

ABSTRACT
Campylobacteriosis is one of the most important foodborne diseases worldwide and a significant health burden in New Zealand. Campylobacter jejuni is the predominant species worldwide, accounting for approximately 90% of human cases, followed by Campylobacter coli. Most studies in New Zealand have focused on C. jejuni; hence, the impact of C. coli strains on human health is not well understood. The aim of this study was to genotype C. coli isolates collected in the Manawatu region of New Zealand from clinical cases, fresh poultry meat, ruminant feces, and environmental water sources, between 2005 and 2014, to study their population structure and estimate the contribution of each source to the burden of human disease. Campylobacter isolates were identified by PCR and typed by multilocus sequence typing. C. coli accounted for 2.9% (n = 47/1,601) of Campylobacter isolates from human clinical cases, 9.6% (n = 108/1,123) from poultry, 13.4% (n = 49/364) from ruminants, and 6.4% (n = 11/171) from water. Molecular subtyping revealed 27 different sequence types (STs), of which 18 belonged to clonal complex ST-828. ST-1581 was the most prevalent C. coli sequence type isolated from both human cases (n = 12/47) and poultry (n = 44/110). When classified using cladistics, all sequence types belonged to clade 1 except ST-7774, which belonged to clade 2. ST-854, ST-1590, and ST-4009 were isolated only from human cases and fresh poultry, while ST-3232 was isolated only from human cases and ruminant sources. Modeling indicated ruminants and poultry as the main sources of C. coli human infection.
IMPORTANCE We performed a molecular epidemiological study of Campylobacter coli infection in New Zealand, one of few such studies globally. This study analyzed the population genetic structure of the bacterium and included a probabilistic source attribution model covering different animal and water sources. The results are discussed in a global context.
INTRODUCTION
The genus Campylobacter currently includes 26 species and 11 subspecies (1). Campylobacter jejuni is the predominant species isolated from cases of campylobacteriosis worldwide, followed by Campylobacter coli (2–4). In 2006, New Zealand had the highest annual campylobacteriosis notification rate in the world, with >380 cases per 100,000 population (5). In 2014, the rate was 150 cases per 100,000 population, and, despite the observed 60% decrease compared to the 2006 rate, campylobacteriosis remained the most commonly notified gastrointestinal infection, accounting for ∼45% of all notifications (6). In comparison, campylobacteriosis was the third most important bacterial foodborne disease in the United States (7), with an incidence of laboratory-confirmed cases of 13.5 cases per 100,000 population in 2014 (8). Estimates in the United States indicate that C. coli accounts for ∼9% of campylobacteriosis cases (9). Considering an estimated incidence of ∼845,000 cases of campylobacteriosis per year (7), the incidence of C. coli infections therefore amounts to ∼84,000 cases per year in the United States alone.
Most epidemiological studies of campylobacteriosis that applied bacterial identification in New Zealand focused on C. jejuni (10, 11). Consumption of chicken meat is considered the main risk factor for C. jejuni infections in New Zealand and in a number of other countries (10, 12). However, there is little information about the sources and risk factors for C. coli infections. Extrapolating source attribution data from C. jejuni to C. coli is problematic, as different Campylobacter species may have different eco-epidemiological features and infection risk factors may also differ (13). For instance, in a case-control study in the Netherlands (14), the main risk factor for C. jejuni enteritis was consumption of chicken meat, whereas C. coli infections had different risk factors: swimming in open water or sea and consumption of game and tripe. Molecular subtyping by means of multilocus sequence typing (MLST) plays an important role in measuring the contributions of different infection sources to the burden of human disease caused by C. jejuni, and the same MLST scheme can be applied to C. coli (15). MLST was applied to C. coli isolates from humans and several other animal sources and water in studies in Scotland (16), Denmark (17), and Switzerland (12). Sheppard et al. (18, 19) carried out phylogenetic analyses of C. coli from Scotland using concatenated sequences obtained by MLST and showed that C. coli could be divided into three distinct clades. All the C. coli isolates from human clinical cases belonged to clade 1, which was dominated by strains isolated from farm animal sources, whereas clades 2 and 3 were mainly composed of environmental water strains. Hence, unlike the case-control study in the Netherlands (where environmental water sources appeared to be a major source of C. coli infection), this study indicated a major role of animal sources in the transmission of C. coli to humans in Scotland.
Most cases of human campylobacteriosis in New Zealand are attributable to C. jejuni. However, the relative contribution of C. coli and the source of these infections had not been previously examined. Therefore, the objectives of this study were, first, to assess the relative contribution of C. coli to campylobacteriosis morbidity by identifying to the species level 1,601 Campylobacter clinical isolates collected between 2005 and 2014 from human cases in one New Zealand region and, second, to analyze the genetic relatedness of the clinical C. coli isolates to isolates obtained from poultry meat, farmed ruminants, and environmental water sources in the same study area and time frame using MLST, in order to infer possible transmission pathways of this important pathogen.
MATERIALS AND METHODS
Isolates and molecular typing. (i) Collection of human clinical Campylobacter spp. and identification of C. coli.
