

ABSTRACT
Although Mn2+ is the most abundant substrate of versatile peroxidases (VPs), repression of Pleurotus ostreatus vp1 expression occurred in Mn2+-sufficient medium. This seems to be a biological contradiction. The aim of this study was to explore the mechanism of direct oxidation by VP1 under Mn2+-deficient conditions, as it was found to be the predominant enzyme during fungal growth in the presence of synthetic and natural substrates. The native VP1 was purified and characterized using three substrates, Mn2+, Orange II (OII), and Reactive Black 5 (RB5), each oxidized by a different active site in the enzyme. While the pH optimum for Mn2+ oxidation is 5, the optimum pH for direct oxidation of both dyes was found to be 3. Indeed, effective in vivo decolorization occurred in media without addition of Mn2+ only under acidic conditions. We have determined that Mn2+ inhibits in vitro the direct oxidation of both OII and RB5 while RB5 stabilizes both Mn2+ and OII oxidation. Furthermore, OII was found to inhibit the oxidation of both Mn2+ and RB5. In addition, we could demonstrate that VP1 can cleave OII in two different modes. Under Mn2+-mediated oxidation conditions, VP1 was able to cleave the azo bond only in asymmetric mode, while under the optimum conditions for direct oxidation (absence of Mn2+ at pH 3) both symmetric and asymmetric cleavages occurred. We concluded that the oxidation mechanism of aromatic compounds by VP1 is controlled by Mn2+ and pH levels both in the growth medium and in the reaction mixture.
IMPORTANCE VP1 is a member of the ligninolytic heme peroxidase gene family of the white rot fungus Pleurotus ostreatus and plays a fundamental role in biodegradation. This enzyme exhibits a versatile nature, as it can oxidize different substrates under altered environmental conditions. VPs are highly interesting enzymes due to the fact that they contain unique active sites that are responsible for direct oxidation of various aromatic compounds, including lignin, in addition to the well-known Mn2+ binding active site. This study demonstrates the limits of versatility of P. ostreatus VP1, which harbors multiple active sites, exhibiting a broad range of enzymatic activities, but they perform differently under distinct conditions. The versatility of P. ostreatus and its enzymes is an advantageous factor in the fungal ability to adapt to changing environments. This trait expands the possibilities for the potential utilization of P. ostreatus and other white rot fungi.
INTRODUCTION
Versatile peroxidases (VPs) from white rot fungi (WRF) such as Pleurotus ostreatus have received much attention in recent years due to their diverse ability to oxidize both low- and high-redox-potential compounds, including lignin, in both Mn2+-mediated and Mn2+-independent modes of action (1). VPs (EC 1.11.1.16) are ligninolytic enzymes and, together with manganese peroxidases (MnPs) (EC 1.11.1.13), comprise the ligninolytic heme peroxidase gene family of P. ostreatus (2). Mn2+ has a dual role in this gene family. First, it is an inducer of mnp genes or a repressor of vps transcription, and second, it is considered the best known substrate for both MnP and VP enzymes (3, 4). This implies that the role of Mn2+ in this system depends on the environmental conditions.
Degradation of lignin by P. ostreatus was shown to be enhanced by the addition of Mn2+, even though significant degradation can also occur under nonamended Mn2+ conditions (5). Therefore, most of the studies in this field were performed under Mn2+-amended conditions. In recent years, VPs were found to be the most frequently expressed genes in fungi growing on natural substrates as well as on synthetic media, and interest in their function has risen considerably (4, 6, 7). The potential of lignin-modifying enzymes to act in both Mn2+-mediated and Mn2+-independent modes of action is highly advantageous for niches where continuous decomposition occurs despite the variability of Mn2+ levels in the environment (8). Another factor whose variations in soil and wood environments can significantly affect enzymatic activity is the pH level. Therefore, the versatility of enzymes, and especially ligninolytic enzymes, to act in a broad range of pH and Mn2+ levels can be very beneficial for a fungus growing on lignocellulosic substrates. Ligninolytic enzymes that act in an Mn2+-independent mode are laccases (copper-containing oxidases), lignin peroxidases (LiPs) (EC1.11.1.14), and VPs. VPs are less common among the WRF and have been described mainly in Pleurotus, Bjerkandera, and a few other genera (9, 10). Since VPs have been discovered, they have been shown to have properties of both LiP and MnP and have been designated hybrid enzymes (11, 12). It is assumed that VPs have a function similar to that of LiPs, as the latter are not found in Pleurotus spp. VPs are capable of oxidizing Mn2+ via a mechanism similar to that described for MnPs (13). In addition, they directly oxidize low- and high-redox-potential substrates due to two additional active sites via a mechanism similar to that described for LiPs (1). The first is the exposed heme edge that provides VPs the capability to directly oxidize phenols, amines, and small dye compounds in the absence of Mn2+ (14). This site is typical of some other peroxidases, such as LiP, Coprinus cinereus peroxidase (CiP), and horseradish peroxidase (HRP) (EC 1.11.1.7) (14–17).
The second site for direct oxidation is the exposed tryptophanyl radical (Trp-164) that is located on the enzyme surface, as found in LiP (18). This site is responsible for the direct oxidation of low- and high-redox-potential compounds, including those that LiP can oxidize only in the presence of redox mediators (14, 18, 19). It was suggested that this site acts as a substrate intermediate protein radical center and initiates a long-range electron transfer (LRET) pathway leading to the heme (15, 20, 21). Recently, this mechanism was described as being responsible for lignin oxidation by VP (22). In glucose-peptone (GP) medium, all nine P. ostreatus peroxidases were found to be expressed. Mn2+ amendment to GP-grown cultures resulted in dramatic differences in the transcript abundance of the MnP/VP-encoding genes (4, 23). The vp1 gene was extremely repressed, while both mnp3 and mnp2 were highly induced (4). This fact poses an apparent biological contradiction, as Mn2+ is considered to be the classic substrate of both MnP and VP (12). In addition, as we have previously shown, in Mn2+-deficient GP (medium without addition of Mn2+), vp1 was the predominantly expressed gene in both trophophase and idiophase (7). This enzyme was found to play a key role in oxidation of dyes as well as aromatic substrates, in addition to Mn2+ (7), while under Mn2+ sufficient conditions, redundancy among the ligninolytic heme peroxidase genes has been demonstrated (4). In addition, when P. ostreatus was grown on a lignocellulosic substrate (poplar chips), VP1 was the most abundant protein in the secretome (6). Moreover, molecular phylogenetic analysis of peroxidases from different Pleurotus species revealed that P. ostreatus VP1 groups together with VPs from other Pleurotus spp., suggesting that VP1 is the predominant peroxidase in the genus Pleurotus (1). P. ostreatus VP1 and P. eryngii VPL2, the most intensively studied ligninolytic enzyme, share 96% identity in their sequences. The crystal structures of these enzymes have been solved (2, 14, 24). The current study focuses on the Mn2+-independent reactions with regard to the complex versatile nature of P. ostreatus VP1. We emphasized the effects of Mn2+ and pH levels on VP1 activity while addressing the potential ecological advantages of what seems to be the biological contradiction of Mn2+ function in the regulation of this enzyme.
MATERIALS AND METHODS
Fungal strains and growth conditions.
P. ostreatus monokaryon strain PC9 (Spanish Type Culture Collection accession number CECT20311), which is a protoclone derived by dedikaryotization of the commercial dikaryon strain N001 (Spanish Type Culture Collection accession number CECT20600), and its derivative, the homokaryon Δvp1 strain (carboxin resistant; designed by inactivation/knockout of the vp1 gene) (25), were used throughout this study. Fungal strains were grown and maintained in nonamended Mn2+ glucose-peptone (GP) medium as described previously (7). Mn2+ concentration in this medium was determined by atomic absorption spectroscopy and was found to be less than 0.1 μM.
When required, 1.5% (wt/vol) agar was added to the appropriate medium, as were the azo dyes Orange II (OII) and Reactive Black 5 (RB5) (Sigma-Aldrich), which were added to a final concentration of 100 mg/liter. The pH levels of the media during fungal growth on agar plates were measured using a Sentek P17 flat-head combination electrode (Sentek, Essex, United Kingdom).
The same dyes were used in the enzymatic assays.
Purification of VP1.
The culture filtrate of P. ostreatus was collected after 10 days of incubation on GP medium. The collected sample was saturated with ammonium sulfate (70%, pH 6.0) overnight with stirring at 4°C. The protein was precipitated by centrifugation (15,000 × g for 15 min at 4°C) using a Thermo Sorvall RC3C centrifuge (Wilmington, DE, USA). The supernatant was discarded (since no activity was measured in this fraction), and the precipitant was fluidized with water and dialyzed (overnight at 4°C with stirring) against 10 mM sodium acetate buffer, pH 4.5, using a dialysis bag consisting of a membrane with a pore size of 12,000 to 14,000 Da (Spectrum, Gardena, CA, USA). The resulting suspension was centrifuged (as described above) and filtered using a 0.45-μm-pore-size vacuum filter.
