

ABSTRACT
In the age of ever-increasing “-omics” studies, the accurate and statistically robust determination of microbial cell numbers within often-complex samples remains a key task in microbial ecology. Microscopic quantification is still the only method to enumerate specific subgroups of microbial clades within complex communities by, for example, fluorescence in situ hybridization (FISH). In this study, we improved an existing automatic image acquisition and cell enumeration system and adapted it for usage at high seas on board an oceanographic research ship. The system was evaluated by testing settings such as minimal pixel area and image exposure times ashore under stable laboratory conditions before being brought on board and tested under various wind and wave conditions. The system was robust enough to produce high-quality images even with ship heaves of up to 3 m and pitch and roll angles of up to 6.3°. On board the research ship, on average, 25% of the images acquired from plankton samples on filter membranes could be used for cell enumeration. Automated enumeration was highly correlated with manual counts (r2 > 0.9). Even the smallest of microbial cells in the open ocean, members of the alphaproteobacterial SAR11 clade, could be confidently detected and enumerated. The automated image acquisition and cell enumeration system developed here enables an accurate and reproducible determination of microbial cell counts in planktonic samples and allows insight into the abundance and distribution of specific microorganisms already on board within a few hours.
IMPORTANCE In this research article, we report on a new system and software pipeline, which allows for an easy and quick image acquisition and the subsequent enumeration of cells in the acquired images. We put this pipeline through vigorous testing and compared it to manual microscopy counts of microbial cells on membrane filters. Furthermore, we tested this system at sea on board a marine research vessel and counted bacteria on board within a few hours after the retrieval of water samples. The imaging and counting system described here has been successfully applied to a number of laboratory-based studies and allowed the quantification of thousands of samples and FISH preparations (see, e.g., H. Teeling, B. M. Fuchs, D. Becher, C. Klockow, A. Gardebrecht, C. M. Bennke, M. Kassabgy, S. Huang, A. J. Mann, J. Waldmann, M. Weber, A. Klindworth, A. Otto, J. Lange, J. Bernhardt, C. Reinsch, M. Hecker, J. Peplies, F. D. Bockelmann, U. Callies, G. Gerdts, A. Wichels, K. H. Wiltshire, F. O. Glöckner, T. Schweder, and R. Amann, Science 336:608–611, 2012, http://dx.doi.org/10.1126/science.1218344). We adjusted the standard image acquisition software to withstand ship movements. This system will allow for more targeted sampling of the microbial community, leading to a better understanding of the role of microorganisms in the global oceans.
INTRODUCTION
The exact quantification of cells is fundamental to microbial ecology and hence for understanding the interaction of microorganisms with biotic and abiotic factors. Still, the counting of microbial cells on membrane filters using an epifluorescence microscope remains the method of choice (1–3) for the enumeration of picoplankton cells, although it is rather time-consuming and relies on the experience of the individual person counting. Additionally, manual counting of an entire membrane filter is not practical within a given time frame, and therefore, only a small part is analyzed (usually 12 to 20 fields of view [FOVs]) (3, 4). Consequently, several tools have been developed over the past 2 decades to automatically enumerate microbial cells by means of image acquisition and subsequent image analysis (e.g., references 5–13). Most of these methods automate only the post-image processing; the image acquisition is still done manually. One of the first fully motorized microscope systems for automated image acquisition was presented by Pernthaler and coworkers in 2003 (7). It was applied to several thousands of sample preparations for high-throughput analysis of fluorescence in situ hybridization (FISH) assays, along a transect across the Atlantic Ocean (14). Further development of this system enabled, for example, the autonomous recognition of the shape of the filter pieces (SamLoc [15]) or sped up the image acquisition time by using light-emitting diode (LED) illumination, which removed the need for manual shutter opening and closing during optical filter changes.
The present study consists of two parts. The first part focuses on the adaptation and further development of the existing counting system of Zeder and Pernthaler (16) to minimize human intervention during the counting process. This system was tested and applied in a stable laboratory environment and compared to manual microscopic counts of microbial cells on membrane filters to verify the quality and reproducibility of the obtained data.
In a next step, we brought this system on board an oceanic research vessel to further develop the automatic microscopy method while being at sea. So far, shipboard cell enumeration has been done either manually with an epifluorescence microscope or by flow cytometry. The flow cytometry method, however, is predominantly used to determine total picoplankton, picoeukaryote, and nanoflagellate abundance. While pigmented microorganisms, like Synechococcus and Prochlorococcus, can be enumerated separately, heterotrophic nonpigmented microorganisms can be counted only as bulk (17, 18). Bringing microscopy systems to sea on board a research vessel would facilitate the counting of nonpigmented microorganisms that have been specifically labeled with oligonucleotide probes after fluorescence in situ hybridization (FISH). This bears some major challenges that need to be overcome. Mass acceleration of the microscope components due to the ship movement up to the meter range results in torsion of the instrument and greatly influences the autofocus and image focus on a nanometer-to-micrometer scale. This is even more true for automated microscope systems with their heavy motorized components, e.g., the stage, which are susceptible to torsional stress. Most of the time, microscopes on board a research vessel are mounted on vibration-free flagstones to minimize the motion of the stage and to reduce unstable focus conditions.
The motivation to develop an onboard automatic microscope counting system is to enable specific picoplankton counts after FISH on-site within a few hours after the retrieval of water samples. So far, the only mention of an automated microscope has been in a grant application in 1989 by Michael Sieracki (NSF, grant 8813356). In order to use our lab system on board a research vessel, we had to account for the ship's constant motion while at sea. Hence, we improved the stacking and focusing routine developed by Zeder and Pernthaler (16) to obtain high-quality (HQ) images independently of the ship's movement. At first, the quick stack with extended depth of focus (QEDF) was developed and further improved by in-depth focusing (FQEDF). Here, we present the first successful employment of an automatic cell enumeration system on board a research vessel, which was quality checked and verified by manual counts on board, resulting in the first onboard data set of the distribution of the alphaproteobacterial clade SAR11.