The study used Campylobacter spp. isolated from human feces in the Manawatu Health District of New Zealand between March 2005 and December 2014. The region had a population of ∼223,000 and its main city, Palmerston North, had a population of ∼80,000 (20, 21). All the human fecal specimens that were submitted to the main medical laboratory (MedLab Central, Palmerston North) during the sampling period and tested positive for Campylobacter spp. by enzyme-linked immunosorbent assay (ELISA; ProSpecT, Remel, USA) were delivered for bacterial culture to the Molecular Epidemiology and Public Health laboratory (mEpiLab) of Massey University (MU). The feces were delivered weekly on Amies charcoal transport swabs (Copan, Brescia, Italy). Fecal swabs were first streaked on modified cefoperazone charcoal deoxycholate agar (mCCDA) plates (Fort Richard, Auckland, New Zealand) and then immersed in 3 ml of Bolton broth (BB) (Lab M, Bury, United Kingdom) in a loose-capped bijou bottle and incubated at 42°C microaerobically (85% N2, 10% CO2, and 5% O2) for 48 h (MACS-VA500 microaerophilic workstation; Don Whitley Scientific, West Yorkshire, United Kingdom). A single colony resembling Campylobacter spp. on mCCDA was subcultured onto Columbia horse blood agar (BA) (Fort Richard, Auckland, New Zealand) and incubated microaerophilically at 42°C for 48 h. If the mCCDA plate was negative for colonies resembling Campylobacter spp., a subculture was made from BB onto another mCCDA plate and the plate was incubated in a microaerobic atmosphere as described above. Pure cultures were frozen in 15% glycerol broth (Oxoid, United Kingdom) at −80°C for future reference.
Campylobacter identification to species level was done by molecular methods. Briefly, DNA was extracted by boiling a freshly grown culture of frozen isolates for 10 min in 1 ml of 2% Chelex (Bio-Rad, USA) in sterile Milli-Q water, followed by centrifugation (12,470 × g for 3 min) to remove cell debris and the Chelex. The supernatant was transferred to a fresh tube and used for PCR for species confirmation and multilocus sequence typing. Isolates were first identified through the use of the mapA gene for the detection of C. jejuni (22), employing the following primers: mapA-F (5′-CTTGGCTTGAAATTTGCTTG-3′) and mapA-R (5′-GCTTGGTGCGGATTGTAAA-3′). All the non-jejuni isolates were tested by a Campylobacter genus-specific PCR using the following primers: C412-F (5′-GGATGACACTTTTCGGAGC-3′) and C1288-R (5′-CATTGTAGCACGTGTGTC-3′) (23). Subsequently, all the non-jejuni Campylobacter spp. were subject to a C. coli species-specific PCR for detection of the ceuE gene (24). Primers were ceuE-F (5′-AATTGAAAATTGCTCCAACTATG-3′) and ceuE-R (5′-TGATTTTATTATTTGTAGCAGCG-3′). Amplification was performed in a 20-μl reaction volume containing 0.2 μl of Platinum Taq polymerase (Invitrogen, USA), 2 μl of 10× PCR buffer (Invitrogen), 1 μl of deoxynucleoside triphosphate (dNTP; Bioline, United Kingdom) (2 mM), 0.6 μl of MgCl2, 2 μl of each forward and reverse primer (2 pmol/μl), 6.2 μl of H2O, and 2 μl of DNA. The reactions were carried out in a thermocycler (SensoQuest, Germany) with denaturation at 96°C for 2 min, followed by 35 cycles of 96°C for 30 s, 58°C for 30 s, and 72°C for 30 s and a final extension of 72°C for 2 min. Electrophoresis in a 1% agarose gel plus staining with ethidium bromide was used to visualize PCR products. The presence of an ∼462-bp product indicated C. coli (25).
(ii) Collection of poultry meat samples.
Over the same sampling period (2005 to 2014), fresh whole poultry carcasses (chicken and turkey samples) from different poultry suppliers were sampled each month from supermarkets in Palmerston North. Carcasses were washed and massaged in 200 ml of sterile buffered peptone water (BPW) (Difco, USA). The liquid from the wash was centrifuged (16,000 × g and 4°C for 30 min) and the resultant pellet resuspended in 5 ml of BPW. This suspension was added to 90 ml of BB and incubated microaerophilically as described above, after which it was subcultured onto mCCDA and incubated for 2 days. Two colonies resembling Campylobacter spp. on mCCDA were subcultured to separate BA and incubated microaerophilically at 42°C for 2 days, and pure cultures were frozen at −80°C. Identification of C. jejuni and C. coli proceeded as for the human clinical isolates.
(iii) Collection of ruminant and water samples.
Water samples were filtered through a sterile 0.45-μm filter (Millipore, USA), and the filter was transferred to 20 ml of BB and incubated at 42°C microaerobically for 2 days; then it was subcultured onto mCCDA and incubated for 2 days. A single colony resembling Campylobacter spp. on mCCDA was subcultured onto BA and incubated microaerophilically at 42°C for 2 days. Eleven C. coli isolates from environmental water (recreational waterways and pretreatment drinking water; nine from Manawatu-Tararua area and two [ST-3302 and ST-7774] from the North Island of New Zealand between 2006 and 2014) and 49 C. coli isolates from ruminant feces (nine different farms in the Manawatu-Tararua area between 2006 and 2011) were collected as part of the same long-term monitoring program. These were kept frozen in our laboratory for future reference and were also included in this study.
Isolates from pig meat were not included in the study due to the low prevalence of Campylobacter organisms from this source (more than 650 pig meat samples were previously collected, from which only one C. coli isolate was identified).
C. coli subtyping by MLST.
All C. coli isolates were subjected to MLST using seven housekeeping genes based on the method described by Dingle et al. (26). Each amplification reaction was performed in a 20-μl reaction volume containing 2 μl of the DNA preparation, 5 pmol of both forward and reverse amplification primers, and 14 μl of the PCR master mix (200 μl of 10× PCR buffer, 100 μl of dNTP, 60 μl of MgCl2, 20 μl of Platinum Taq polymerase, and 1,020 μl of H2O). Sequencing reactions were performed using 2 μl of the PCR product, 3.2 pmol of forward primer, 1 μl of ABI BigDye (Applied Biosystems, USA), 2 μl of 5× buffer, and H2O to a total volume of 10 μl. Products were sequenced at the Institute of Environmental Science and Research (ESR, Porirua, New Zealand) on an ABI 3130XL automated DNA sequencer using ABI BigDye v3.1.