The dialyzed sample was resolved on a HiTrap Q anion exchange column (5 ml) (GE) using fast protein liquid chromatography (FPLC; Äkta, GE). Fractions of 5 ml were collected at a flow rate of 4 ml/min. Proteins were eluted using six steps of 0 to 100% of dialysis buffer with 1 M NaCl. Fractions containing VP activities were collected at the 12% step, pooled, and dialyzed with the same dialysis buffer using a centrifugal filter device with a cutoff of 10,000 Da (Millipore, Carrigtwohill, Co. Cork, Ireland). At the last purification step the dialyzed sample was loaded onto a Mono Q anion exchange column (1 ml) (GE). The enzyme was eluted using the dialysis buffer with a linear gradient of the same buffer containing 1 M NaCl. Fractions of 0.5 ml were collected at a flow rate of 1 ml/min. The fractions containing VP activities were collected, pooled, and dialyzed as described above and stored at −20°C.
The visible spectra (270 to 700 nm) of the purified VP1 (dissolved in sterile distilled water) were obtained using an Evolution 300 UV-visible spectrophotometer (Thermo Scientific, Madison, WI, USA). The molarity of the enzyme was estimated using the ε406 of this enzyme as reported previously (2).
Enzymatic activity assays.
Oxidation of phenol red in the absence and presence of 100 μM Mn2+ (added as MnSO4; Difco) (ε610 = 22.0 mM−1 cm−1) was determined in 20 mM sodium succinate buffer at pH 4.5. The reaction mixtures contained 0.01% (wt/vol) phenol red and were initiated by adding 100 μM H2O2. The total reaction volume was 1 ml. Activity in the absence of H2O2 was measured to establish specific peroxidase activity. Activity was deduced after the subtraction of the value corresponding to its corresponding reaction in the absence of H2O2. Activity was determined in a 1-cm cell using a BioMate 3 spectrophotometer (Thermo Scientific, Madison, WI, USA). An enzyme unit was defined as the amount of enzyme producing 1 μmol of product per min.
The standard enzyme activities of the purified VP1 were determined as follows (unless otherwise mentioned). Oxidation of 100 μM Mn2+ (added as MnSO4) was measured by monitoring the production of Mn3+-chelate. The Mn3+-tartrate complex (ε238 = 6.5 mM−1 cm−1) was determined in 100 mM sodium tartrate at pH 5. The oxidations of 25 μM RB5 (ε598 = 30.0 mM−1 cm−1) and 25 μM OII (ε483 = 18.2 mM−1 cm−1) were determined in 100 mM sodium tartrate at pH 3. The reactions were initiated by adding 100 μM H2O2. The enzymatic activity assays of the purified VP1 were conducted in a volume of 250 μl in microtiter plates using a Synergy HTX multimode microplate reader (BioTek, Winooski, VT, USA).
All enzymatic assays were carried out at 25°C using 0.05 μM VP1.
Protein concentration was determined using the Bio-Rad protein assay with bovine serum albumin as a standard.
Gel electrophoresis.
To estimate the purity level of the collected fractions, samples were separated on a NuPAGE 4 to 12% bis-Tris gel in MES (morpholineethanesulfonic acid)-SDS running buffer (Invitrogen). Only fractions with one band were collected and maintained at 4°C. The samples subsequently were analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS; as detailed below). Samples that contained the highest purity levels were pooled. The pooled sample was separated on SDS-PAGE under the aforementioned conditions.
In-gel proteolysis and mass spectrometry analysis.
The proteins in the gel were reduced with 2.8 mM dithiothreitol (DTT) (60°C for 30 min), modified with 8.8 mM iodoacetamide in 100 mM ammonium bicarbonate (in the dark at room temperature for 30 min), and digested in 10% acetonitrile and 10 mM ammonium bicarbonate with modified trypsin (Promega) at a 1:10 enzyme-to-substrate ratio overnight at 37°C.
The resulting tryptic peptides were resolved by reverse-phase chromatography on 0.075- by 200-mm fused silica capillaries (J&W) packed with Reprosil reverse-phase material (Maisch GmbH, Germany). The peptides were eluted with a linear 30-min gradient of 5% to 28% acetonitrile with 0.1% formic acid in water, 15-min gradient of 28% to 95% acetonitrile with 0.1% formic acid in water, and 15-min gradient at 95% acetonitrile with 0.1% formic acid in water at flow rates of 0.15 μl/min. Mass spectrometry was performed by a Q-Exactive plus mass spectrometer (QE, Thermo) in positive mode using repetitively full MS scan followed by high-energy collision dissociation (HCD) of the 10 most dominant ions selected from the first MS scan.
The mass spectrometry data were analyzed using Proteome Discoverer 1.4 software with the Sequest (Thermo) algorithm against the P. ostreatus PC9 database, which is available at the JGI website (http://genome.jgi-psf.org/PleosPC9_1). Identified peptides were filtered according to top rank, mass accuracy, a minimum of two peptides per protein, and 1% false discovery rate.
Thermal and pH stability.
Thermal stability of the enzyme was tested by incubating the purified enzyme (0.05 μM) in 10 mM tartrate buffer (pH 5 or pH 3 for testing Mn2+ oxidation and direct oxidations of both OII and RB5, respectively) at a temperature range of 25 to 70°C for 6 h. The residuals of the standard activities (as described above in the “Enzymatic activity assays” section) of VP1 on the three substrates were determined.
To study the pH stability, 0.05 μM VP1 was dissolved in Britton-Robinson (B&R) buffer at a pH range of 2 to 9 and kept at 4°C for different time periods. Residual standard activities of VP1 on the three substrates were determined after 6 and 24 h.
Top and pHop studies.
Optimum pHs (pHopt) for oxidation of Mn2+, OII, and RB5 were determined using 100 mM sodium tartrate buffer in a range of 2.5 to 7.0. The standard reactions carried out at 25°C at the different pHs. To determine the optimum temperature (Topt) for oxidation of the three substrates, the standard reactions were carried out at different temperatures in the range of 25 to 50°C.
Kinetic constants on selected substrates.
The steady-state enzyme kinetics of the purified VP1 was studied using H2O2, Mn2+, RB5, and OII as the substrates. Oxidation of Mn2+ (added as MnSO4) in the range of 10 to 500 μM was measured by monitoring the production of Mn3+-chelate in 100 mM sodium tartrate at pH 5. The oxidations of RB5 in the range of 2.5 to 35 μM and OII in the range of 2.5 to 100 μM were determined in 100 mM sodium tartrate at pH 3.
In the reactions of mixed substrates, the λmax of both relevant substrates was monitored. The reactions were initiated by adding 100 μM H2O2. In addition, Mn2+ oxidation was measured at different H2O2 concentrations (10 to 500 µM). A reaction without an enzyme was carried out as a control for each condition.
All enzymatic activity assays were conducted in a volume of 250 μl in microtiter plates using a Synergy HTX multimode microplate reader (BioTek, Winooski, VT, USA). Steady-state kinetic constants were calculated from the oxidation of increasing substrate concentrations at the defined range. The affinity Michaelis constant (Km) and maximal reaction velocity (Vmax) were calculated from Lineweaver-Burk plots. The enzyme catalytic constant (Kcat) was obtained by dividing the Vmax by the enzyme concentration. The catalytic efficiency value was calculated as Kcat/Km.
In the reaction mixtures 0.05 μM VP1 was used, except in the studies of RB5 oxidation, where 0.01 μM VP1 was used.
OII degradation product analysis.
Samples were analyzed on an LC-MS system, which consisted of a Dionex ultimate 3000 high-performance liquid chromatograph (HPLC) coupled to an LTQ Orbitrap discovery hybrid FT mass spectrometer equipped with an electrospray ionization source (Thermo Fisher Scientific Inc.). HPLC separations were carried out using an Acclaim rapid-separation LC C18 column (2.1 by 150 mm; 2.2-μm volume; Dionex). The mass spectrometer was operated in negative ionization mode (unless otherwise mentioned), and ion source parameters were the following: spray voltage, 3.5 kV; capillary temperature, 300°C; ion-transfer optic parameters were optimized using the automatic tune option; sheath gas rate (arbitrary units), 35; and auxiliary gas rate (arbitrary units), 15. Mass spectra were acquired in the m/z 150 to 2,000 range. The LC-MS system was controlled and data were analyzed using Chromeleon and Xcalibur software (Thermo Fisher Scientific Inc.).
RESULTS
VP1 purification.