MATERIALS AND METHODS
Adaptation of existing counting system and further development. (i) Sampling: land-based image acquisition.
Marine surface water samples were collected twice a week from 1 m below the sea surface between 1 January and 31 May 2011 with the research vessel Ade at station Kabeltonne, Helgoland Roads, North Sea (54°11′30″N 7°54′00″E). All samples were fixed for 1 h at room temperature by adding a 37% formaldehyde solution (Sigma-Aldrich, Taufkirchen, Germany) to a final concentration of 1%. Filtration was done using a reusable Nalgene bottle top filtration device (catalog no. DS0320-2545), and 10 ml of surface water was filtered through 0.2-μm-pore-size polycarbonate membrane filters (catalog no. WH7060-4702; GE Healthcare, Freiburg, Germany) equipped with cellulose nitrate support filters (pore size, 0.45 μm; model 11306-47-N; Sartorius). Filtration was performed by applying a gentle vacuum of <20 kPa. After drying, filters were stored at −20°C until further analysis.
(ii) Fluorescence in situ hybridization and DAPI staining.
Filters were cut and subsequently mounted on glass slides using a mixture of glycerin–phosphate-buffered saline (PBS) mounting solutions (CitiFluor AF1 [CitiFluor Ltd., London, United Kingdom] and Vectashield [Vector Laboratories, Inc., Burlingame, CA, USA]) containing the nucleic acid dye DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich, Steinheim, Germany) at a final concentration of 1 μg ml−1. The samples that were used for community analysis and the estimation of the minimal object size for bacterial cells were additionally stained by fluorescence in situ hybridization and underwent catalyzed reporter deposition (CARD-FISH), according to Thiele et al. (19). The oligonucleotide probes used to target the whole Bacteria clade and the Bacteroidetes clade are listed in Table 1.
TABLE 1.
Oligonucleotide probes used in this studya
| Probe name | Target group | Probe sequence (5′→3′) | Length (nt)b | FA (%)c | Reference |
|---|---|---|---|---|---|
| CF319a | Bacteroidetes | TGGTCCGTGTCTCAGTAC | 18 | 35 | 29 |
| SAR11-152R | SAR11 clade | ATTAGCACAAGTTTCCYCGTGT | 22 | 25 | 30 |
| SAR11-441R | SAR11 clade | TACAGTCATTTTCTTCCCCGAC | 22 | 25 | 30 |
| SAR11-441R(modif) | SAR11 clade | TACCGTCATTTTCTTCCCCGAC | 22 | 25 | 31 |
| SAR11-487(modif) | SAR11 clade | CGGACCTTCTTATTCGGG | 18 | 25 | 31 |
| SAR11-487-H3d | SAR11 clade | CGGCTGCTGGCACGAAGTTAGC | 22 | 25 | 31 |
| SAR11-542R | SAR11 clade | TCCGAACTACGCTAGGTC | 18 | 25 | 30 |
| SAR11-732R | SAR11 clade | GTCAGTAATGATCCAGAAAGYTG | 23 | 25 | 30 |
| EUB338 I | Bacteria | GCTGCCTCCCGTAGGAGT | 18 | 35 | 32 |
| EUB338 II | Supplement to EUB338 | GCAGCCACCCGTAGGTGT | 18 | 35 | 33 |
| EUB338 III | Supplement to EUB338 | GCTGCCACCCGTAGGTGT | 18 | 35 | 33 |
All Bacteria-specific probes (EUB338 I, EUB338 II, EUB338 III) were applied together as a mix. This applied also to all SAR11-specific probes.
nt, nucleotides.
FA, formamide concentration (vol/vol) in the hybridization buffer.
Unlabeled helper oligonucleotide to probe SAR11-487(modif).
(iii) Refined image acquisition system.
Image acquisition was done using a multipurpose fully automated microscope imaging system (MPISYS) (see Fig. S1A to G in the supplemental material) on a Zeiss AxioImager.Z2 microscopic stand (Carl Zeiss MicroImaging GmbH, Göttingen, Germany) with a cooled charged-coupled-device (CCD) camera (AxioCam MRm; Carl Zeiss) and a Colibri LED light source (Carl Zeiss) with three light-emitting diodes (UV-emitting LED, 365 ± 4.5 nm for DAPI; blue-emitting LED, 470 ± 14 nm for the tyramide Alexa Fluor 488; red-emitting LED, 590 ± 17.5 nm for the tyramide Alexa Fluor 594), combined with the HE-62 multifilter module (Carl Zeiss). This module consists of a triple emission filter TBP 425 (± 25), 527 (± 27), LP615, including a triple beam splitter of TFT 395/495/610.
The imaging software initially acquired an overview image of the microscope stage and object slides in bright-field illumination with a 1× objective (Carl Zeiss) (see Fig. S1B to D in the supplemental material) (SamLoc [15]). Subsequently, the SamLoc software was used to define coordinates for image acquisition based on user-defined grids with FOVs, with a minimum distance of 250 μm between FOVs (see Fig. S1E in the supplemental material). This distance takes into account the frame size (FOV width) of the CCD camera with the 63× objective (Carl Zeiss), which is 141 by 105 μm. To avoid any overlap of FOVs, a minimum distance of one-and-a-half times the diagonal of the CCD camera frame was found to be a reliable distance between two imaged FOVs. After defining the coordinates, an FOV coordinate list was generated, which was subsequently used by the MPISYS software during the automatic image acquisition. This system was evaluated for planktonic samples, in which 55 FOVs were adequate for cell enumeration; however, the number of FOVs needs to be newly evaluated for samples from other habitats.