Sequence data were collated and submitted for allele and sequence type (ST) designation with their corresponding clonal complex (CC) online using the Campylobacter PubMLST database (http://pubmlst.org/campylobacter/). Isolates yielding novel STs or alleles that did not give clear sequences were reamplified and sequenced using the same protocol. If the sequence products were not readable, they were resequenced with the reverse primer and, if this failed, the locus was reamplified and resequenced. New MLSTs were submitted to the online database.
Data analysis. (i) Assessment of C. coli population structure and differentiation.
The relatedness between human, poultry, ruminant, and environmental water C. coli isolates was assessed using multiple methods. Minimum spanning trees were implemented to visualize allelic differences between STs of isolates from the different sources using the pairwise Hamming distance matrix. The trees were calculated by Prim's algorithm (27) as implemented in the Bionumerics software (Applied Maths).
A maximum likelihood (ML) method based on the Kimura 2-parameter model was used to infer the evolutionary history (28) using the concatenated DNA sequences composed of 3,309 nucleotide positions across the seven loci. Initial trees for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms (29) to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach. Previous phylogenetic work done in Scotland identified 3 C. coli clades of closely related STs associated with different sources (30); we examined the position of our STs in the same clades. Therefore, the ML method was implemented using 36 nucleotide sequences representing the different STs found in this study (n = 27) with the addition of 9 STs taken at random (4 STs from clade 2 [ST-3304, ST-3122, ST-3180, and ST-3182] and 5 STs from clade 3 [ST-2485, ST-2681, ST-3109, ST-3123, and ST-3124]) from two clades described in Scotland and not present in our study. Clade 1 had 10 shared STs with the Scottish study, so no additional STs were sampled from this clade. These analyses were conducted using MEGA6 software (31).
A different approach to assess genetic relatedness between C. coli utilized permutational multivariate analysis of variance (PERMANOVA) (32, 33). This analysis was implemented using PERMANOVA+, an “add in” to the PRIMER 6 software (34). Finally, the genetic diversities of C. coli from the different sources were compared using Simpson's and Shannon's diversity indices and their 95% bootstrap credible intervals (CrIs) (calculated using PAST software, version 2.17c) (35) and rarefaction analysis (performed using the package ‘vegan’ in R, version 3.1.3) (36).
(ii) C. coli source attribution.
Although human-to-human spread may be implicated in some outbreaks (37), we assume that human-to-human spread as a cause of sporadic cases is negligible in common with other source attribution studies and therefore do not consider anthroponotic spread (11, 30). The relative contributions of different C. coli sources to the human disease burden were estimated using a number of tests. The similarity between the frequency distribution of human C. coli STs and those of the different sources was estimated using proportional similarity indices (PSI) and their bootstrap CrIs, as previously described (38). The PSI measures the area of intersection between two frequency distributions (39) and ranges between 0 and 1, where 0 indicates no similarity and 1 indicates identical frequency distributions. The PSI is calculated by the equation PSI = 1 − 0.5 ∑i | pi − qi ∣ = ∑i min (pi,qi), where pi and qi are the proportions of strains that belong to type i out of all strains typed from sources P and Q (40). Calculations were performed using R, version 3.1.3.
The asymmetric island model was used to probabilistically assign each human isolate to one of the source populations (poultry, ruminants, or environmental water) (41).
RESULTS
Proportion of C. coli in ELISA-positive fecal specimens and ST diversity.
A total of 2,009 Campylobacter ELISA-positive human fecal specimens were submitted by the medical laboratory for culture to mEpiLab between March 2005 and December 2014, and 1,601 (∼80%) were Campylobacter positive by culture. The dominant species was C. jejuni, accounting for 1,552/1,601 (∼96%) isolates, followed by C. coli, accounting for 47/1,601 isolates (∼2.9%). Two isolates (<0.2%) were not identified as either C. jejuni or C. coli and were not further analyzed. The annual numbers of C. coli isolates were 9 (2005), 6 (2006), 2 (2007), 6 (2008), 0 (2009), 4 (2010), 2 (2011), 6 (2012), 7 (2013), and 5 (2014). A total of 1,123 Campylobacter isolates were isolated from poultry (chicken, n = 1,074; turkey, n = 49, of which 6 were C. coli) meat samples. C. jejuni accounted for 980/1,123 (87.3%) and C. coli for 108/1,123 (9.6%) of the isolates. The Campylobacter species remained unidentified in 29/1,123 (<3%) isolates. The proportion of C. coli was significantly higher in the poultry meat samples than in human feces (chi-square, P < 0.01).
Two hundred fifteen C. coli isolates from all sources were subtyped by MLST (47 from human feces, 49 from ruminant feces [32 sheep feces and 17 cattle feces], 108 from poultry meat, and 11 from environmental water). There were 27 different C. coli STs. Eighteen STs (66%) belonged to the ST-828 CC, which was the predominant CC in our data and accounted for 54% (116/215) of the isolates. The remaining STs could not be classified into any known CC. Three previously unidentified STs (ST-7767, ST-7774, and ST-7776) were detected. ST-7767 and ST-7776 were isolated from poultry and ST-7774 from environmental water. The most prevalent sequence type, ST-1581, which was identified in 60/215 (28%) of the isolates (12 from human feces, 44 from poultry meat, 4 from ruminant feces, and 0 from water), is not currently assigned to any CC. The second most prevalent ST was ST-3072 (25/215 [∼11.6%]), and the third most prevalent STs were ST-3222 and ST-2397 (with 21 isolates each). These were not predominant STs in other countries (12, 16, 17, 30).