P. ostreatus VP1 (JGI protein identifier 137757; JGI model name fjr_estExt_fgenesh1_kg.C_80157; PDB entry 4BLK_A) was purified from 10-day-old cultures using a protocol consisting of ammonium sulfate precipitation followed by two steps utilizing anion exchange columns. During the purification process we measured the oxidation of phenol red in the absence and presence of Mn2+ in the reaction mixture (Table 1). As VP exhibits both Mn2+-dependent and Mn2+-independent activities, only fractions in which an increase in the specific activity of both types of reactions was evident were used for further analysis. Specific activity of the purified enzyme for direct oxidation of phenol red was 12.33 U/mg, and it was 61.48 U/mg for Mn2+-mediated oxidation of phenol red. The purified enzyme was resolved as a single band by SDS-PAGE (4 to 12%) (Fig. 1). The visualized band was identified, by LC-MS, as VP1 with a purity level of 95% (calculated on the basis of peptide spectrum match [PMS] values). All 25 identified peptides (Table 2) cover more than 67% of the mature VP1. Peptides 1 and 19 are common to MnP3 and VP3, respectively, but the presence of these enzymes can be ruled out. Under these growth conditions the expression level of mnp3 is negligible (7), and the purification process should eliminate MnP3 even if it is produced. Peptides 20 and 21, which are unique to VP1, contain peptide 19. Moreover, technically, one peptide is not enough to identify a protein. Other than the 25 peptides (Table 2), no other MnP or VP peptides were identified within the sample. The purified enzyme displayed a typical spectrum with a high Reinheitszahl ratio (Rz; A406/A280) of 5.4 (Fig. 1).
TABLE 1.
Purification process for P. ostreatus VP1a
| Purification step | Vol (ml) | Protein concn (mg/ml) | Total protein (mg) | Activityb (U/ml) |
Total activity (U) |
Sp act (U/mg) |
Yield (%) |
Purification (fold) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Direct | Mn2+ mediated | Direct | Mn2+ mediated | Direct | Mn2+ mediated | Direct | Mn2+ mediated | Direct | Mn2+ mediated | ||||
| Crude | 1,340.00 | 0.07 | 93.80 | 0.13 | 0.76 | 177.85 | 1,018.40 | 1.90 | 10.86 | 100.00 | 100.00 | 1.00 | 1.00 |
| (NH4)2SO4c | 200.00 | 0.14 | 28.80 | 0.65 | 3.50 | 130.00 | 700.00 | 4.51 | 24.31 | 73.09 | 68.74 | 2.38 | 2.24 |
| HiTrap Q | 19.80 | 0.55 | 10.97 | 5.78 | 31.87 | 114.50 | 631.06 | 10.44 | 57.55 | 64.38 | 61.97 | 5.51 | 5.30 |
| Mono Q | 3.81 | 0.81 | 3.10 | 10.03 | 50.00 | 38.23 | 190.50 | 12.33 | 61.48 | 21.49 | 18.71 | 6.50 | 5.66 |
Direct oxidation or Mn2+-mediated oxidation was calculated from the oxidation values measured in the absence or presence of Mn2+ in the reaction mixtures (after excluding the activity in the absence of H2O2), respectively.
Enzyme activity values are based on direct oxidation (left) or Mn2+-mediated oxidation (right) of phenol red.
Residual fraction.
FIG 1.
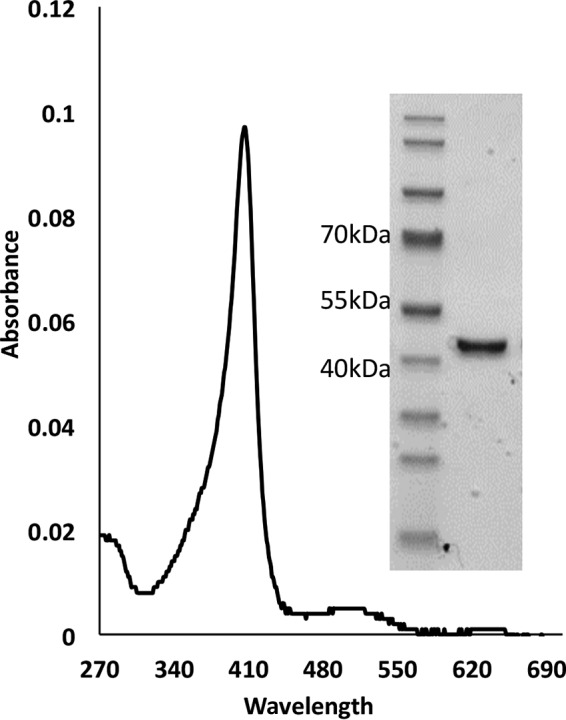
UV-vis spectra and SDS-PAGE of the purified P. ostreatus VP1. VP1 shows a typical UV-visible spectrum (270 to 700 nm) with a Soret peak at 406 nm and a high Rz value (A406/A280) of 5.4. (Inset) VP1 shows a single band on 4 to 12% SDS-PAGE in MES buffer. This band was identified by LC-MS/MS as VP1 with a high purity level of 95% and no evidence of other peroxidases.
TABLE 2.
Identified peptides of VP1
| Peptide no. | Sequence | Note |
|---|---|---|
| 1 | ATCADGR | Also in MnP3 |
| 2 | TANAACCVLFPILDDIQENLFDGAQCGEEVHESLR | Only in VP1 |
| 3 | HNISAGDFIQFAGAVGVSNCPGGVR | Only in VP1 |
| 4 | IPFFLGRPDAVAASPDHLVPEPFDSVDTILAR | Only in VP1 |
| 5 | MGDAGFSAVEVVWLLASHSIAAADK | Only in VP1 |
| 6 | VDPSIPGTPFDSTPGVFDSQFFIETQLK | Only in VP1 |
| 7 | GRLFPGTPDNK | Only in VP1 |
| 8 | GRLFPGTPDNKGEVQSPLQGEIR | Only in VP1 |
| 9 | LFPGTPDNK | Only in VP1 |
| 10 | LFPGTPDNKGEVQSPLQGEIR | Only in VP1 |
| 11 | LFPGTPDNKGEVQSPLQGEIRLQSDHLLAR | Only in VP1 |
| 12 | DPQTACEWQSMVNNQPK | Only in VP1 |
| 13 | DPQTACEWQSMVNNQPKIQNR | Only in VP1 |
| 14 | FAGTMSK | Only in VP1 |
| 15 | FAGTMSKMALLGQDK | Only in VP1 |
| 16 | FAGTMSKMALLGQDKSK | Only in VP1 |
| 17 | GEVQSPLQGEIR | Only in VP1 |
| 18 | GEVQSPLQGEIRLQSDHLLAR | Only in VP1 |
| 19 | LQSDHLLAR | Also in VP3 |
| 20 | LQSDHLLARDPQTACEWQSMVNNQPK | Only in VP1 |
| 21 | LQSDHLLARDPQTACEWQSMVNNQPKIQNR | Only in VP1 |
| 22 | IQNRFAGTMSK | Only in VP1 |
| 23 | MALLGQDK | Only in VP1 |
| 24 | MALLGQDKSK | Only in VP1 |
| 25 | MGDAGFSAVEVVWLLASHSIAAADK | Only in VP1 |
We selected three substrates to further characterize purified VP1, namely, Mn2+, RB5, and OII, each of which is known to be oxidized at a different site: the well-known Mn2+ binding and active site (13), the tryptophanyl radical (Trp164) active site (18), and the heme edge active site (14, 15), respectively.
Activities of VP1 are stable in a wide range of pH and temperature levels.
To study the pH stability of the purified VP1 during oxidation of the three selected substrates, the enzyme was incubated at a wide range of pH values (pH 2 to pH 9) at 4°C for 6 and 24 h. The oxidation of RB5 was more stable than that of Mn2+ and OII at all pH levels studied after 6 h of incubation, but it was more pronounced at the two extreme pHs, 2 and 9 (Fig. 2A). After 24 h, negligible values of oxidation activity were determined when any of the three substrates was used at the two extreme pHs (Fig. 2B), while stable activity levels of at least 80% were measured at the other pH levels.
FIG 2.
