

ABSTRACT
Shiga toxin (Stx)-producing Escherichia coli (STEC) is a major cause of foodborne illness, including the life-threatening complication hemolytic-uremic syndrome. The German outbreak in 2011 resulted in nearly 4,000 cases of infection, with 54 deaths. Two forms of Stx, Stx1 and Stx2, differ in potency, and subtype Stx2a is most commonly associated with fatal human disease. Stx is considered to be an AB5 toxin. The single A (enzymatically active) subunit inhibits protein synthesis by cleaving a catalytic adenine from the eukaryotic rRNA. The B (binding) subunit forms a homopentamer and mediates cellular association and toxin internalization by binding to the glycolipid globotriaosylceramide (Gb3). Both subunits are essential for toxicity. Here we report that unlike other AB5 toxin family members, Stx is produced by STEC as unassembled A and B subunits. A preformed AB5 complex is not required for cellular toxicity or in vivo toxicity to mice, and toxin assembly likely occurs at the cell membrane. We demonstrate that disruption of A- and B-subunit association by use of A-subunit peptides that lack enzymatic activity can protect mice from lethal doses of toxin. Currently, no treatments have been proven to be effective for hemolytic-uremic syndrome. Our studies demonstrate that agents that interfere with A- and B-subunit assembly may have therapeutic potential. Shiga toxin (Stx) produced by pathogenic Escherichia coli is considered to be an AB5 heterohexamer; however, no known mechanisms ensure AB5 assembly. Stx released by E. coli is not in the AB5 conformation and assembles at the receptor interface. Thus, unassembled Stx can impart toxicity. This finding shows that preventing AB5 assembly is a potential treatment for Stx-associated illnesses.
IMPORTANCE Complications due to Shiga toxin are frequently fatal, and at present, supportive care is the only treatment option. Furthermore, antibiotic treatment is contraindicated due to the ability of antibiotics to amplify bacterial expression of Shiga toxin. We report, contrary to prevailing assumptions, that Shiga toxin produced by STEC circulates as unassembled A and B subunits at concentrations that are lethal to mice. Similar to the case for anthrax toxin, assembly occurs on receptors expressed on the surfaces of mammalian target cells. Disruption of Shiga toxin assembly by use of A-subunit peptides that lack enzymatic activity protects mice from lethal challenge with Shiga toxin, suggesting a new approach for development of therapeutics.
INTRODUCTION
Shiga toxin (Stx)-producing Escherichia coli (STEC) is responsible for an estimated 265,000 cases of foodborne illness annually (1). The pathogen is acquired through contaminated food or water, typically undercooked ground beef, contaminated produce, untreated well water, or unpasteurized juices and milk (2). After ingestion, bacteria colonize the intestinal mucosa and produce the primary virulence factor responsible for disease progression, i.e., Stx (3). Symptoms include watery, often bloody diarrhea and usually resolve over the course of a few days. However, about 10% of cases result in serious complications, including neurological dysfunction and hemolytic-uremic syndrome (HUS). HUS is characterized by hemolytic anemia, acute renal failure, and thrombocytopenia and can be fatal (4–6). Long-term sequelae may result in chronic renal failure.
Considered to be members of the AB5 toxin family, both Stx1 and Stx2a contain an enzymatically active A subunit, which inhibits protein synthesis by cleaving adenine 4324 in the 28S portion of the ribosomal 60S subunit (7). The A monomer associates with a homopentamer of B subunits by noncovalent interactions. The B pentamer binds to its glycolipid receptor, globotriaosylceramide (Gb3), and mediates internalization of the A subunit into target cells (8–10).
Other AB5 toxins, such as cholera toxin and pertussis toxin, have dedicated secretion systems that recognize and secrete only the assembled holotoxin past the bacterial outer membrane (11, 12). Stx does not possess a specific secretion system and uses a unique mechanism to ensure passage past the outer membrane. The genes for the Stx A and B subunits are carried in the late gene region of lysogenic bacteriophages (13, 14). The late gene functions are responsible for viral replication, assembly, and host cell lysis, as well as Stx expression (Fig. 1A). Stx is transcriptionally silent until viral lytic replication of the bacteriophage is triggered by the bacterial SOS stress response (15). Unlike that of other AB5 toxins, egress of Stx from the bacterial cell relies solely on bacterial cell lysis. As a result, there is no proofreading step to ensure holotoxin release, and both phage and toxin are released when bacteria lyse.
FIG 1.
