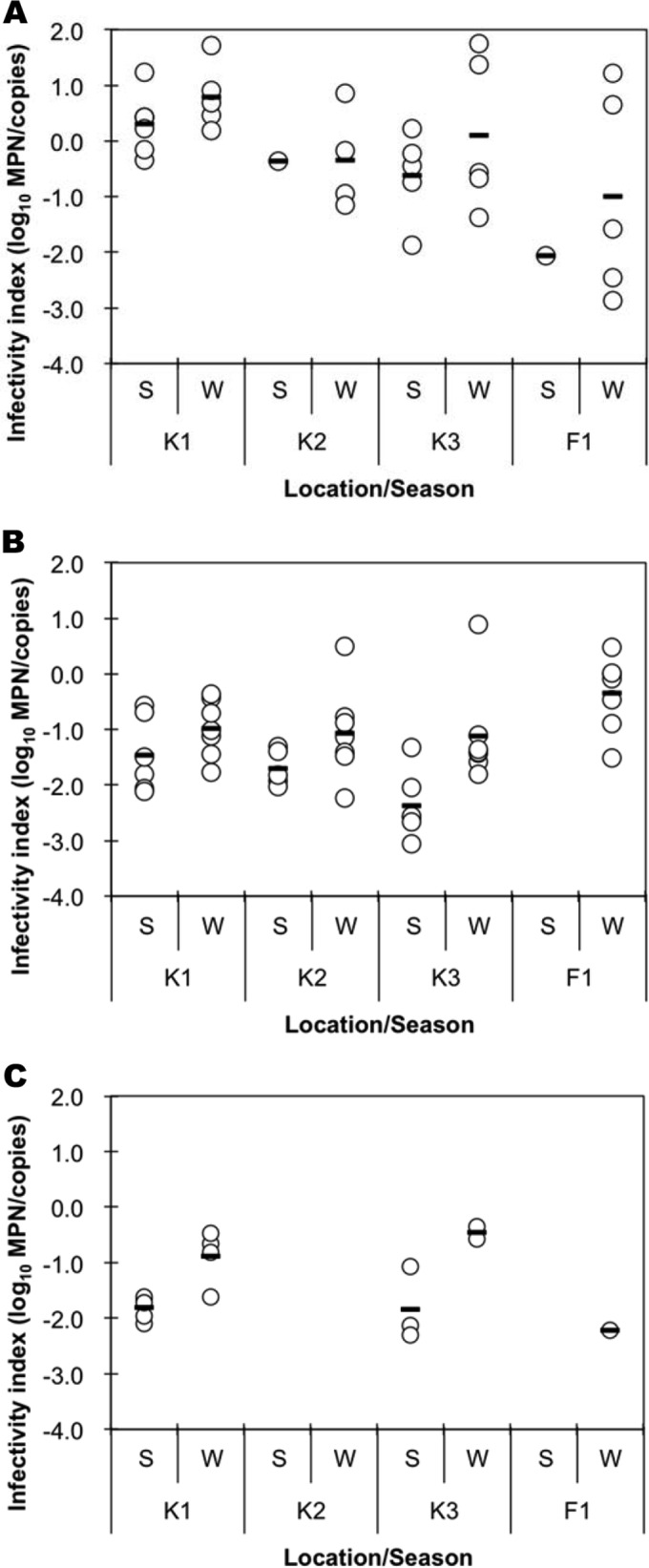
Infectivity indices of GI (A), GII (B), and GIII (C) FRNAPHs in the surface water samples. The infectivity index was defined as the difference between the log10-transformed number of infectious FRNAPHs (MPN) and the total number (copies) for each FRNAPH genotype. The circles represent samples that were positive in both the IC–RT-PCR–MPN and RT-qPCR assays. The bars indicate the mean values, excluding the negative data. S and W on the horizontal axis indicate samples collected during the summer (from May to September, n = 6) and winter (from October to March, n = 7) seasons, respectively. If the IC–RT-PCR–MPN and/or RT-qPCR assays showed negative results, the corresponding data were not plotted.