C. coli population structure and differentiation between sources.
The minimum spanning tree visualizing the ST clusters is shown in Fig. 1. Except for the environmental water isolates, the diagram shows no distinctive partition of C. coli STs between the sources, with most of the highly abundant STs represented in at least three sources. The diagram also shows a large cluster of closely related STs. However, four environmental water C. coli STs (ST-7774, ST-3302, ST-3301, and ST-1243) appear dissociated from this cluster, and two of these STs (ST-3301 and ST-3302) are single-locus variants.
FIG 1.

Minimum spanning tree of C. coli STs from 4 different sources. Each node represents an ST; its size is proportional to the frequency of isolation and the colors represent the different source types. The thickness of the connecting lines is proportional to the similarities between STs, with the thickest connector linking single-locus variants. The shaded area represents members of the ST-828 CC.
Ten out of 27 STs found in this study were previously reported by Sheppard et al. in Scotland (30) and belong to clade 1. Hence, in order to compare the phylogenetic relatedness of C. coli from New Zealand and Scotland, we generated an ML diagram with the addition of 9 Scottish STs belonging to clades 2 and 3 but not found in our study (Fig. 2). Interestingly, 26/27 of the New Zealand STs clustered within the Scottish clade 1, only one ST of an environmental water C. coli clustered in clade 2 (ST-7774), and no STs clustered in clade 3 (Fig. 2). The frequencies of the different STs along with their clade designation are provided in supplemental material (see Table S1).
FIG 2.
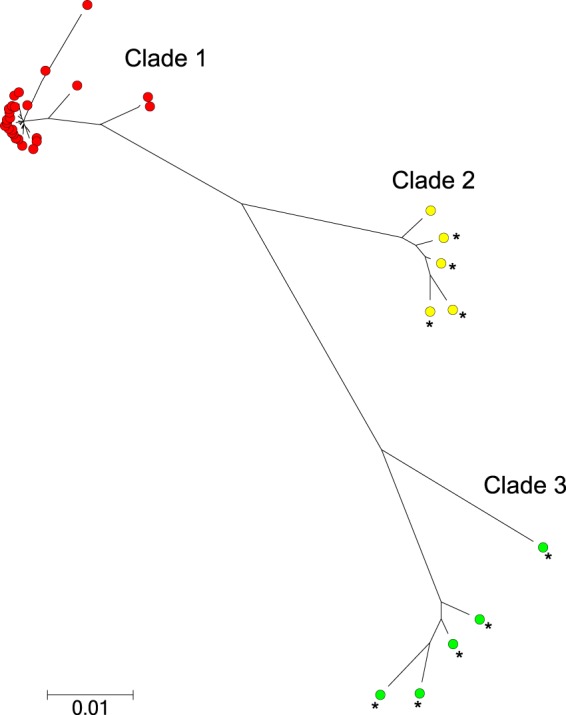
Molecular phylogenetic analysis by maximum likelihood method. The 27 STs found in this study and 9 STs from clades 2 and 3 from Sheppard et al. (30) were used. Clade 1 is indicated in red, clade 2 in yellow, and clade 3 in green. C. coli STs in the work of Sheppard et al. (30) are indicated with an asterisk. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The scale bar represents a genetic distance of 0.01 (i.e., 1% of the nucleotides differ).
PERMANOVA was used to formally test for significant population differentiation, comparing mean pairwise Hamming distances of C. coli isolates from different sources. The pseudo-F statistic was 8.92 (P = 0.001), indicating that at least one of the populations differed from the other three. A nonmetric multidimensional scaling (NMDS) plot was used to assess which populations differed (see Fig. S1 in the supplemental material). NMDS together with the minimum spanning tree indicated that most C. coli isolates from water sources were differentiated from the strains circulating in the other sources, although this was not obvious from the results of the maximum likelihood phylogenetic analysis.
Source attribution for human infections.
The proportional similarity indices (PSI) and their 95% CrIs are reported in Table 1. The PSI between ruminant and human C. coli isolates (PSI = 0.50; 95% CrI = 0.31 to 0.59) was similar to that observed between poultry and human isolates (PSI = 0.46; 95% CrI = 0.30 to 0.56), and the 95% CrI overlapped. However, in relation to human C. coli, the ruminant and poultry PSI were significantly greater than PSI obtained with environmental water C. coli (PSI = 0.10; 95% CrI = 0 to 0.17).
TABLE 1.
Numerical indices measuring diversities, PSI, and asymmetric island model in C. coli
| Source | Value for index (95% CrI) |
|||
|---|---|---|---|---|
| PSI | Asymmetric island model output | Simpson's index | Shannon's index | |
| Humans | NAa | NA | 0.87 (0.79–0.89) | 2.32 (1.89–2.41) |
| Ruminants | 0.50 (0.31–0.59) | 0.55 (0.31–0.79) | 0.79 (0.69–0.84) | 1.88 (1.49–2.03) |
| Poultry | 0.46 (0.30–0.56) | 0.38 (0.16–0.58) | 0.78 (0.70–0.83) | 1.93 (1.66–2.06) |
| Environmental water | 0.10 (0.00–0.17) | 0.07 (0.00–0.24) | 0.81 (0.56–0.83) | 1.80 (0.99–1.85) |
NA, not available.