Next, so-called channels were defined within the MPISYS software according to the fluorescent dyes used (e.g., DAPI, Alexa Fluor 488, and Alexa Fluor 594) (see Fig. S1F in the supplemental material). The channels are user-specified settings for image acquisition (see Fig. S1G in the supplemental material). For each channel, the exposure time (constant or variable), focusing procedure, and number of images per z-stack for the compensation of filter unevenness were selected (20). Notably, for each channel, only one exposure time, either constant or variable, could be selected. If the user requested different exposure times for the same excitation setting, several channels were defined, selecting similar excitation settings but different exposure times.
The algorithm for the focusing routine was adapted from that of Zeder and Pernthaler (16). Focusing was done for each FOV in the first acquired channel and then set as a fixed focal position for the other channels. A z-stack of seven layers per FOV and exposure time was recorded, and subsequently, a single extended depth of field image (EDF) was created (see Fig. S2A in the supplemental material). The EDF was used to compensate for the unevenness of a sample (like, for example, wrinkles on the filter piece) by taking multiple images corresponding to the different focal planes and subsequently creating a single in-focus image (21). For this, the wavelet-based extended-focus functionality of the AxioVision software (Carl Zeiss), which is in compliance with the EDF algorithm, was used.
Images were acquired using a 63× magnification and 1.4 numerical aperture oil immersion plan apochromatic objective (Carl Zeiss). The EDFs were saved in .tiff format and were subsequently loaded into the Automated Cell Measuring and Enumeration tool 2.0 (ACMEtool2.0) program (http://www.technobiology.ch/index.php?id=acmetool) (see Fig. S1H to L in the supplemental material). The images obtained using the above-mentioned CCD camera and a 63× oil objective with numerical aperture of 1.4 have a pixel size of 0.1015 μm pixel−1.
(iv) Image selection and cell determination and enumeration using ACMEtool2.0.
All images taken with MPISYS were manually inspected in ACMEtool2.0 (see Fig. S1H in the supplemental material), and low-quality images, such as images with over- or underexposed parts, areas out of focus (unevenness), or too many aggregates, large phytoplankton cells, debris, and particles, were excluded from further analysis to ensure high data quality (see Fig. S3 to S6 in the supplemental material) (20). In a second step, a so-called metafile was calculated from the remaining HQ images for faster image processing (see Fig. S1I in the supplemental material). This metafile contained the coordinates of all recognized objects of each image of all channels and stored parameter values, like object area, circularity, mean gray value, and signal-to-background ratio. These parameters were used to define logical selection rules and stored in “set” and “subset” definitions. To enumerate FISH-positive cells, the following sets were defined. One set was defined to identify all blue fluorescent DAPI-stained cells under UV illumination. A second set was defined to determine which cells show green fluorescence (the FISH signal) under blue excitation. The third set was defined to detect all red-emitting objects under green excitation (see Fig. S3A to F in the supplemental material). Due to the high productivity of the sampling site at Helgoland Roads, this was needed to discriminate against autofluorescence signals of debris and small algae (sample image material is provided in Fig. S4A and B and S5A and B in the supplemental material). Consequently, FISH-positive cells were identified as the logical combination of the three sets: blue positive, green positive, and red negative.
Finally, after optimal selection of parameters and manual cross-checks, cells were counted automatically by the program (see Fig. S1J in the supplemental material), and cell numbers were exported in a tab-delimited report file (see Fig. S1K in the supplemental material). Furthermore, ACMEtool2.0 provides a summary file in which the number of analyzed FOVs per sample and total counted cells were given. For a detailed overview, an FOV report of the numbers of counted cells per FOV was provided. This report also gives a detailed overview of the cell distribution on the filter.
Onboard automatic counting system. (i) Sampling and sample preparation on board.
Planktonic seawater samples were taken during the Atlantic Meridional Transect (AMT) 22 on the research vessel RRV James Cook (Southampton, United Kingdom, to Punta Arenas, Chile, 10 October to 24 November 2012). Samples were taken using a Sea Bird CTD with carousel water sampler (Sea Bird Electronics, Inc., USA), which was deployed twice daily at predawn and solar noon intervals. A total of 50 stations were sampled at a 20-m water depth. From each sample, 100 ml of seawater was fixed with 37% formaldehyde in a final concentration of 1% for 1 to 2 h at room temperature. Subsequently, triplicate 20-ml subsamples were filtered onto 47-mm polycarbonate membrane filters with a 0.2-μm pore size using a vacuum of 20 kPa. These filters were dried and stored at −20°C until further analysis.
All stations were analyzed using CARD-FISH directly on board the research ship within a few hours after the retrieval of samples. The filters were processed according to the method of Thiele et al. (19) and subsequently mounted on glass slides using a DAPI-amended mixture of CitiFluor AF1 and Vectorshield. The oligonucleotide probes used to target the alphaproteobacterial clade SAR11 are listed in Table 1. Cell enumeration and community analyses were done directly on board using a fully motorized Axioplan 2 microscope (Carl Zeiss) for automatic image acquisition and the ACMEtool2.0 software for image analysis. Manual verification of the automatically obtained cell counts was done after completion of automated image acquisition to prevent bleaching of the stained cells. The typical manual inspection period is in the range of seconds to minutes, which affects the signal strength of the stained cells. In contrast, exposure times in the range of milliseconds do not negatively influence the signal intensities (data not shown).
(ii) Automated image acquisition on board.