Regarding GI FRNAPHs, the infectivity index tended to be above 0.0 log10, which can be interpreted as indicating that the number of infectious viruses was higher than the total number of viruses. On the other hand, for GII and GIII FRNAPHs, the infectivity indices were lower than 0.0 log10 for the majority of the samples.
The infectivity indices tended to be higher during the winter season (from October to April) than during the summer season (from May to September). However, significant differences in the indices were seen only for GII FRNAPHs at K3 and GIII FRNAPHs at K1 (Student's t test, P = 0.015 and 0.007, respectively). When the sampling sites are compared, the indices showed significant differences for GI FRNAPHs at K2 (0.52 ± 0.59) and F1 (−1.18 ± 1.71) and for GII FRNAPHs at K3 (−1.69 ± 1.00) and F1 (−0.35 ± 0.67) (Tukey's multiple-comparison test, P = 0.02 and 0.004, respectively).
DISCUSSION
FRNAPHs are considered potential viral indicators for water environments or during water purification treatment (1, 2). In some countries or regions, the concentration or reduction of FRNAPHs is set as a parameter to ensure virological water quality (36, 37, 38). However, as indicated in a previous study, the differential persistence of FRNAPH genotypes may lead to the over- or underestimation of viral risks (9). For the further validation of FRNAPHs as potential viral indicators, the differentiation of FRNAPH genotypes is required. Such trials have been performed by applying conventional RT-qPCR assays (7, 19, 20) or RT-PCR-based typing of isolated plaques using RT-PCR- or reverse line blot (RLB)-based methods (4, 5, 6, 13, 17, 24, 39, 40). However, to our knowledge, the quantitative genotyping of infectious FRNAPHs has never been conducted, and thus, the fates of FRNAPH genotypes in the water environment have not been clarified. In the present study, we aimed to quantify infectious FRNAPH genotypes by the IC–RT-PCR and MPN assays. For the detection of low concentrations of FRNAPH genotypes, we first examined FRNAPH recovery methods applied to a large volume of water. Adsorption-elution methods are widely accepted as virus concentration methods; however, their elution efficiencies occasionally become low, even though their adsorption efficiencies are high (27, 41, 42). Additionally, it was suggested that FRNAPH Qβ is inactivated before the elution step when an electronegative membrane is used (26). Thus, in our methods, the membranes were subjected to FRNAPH propagation immediately after the adsorption or entrapment step. In the present study, two types of FRNAPH recovery methods, an adsorption method using an electronegative membrane and an entrapment method using a noncharged membrane, were compared. With the adsorption method using the type HA membrane, MS2 could multiply even if its spiked amount was low (on the order of 100 PFU/sample), but the GA-like phage and Qβ could not multiply well. This indicates that most of GA-like phages and Qβ lost infectivity during the process or could not properly infect their host. Because the method using a GF/B membrane or AlCl3 as a cation solution also resulted in inadequate recovery of Qβ, the interactions of Qβ (and GA-like phage) with the cation solution and the electronegative membrane may cause inactivation of the phages. Consequently, the adsorption method is not preferable for recovering all FRNAPHs from a sample. In contrast, with the entrapment method, all tested FRNAPH strains (genotypes) could multiply even if their spiked amounts were low (on the order of 100 PFU/sample). Our concern regarding this method was that the coagulant might hamper the infection of host bacteria by FRNAPHs. If FRNAPHs are surrounded by other types of FRNAPHs or suspended solids during coagulation, they may be unable to infect. Thus, to facilitate FRNAPH infection, we spiked their host (S. Typhimurium WG49) simultaneously with the coagulant (AlCl3) into the sample. It was also expected that the formation of the FRNAPH-S. Typhimurium WG49 complex would make FRNAPH entrapment by the membrane more likely because the strain WG49 cells are larger than the membrane pore size (0.45 μm). All tested FRNAPH genotypes were also effectively recovered from the lake water samples, which contained suspended solids, even if a genotype was less predominant than other genotypes. Exceptionally, 1 out of 3 trials to detect less-predominant Qβ resulted in negative data. It is possible that the less-predominant Qβ could not multiply in the test tube due to competition to infect S. Typhimurium WG49 and/or surrounding suspended solids. Unequal distribution of Qβ in the stock solution is another possible explanation. Considering that Qβ in the stock solution might form aggregates (43) and was spiked at a low number (1.3 × 101 PFU), it is possible that Qβ was absent from the test tube. Thus, we can conclude that the entrapment method can make FRNAPHs infect their host even in the presence of suspended solids and that the method is potentially effective to recover all infectious FRNAPHs from environmental samples.