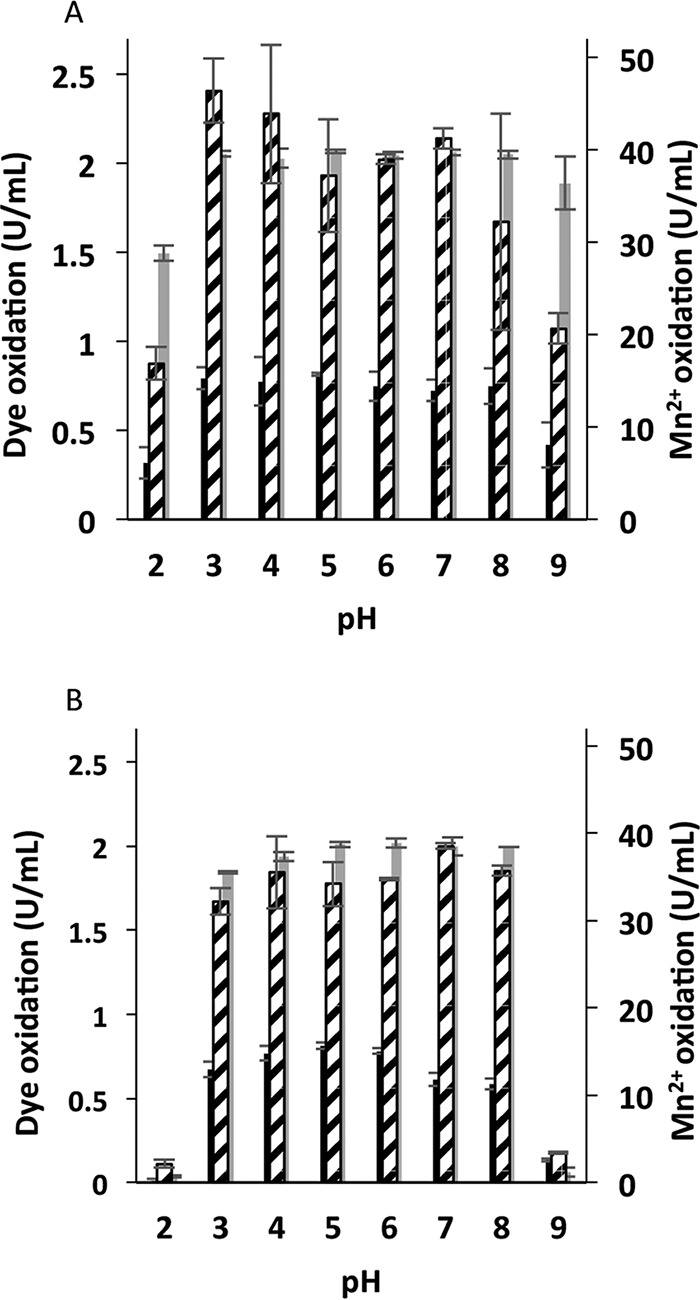
pH stability of P. ostreatus VP1. Residual activities after 6 (A) and 24 (B) h of incubation at 8 different pH levels in the range of 2 to 9 are shown. Activities of oxidation of Mn2+ (broken), OII (black), and RB5 (gray) are displayed in activity units (U/ml). These activities were determined after preincubation of the enzyme in 100 mM B&R buffer at a suitable pH level. Data represent averages from three biological replicates. Bars denote the standard deviations.
The thermal stability of the purified VP1 was studied by measuring the residual activities after incubation at a range of 25 to 70°C for 6 h (Fig. 3). Residual activity of roughly 100% was found after incubation at 25 to 55°C with all three substrates. Stability decreased gradually at temperatures of 60 to 70°C.
FIG 3.
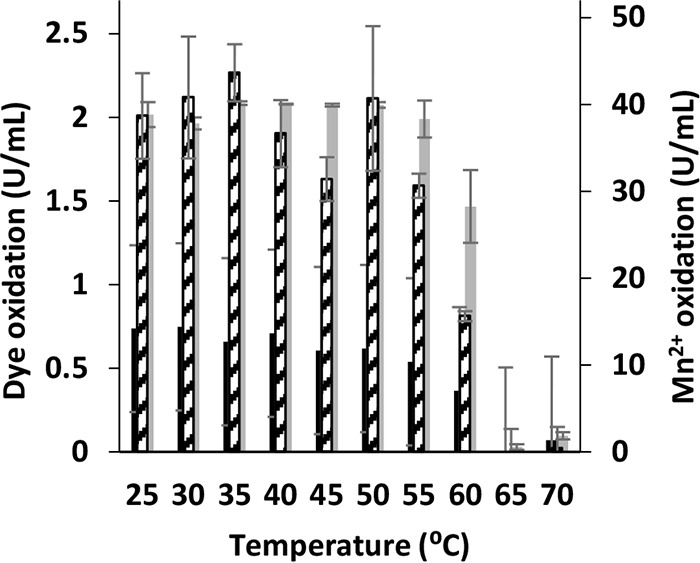
Thermal stability of P. ostreatus VP1. Residual activities after 6 h of incubation at 10 different temperatures in the range of 25 to 70°C are shown. Activities of oxidation of Mn2+ (broken), OII (black), and RB5 (gray) are displayed in activity units (U/ml). Activities were determined after preincubation of the enzyme at a suitable temperature. The enzyme was dissolved in 10 mM sodium tartrate buffer at pH 5 or pH 3 to determine Mn2+ oxidation or direct oxidations of the dyes, respectively. Data represent averages from three biological replicates. Bars denote the standard deviations.
pH optima: in vitro, in vivo, and “in-between.”
The pH optima (pHopt) of purified VP1 for oxidation of the three selected substrates were determined (Fig. 4). The pHopt for Mn2+ oxidation was found to be 5, while those for oxidation of RB5 and OII were found to be 3.5 and 3, respectively. Only at pH levels above 4 was Mn2+ oxidation evident. The high affinity to Mn2+ as a reducing substrate may be decreased at low pH levels. To determine whether the in vitro results reflect the fungal behavior in vivo, P. ostreatus was cultured on solid GP medium with initial pH levels of 3.5 and 6. Decolorizations of OII and RB5 were observed only in the more acidic medium, after 6 and 8 days of incubation, respectively, and became more dominant as incubation time progressed (Fig. 5A, upper). During the incubation period (20 days) the initial pH of the agar medium did not significantly change, and in most cases it stayed below 4 (when the initial pH was 3.5) and above 5.5 (when the initial pH was 6) (Fig. 5B). However, when the PC9 strain was grown on solid GP medium at an initial pH of 6, neither OII nor RB5 was decolorized, even after incubation periods of 30 days. It was concluded that as pure VP1 does not oxidize these dyes directly at pH 6, the same is happening in the fungal culture. Indeed, VP1 has a key role in the process both in vitro and in vivo. This conclusion was verified by conducting the same experiment with a Δvp1 strain which could not decolorize OII and RB5 at all pH levels (Fig. 5A, lower). Under liquid GP medium, decolorization was improved when the initial pH of the medium was 4.5 compared to that at pH 7 (4, 7). However, at an initial pH of 3.5 a significant inhibition in the fungal growth rate was observed. It is challenging to find conditions optimal for both in vivo and in vitro decolorization. Here, we demonstrated that VP1 is responsible for OII and RB5 oxidation and that pH 3.5 is the optimal pH for decolorization reactions both in vivo and in vitro.
FIG 4.
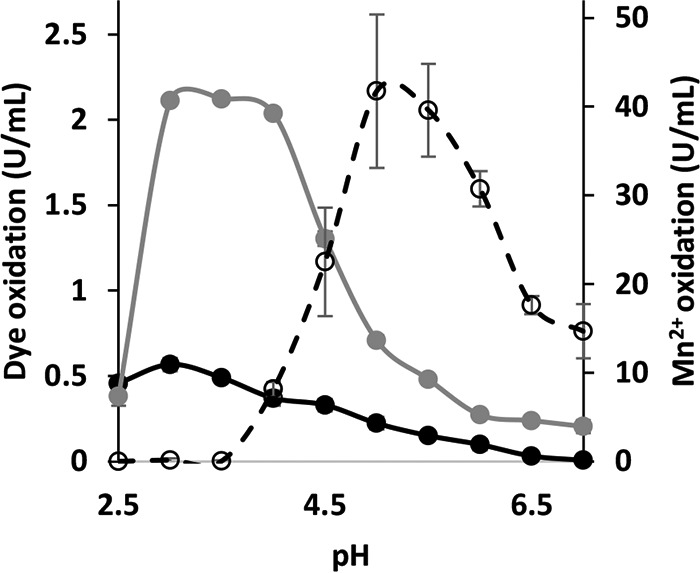
Optimum pH of P. ostreatus VP1 activities. The oxidations of Mn2+, OII, and RB5 were determined using 100 mM sodium tartrate buffer in a range of 2.5 to 7.0. Activities of oxidation of Mn2+ (broken), OII (black), and RB5 (gray) are displayed in activity units (U/ml). Data represent averages from three replicates. Bars denote the standard deviations.
FIG 5.