The Axioplan 2 microscope was equipped with a 63×/1.4 oil plan apochromatic objective lens, a four-slide scanning stage (Märzhäuser, Wetzlar, Germany), LED epifluorescence illumination of 365 nm, 470 −5/+15 nm, and 590 ± 10 nm (KSL 70; Rapp OptoElectronic, Wedel, Germany), a multiband optical filter with beam splitter HC395/495/610, and emission filter HC425 (± 25), 527 (± 25), LP615 (AHF Analysentechnik, Tübingen, Germany), and a cooled CCD camera (Orca C4742-95-12NR; Hamamatsu Photonics, Hamamatsu City, Japan). The pixel size of this microscope and the associated objectives was 0.1068 μm pixel−1. The acquisition of overview images was done via webcam and using the SamLoc software (15). The previously described automated image acquisition software MPISYS was modified for the different hardware requirements and was extended with new functionality to account for the ship's movements. We developed a new quick-stack method with an extended depth of field (QEDF) routine. In comparison to the original stack image acquisition with EDF (see Fig. S2A in the supplemental material), in which positioning and image acquisition were done subsequently for every single image of the stack, QEDF used a live video stream approach (see Fig. S2B in the supplemental material). A video stream was recorded, while the microscopic stage moved from first to last stack position. The image frames of the live stream, which fit the needed stack positions best, were taken for the EDF calculation that followed. However, the time-consuming EDF calculation was postponed until all stacks from all channels were recorded to avoid a focus shift between alterations of channels. Using QEDF, it was possible to acquire multichannel multilayer images on board, but the yield of good-quality images was still low. This is because QEDF is still a pure image acquisition method and relies on the initially found focal position. In order to increase the amount of HQ images, we combined the QEDF stacking algorithm with in-depth focusing, and this was called focused QEDF (FQEDF) (see Fig. S2C in the supplemental material). The live stream image acquisition was done over a distance of 3 times the required stack size. The focal position of the live stream was calculated and represents the center of the required stack. Around this position, image frames were taken, and the FQEDF file was calculated, similar to the procedure with QEDF and EDF. The frame rate for QEDF and FDEQF is nearly exclusively dependent on the exposure time, because live imaging needs to be done in full frame at full resolution. In QEDF and FQEDF, there is continuous movement of the stage during image acquisition but in a range of 1 to 3 μm s−1; therefore, it is slow enough that axial motion blur does not pose a problem.
RESULTS
Adaptation of existing counting system and further development. (i) Influence of exposure time on counting accuracy.
The first step in our automated cell counting routine is the image acquisition of the stained microorganisms. The influence of exposure times on subsequent cell detection and enumeration was tested. We acquired images with various measured exposure times by the autoexposure function of the AxioVision software for every FOV. Here, this variable measured exposure time is called DAPI-auto. The automatically measured exposure times ranged from a minimum of 49 ms to maximum of 255 ms, with a mean exposure time of 74 ± 22 ms. Additionally, from the same set of samples, images were acquired with constant exposure times of 50 ms (termed DAPI-50ms) and 25 ms (DAPI-25ms). Fifty milliseconds was selected as a reference exposure time at the lower limit of DAPI-auto, and 25 ms was randomly chosen to see if the acquisition time could be reduced.
In total, 44 marine DAPI-stained planktonic samples were imaged by using the three different channel definitions. From each sample, 55 FOVs were recorded, and after manual image inspection, >50% (30 ± 10) high-quality FOVs per sample were obtained. These were then processed further for cell enumeration. The two fixed exposure times (DAPI-25ms and DAPI-50ms) resulted in similar cell numbers per FOV (P = 0.685, t test), but the cell numbers differed slightly from the ones derived from various exposure times from the DAPI-auto channel (P = 0.006) (Fig. 1). The DAPI-auto channel occasionally resulted in an overestimation of the cells per FOV, which originated from elevated background fluorescence leading to a low signal-to-noise-ratio. Therefore, two closely located cells appeared to be merged and were consequently counted as one by the algorithm. However, the automatically measured exposure times still often resulted in similar numbers of cells per volume as fixed exposure times, although the conditions of image acquisition differed for each FOV. Additionally, the image acquisition time for 55 FOVs using DAPI-auto settings exceeded those from DAPI-50ms and DAPI-25ms settings by >1 h. Hence, the exposure time was set to 50 ms under stable laboratory conditions but had to be newly evaluated for samples from different habitats. Using fixed exposure times results in faster and standardized image acquisition for all samples from one habitat, leading to high reproducibility and comparability of images. The exposure time of 25 ms was rejected, and 50 ms was chosen instead, since 25 ms was at the lower limit of the image acquisition time with respect to signal-to-background ratio and was not considerably faster than 50 ms.
FIG 1.
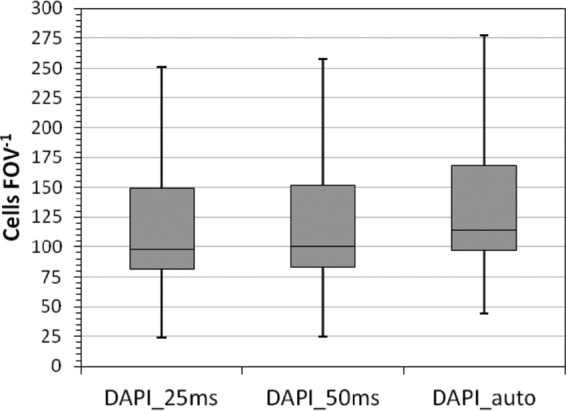
Number of cells per FOV obtained with different exposure time settings. Two constant exposure times (DAPI-25ms and DAPI-50ms) and one varying exposure time (DAPI-auto) were selected.
(ii) Estimation of the minimal object size for bacterial cells.