The asymmetric island model results were consistent with the PSI (Table 1), where ruminants and poultry were identified by both as the main sources for human infection. The model attributed 55% (95% CrI = 31 to 79%) and 38% (95% CrI = 16 to 58%) of the infections to ruminants and poultry sources, respectively, but the 95% CrIs of the estimated contributions of these sources overlapped widely, whereas the environmental water source accounted for only 7% (95% CrI = 0 to 24%) of infections, and the CrI overlapped only slightly with that of the ruminant source. As a whole, these results attributed the majority of the infections to ruminant and poultry sources, whereas exposure to environmental water sources appeared to contribute less to the burden of disease.
Simpson's and Shannon's indices of diversity are reported in Table 1, and rarefaction curves are shown in Fig. 3. Human C. coli isolates showed the greatest number of STs (n = 14), followed by isolates from poultry (n = 12), ruminants (n = 11), and environmental water (n = 7). Some human STs (ST-854, ST-1590, and ST-4009) were found only in poultry, and one (ST-3232) was isolated only from ruminants. The diversity indices of human C. coli STs were the greatest among all the sources (Table 1), with 95% CrIs overlapping. Consistent with the diversity indices, rarefaction curves indicated a greater ST richness of human C. coli than for the other sources (except the environmental water source, the curve of which overlapped the human curve). When the ruminant, poultry, and environmental water data were combined in one rarefaction curve, this curve did not differ from the human rarefaction curve (data not shown).
FIG 3.

Rarefaction curves of the human, poultry, ruminant, and environmental water C. coli STs. The shaded areas represent the 95% CrI. Note that the poultry and ruminant upper boundary of the 95% CrI does not reach the point estimate of the human curve at maximum sample size. The environmental water curve overlaps the human curve.
DISCUSSION
In this paper, we report a molecular source attribution study of C. coli campylobacteriosis in New Zealand, one of few such studies globally. The study analyzed 215 C. coli isolates gathered from 2,009 Campylobacter ELISA-positive human fecal specimens collected between 2005 and 2014. Our study did not include pork meat, as only one C. coli isolate was from this source. Although in some studies, C. coli has been found at a relatively high prevalence in pig feces (42, 43), a low rate of isolation of C. coli from pork meat has also been observed in a study in the greater Washington, DC, area (44). A limitation of this study was an inability to assess variability statistically between years, due to the modest number of C. coli organisms identified each year. However, aggregation of the samples into 3-year intervals did not provide any evidence of temporal changes in dominant STs (data not shown).
C. coli accounted for ∼3% of the human isolates, a relatively low proportion compared to the situation in other countries, where C. coli has been identified in 10% of the cases (13, 45–47). In the last 6 years the average annual number of campylobacteriosis cases in New Zealand was ∼7,000, or 155 cases per 100,000 population (https://surv.esr.cri.nz/surveillance/surveillance.php). By extrapolation to the whole country without accounting for possible regional differences (48), there would be an average of ∼200 C. coli infections per year. Estimation of the true incidence of C. coli infections in the population is hindered by significant underreporting. For example, in New Zealand it has been estimated that for every reported case of acute gastrointestinal illness (from all causes) there could be 222 unreported cases. However, the majority of these cases could be due to viral infections (49). One estimate in the United Kingdom suggested that the number of cases of campylobacteriosis in the population could be 7-fold the number of notifications (50). If we extrapolate the United Kingdom estimate to New Zealand, there could be ∼1,400 (200 × 7) C. coli infections per year, equivalent to an incidence rate of ∼31 cases per 100,000 population. This rate is high compared with an estimate of 8.5 cases per 100,000 population in the United States, even though the relative contribution of C. coli to the campylobacteriosis burden is approximately three times lower in New Zealand than in the United States (9). This is due to the relatively high incidence rate of campylobacteriosis (due to any species) in New Zealand.
A comparison between C. coli populations cycling in New Zealand and elsewhere revealed a number of key features: STs belonging to CC ST-828 were predominant in our study (54% of the isolates), and interestingly, also predominant in Scotland (94% of the isolates) (45) and Switzerland (71%) (12). Ten out of 27 STs identified in the New Zealand study have also been reported in Scotland. What determines the apparent global distribution and predominance of STs belonging to CC ST-828 is not currently well understood and may require more samples and the application of higher-resolution genotyping using whole-genome sequencing to resolve. Three novel STs (ST-7767, ST-7774, and ST-7776) were identified in our study, but none of these were detected in humans. Rarefaction analysis and diversity indices were in broad agreement and indicated that human clinical isolates had the highest ST diversity (Fig. 3; Table 1). However, when the rarefaction curve for human isolates was compared to that for all other sources combined, the rarefaction curves were very similar, providing evidence that the increased diversity in clinical isolates could be due to multiple sources contributing to C. coli-associated campylobacteriosis.
Most human isolates (32/47) belonged to CC ST-828 (see Table S1 in the supplemental material). However, the most prevalent ST in humans and poultry meat (ST-1581) did not belong to any known CC and appeared sporadically but with regularity in humans in the past 10 years. Another ST that appeared with regularity in humans was ST-3072. The remaining 12 STs found in humans either were detected sporadically in a single year or disappeared and reappeared after several years. For example, ST-3232 was detected in 2005 and disappeared and reappeared in 2010. The predominance of ST-1581 in humans has not been reported elsewhere. ST-1581 was detected in Denmark from a clinical case, but it was not the dominant strain (17). In a study in Switzerland, none of the 616 C. coli isolates collected between 2002 and 2012 from poultry, pigs, and humans belonged to ST-1581. In that study, the dominant human ST was ST-827, which belongs to CC ST-828 but was not detected in our study (12). Another study in England did not detect ST-1581 in the 175 human and water C. coli isolates examined (51).