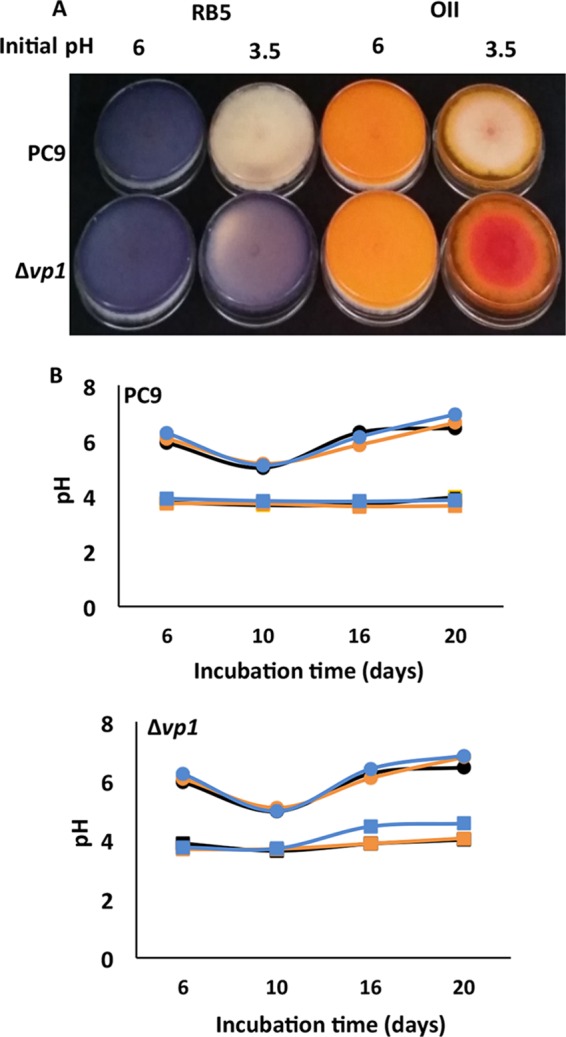
In vivo decolorizations occurred only under acidic pH in the absence of Mn2+. In vivo decolorization of both OII (right) and RB5 (left) (100 mg/liter) by PC9 (upper) and Δvp1 (lower) strains after 15 days of incubation is depicted. (A) The decolorizations were studied in solid GP medium (2% agar) under two different initial pH conditions, 6 and 3.5. (B) pH monitoring of the agar plates during the incubation period of PC9 (upper) and Δvp1 (lower) strains. GP media at initial pHs of 6 (circle) and 3.5 (square) without supplement of dye (black), with OII (orange), and with RB5 (blue) are shown. These represent at least three biological replicates.
The temperature range for activity was found to be wide, as no significant differences in the activity levels were observed when the reactions were carried out between 25 and 50°C.
Analysis of VP1 kinetics reveals interactions between the three catalytic sites.
To date, studies concerning the catalytic properties of VP enzymes have mostly focused on the examination of a single substrate at a time. To address the question of whether interactions between the three oxidation sites of VP1 exist, we compared the kinetic parameters (Km, Vmax, Kcat, and Km/Kcat) of reactions in various combinations of Mn2+, RB5, and OII (Table 3). We found that in some cases the presence of two substrates in the same reaction mixture can alter the efficiency of oxidation of one of the substrates. This is possible thanks to different pH optima. For example, the presence of Mn2+ inhibits the oxidation of RB5, although the oxidation of Mn2+ does not occur at pH 3 (Fig. 4). Conversely, the presence of RB5 in a Mn2+ oxidation reaction mixture improved the reaction efficiency (Kcat/Km).
TABLE 3.
Kinetic constants of P. ostreatus VP1
| Testing substrate | pH | Additional substrate | Km (μM) | Vmax (μmol/s) | Kcat (1/s) | Kcat/Km (1/s · mM) |
|---|---|---|---|---|---|---|
| H2O2a | 5 | 2 ± 10 | 0.03 ± 14.5 | 0.6 ± 290 | 5,961 ± 29,215 | |
| Mn2+ | 5 | 171 ± 9.6 | 0.3 ± 8.8 | 6.2 ± 176 | 80.3 ± 1,038 | |
| Mn2+ | 5 | 25 μM OII | 156 ± 1.7 | 0.2 ± 8.36 | 3.5 ± 167.2 | 28.3 ± 1,068 |
| Mn2+ | 5 | 50 μM OII, pH 5 | Complete inhibition | |||
| Mn2+ | 5 | 25 μM RB5 | 155 ± 8.1 | 7 ± 83.7 | 140 ± 1,674.4 | 1,485 ± 10,862 |
| Mn2+ | 5 | 50 μM RB5 | 55 ± 11.7 | 1.6 ± 8.3 | 31.6 ± 165.5 | 100 ± 2,746 |
| OII | 3 | 35.8 ± 1.8 | 0.04 ± 0.26 | 0.83 ± 5.1 | 15.7 ± 142.7 | |
| OII | 3 | 50 μM Mn2 | 58.7 ± 10 | 0.37 ± 0.05 | 7.3 ± 0.94 | 126.4 ± 12.4 |
| OII | 3 | 500 μM Mn2+ | Complete inhibition | |||
| OII | 3 | 25 μM RB5 | 210.7 ± 24 | 2 ± 0.14 | 40 ± 2.7 | 26.6 ± 202 |
| RB5 | 3 | 24.8 ± 5.2 | 0.54 ± 0.04 | 54 ± 3.5 | 2,224.4 ± 365 | |
| RB5 | 3 | 50 μM Mn2+ | 40.2 ± 7.8 | 0.65 ± 0.12 | 65.4 ± 12 | 1,631 ± 40 |
| RB5 | 3 | 500 μM Mn2 | Complete inhibition | |||
| RB5 | 3 | 25 μM OII | Complete inhibition |
In this reaction, Mn2+ was oxidized at a different concentration of H2O2.
When the oxidation of Mn2+ was studied, the presence of 25 μM OII did not significantly affect the reaction rate, but when the concentration of OII was doubled (to 50 μM), significant inhibition was observed (Fig. 6). It seems that the generated Mn3+-chelates do not function as oxidants of OII during the first 25 s at the conditions relevant for Mn2+ oxidation (pH 5), as OII (measured at 483 nm) is not decolorized.
FIG 6.
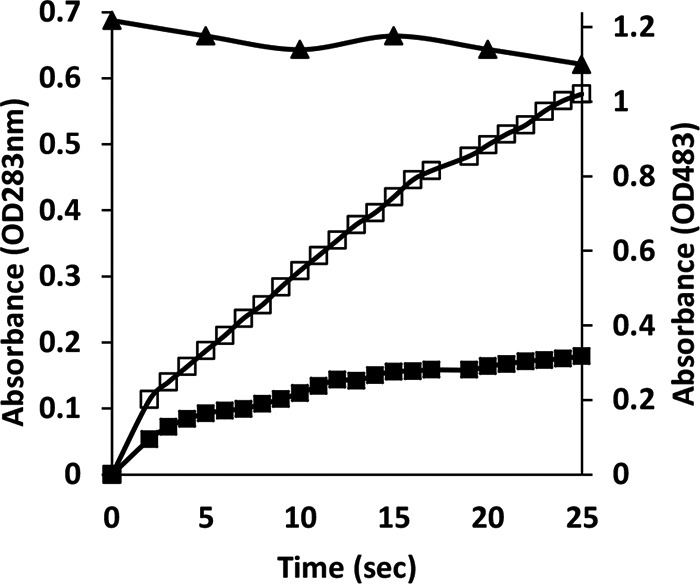
Orange II inhibits Mn2+ oxidation by P. ostreatus VP1. Oxidation of 100 μM Mn2+ (expressed in values corresponding to the optical density at 238 nm [OD238]) in the absence of OII (open black squares). In the presence of 50 μM OII, oxidation of Mn2+ (filled black squares) and OII (black triangles) was monitored with the same reaction mixture. The reaction was repeated three independent times.
When oxidation of OII was monitored, a substrate inhibition phenomenon was observed (Fig. 7A). In the presence of 0.05 mM Mn2+ this phenomenon was less pronounced and inhibition was observed (Fig. 7B and Table 3), resulting in complete inhibition in the presence of 0.5 mM Mn2+ (Table 3). In contrast, in the presence of RB5, the kinetic parameters of Mn2+ oxidation improved (Table 3).
FIG 7.

Effect of Mn2+ on Michaelis-Menten saturation curves of direct oxidation (OII or RB5) by VP1. Data represent oxidation of OII in the absence (A) and presence (B) of 0.05 μM Mn2+ and oxidation of RB5 in the absence (C) and presence (D) of 0.05 μM Mn2+. Each reaction was repeated three independent times.
In the case of RB5 oxidation, the substrate inhibition phenomenon was also observed, similar to that described in the case of OII (Fig. 7C). The presence of 0.05 mM Mn2+ also resulted in inhibition (Fig. 7D and Table 3), and when the Mn2+ concentration was increased to 0.5 mM, the reactions were completely inhibited (Table 3). RB5 oxidation was also completely abolished in the presence of OII in the reaction mixture (Table 3) concomitantly with oxidation of OII.
The mechanisms of direct and Mn2+-mediated OII degradation by VP1: two options of dye cleavage.