For the quantitative genotyping of FRNAPHs in the surface water samples, an IC–RT-PCR–MPN assay was performed in this study. For recovering FRNAPHs from 100 ml of a sample, the entrapment method was employed. In most samples, the total number of infectious FRNAPH genotypes was comparable to the number of infectious F+ coliphages determined by the conventional plaque assay. This indicates that the novel IC–RT-PCR–MPN-based quantification assay can quantify FRNAPH genotypes with accuracy that is comparable to that of the conventional assay. However, in some samples collected at F1 during summer months (June to September), the IC–RT-PCR–MPN assay resulted in negative data, even though the plaque assay indicated sufficiently high concentrations of F+ coliphages (>4.0 × 100 PFU/50 ml) (Fig. 2). Our plaque assay potentially detects not only FRNAPHs but also FDNAPHs. FDNAPHs have been reported to possess superior stability over other F+ coliphages under warm conditions and to be prevalent in surface water during the summer season (8, 14). Further, FDNAPHs are more prevalent among F+ coliphages in raw wastewater than in treated wastewater (6). Because F1 is a point supposedly impacted by insufficiently treated wastewater, it is likely that FDNAPHs were the predominant type of F+ coliphages at this point during the summer season. It may be possible to specifically detect FDNAPHs using IC-PCR. However, the detection of FDNAPHs was beyond the scope of this study. FDNAPHs are rod-shaped viruses, and thus, they are not considered good enteric viral indicators, unlike FRNAPHs.
In previous studies, typing of infectious FRNAPHs in environmental samples was conducted by plaque isolation, followed by RT-PCR or RLB (4, 5, 6, 13, 17, 24, 39, 40). These assays require an overnight incubation process for plaque formation, a plaque isolation and purification process, and a typing process using RT-PCR or RLB after RT-PCR. Besides, many isolates are required for quantification. In contrast, our IC–RT-PCR–MPN assay requires only overnight incubation and RT-PCR processes. Thus, our IC–RT-PCR–MPN assay enables less time-consuming and labor-intensive quantification and genotyping of infectious FRNAPHs. If multiplex RT-PCR is applied, the assay may be further simplified (31).
In the present study, surface water samples were collected from four different locations. K1, K2, and K3 are located on a river receiving treated municipal wastewater, and F1 receives not fully treated wastewater. At the locations receiving treated wastewater (K1 to K3), GI FRNAPHs tended to be predominant among the FRNAPH genotypes, while at the location receiving insufficiently treated wastewater (F1), GII FRNAPHs tended to be predominant. These trends are consistent with findings from previous studies targeting wastewater samples (5, 6, 7). For instance, our previous study showed that GII is the predominant FRNAPH genotype in influent wastewater, while GI is the predominant FRNAPH genotype in effluents (7). In the previous study, the reduction of GI FRNAPHs by wastewater treatments (0.5 log10) was significantly lower than the reduction of GII (2.0 log10) and GIII (3.4 log10) FRNAPHs (7). These observations suggest that GI and GII FRNAPHs are good indicators of viral contaminations originating from untreated wastewater and treated municipal wastewater, respectively. Considering that GIII FRNAPHs are more susceptible to wastewater treatments than GII FRNAPHs (7), the former may be a better indicator of the contamination of untreated wastewater.
The efficiency of recovery of GIV FRNAPHs by our entrapment method is unknown because the GIV FRNAPH strain was not used for the optimization of the method. However, in this study, GIV FRNAPHs were detected at a location where treated municipal wastewater is discharged (K2). Previous studies have suggested that GIV is a rare FRNAPH genotype in water environments where human waste is the main contamination source (4, 5, 6, 7, 19, 39, 44). Consistent with these previous studies, the GIV FRNAPH concentrations (on the order of 10−1 MPN/50 ml) were 3-log10 lower than those of the predominant GI FRNAPHs. Besides, our results suggest that GIV FRNAPHs were also effectively recovered by the entrapment method without inactivation. Further, as discussed above, even nonpredominant genotypes can be quantified by our assay.