There were 10 STs shared between our study and a study conducted in Scotland (30). These STs belonged to clade 1 as defined by Sheppard et al. (30), and in the ML tree they clustered with 16 other STs that were not reported in Scotland (Fig. 2). Conversely, a previously unreported ST (ST-7774) isolated from environmental water clustered with Scottish STs belonging to clade 2, and none clustered with the Scottish clade 3. The finding of human and animal C. coli sources harboring STs from clade 1 is consistent with the findings in Scotland. However, in Scotland all the STs found in environmental water sources clustered in clades 2 and 3 rather than clade 1 (30), whereas in our study we did not find any clade 3 STs in environmental water. Further, the C. coli STs isolated from water samples collected in northwest England also belonged to clades 2 and 3 (51). It is possible that increasing the sample size from environmental water sources will result in the identification of clade 2 and clade 3 STs in New Zealand.
In spite of the fact that the ML analysis did not well differentiate the environmental water C. coli from isolates obtained from other sources, the PERMANOVA results indicated a significant differentiation of these isolates from C. coli cycling in humans, poultry, and ruminants. The PSI was calculated to evaluate the similarity between ST frequency distributions and the highest PSI was observed between STs from humans and ruminant sources, but the CrI overlapped widely with the CrI of the PSI between human and poultry C. coli. The asymmetric island model results supported the PSI and indicated that ruminant and poultry sources were the largest contributor to human infections, again with overlapping 95% CrIs (Table 1). In contrast, up to 76% of C. jejuni infections were attributed to poultry sources in New Zealand and ruminants were the second source, accounting for up to 20% of the cases (10). In Scotland, the major source of C. coli campylobacteriosis, based on the asymmetric island model, was poultry (57%), and 41% of cases were attributed to the ruminant source (30).
Although it is possible that a mutation could occur during culture, and this could result in a change in an allele and the ST, such events are likely to occur at very low frequencies due to the low intrinsic rate of mutation in Campylobacter housekeeping genes (52). In fact, only one new ST occurred as a singleton in our study (ST-7776), and the inclusion and exclusion of this isolate in the analyses did not affect the conclusions.
In summary, our results indicate that ∼3% of human campylobacteriosis cases in the study area could be attributed to infection with C. coli. This relative contribution of C. coli appears to be smaller than the contribution estimated in other countries. However, calculations suggest that the incidence rate of C. coli infection remained higher than in other countries due to the higher incidence of campylobacteriosis in general. Future studies should assess whether these results can be generalized to the whole country given that specific demographic differences between the Manawatu and other New Zealand regions may affect source attribution studies (48). The population genetic structure of C. coli in the study area is reminiscent of the structure described in the United Kingdom, with the predominance of CC ST-828 and the presence of many shared STs. As in Scotland, our source attribution analysis identified ruminants and poultry as the main infection sources, as well as a smaller contribution from surface water sources. Unlike the situation with C. jejuni, our results suggest that the ruminant sources might have a greater relative contribution to C. coli infection burden than poultry. These results highlight the need to consider each Campylobacter species separately when designing public health interventions.
Supplementary Material
ACKNOWLEDGMENTS
The isolates examined in this study were collected as part of the campylobacteriosis surveillance program funded by Ministry of Primary Industries, New Zealand.
We thank Rukhshana Akhter and Lynn Rogers from the mEpiLab team for their laboratory work contribution and Phil Carter from ESR for help with sequence typing.
Funding Statement
The isolates examined in this study were collected as part of the campylobacteriosis surveillance program funded by Ministry of Primary Industries, New Zealand.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00934-16.
REFERENCES
- 1.Euzeby JP. List of prokaryotic names with standing in nomenclature. Genus Campylobacter. http://www.bacterio.net/campylobacter.html. [Google Scholar]
- 2.Horrocks SM, Anderson RC, Nisbet DJ, Ricke SC. 2009. Incidence and ecology of Campylobacter jejuni and coli in animals. Anaerobe 15:18–25. doi: 10.1016/j.anaerobe.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 3.WHO. 2011. Campylobacter. WHO, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs255/en/. [Google Scholar]
- 4.Moore JE, Corcoran D, Dooley JS, Fanning S, Lucey B, Matsuda M, McDowell DA, Megraud F, Millar BC, O'Mahony R, O'Riordan L, O'Rourke M, Rao JR, Rooney PJ, Sails A, Whyte P. 2005. Campylobacter. Vet Res 36:351–382. doi: 10.1051/vetres:2005012. [DOI] [PubMed] [Google Scholar]
- 5.ESR. 2006. Notifiable and other diseases in New Zealand: annual report 2006. Institute of Environmental Science and Research Ltd, Porirua, New Zealand. [Google Scholar]
- 6.ESR. 2014. Notifiable diseases in New Zealand: annual report 2014. Institute of Environmental Science and Research Ltd, Porirua, New Zealand. [Google Scholar]