To better understand the effects of pH and Mn2+ on OII oxidation mechanisms by VP1, transformation products (TPs) produced under four different reaction conditions were identified by LC-MS. These included (i) absence of Mn2+ at pH 3, representing optimal direct oxidation, (ii) the presence of Mn2+ at pH 5, representing optimal Mn2+-mediated oxidation, as well as the absence (iii) or presence (iv) of Mn2+ at pH 4.5, a point where both activities are at a median level (Fig. 4). In general, the direct oxidation at pH 3 was found to be more efficient. Under these conditions, VP1 degraded 60% of OII (50 μM) within the first minute and up to 90% within 6 min. Six TPs were identified in reactions carried out in the absence of Mn2+ at pH 3, and their nature indicates the occurrence of both symmetric and asymmetric cleavages of the azo bond (Table 4 and Fig. 8). In contrast, in the presence of Mn2+ at pH 5, four TPs (three of them common to both reaction types) were identified, indicating that only asymmetric cleavage near the naphthalene moiety (Table 5) had occurred. To verify that this is not a consequence of more advanced oxidation of OII, we conducted a time course experiment (data not shown). The kinetics of TP formation clearly showed that 3-aminonaphthalene-2,6-dione (product no. 7), which resulted from symmetric cleavage, had accumulated during reaction i and was not identified in reaction ii (Fig. 9). From these results we concluded that in the presence of Mn2+ at pH 5, OII oxidation occurs only via one mechanism.
TABLE 4.
Identified OII transformation products resulting from VP1 activity in the absence of Mn2+ in the reaction mixturea
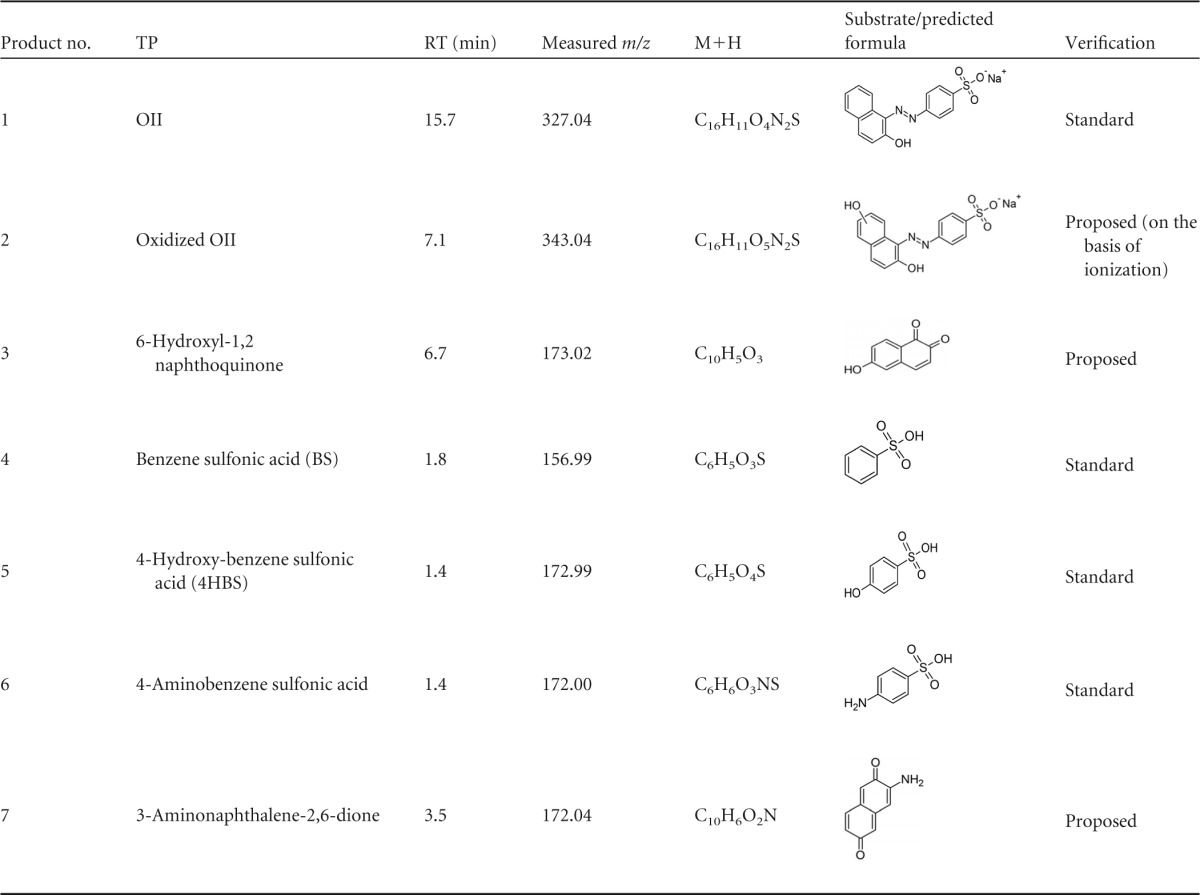
TPs were identified using negative electrospray ionization (nESI) mode. RT, retention time.
FIG 8.

Proposed pathway for OII oxidation by purified P. ostreatus VP1 in the absence of Mn2+. The reaction mixtures contained sodium tartrate, pH 3, 50 μM OII, 0.05 μM enzyme, and 0.1 mM H2O2. Samples were analyzed after 1, 5, 10, and 20 min of incubation. All of the transformation products (TPs) were identified in all samples but in different relative amounts (data not shown). Under these conditions, evidence of asymmetric and symmetric cleavages were found. The brackets indicate TPs with proposed structures (the TPs are numbered as described in Table 4).
TABLE 5.
Identified OII transformation products resulting from VP1 activity in the presence of Mn2+ in reaction mixturea
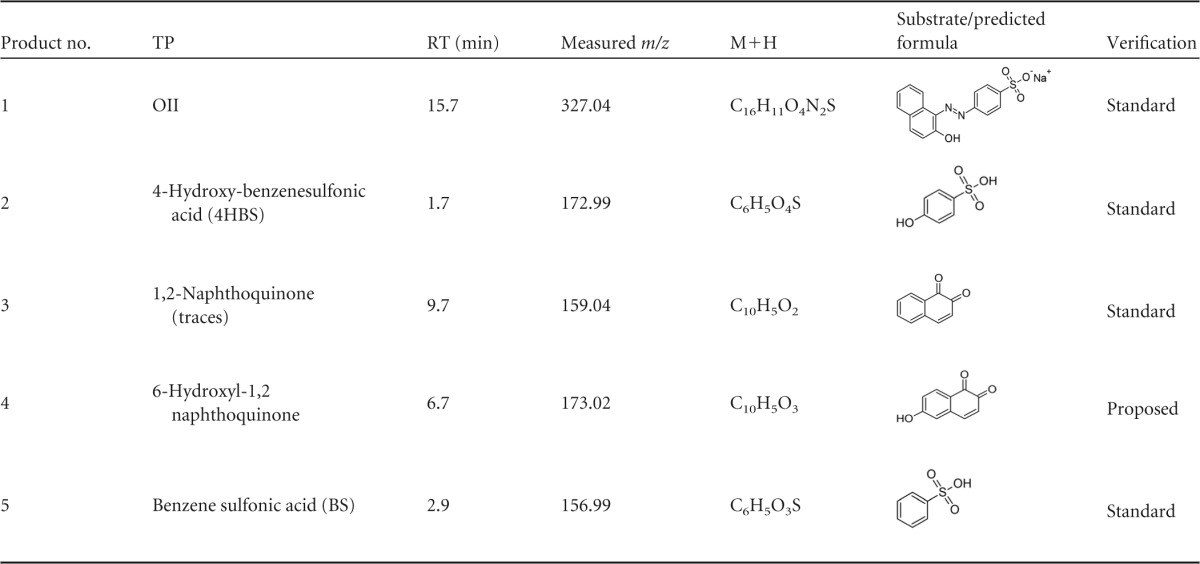
TPs were identified using nESI mode except for the 1,2-naphthoquinone which identified only using positive electrospray ionization (pESI).
FIG 9.

OII transformation product (TP) accumulation during in vitro reaction with purified P. ostreatus VP1 in the absence of Mn2+. The TPs are numbered as described in Table 4. (A) TP3, 6-hydroxyl-1,2 naphthoquinone; (B) TP4, BS (B); (C) TP5, 4HBS; (D) TP7, 3-aminonaphthalene-2,6-dione.
When OII was oxidized either in the absence or presence of Mn2+ at pH 4.5 (reactions iii and iv, respectively), the four TPs which were identified were the same as those in the presence of Mn2+ at pH 5 (reaction ii). These TPs can be produced only by asymmetric cleavage, demonstrating that in addition to Mn2+ deficiency, acidic pH is also required for the symmetric cleavage mechanism (Table 5).