When we compared the two FRNAPH genotype quantification assays, i.e., the conventional RT-qPCR and IC–RT-qPCR–MPN assays, the infectivity index (MPN/copies) for GI FRNAPHs tended to be above 0.0 log10 (Fig. 4). The number of infectious particles cannot be larger than that of a gene. Considering that the RNA extraction and RT-qPCR processes were supposedly conducted with sufficient efficiencies, the result is probably due to a low efficiency of recovery of GI FRNAPHs by the adsorption-elution method. Previous studies have mentioned that the recovery efficiency of a method depends on the virus type (45, 46), and thus, it is possible that the method was not effective for GI FRNAPHs in our samples.
At some sampling locations, the infective FRNAPH genotypes showed winter seasonality. We also found that the infectivity index of all FRNAPH genotypes tended to be higher during the winter season under an assumption that the recovery efficiencies of the adsorption-elution method are consistent for the samples. The low temperature and low sunlight intensity conditions during the winter season, which make viral infectivity more conserved (8, 47, 48), might have contributed to the results. Such tendencies are more clearly seen at F1. Another study (S. Hanamoto, unpublished data) has suggested that surface water at F1 is severely affected by sunlight exposure, particularly during the summer season, which is consistent with our observations.
In this study, we developed a method for recovering FRNAPHs without inactivation. The method, coupled with the MPN approach, was effective for the quantitative genotyping of infectious FRNAPHs in surface water. Our approach effectively characterized the FRNAPH genotypes in water environments regardless of their predominance, in terms of their occurrence and seasonality. The comparability of the data to those obtained by the conventional RT-qPCR assay makes it possible to characterize FRNAPH genotypes in terms of their stability in water.
ACKNOWLEDGMENTS
This work was supported by a grant-in-aid for JSPS Fellows (grant 14J03643) from the Japan Society for the Promotion of Science.
REFERENCES
- 1.IAWPRC Study Group on Health Related Water Microbiology. 1991. Bacteriophages as model viruses in water quality control. Water Res 25:529–545. doi: 10.1016/0043-1354(91)90126-B. [DOI] [Google Scholar]
- 2.Vergara GG, Goh SG, Rezaeinejad S, Chang SY, Sobsey MD, Gin KY. 2015. Evaluation of FRNA coliphages as indicators of human enteric viruses in a tropical urban freshwater catchment. Water Res 79:39–47. doi: 10.1016/j.watres.2015.04.022. [DOI] [PubMed] [Google Scholar]
- 3.Furuse K. 1987. Distribution of coliphages in the environment: general considerations, p 87–124. In Goyal SM, Gerba CP, Bitton G (ed), Phage ecology. John Wiley & Sons, New York, NY. [Google Scholar]
- 4.Haramoto E, Kitajima M, Katayama H, Asami M, Akiba M, Kunikane S. 2009. Application of real-time PCR assays to genotyping of F-specific phages in river water and sediments in Japan. Water Res 43:3759–3764. doi: 10.1016/j.watres.2009.05.043. [DOI] [PubMed] [Google Scholar]
- 5.Haramoto E, Otagiri M, Morita H, Kitajima M. 2012. Genogroup distribution of F-specific coliphages in wastewater and river water in the Kofu basin in Japan. Lett Appl Microbiol 54:367–373. doi: 10.1111/j.1472-765X.2012.03221.x. [DOI] [PubMed] [Google Scholar]
- 6.Haramoto E, Fujino S, Otagiri M. 2015. Distinct behaviors of infectious F-specific RNA coliphage genogroups at a wastewater treatment plant. Sci Total Environ 520:32–38. doi: 10.1016/j.scitotenv.2015.03.034. [DOI] [PubMed] [Google Scholar]