One major aspect of cell detection using the ACMEtool2.0 software is to define the set and subset definitions, which heavily rely on the signal-to-background ratio and on the size (in pixels) of the objects. A specific threshold in the object size is necessary to be able to distinguish between bacterial cells and other objects, like viruses and autofluorescent particles, which tend to be smaller (22, 23). To determine the minimal object size needed for the accurate detection of only bacterial cells, 33 marine planktonic samples were DAPI stained to determine the total object abundance and hybridized with probes specific for Bacteria (EUB I to III) and Bacteroidetes (CF319a). The bacterial probe mixture of EUB I to III was used, since it detects mainly all bacterial cells in planktonic waters, assuming a 100% detection rate.
After image acquisition and quality checking, various thresholds of 10 to 50 pixels (equivalent to 0.1 to 0.5 μm2) for the object size were tested (Fig. 2A and B). The lowest area consisting of 10 pixels (0.1 μm2) was assumed to detect 100% of the objects stained by DAPI. Increasing thresholds (>15 pixels, >0.15 μm2) for the object size logically resulted in decreased numbers of detected objects. This decrease in numbers represents a fraction of smaller particles, such as virus-like or other particles, which are picked up when using lower thresholds (<15 pixels, <0.15 μm2). By removing these particles from the total object counts, the accuracy of the number of bacterial cells, defined as an object containing both a DAPI and an EUB I to III (Bacteria-specific probe) signal, increased. Small objects (e.g., viruses) that contained only a DAPI but no FISH signals were no longer included.
FIG 2.

Estimation of the minimal object area size in pixels for the accurate enumeration of bacterial cells. Relative object counts of (A) DAPI-stained (30 ms) and EUB I- to III-stained (100 ms) and (B) DAPI-stained (30 ms) and CF319a-stained (150 ms) cells as the percentage of total counts from the smallest pixel area measured (10 pixels). The optimal pixel area size was determined by testing fixed object area sizes from 10 to 50 pixels from DAPI- and FISH-stained cells. Error bars indicate standard deviations (n = 33).
If, however, the threshold was set too high, for example, at ≥30 pixels, there was a decreased detection rate of both total cells (DAPI, 10%) and hybridized cells (EUB I to III and CF319a, 19% [Fig. 2A] and 35% [Fig. 2B], respectively). For the automated detection of bacterial cells, the optimum object size for our samples was found to be in the range of 17 to 20 pixels or 0.18 to 0.21 μm2.
(iii) Comparison of manual versus automated cell counting.
To further evaluate the automated counting process, bacterial cells in a subset of 22 of the above-mentioned 44 stained and hybridized water samples were enumerated both manually and automatically. Student's two-sample t test was used to compare the distribution of bacterial counts done by manual analysis with the distribution of bacterial counts done by automatic analysis. The obtained P value was >0.05, revealing that the distribution of bacterial counts does not differ between manual and automatic analyses. Additionally, regression analysis of the obtained manual and automatic counts revealed r2 values of >0.98 (Fig. 3A and B), indicating a slight underestimation by automated counting. However, the coefficients of variation (CVs) for automatic and manual counts differed greatly. The CV per sample is determined by dividing the standard deviation by the mean of cell counts from all fields of view per sample, whereas the average CV is defined by the mean of CVs across all 22 samples. Student's two-sample t test was used to compare the CVs per sample across all 22 samples done by manual analysis with the CVs per sample across all 22 samples done by automatic analysis. The obtained P value was 0.000234, thus being smaller than the significance level of 0.001, revealing significant differences between the CVs per sample across all 22 samples acquired by automatic counting compared to manual analysis. This means that the average CV was higher for manual counting (19% ± 7%) than for automatic counting (12% ± 3%). Hence, with the automatic system, the lowest CV per sample accounted for a minimum of 7% variance within one sample. In contrast, 10% variance per sample was received with manual analysis. Moreover, the highest CV per sample obtained by manual counting was 35%, whereas the automatic analysis resulted in the highest CV per sample of about 24%.
FIG 3.

Manual versus automatic cell counts ashore. Cell enumeration was done after CARD-FISH with the oligonucleotide probe CF319a. (A) DAPI counts per volume. (B) FISH-positive cell counts per volume. The regression coefficient (r2) and formula are depicted at the lower right side of each graph.
Onboard automatic counting system. (i) Modifications in focusing routine for onboard usage.
Based on the promising results of part I, in which we successfully established the automatic counting system inside a stable laboratory environment, an adaptation for shipboard usage was addressed. To address the complexity of the ship's constant motions while being at sea, the stacking and focusing routine to obtain high-quality images was improved. At first, the quick stack with extended depth of focus (QEDF) was developed and further improved by in-depth focusing (FQEDF) (Table 2).
TABLE 2.
Overview of improvements in stacking and focusing routines to obtain HQ images on board a research vessel
| Method | High-quality image yield (%) | Criteria fulfilled (minimum 10 high-quality images) (%) |
|---|---|---|
| EDF | 0–25 | 19 |
| QEDF | 4–26 | 62 |
| FQEDF | 11–35 | 97 |
On board, the average performance of using standard stacking (stacking seven z-layers) and the EDF method yielded, on average, only 10% HQ images, with a range from 0 to 25%. In contrast, in the laboratory, an average of >50% HQ images was generated. It was reported previously (4), consistent with our own experience, that a minimum of 10 HQ images is required to obtain statistically relevant counts of the bacterial cell abundance in the downstream image analysis. By using the standard stacking and EDF method, this criterion was fulfilled on board in only 19% of the cases (n = 32; see Table S1 in the supplemental material).