DISCUSSION
During the past decade, significant progress was made in utilizing the heterologous expression of VP enzymes followed by a refolding step of the produced proteins, mainly in Escherichia coli (2, 26). This facilitated the study of structure-function relationships and the role of specific amino acids in the proteins (2, 22, 27). Nonetheless, the heterologously produced enzymes still do not precisely exhibit all the activity-related properties observed in the native enzymes (6). Some of these differences can be attributed to insufficient posttranslational modifications, such as glycosylation and phosphorylation, in the prokaryotic expression systems. Our goal was to study the functionality of VP1 under conditions of Mn2+ deficiency both in vivo and in vitro. We therefore focused on the use of the native enzyme as produced by the WRF. Our previous results showed that VP1 is the main ligninolytic enzyme responsible for the oxidation of aromatic substrates in GP medium (7). Here, we purified the native enzyme and characterized the reaction mechanisms from GP medium. Three substrates, Mn2+, RB5, and OII, were studied, representing oxidation by the three sites of the enzyme: (i) the well-known Mn2+ binding and active site (13), (ii) the tryptophanyl radical (Trp164) active site (18), and (iii) the heme edge active site (14), respectively. We studied the interactions among these sites and determined that Mn2+ inhibited the direct oxidation of both RB5 and OII, even though this reaction occurred at a very low pH, when Mn2+ oxidation was undetectable. Similar results were demonstrated with VP from Bjerkandera adjusta (28). OII was found to inhibit the oxidation of Mn2+ and RB5, while RB5 acts as an activator in the oxidation of OII and Mn2+. Taken together, and based on the almost complete sequence identity along with the highly similar three-dimensional structures of the heterologous VPs (2, 14, 24), we concluded that the Mn2+ binding and active site and the heme edge active site are very close to each other and to the heme access channel of the H2O2 molecule. In addition, the Trp-164 active site is located 180° from the Mn2+ active site. Hence, the presence of Mn2+ or OII can block the access of H2O2 that is required for the shuttling of electrons and by doing so can interfere with the activities. At the same time, the presence of RB5 in the system can stabilize the structure and the oxidation by the other active sites.
In addition, we could demonstrate that VP1 can cleave OII in two different modes. Under Mn2+ oxidation optimum conditions, VP1 was able to cleave the azo-bond form only in an asymmetric mode, while under the optimum conditions for direct oxidation (absence of Mn2+ at pH 3) both symmetric and asymmetric cleavages occurred. Moreover, we have shown here that our findings under in vitro conditions correspond to those observed in vivo. Only under very acidic conditions was the direct oxidation of both OII and RB5 detected. By using the Δvp1 strain, we showed that VP1 is indeed responsible for these activities. The biological significance of these results is that Mn2+ is a substrate (at pH 5) of VP1 and also inhibits the direct oxidation of aromatic substrates (at pH 3). These results shed light on the aforementioned biological contradiction and may explain the repression of vp1 expression in Mn2+-sufficient medium, where a number of mnp genes are induced and the enzymes react in a Mn2+-mediated mode (4).
As VPs have features of both MnPs and LiPs, they seem to share at least some regulatory features, as suggested for Phanerochaete chrysosporium. In this model fungus, in an ambient atmosphere, both MnP and LiP expression require starving culture conditions. In addition to that, the presence of Mn2+ is required for MnP expression, and Mn2+ deficiency is required for LiP expression (29, 30). The fact that the optimum pH of direct oxidations of the most robust ligninolytic enzymes, LiP and VP, is around 3 and is different from that of MnP (about 5) calls for an explanation, as they cannot act in concert. It was suggested that white rot fungi secrete oxalic and other organic acids to chelate Mn3+ and overcome the fact that Mn3+ generated by MnP is quite unstable in aqueous media (31, 32). This creates an environmental pH of around 5, which is optimal for Mn2+ oxidation. The optimum pH for direct oxidation by VP and LiP is lower, an important characteristic for reaching high redox potential, which enables the direct oxidation of nonphenolic aromatic substrates (33).
The different pH optima for these enzymes may have environmental significance. Fujii et al. showed that the humus (FH) layer is more acidic than the overlying litter (L) layer (34). In addition, Mn concentration in the FH layer was lower than that in the L layer. It was suggested that the HF layer is optimal (i.e., low pH and low Mn level) for LiP activity (34). Another example is the case of brown rot fungi that excrete oxalate to considerably lower the pH, especially in the immediate vicinity of fungal hyphae. This serves as a reduction agent of iron via a nonenzymatic decay pathway by the Fenton reaction (35, 36). If this also occurs in the case of WRF, it would be expected that very close to the fungal hyphae the pH will be considerably lower than 5, and it will still be acidic, albeit higher, at some distance. A biological explanation for these findings could be that different ligninolytic enzymes (such as MnP, VP, LiP, and laccase) with different pH optima can act simultaneously. In the case of VP, the adaptability of the enzyme allows adjustment to the pH conditions in the environment. Recently, Saez-Jimenez et al. reported on the ability of VP from P. eryngii to oxidize water-soluble lignins. It was concluded that the main reaction implies direct electron transfer to a surface Trp-164 residue responsible for lignin oxidation (22), and the highest activity was at pH 3.
Taken together, the results of this study demonstrate the limits of the versatility of P. ostreatus VP1, which harbors multiple active sites, exhibiting a broad range of enzymatic activities. In many cases, a broad range can imply overlapping activities, yet as shown here, in the case of VP1 the broad range provides the capacity to catalyze oxidative reactions under a broad spectrum of environmental conditions, such as pH and Mn2+ levels.
We propose that under certain conditions (acidic pH), not only is Mn2+ not oxidized by VP1 but it also inhibits the capacity of this enzyme to directly oxidize aromatic substances.
The versatility of P. ostreatus and its enzymes is an advantageous factor in the fungal ability to adapt to changing environments. This trait expands the possibilities for the potential utilization of P. ostreatus and other WRF.
ACKNOWLEDGMENTS
We thank Julius Ben-Ari for his help with the identification of OII TPs. We are also grateful to the Smoler Proteomics Center at the Technion for their help with the in-gel proteolysis and mass spectrometry analyses.
This research was partially supported by the Israel Science Foundation.