- 7.Hata A, Kitajima M, Katayama H. 2013. Occurrence and reduction of human viruses, F-specific RNA coliphage genogroups and microbial indicators at a full-scale wastewater treatment plant in Japan. J Appl Microbiol 114:545–554. doi: 10.1111/jam.12051. [DOI] [PubMed] [Google Scholar]
- 8.Long SC, Sobsey MD. 2004. A comparison of the survival of F+RNA and F+DNA coliphages in lake water microcosms. J Water Health 2:15–22. [PubMed] [Google Scholar]
- 9.Muniesa M, Payan A, Moce-Llivina L, Blanch AR, Jofre J. 2009. Differential persistence of F-specific RNA phage subgroups hinders their use as single tracers for faecal source tracking in surface water. Water Res 43:1559–1564. doi: 10.1016/j.watres.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 10.Schaper M, Durán AE, Jofre J. 2002. Comparative resistance of phage isolates of four genotypes of f-specific RNA bacteriophages to various inactivation processes. Appl Environ Microbiol 68:3702–3707. doi: 10.1128/AEM.68.8.3702-3707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaper M, Jofre J, Uys M, Grabow WO. 2002. Distribution of genotypes of F-specific RNA bacteriophages in human and non-human sources of faecal pollution in South Africa and Spain. J Appl Microbiol 92:657–667. doi: 10.1046/j.1365-2672.2002.01600.x. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Griffiths MW. 2013. Comparative persistence of subgroups of F-specific RNA phages in river water. Appl Environ Microbiol 79:4564–4567. doi: 10.1128/AEM.00612-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love DC, Vinjé J, Khalil SM, Murphy J, Lovelace GL, Sobsey MD. 2008. Evaluation of RT-PCR and reverse line blot hybridization for detection and genotyping F+ RNA coliphages from estuarine waters and molluscan shellfish. J Appl Microbiol 104:1203–1212. doi: 10.1111/j.1365-2672.2007.03646.x. [DOI] [PubMed] [Google Scholar]
- 14.Cole D, Long SC, Sobsey MD. 2003. Evaluation of F+ RNA and DNA coliphages as source-specific indicators of fecal contamination in surface waters. Appl Environ Microbiol 69:6507–6514. doi: 10.1128/AEM.69.11.6507-6514.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim MY, Kim JM, Ko G. 2010. Disinfection kinetics of murine norovirus using chlorine and chlorine dioxide. Water Res 44:3243–3351. doi: 10.1016/j.watres.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Pecson BM, Martin LV, Kohn T. 2009. Quantitative PCR for determining the infectivity of bacteriophage MS2 upon inactivation by heat, UV-B radiation, and singlet oxygen: advantages and limitations of an enzymatic treatment to reduce false-positive results. Appl Environ Microbiol 75:5544–5554. doi: 10.1128/AEM.00425-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentry-Shields J, Myers K, Pisanic N, Heaney C, Stewart J. 2015. Hepatitis E virus and coliphages in waters proximal to swine concentrated animal feeding operations. Sci Total Environ 505:487–493. doi: 10.1016/j.scitotenv.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langlet J, Ogorzaly L, Schrotter J-C, Machinal C, Gaboriaud F, Duval JFL, Gantzer C. 2009. Efficiency of MS2 phage and Qβ phage removal by membrane filtration in water treatment: applicability of real-time RT-PCR method. J Membr Sci 326:111–116. doi: 10.1016/j.memsci.2008.09.044. [DOI] [Google Scholar]