- 7.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M-A, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.CDC. 2015. Preliminary incidence and trends of infection with pathogens transmitted commonly through food—Foodborne Diseases Active Surveillance Network, 10 U.S. sites, 2006–2014. CDC, Atlanta, GA. [PMC free article] [PubMed] [Google Scholar]
- 9.CDC. 2012. National antimicrobial resistance system, enteric bacteria, human isolates final report 2010. CDC, Atlanta, GA. [Google Scholar]
- 10.Mullner P, Spencer SE, Wilson DJ, Jones G, Noble AD, Midwinter AC, Collins-Emerson JM, Carter P, Hathaway S, French NP. 2009. Assigning the source of human campylobacteriosis in New Zealand: a comparative genetic and epidemiological approach. Infect Genet Evol 9:1311–1319. doi: 10.1016/j.meegid.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Müllner P, Collins-Emerson JM, Midwinter AC, Carter P, Spencer SE, van der Logt P, Hathaway S, French NP. 2010. Molecular epidemiology of Campylobacter jejuni in a geographically isolated country with a uniquely structured poultry industry. Appl Environ Microbiol 76:2145–2154. doi: 10.1128/AEM.00862-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kittl S, Heckel G, Korczak BM, Kuhnert P. 2013. Source attribution of human Campylobacter isolates by MLST and fla-typing and association of genotypes with quinolone resistance. PLoS One 8:e81796. doi: 10.1371/journal.pone.0081796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillespie IA, O'Brien SJ, Frost JA, Adak GK, Horby P, Swan AV, Painter MJ, Neal KR. 2002. A case-case comparison of Campylobacter coli and Campylobacter jejuni infection: a tool for generating hypotheses. Emerg Infect Dis 8:937–942. doi: 10.3201/eid0809.010817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doorduyn Y, Van Den Brandhof WE, Van Duynhoven YT, Breukink BJ, Wagenaar JA, Van Pelt W. 2010. Risk factors for indigenous Campylobacter jejuni and Campylobacter coli infections in the Netherlands: a case-control study. Epidemiol Infect 138:1391–1404. doi: 10.1017/S095026881000052X. [DOI] [PubMed] [Google Scholar]
- 15.Dingle KE, Colles FM, Falush D, Maiden MC. 2005. Sequence typing and comparison of population biology of Campylobacter coli and Campylobacter jejuni. J Clin Microbiol 43:340–347. doi: 10.1128/JCM.43.1.340-347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roux F, Sproston E, Rotariu O, Macrae M, Sheppard SK, Bessell P, Smith-Palmer A, Cowden J, Maiden MC, Forbes KJ, Strachan NJ. 2013. Elucidating the aetiology of human Campylobacter coli infections. PLoS One 8(5):e64504. doi: 10.1371/journal.pone.0064504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Litrup E, Torpdahl M, Nielsen EM. 2007. Multilocus sequence typing performed on Campylobacter coli isolates from humans, broilers, pigs and cattle originating in Denmark. J Appl Microbiol 103:210–218. doi: 10.1111/j.1365-2672.2006.03214.x. [DOI] [PubMed] [Google Scholar]
- 18.Sheppard SK, Didelot X, Jolley KA, Darling AE, Pascoe B, Meric G, Kelly DJ, Cody A, Colles FM, Strachan NJ, Ogden ID, Forbes K, French NP, Carter P, Miller WG, McCarthy ND, Owen R, Litrup E, Egholm M, Affourtit JP, Bentley SD, Parkhill J, Maiden MC, Falush D. 2013. Progressive genome-wide introgression in agricultural Campylobacter coli. Mol Ecol 22:1051–1064. doi: 10.1111/mec.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheppard SK, McCarthy ND, Falush D, Maiden MCJ. 2008. Convergence of Campylobacter species: implications for bacterial evolution. Science 320:237–239. doi: 10.1126/science.1155532. [DOI] [PubMed] [Google Scholar]
- 20.Statistics New Zealand. 2013. 2013 Census QuickStats about a place: Palmerston North City. Number of people counted. Statistics New Zealand, Wellington, New Zealand: http://www.stats.govt.nz/Census/2013-census/profile-and-summary-reports/quickstats-about-a-place.aspx?request_value=14263&parent_id=14181&tabname=&gclid=CI_Go_-I2cYCFUtwvAodd44Peg#14263. [Google Scholar]
- 21.Department of Internal Affairs, New Zealand Government. 2013. Manawatu-Wanganui Regional Council. Community statistics as at the 2013 census. Department of Internal Affairs, New Zealand Government, Wellington, New Zealand: http://www.localcouncils.govt.nz/lgip.nsf/wpg_url/Profiles-Councils-Manawatu-Wanganui-Regional-Council-main. [Google Scholar]
- 22.Stucki U, Frey J, Nicolet J, Burnens AP. 1995. Identification of Campylobacter jejuni on the basis of a species-specific gene that encodes a membrane protein. J Clin Microbiol 33:855–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inglis GD, Kalischuk LD. 2003. Use of PCR for direct detection of Campylobacter species in bovine feces. Appl Environ Microbiol 69:3435–3447. doi: 10.1128/AEM.69.6.3435-3447.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez I, Grant KA, Richardson PT, Park SF, Collins MD. 1997. Specific identification of the enteropathogens Campylobacter jejuni and Campylobacter coli by using a PCR test based on the ceuE gene encoding a putative virulence determinant. J Clin Microbiol 35:759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denis M, Soumet C, Rivoal K, Ermel G, Blivet D, Salvat G, Colin P. 1999. Development of a m-PCR assay for simultaneous identification of Campylobacter jejuni and C. coli. Lett Appl Microbiol 29:406–410. doi: 10.1046/j.1472-765X.1999.00658.x. [DOI] [PubMed] [Google Scholar]