QEDF enabled faster stacking of images than the EDF stacking. This increased the total number of FOVs per sample and yielded an average of 13% (4 to 26%) HQ images, slightly more than that with EDF. Additionally, the number of samples with ≥10 HQ images was much higher (62% compared to 19%, n = 21; see Table S2 in the supplemental material). However, QEDF is still a pure-image acquisition method and depends on the initially found focal position. FQEDF, however, combines the fast image stacking acquisition with a refocusing approach; it can to an extent follow the movement caused by the ship. For example, FQEDF produced up to 20% HQ images for DAPI and the specific FISH probe, with a maximum roll angle of 6.3°, a maximum pitch angle of 5°, and a maximum ship heave of 3 m. In contrast, QEDF yielded only 10% HQ DAPI images and 20% HQ FISH images, with maximum pitch and roll angles of 5° and 3.8°, respectively, and a ship heave of 2.3 m. With the regular EDF calculation algorithm, 13% HQ DAPI images and 5% HQ FISH images were acquired, with maximum pitch and roll angles of 4.2° and 8.8°, as well as a heave of up to 2.5 m (see Table S1 in the supplemental material).
The FQEDF stacking and imaging algorithm allowed for onboard automated multichannel and multilayer image acquisition. On average, the FQEDF stacking and imaging algorithm yielded 24% (range, 11% to 53%) HQ images. The statistical criteria for ≥10 HQ images per sample and channel were achieved in 97% of all cases (n = 58; see Table S1 in the supplemental material).
(ii) Onboard automated and manual microscopy.
Water samples from >50 stations were processed directly after sampling with CARD-FISH, using a mix of six oligonucleotide probes specific for the SAR11 clade (Table 1). All samples were analyzed on board using the FQEDF algorithm of the automatic microscope and the cell enumeration system ACMEtool2.0. Subsequently, manual counts were obtained on board directly after the image acquisition and compared to the automatically produced counts. Image acquisition was done using fixed exposure times, as described in part I, to keep the analyses comparable. The optimal exposure time for DAPI in the oligotrophic open ocean waters could be set to 30 ms and for SAR11 to 150 ms.
The automatically obtained total cell abundances and SAR11-specific cell counts were manually validated directly on board and were in good agreement with the automatic counts (Fig. 4A and B). Regression analysis revealed r2 values of >0.90 for all analyses. Student's two-tailed test provided P values of >0.05 (DAPI, P = 0.679; SAR11, P = 0.317), indicating that there were no differences between the onboard manual and automatic cell enumeration methods.
FIG 4.

Manual versus automatic cell counts obtained onboard. Cell enumeration was done after CARD-FISH with the oligonucleotide probe SAR11. (A) DAPI counts per volume. (B) FISH-positive cell counts per volume. The regression coefficient (r2) and formula are depicted at the lower right side of each graph.
DISCUSSION
In this study, we evaluated and further improved an automated image acquisition and counting system for the enumeration of microbial cells in plankton samples on board a research ship.
The imaging and counting system described here has been successfully applied to a number of laboratory-based studies and allowed the quantification of thousands of samples and FISH preparations (see, e.g., reference 24). The newly developed onboard focusing routine allows for a detailed on-site analysis of the microbial community. We improved the standard image acquisition algorithm to withstand ship movements (pitch and roll angles up to 6.3° and ship heaves of up to 3 m). Onboard, the focal position is not stable; therefore, a fast method was needed in order to acquire the image stacks for the extended depth of field method before the focus is lost: QEDF. However, QEDF is still a pure image acquisition method, relying on the initially found focal position. FQEDF combines the fast image stack acquisition with a refocusing approach. It not only tries to be faster than focus loss, but it can follow the focus within certain limits. The frame rate for QEDF or FQEDF is nearly exclusively dependent on the exposure time, because for this, the live imaging needs to be done in full frame at full resolution. For the initial focus routine, we can go with subframes and pixel binning up to 32 frames per second. For EDF, the stage is not in motion during image acquisition. It moves to a certain stack position and stops, the image is acquired, and it moves on. In QEDF and FQEDF, the stage moves continuously during live image acquisition but in the range of 1 to 3 μm s−1, which is slow enough that axial motion blur is not a problem.
With the FQEDF routine, one in four images was suitable for further quantification with the ACMEtool2.0. Since the system can run autonomously for many hours, the output is sufficient to have an in-depth analysis of the samples on site. The ability to quickly receive an insight into both the total cell count and relative abundance of specific microbial groups (FISH) at a precise location can lead to more targeted sampling approaches. This will enable more hypothesis-driven analyses of the microbial community, which is particularly relevant for sampling done at sites that are not or that cannot be sampled regularly. Furthermore, this system could outcompete the routinely used aquatic cell enumeration system of flow cytometry. Flow cytometry is not well suited for the quantification of a large number of samples after FISH, for several reasons. FISH is best done on filters; however, flow cytometry requires cells to be suspended in a liquid. The transfer of cells from filter into liquid is feasible but always at the cost of cell loss (25). Additionally, FISH in suspension is not applicable to marine planktonic samples containing small cells, like those of SAR11, since the centrifugation steps involved in the protocol with standard benchtop centrifuges are insufficient for the sedimentation of these tiny cell types.
As a general recommendation, the automated microscope should ideally be located at or near the center of the ship's movement to minimize the influence of pitch, roll, and heave. In modern research ships, this often would be where the labs used for gravimetric analyses are located. For future use on board, we would propose also a lightweight slide scanning stage, which would be less susceptible to ship movements and mass acceleration, hence increasing the yield of high-quality images suitable for ACME analysis. Additionally, it is important to test the optimum exposure times for each sample type, as differences in staining or sample preparation might interfere with optimal image quality for downstream ACME analyses.
Under optimal shipboard and sea weather conditions, the system has the capacity to acquire images from a single filter piece consisting of 55 FOVs and two fluorescent channels (DAPI and FISH) in approximately 15 min, which adds up to a potential throughput of around 50 filters in 12 h. The subsequent manual inspection and ACME tool analysis take up another 2 to 3 h for the 50 filters. The final results can be obtained after ∼15 h from 50 filters, while manual counting of the same amount of 55 FOVs on only a single filter takes a minimum of about 2 h. Naturally, under bad weather conditions, these performance values will deteriorate with increasing wave impact.