- 19.Ogorzaly L, Tissier A, Bertrand I, Maul A, Gantzer C. 2009. Relationship between F-specific RNA phage genogroups, faecal pollution indicators and human adenoviruses in river water. Water Res 43:1257–1264. doi: 10.1016/j.watres.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Ogorzaly L, Bertrand I, Paris M, Maul A, Gantzer C. 2010. Occurrence, survival, and persistence of human adenoviruses and F-specific RNA phages in raw groundwater. Appl Environ Microbiol 76:8019–8025. doi: 10.1128/AEM.00917-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravva SV, Sarreal CZ, Cooley MB. 2015. Male-specific coliphages for source tracking fecal contamination in surface waters and prevalence of Shiga-toxigenic Escherichia coli in a major produce production region of the Central Coast of California. Environ Sci Process Impacts 17:1249–1256. doi: 10.1039/C4EM00537F. [DOI] [PubMed] [Google Scholar]
- 22.Rezaeinejad S, Vergara GG, Woo CH, Lim TT, Sobsey MD, Gin KY. 2014. Surveillance of enteric viruses and coliphages in a tropical urban catchment. Water Res 58:122–131. doi: 10.1016/j.watres.2014.03.051. [DOI] [PubMed] [Google Scholar]
- 23.Hsu FC, Shieh YS, van Duin J, Beekwilder MJ, Sobsey MD. 1995. Genotyping male-specific RNA coliphages by hybridization with oligonucleotide probes. Appl Environ Microbiol 61:3960–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nappier SP, Aitken MD, Sobsey MD. 2006. Male-specific coliphages as indicators of thermal inactivation of pathogens in biosolids. Appl Environ Microbiol 72:2471–2475. doi: 10.1128/AEM.72.4.2471-2475.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katayama H, Shimasaki S, Ohgaki S. 2002. Development of a virus concentration method and its application to detection of enterovirus and Norwalk virus from coastal seawater. Appl Environ Microbiol 68:1033–1039. doi: 10.1128/AEM.68.3.1033-1039.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katayama H, Shimasaki S, Ohgaki S. 2002. Development of virus concentration method using negatively charged membrane by alkaline elution after acid rinse. J Jap Soc Water Environ 25:469–475. (In Japanese.) doi: 10.2965/jswe.25.469. [DOI] [Google Scholar]
- 27.Hata A, Katayama H, Kojima K, Sano S, Kasuga I, Kitajima M, Furumai H. 2014. Effects of rainfall events on the occurrence and detection efficiency of viruses in river water impacted by combined sewer overflows. Sci Total Environ 468-469: 757–763. [DOI] [PubMed] [Google Scholar]
- 28.Matsushita T, Shirasaki N, Tatsuki Y, Matsui Y. 2013. Investigating norovirus removal by microfiltration, ultrafiltration, and precoagulation-microfiltration processes using recombinant norovirus virus-like particles and real-time immuno-PCR. Water Res 47:5819–5827. doi: 10.1016/j.watres.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Shirasaki N, Matsushita T, Matsui Y, Urasaki Y, Kimura M, Ohno K. 2014. Virus removal by an in-line coagulation–ceramic microfiltration process with high-basicity polyaluminum coagulation pretreatment. Water Sci Technol Water Suppl 14:429–437. doi: 10.2166/ws.2013.218. [DOI] [Google Scholar]
- 30.Friedman SD, Cooper EM, Casanova L, Sobsey MD, Genthner FJ. 2009. A reverse transcription-PCR assay to distinguish the four genogroups of male-specific (F+) RNA coliphages. J Virol Methods 159:47–52. doi: 10.1016/j.jviromet.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 31.Wolf S, Hewitt J, Rivera-Aban M, Greening GE. 2008. Detection and characterization of F+ RNA bacteriophages in water and shellfish: application of a multiplex real-time reverse transcription PCR. J Virol Methods 149:123–128. doi: 10.1016/j.jviromet.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Mooijman KA, Bahar M, Muniesa M, Havelaar AH. 2002. Optimisation of ISO 10705-1 on enumeration of F-specific bacteriophages. J Virol Methods 103:129–136. doi: 10.1016/S0166-0934(02)00004-6. [DOI] [PubMed] [Google Scholar]
- 33.Blodgett R. 2010. Bacteriological analytical manual, appendix 2. Most probable number from serial dilutions. http://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm109656.htm Accessed 23 April 2016.