- 26.Dingle KE, Colles FM, Wareing DR, Ure R, Fox AJ, Bolton FE, Bootsma HJ, Willems RJ, Urwin R, Maiden MC. 2001. Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol 39:14–23. doi: 10.1128/JCM.39.1.14-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 29.Gascuel O. 1997. BIONJ an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol 14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 30.Sheppard SK, Dallas JF, Wilson DJ, Strachan NJC, McCarthy ND, Jolley KA, Colles FM, Rotariu O, Ogden ID, Forbes KJ, Maiden MCJ. 2010. Evolution of an agriculture-associated disease causing Campylobacter coli clade: evidence from national surveillance data in Scotland. PLoS One 5(12):e15708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 33.McArdle BH, Anderson J. 2001. Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecol 82:290–297. doi: 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2. [DOI] [Google Scholar]
- 34.Anderson MJ, Gorley RN, Clarke KR. 2008. PERMANOVA+ for PRIMER: guide to software and statistical methods. PRIMER-E, Plymouth, United Kingdom. [Google Scholar]
- 35.Hammer Ø, Harper DAT, Ryan PD. 2001. PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4:9. [Google Scholar]
- 36.R Core Team. 2005. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 37.Mughini-Gras L, Smid JH, Wagenaar JA, De Boer A, Havelaar AH, Friesema IH, French NP, Graziani C, Busani L, Van Pelt W. 2014. Campylobacteriosis in returning travellers and potential secondary transmission of exotic strains. Epidemiol Infect 142:1277–1288. doi: 10.1017/S0950268813002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garrett N, Devane ML, Hudson JA, Nicol C, Ball A, Klena JD, Scholes P, Baker MG, Gilpin BJ, Savill MG. 2007. Statistical comparison of Campylobacter jejuni subtypes from human cases and environmental sources. J Appl Microbiol 103:2113–2121. doi: 10.1111/j.1365-2672.2007.03437.x. [DOI] [PubMed] [Google Scholar]
- 39.Rosef O, Kapperud G, Lauwers S, Gondrosen B. 1985. Serotyping of Campylobacter jejuni, Campylobacter coli, and Campylobacter laridis from domestic and wild animals. Appl Environ Microbiol 49:1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feinsinger P, Spears EE, Poole RW. 1981. A simple measure of niche breadth. Ecology 62:27–32. doi: 10.2307/1936664. [DOI] [Google Scholar]
- 41.Wilson DJ, Gabriel E, Leatherbarrow AJ, Cheesbrough J, Gee S, Bolton E, Fox A, Fearnhead P, Hart CA, Diggle PJ. 2008. Tracing the source of campylobacteriosis. PLoS Genet 4:e1000203. doi: 10.1371/journal.pgen.1000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller WG, Englen MD, Kathariou S, Wesley IV, Wang G, Pittenger-Alley L, Siletz RM, Muraoka W, Fedorka-Cray PJ, Mandrell RE. 2006. Identification of host-associated alleles by multilocus sequence typing of Campylobacter coli strains from food animals. Microbiology 152:245–255. doi: 10.1099/mic.0.28348-0. [DOI] [PubMed] [Google Scholar]
- 43.Wright SL, Carver DK, Siletzky RM, Romine S, Morrow WEM, Kathariou S. 2008. Longitudinal study of prevalence of Campylobacter jejuni and Campylobacter coli from turkeys and swine grown in close proximity. J Food Prot 71:1791–1796. [DOI] [PubMed] [Google Scholar]
- 44.Zhao C, Ge B, De Villena J, Sudler R, Yeh E, Zhao S, White DG, Wagner D, Meng J. 2001. Prevalence of Campylobacter spp., Escherichia coli, and Salmonella serovars in retail chicken, turkey, pork, and beef from the Greater Washington, DC, area. Appl Environ Microbiol 67:5431–5436. doi: 10.1128/AEM.67.12.5431-5436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheppard SK, Dallas JF, Strachan NJ, MacRae M, McCarthy ND, Wilson DJ, Gormley FJ, Falush D, Ogden ID, Maiden MC, Forbes KJ. 2009. Campylobacter genotyping to determine the source of human infection. Clin Infect Dis 48:1072–1078. doi: 10.1086/597402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schielke A, Rosner BM, Stark K. 2014. Epidemiology of campylobacteriosis in Germany—insights from 10 years of surveillance. BMC Infect Dis 14:30. doi: 10.1186/1471-2334-14-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cody AJ, McCarthy NM, Wimalarathna HL, Colles FM, Clark L, Bowler IC, Maiden MC, Dingle KE. 2012. A longitudinal 6-year study of the molecular epidemiology of clinical Campylobacter isolates in Oxfordshire, United Kingdom. J Clin Microbiol 50:3193–3201. doi: 10.1128/JCM.01086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bolwell CF, Gilpin BJ, Campbell D, French NP. 2015. Evaluation of the representativeness of a sentinel surveillance site for campylobacteriosis. Epidemiol Infect 143:1990–2002. doi: 10.1017/S0950268814003173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lake RJ, Adlam SB, Perera S, Campbell DM, Baker MG. 2010. The disease pyramid for acute gastrointestinal illness in New Zealand. Epidemiol Infect 138:1468–1471. doi: 10.1017/S0950268810000397. [DOI] [PubMed] [Google Scholar]
- 50.Tam CC, Rodrigues LC, Viviani L, Dodds JP, Evans MR, Hunter PR, Gray JJ, Letley LH, Rait G, Tompkins DS, O'Brien SJ. 2012. Longitudinal study of infectious intestinal disease in the UK (IID2 study): incidence in the community and presenting to general practice. Gut 61:69–77. doi: 10.1136/gut.2011.238386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sopwith W, Birtles A, Matthews M, Fox A, Gee S, James S, Kempster J, Painter M, Edwards-Jones V, Osborn K, Regan M, Syed Q, Bolton E. 2010. Investigation of food and environmental exposures relating to the epidemiology of Campylobacter coli in humans in Northwest England. Appl Environ Microbiol 76:129–135. doi: 10.1128/AEM.00942-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson DJ, Gabriel E, Leatherbarrow AJ, Cheesbrough J, Gee S, Bolton E, Fox A, Hart CA, Diggle PJ, Fearnhead P. 2009. Rapid evolution and the importance of recombination to the gastroenteric pathogen Campylobacter jejuni. Mol Biol Evol 26:385–397. doi: 10.1093/molbev/msn264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.