The bottleneck and critical step in our workflow is the image quality assessment, which needs manual inspection of thousands of individual images. This is necessary to weed out low-quality images, like out-of-focus images, images with over- or underexposed parts, or images with too many aggregates, large phytoplankton cells, or debris (see Fig. S4 to S6 in the supplemental material). This process cannot yet be automated in a confident manner. Although the manual image control step is still quite laborious, the entire workflow is considerably faster and more objective than manual microscopic counting.
The cell counts obtained from the automated enumeration program ACMEtool2.0 were not significantly different from manually obtained cell counts. Furthermore, with our automated system, a larger number of FOVs are processed, and consequently, more cells are examined, leading to more statistically significant cell quantifications. Ultimately, human errors during counting are minimized, and hence, the standardized counting routine allows for a direct comparison of different sampling campaigns. Original image files can be archived and reanalyzed anytime with improved or different image analysis tools.
In principle, this pipeline can be applied to a wide range of microbial habitats and environments, not just planktonic samples. In the case of sediments, cells have to be detached from the substratum by sonication or other methods and diluted in buffer before being brought onto a filter (26, 27). Additionally, Bižić-Ionescu and coworkers (28) successfully adapted the autonomous cell enumeration system to quantify specific cell types after FISH in aggregate samples. Many samples are characterized by intense autofluorescence, which may render fluorescent staining of cells impossible. One solution in the future might be to use multispectral imaging to distinguish between specific signals and background. It should be technically feasible to implement this step into our imaging and counting pipeline. Our system consists of a two-component pipeline, the autonomous image acquisition followed by cell enumeration with ACMEtool, and these components can also be used independently. For example, ACMEtool has the potential to be used to enumerate objects from high-quality confocal laser scanning or even superresolution structured illumination micrographs, as it works with standard image file formats as input (e.g., 8-bit .tiff).
In summary, the system and software pipeline we present here generates fast and reliable cell counts both in the laboratory and on board a research vessel. This system will allow for more targeted sampling of the microbial community, leading to a better understanding of the role of microorganisms in difficult-to-assess sites.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Max Planck Society, the Helmholtz Society, and the Natural Environment Research Council (NERC). The German Federal Ministry of Education and Research (BMBF) supported this study by funding the Microbial Interactions in Marine Systems project (MIMAS, project 03F0480A; http://www.mimas-project.de).
We thank the captain and crew of the Ade for help with sampling at the island Helgoland. We are also grateful to the captain and crew of the RRS James Cook for assistance in sampling during the AMT 22 cruise. We greatly appreciate the substantial contribution of Marcus Blohs (University of Applied Sciences Lausitz, Germany) for microscopy. We are also grateful to Rudolf Amann from the Max Planck Institute for Marine Microbiology, Bremen, Germany, for many fruitful discussions.
This study is a contribution to the international IMBER project and was supported by the United Kingdom Natural Environment Research Council National Capability funding to Plymouth Marine Laboratory and the National Oceanography Centre, Southampton, United Kingdom.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03931-15.
REFERENCES
- 1.Porter FG, Feig YS. 1980. The use of DAPI for identifying and counting aquatic microflora. Limnol Oceanogr 1980:943–948. [Google Scholar]
- 2.Hobbie JE, Daley RJ, Jasper S. 1977. Use of nuclepore filters for counting bacteria by fluorescence microscopy. Appl Environ Microbiol 33:1225–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kepner RL, Pratt JR. 1994. Use of fluorochromes for direct enumeration of total bacteria in environmental samples: past and present. Microbiol Rev 58:603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seo EY, Ahn TS, Zo YG. 2010. Agreement, precision, and accuracy of epifluorescence microscopy methods for enumeration of total bacterial numbers. Appl Environ Microbiol 76:1981–1991. doi: 10.1128/AEM.01724-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bloem J, Veninga M, Shepherd J. 1995. Fully automatic determination of soil bacterium numbers, cell volumes, and frequencies of dividing cells by confocal laser scanning microscopy and image analysis. Appl Environ Microbiol 61:926–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singleton S, Cahill JG, Watson GK, Allison C, Cummins D, Thurnheer T, Guggenheim B, Gmür R. 2001. A fully automated microscope bacterial enumeration system for studies of oral microbial ecology. J Immunoassay Immunochem 22:253–274. doi: 10.1081/IAS-100104710. [DOI] [PubMed] [Google Scholar]
- 7.Pernthaler J, Pernthaler A, Amann R. 2003. Automated enumeration of groups of marine picoplankton after fluorescence in situ hybridization. Appl Environ Microbiol 69:2631–2637. doi: 10.1128/AEM.69.5.2631-2637.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abramoff MD, Magelhaes PJ, Ram S. 2004. Image processing with ImageJ. Biophotonics Int 11:36–42. [Google Scholar]