- 34.Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. 2004. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol 2(12):e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitajima M, Tohya Y, Matsubara K, Haramoto E, Utagawa E, Katayama H, Ohgaki S. 2008. Use of murine norovirus as a novel surrogate to evaluate resistance of human norovirus to free chlorine disinfection in drinking water supply system. Environ Eng Res 45:361–370. (In Japanese.) [Google Scholar]
- 36.Smeets PWMH, Medema GJ, van Dijk JC. 2009. The Dutch secret: how to provide safe drinking water without chlorine in the Netherlands. Drink Water Eng Sci 2:1–14. doi: 10.5194/dwes-2-1-2009. [DOI] [Google Scholar]
- 37.California Department of Public Health. 2014. Regulations related to recycled water. http://www.waterboards.ca.gov/drinking_water/certlic/drinkingwater/documents/lawbook/RWregulations_20140618.pdf Accessed 22 February 2016.
- 38.Sanz LA, Gawli BM. 2014. Water reuse in Europe, relevant guidelines, needs for and barriers to innovation. http://publications.jrc.ec.europa.eu/repository/handle/JRC92582 Accessed 22 February 2016.
- 39.Haramoto E, Kitajima M, Kishida N, Katayama H, Asami M, Akiba M. 2012. Occurrence of viruses and protozoa in drinking water sources of Japan and their relationship to indicator microorganisms. Food Environ Virol 4:93–101. doi: 10.1007/s12560-012-9082-0. [DOI] [PubMed] [Google Scholar]
- 40.Vinjé J, Oudejans SJ, Stewart JR, Sobsey MD, Long SC. 2004. Molecular detection and genotyping of male-specific coliphages by reverse transcription-PCR and reverse line blot hybridization. Appl Environ Microbiol 70:5996–6004. doi: 10.1128/AEM.70.10.5996-6004.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cashdollar JL, Brinkman NE, Griffin SM, McMinn BR, Rhodes ER, Varughese EA, Grimm AC, Parshionikar SU, Wymer L, Fout GS. 2013. Development and evaluation of EPA method 1615 for detection of enterovirus and norovirus in water. Appl Environ Microbiol 79:215–223. doi: 10.1128/AEM.02270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibbons CD, Rodríguez RA, Tallon L, Sobsey MD. 2010. Evaluation of positively charged alumina nanofibre cartridge filters for the primary concentration of noroviruses, adenoviruses and male-specific coliphages from seawater. J Appl Microbiol 109:635–641. doi: 10.1111/j.1365-2672.2010.04691.x. [DOI] [PubMed] [Google Scholar]
- 43.Langlet J, Gaboriaud F, Duval JF, Gantzer C. 2008. Aggregation and surface properties of F-specific RNA phages: implication for membrane filtration processes. Water Res 42:2769–2777. doi: 10.1016/j.watres.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Ogorzaly L, Gantzer C. 2006. Development of real-time RT-PCR methods for specific detection of F-specific RNA bacteriophage genogroups: application to urban raw wastewater. J Virol Methods 138:131–139. doi: 10.1016/j.jviromet.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Hamza IA, Jurzik L, Stang A, Sure K, Uberla K, Wilhelm M. 2009. Detection of human viruses in rivers of a densely populated area in Germany using a virus adsorption elution method optimized for PCR analyses. Water Res 43:2657–2668. doi: 10.1016/j.watres.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Victoria M, Guimarães F, Fumian T, Ferreira F, Vieira C, Leite J, Miagostovich M. 2009. Evaluation of an adsorption-elution method for detection of astrovirus and norovirus in environmental waters. J Virol Methods 156:73–76, 2009. doi: 10.1016/j.jviromet.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Bertrand I, Schijven JF, Sánchez G, Wyn-Jones P, Ottoson J, Morin T, Muscillo M, Verani M, Nasser A, de Roda Husman AM, Myrmel M, Sellwood J, Cook N, Gantzer C. 2012. The impact of temperature on the inactivation of enteric viruses in food and water: a review. J Appl Microbiol 112:1059–1074. doi: 10.1111/j.1365-2672.2012.05267.x. [DOI] [PubMed] [Google Scholar]
- 48.Love DC, Silverman A, Nelson KL. 2010. Human virus and bacteriophage inactivation in clear water by simulated sunlight compared to bacteriophage inactivation at a southern California beach. Environ Sci Technol 44:6965–6970. doi: 10.1021/es1001924. [DOI] [PubMed] [Google Scholar]