REFERENCES
- 1.Knop D, Yarden O, Hadar Y. 2015. The ligninolytic peroxidases in the genus Pleurotus: divergence in activities, expression, and potential applications. Appl Microbiol Biotechnol 99:1025–1038. doi: 10.1007/s00253-014-6256-8. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Fueyo E, Ruiz-Duenas FJ, Jesus Martinez M, Romero A, Hammel KE, Javier Medrano F, Martinez AT. 2014. Ligninolytic peroxidase genes in the oyster mushroom genome: heterologous expression, molecular structure, catalytic and stability properties, and lignin-degrading ability. Biotechnol Biofuels 7:2. doi: 10.1186/1754-6834-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen R, Yarden O, Hadar Y. 2002. Lignocellulose affects Mn2+ regulation of peroxidase transcript levels in solid-state cultures of Pleurotus ostreatus. Appl Environ Microbiol 68:3156–3158. doi: 10.1128/AEM.68.6.3156-3158.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salame TM, Yarden O, Hadar Y. 2010. Pleurotus ostreatus manganese-dependent peroxidase silencing impairs decolourization of Orange II. Microb Biotechnol 3:93–106. doi: 10.1111/j.1751-7915.2009.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerem Z, Hadar Y. 1995. Effect of manganese on preferential degradation of lignin by Pleurotus ostreatus during solid-state fermentation. Appl Environ Microbiol 61:3057–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez-Fueyo E, Castanera R, Ruiz-Duenas FJ, Lopez-Lucendo MF, Ramirez L, Pisabarro AG, Martinez AT. 2014. Ligninolytic peroxidase gene expression by Pleurotus ostreatus: differential regulation in lignocellulose medium and effect of temperature and pH. Fungal Genet Biol 72:150–161. doi: 10.1016/j.fgb.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Knop D, Ben-Ari J, Salame TM, Levinson D, Yarden O, Hadar Y. 2014. Mn2+-deficiency reveals a key role for the Pleurotus ostreatus versatile peroxidase (VP4) in oxidation of aromatic compounds. Appl Microbiol Biotechnol 98:6795–6804. doi: 10.1007/s00253-014-5689-4. [DOI] [PubMed] [Google Scholar]
- 8.Vandecasteele B, Willekens K, Zwertvaegher A, Degrande L, Tack FMG, Du Laing G. 2013. Effect of composting on the Cd, Zn and Mn content and fractionation in feedstock mixtures with wood chips from a short-rotation coppice and bark. Waste Manag 33:2195–2203. doi: 10.1016/j.wasman.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 9.Carabajal M, Kellner H, Levin L, Jehmlich N, Hofrichter M, Ullrich R. 2013. The secretome of Trametes versicolor grown on tomato juice medium and purification of the secreted oxidoreductases including a versatile peroxidase. J Biotechnol 168:15–23. doi: 10.1016/j.jbiotec.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Fueyo E, Ruiz-Duenas FJ, Miki Y, Martinez MJ, Hammel KE, Martinez AT. 2012. Lignin-degrading peroxidases from genome of selective ligninolytic fungus Ceriporiopsis subvermispora. J Biol Chem 287:16903–16916. doi: 10.1074/jbc.M112.356378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camarero S, Sarkar S, Ruiz-Duenas FJ, Martinez MJ, Martinez AT. 1999. Description of a versatile peroxidase involved in the natural degradation of lignin that has both manganese peroxidase and lignin peroxidase substrate interaction sites. J Biol Chem 274:10324–10330. doi: 10.1074/jbc.274.15.10324. [DOI] [PubMed] [Google Scholar]
- 12.Martinez MJ, Ruiz-Duenas FJ, Guillen F, Martinez AT. 1996. Purification and catalytic properties of two manganese peroxidase isoenzymes from Pleurotus eryngii. Eur J Biochem 237:424–432. doi: 10.1111/j.1432-1033.1996.0424k.x. [DOI] [PubMed] [Google Scholar]
- 13.Ruiz-Duenas FJ, Morales M, Perez-Boada M, Choinowski T, Martinez MJ, Piontek K, Martinez AT. 2007. Manganese oxidation site in Pleurotus eryngii versatile peroxidase: a site-directed mutagenesis, kinetic, and crystallographic study. Biochemistry 46:66–77. doi: 10.1021/bi061542h. [DOI] [PubMed] [Google Scholar]
- 14.Morales M, Mate MJ, Romero A, Jesus Martinez M, Martinez AT, Ruiz-Duenas FJ. 2012. Two oxidation sites for low redox potential substrates: a direct mutagenesis, kinetic, and, crystallographic study on Pleurotus eryngii versatile peroxidase. J Biol Chem 287:41053–41067. doi: 10.1074/jbc.M112.405548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doyle WA, Blodig W, Veitch NC, Piontek K, Smith AT. 1998. Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry 37:15097–15105. doi: 10.1021/bi981633h. [DOI] [PubMed] [Google Scholar]
- 16.Hofrichter M, Ullrich R, Pecyna MJ, Liers C, Lundell T. 2010. New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol 87:871–897. doi: 10.1007/s00253-010-2633-0. [DOI] [PubMed] [Google Scholar]
- 17.Smith AT, Veitch NC. 1998. Substrate binding and catalysis in heme peroxidases. Curr Opin Chem Biol 2:269–278. doi: 10.1016/S1367-5931(98)80069-0. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Boada M, Ruiz-Duenas FJ, Pogni R, Basosi R, Choinowski T, Martinez MJ, Piontek K, Martinez AT. 2005. Versatile peroxidase oxidation of high redox potential aromatic compounds: site-directed mutagenesis, spectroscopic and crystallographic investigation of three long-range electron transfer pathways. J Mol Biol 354:385–402. doi: 10.1016/j.jmb.2005.09.047. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz-Duenas FJ, Morales M, Mate MJ, Romero A, Marinez MJ, Smith AT, Martinez AT. 2008. Site-directed mutagenesis of the catalytic tryptophan environment in Pleurotus eryngii versatile peroxidase. Biochemistry 47:1685–1695. doi: 10.1021/bi7020298. [DOI] [PubMed] [Google Scholar]
- 20.Blodig W, Smith AT, Doyle WA, Piontek K. 2001. Crystal structures of pristine and oxidatively processed lignin peroxidase expressed in Escherichia coli and of the W171F variant that eliminates the redox active tryptophan 171. Implications for the reaction mechanism. J Mol Biol 305:851–861. [DOI] [PubMed] [Google Scholar]
- 21.Smith AT, Doyle WA, Dorlet P, Ivancich A. 2009. Spectroscopic evidence for an engineered, catalytically active Trp radical that creates the unique reactivity of lignin peroxidase. Proc Natl Acad Sci U S A 106:16084–16089. doi: 10.1073/pnas.0904535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saez-Jimenez V, Baratto MC, Pogni R, Rencoret J, Gutierrez A, Ignacio Santos J, Martinez AT, Javier Ruiz-Duenas F. 2015. Demonstration of lignin-to-peroxidase direct electron transfer a transient-state kinetics, directed mutagenesis, EPR and NMR study. J Biol Chem 290:23201–23213. doi: 10.1074/jbc.M115.665919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen R, Hadar Y, Yarden O. 2001. Transcript and activity levels of different Pleurotus ostreatus peroxidases are differentially affected by Mn2+. Environ Microbiol 3:312–322. doi: 10.1046/j.1462-2920.2001.00197.x. [DOI] [PubMed] [Google Scholar]
- 24.Ruiz-Duenas FJ, Morales M, Garcia E, Miki Y, Martinez MJ, Martinez AT. 2009. Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J Exp Bot 60:441–452. doi: 10.1093/jxb/ern261. [DOI] [PubMed] [Google Scholar]
- 25.Salame TM, Knop D, Tal D, Levinson D, Yarden O, Hadar Y. 2012. Predominance of a versatile-peroxidase-encoding gene, mnp4, as demonstrated by gene replacement via a gene targeting system for Pleurotus ostreatus. Appl Environ Microbiol 78:5341–5352. doi: 10.1128/AEM.01234-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Boada M, Doyle WA, Ruiz-Duenas FJ, Martinez MJ, Martinez AT, Smith AT. 2002. Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimisation of in vitro folding. Enzyme Microb Technol 30:518–524. doi: 10.1016/S0141-0229(02)00008-X. [DOI] [Google Scholar]
- 27.Saez-Jimenez V, Fernandez-Fueyo E, Javier Medrano F, Romero A, Martinez AT, Ruiz-Duenas FJ. 2015. Improving the pH-stability of versatile peroxidase by comparative structural analysis with a naturally-stable manganese peroxidase. PLoS One 10:e0140984. doi: 10.1371/journal.pone.0140984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ertan H, Siddiqui KS, Muenchhoff J, Charlton T, Cavicchioli R. 2012. Kinetic and thermodynamic characterization of the functional properties of a hybrid versatile peroxidase using isothermal titration calorimetry: insight into manganese peroxidase activation and lignin peroxidase inhibition. Biochimie 94:1221–1231. doi: 10.1016/j.biochi.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 29.Brown JA, Glenn JK, Gold MH. 1990. Manganese regulates expression of manganese peroxidase by Phanerochaete chrysosporium. J Bacteriol 172:3125–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothschild N, Levkowitz A, Hadar Y, Dosoretz CG. 1999. Manganese deficiency can replace high oxygen levels needed for lignin peroxidase formation by Phanerochaete chrysosporium. Appl Environ Microbiol 65:483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kishi K, Wariishi H, Marquez L, Dunford HB, Gold MH. 1994. Mechanism of manganese peroxidase compound-II reduction–effect of organic-acid chelators and pH. Biochemistry 33:8694–8701. doi: 10.1021/bi00195a010. [DOI] [PubMed] [Google Scholar]
- 32.Kuan IC, Tien M. 1993. Stimulation of Mn-peroxidase activity–a possible role for oxalate in lignin biodegradation. Proc Natl Acad Sci U S A 90:1242–1246. doi: 10.1073/pnas.90.4.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong DWS. 2009. Structure and action mechanism of ligninolytic enzymes. Appl Biochem Biotechnol 157:174–209. doi: 10.1007/s12010-008-8279-z. [DOI] [PubMed] [Google Scholar]
- 34.Fujii K, Uemura M, Hayakawa C, Funakawa S, Kosaki T. 2013. Environmental control of lignin peroxidase, manganese peroxidase, and laccase activities in forest floor layers in humid Asia. Soil Biol Biochem 57:109–115. doi: 10.1016/j.soilbio.2012.07.007. [DOI] [Google Scholar]
- 35.Arantes V, Goodell B. 2014. Current understanding of brown-rot fungal biodegradation mechanisms: a review, p 3–21. In Schultz TP, Goodell B, Nicholas DD (ed), Deterioration and protection of sustainable biomaterials. ACS Symposium Series, no. 1158. American Chemical Society, Washington, DC. [Google Scholar]
- 36.Arantes V, Milagres AMF, Filley TR, Goodell B. 2011. Lignocellulosic polysaccharides and lignin degradation by wood decay fungi: the relevance of nonenzymatic Fenton-based reactions. J Indust Microbiol Biotechnol 38:541–555. doi: 10.1007/s10295-010-0798-2. [DOI] [PubMed] [Google Scholar]