- 9.Selinummi J, Seppälä J, Yli-Harja O, Puhakka JA. 2005. Software for quantification of labeled bacteria from digital microscope images by automated image analysis. Biotechniques 39:859–863. doi: 10.2144/000112018. [DOI] [PubMed] [Google Scholar]
- 10.Thiel R, Blaut M. 2005. An improved method for the automated enumeration of fluorescently labelled bacteria in human faeces. J Microbiol Methods 61:369–379. doi: 10.1016/j.mimet.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Z, Pons MN, Raskin L, Zilles JL. 2007. Automated image analysis for quantitative fluorescence in situ hybridization with environmental samples. Appl Environ Microbiol 73:2956–2962. doi: 10.1128/AEM.02954-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daims H. 2009. Use of fluorescence in situ hybridization and the daime image analysis program for the cultivation-independent quantification of microorganisms in environmental and medical samples. Cold Spring Harb Protoc 2009:pdb.prot5253. doi: 10.1101/pdb.prot5253. [DOI] [PubMed] [Google Scholar]
- 13.Morono Y, Terada T, Masui N, Inagaki F. 2009. Discriminative detection and enumeration of microbial life in marine subsurface sediments. ISME J 3:503–511. doi: 10.1038/ismej.2009.1. [DOI] [PubMed] [Google Scholar]
- 14.Schattenhofer M, Fuchs BM, Amann R, Zubkov MV, Tarran GA, Pernthaler J. 2009. Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ Microbiol 11:2078–2093. doi: 10.1111/j.1462-2920.2009.01929.x. [DOI] [PubMed] [Google Scholar]
- 15.Zeder M, Ellrott A, Amann R. 2011. Automated sample area definition for high-throughput microscopy. Cytometry A 79:306–310. doi: 10.1002/cyto.a.21034. [DOI] [PubMed] [Google Scholar]
- 16.Zeder M, Pernthaler J. 2009. Multispot live-image autofocusing for high-throughput microscopy of fluorescently stained bacteria. Cytometry A 75:781–788. doi: 10.1002/cyto.a.20770. [DOI] [PubMed] [Google Scholar]
- 17.Zubkov MV, Burkill PH, Topping JN. 2006. Flow cytometric enumeration of DNA-stained oceanic planktonic protists. J Plankton Res 29:79–86. doi: 10.1093/plankt/fbl059. [DOI] [Google Scholar]
- 18.Hill PG, Heywood JL, Holland RJ, Purdie DA, Fuchs BM, Zubkov MV. 2012. Internal and external influences on near-surface microbial community structure in the vicinity of the Cape Verde islands. Microb Ecol 63:139–148. doi: 10.1007/s00248-011-9952-2. [DOI] [PubMed] [Google Scholar]
- 19.Thiele S, Fuchs B, Amann R. 2011. Identification of microorganisms using the ribosomal RNA approach and fluorescence in situ hybridization, p 171–189. In Wilderer P. (ed), Treatise on water science. Academic Press, Oxford, United Kingdom. [Google Scholar]
- 20.Zeder M, Kohler E, Pernthaler J. 2010. Automated quality assessment of autonomously acquired microscopic images of fluorescently stained bacteria. Cytometry A 77:76–85. doi: 10.1002/cyto.a.20810. [DOI] [PubMed] [Google Scholar]
- 21.Forster B, Van de Ville D, Berent J, Sage D, Unser M. 2004. Complex wavelets for extended depth-of-field: a new method for the fusion of multichannel microscopy images. Microsc Res Tech 65:33–42. doi: 10.1002/jemt.20092. [DOI] [PubMed] [Google Scholar]
- 22.Fuhrman JA. 1999. Marine viruses and their biogeochemical and ecological effects. Nature 399:541–548. doi: 10.1038/21119. [DOI] [PubMed] [Google Scholar]
- 23.Levin PA, Angert ER. 2015. Small but mighty: cell size and bacteria. Cold Spring Harb Perspect Biol 7:a019216. doi: 10.1101/cshperspect.a019216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J, Weber M, Klindworth A, Otto A, Lange J, Bernhardt J, Reinsch C, Hecker M, Peplies J, Bockelmann FD, Callies U, Gerdts G, Wichels A, Wiltshire KH, Glöckner FO, Schweder T, Amann R. 2012. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336:608–611. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 25.Sekar R, Fuchs BM, Amann R, Pernthaler J. 2004. Flow sorting of marine bacterioplankton after fluorescence in situ hybridization. Appl Environ Microbiol 70:6210–6219. doi: 10.1128/AEM.70.10.6210-6219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Velji MI, Albright LJ. 1986. Microscopic enumeration of attached marine bacteria of seawater, marine sediment, fecal matter, and kelp blade samples following pyrophosphate and ultrasound treatments. Can J Microbiol 32:121–126. doi: 10.1139/m86-024. [DOI] [Google Scholar]
- 27.Tischer K, Zeder M, Klug R, Pernthaler J, Schattenhofer M, Harms H, Wendeberg A. 2012. Fluorescence in situ hybridization (CARD-FISH) of microorganisms in hydrocarbon contaminated aquifer sediment samples. Syst Appl Microbiol 35:526–532. doi: 10.1016/j.syapm.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Bižić-Ionescu M, Zeder M, Ionescu D, Orlić S, Fuchs BM, Grossart H, Amann R. 2014. Comparison of bacterial communities on limnic versus coastal marine particles reveals profound differences in colonization. Environ Microbiol 17:3500–3514. doi: 10.1111/1462-2920.12466. [DOI] [PubMed] [Google Scholar]
- 29.Manz W, Amann R, Ludwig W, Vancanneyt M, Schleifer KH. 1996. Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum Cytophaga-Flavobacter-Bacteroides in the natural environment. Microbiology 142:1097–1106. doi: 10.1099/13500872-142-5-1097. [DOI] [PubMed] [Google Scholar]
- 30.Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418:630–633. doi: 10.1038/nature00917. [DOI] [PubMed] [Google Scholar]
- 31.Gómez-Pereira PR, Hartmann M, Grob C, Tarran GA, Martin AP, Fuchs BM, Scanlan DJ, Zubkov MV. 2013. Comparable light stimulation of organic nutrient uptake by SAR11 and Prochlorococcus in the North Atlantic subtropical gyre. ISME J. 7:603–614. doi: 10.1038/ismej.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. 1990. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56:1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daims H, Brühl A, Amann R, Schleifer KH, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.