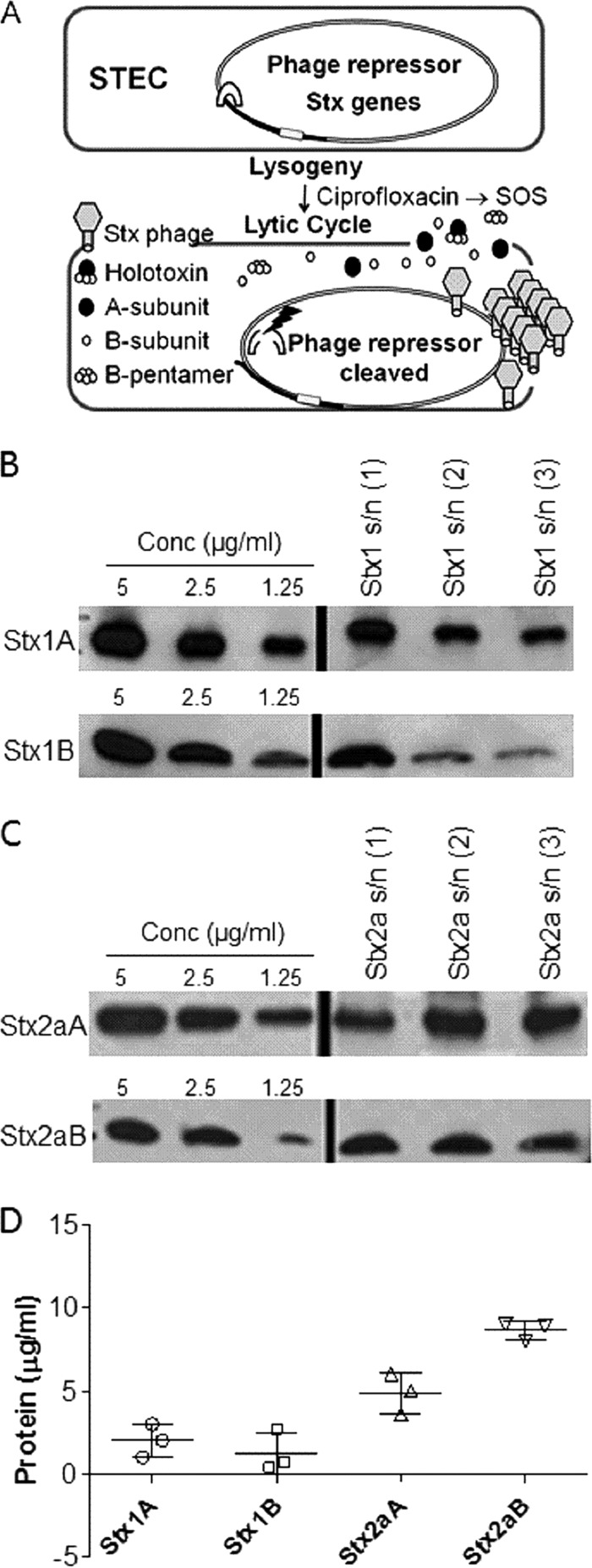
Characterization of Stx released by phage lysis. (A) Summary of regulation of Stx expression in E. coli. The A and B subunits of Stx are encoded on lysogenic bacteriophages. Stx is not expressed by lysogenic bacteria. DNA damage or stalled replication (e.g., via ciprofloxacin treatment) can activate the bacterial SOS response, resulting in cleavage of the phage repressor. Loss of repression leads to induction of the lytic cycle, during which new phage particles and Stx are released following phage-mediated lysis. (B) Western blot of purified Stx1 standards and three independent supernatants (s/n) from C600::H19B. (C) Western blot of purified Stx2a standards and three independent supernatants from C600::933W. (D) Amounts of toxin (means ± standard deviations; n = 3) in culture supernatants were determined from standard curve dilution series of purified Stx subunits. An A:B subunit ratio of 1:3 was obtained for Stx1, and a ratio of 1:8 was obtained for Stx2a.
Despite its sharing of overall structure with other AB5 toxins, several studies suggest that Stx is less likely to form an AB5 complex. Multiple oligomeric forms of Stx1 and Stx2a were observed at concentrations of ≤50 μM (16–18), and Stx2a was found to possess a conserved amino acid that destabilized pentamer formation (16). In addition, AB5 toxins, including cholera toxin and subtilase cytotoxin, display hydrophobic residues in the upper portion of the central pore of the B pentamer which play a critical role in the assembly of the A subunit with the B pentamer (19). In the case of Stx, especially Stx2a, this hydrophobic patch is much smaller and contains hydrophilic residues near the central pore of the B subunits, further suggesting a decreased tendency of Stx to maintain an intact AB5 conformation.
With the lack of a proofreading step for toxin assembly and the presence of structural motifs that negatively affect AB5 assembly, we explored the hypothesis that Stx released by phage expression may not preassemble as an AB5 holotoxin and that toxin assembly occurs at the cell surface.
MATERIALS AND METHODS
Purified holotoxin and subunits.
Purified Stx1, Stx2a, and Stx1 A subunit were obtained from BEI Resources, and cholera toxin was obtained from List Biological. Molarity was calculated using the mass predicted from the mature protein sequence (e.g., accounting for proteolytic processing and removal of the signal peptide).
The Stx1 A subunit was also prepared by transforming expression plasmids encoding the Stx1 A subunit into E. coli DH5α (20), followed by purification using AffiGel Blue chromatography, Q Sepharose Fast Flow ion-exchange chromatography (GE Healthcare, Uppsala, Sweden), and Superdex 75 HiLoad 26/60 size exclusion chromatography (GE Healthcare). The purity of the Stx1 A subunit was verified by the presence of a single band at 32 kDa on Coomassie-stained SDS-PAGE gels. Preparations of Stx1 and Stx2 B subunits were described previously (16, 21, 22).
Preparation of Stx supernatants.
Stx expression in E. coli C600 strains lysogenized with the Stx-producing phages H19B (Stx1) and 933W (Stx2a), originally obtained from A. O'Brien (13), was induced with ciprofloxacin (20 ng ml−1) as previously described (20, 21). Bacterial cells were removed by centrifugation and filter sterilized using Millipore Stericup 0.22-μm filters with a 250-ml capacity.
Western blots.
Antibodies used included rabbit polyclonal anti-Stx1 (Thermo Scientific, Rockford, IL), mouse anti-Stx2a A subunit (11E10; BEI Resources), rabbit anti-Stx2 B subunit (BEI Resources), and mouse anti-Stx1 B subunit (BEI Resources). For denaturing gels, electrophoresis and blotting were performed as previously described (21). Briefly, purified Stx1 A-subunit, Stx1 B-subunit, Stx2a A-subunit, Stx2a B-subunit, and supernatant samples were boiled in nonreducing SDS sample loading buffer for 5 min and resolved in 4 to 15% polyacrylamide-Tris-glycine gels (Bio-Rad). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes via wet electroblotting. Blots were blocked and then incubated overnight at 4°C with primary antibody. The signal was developed with an appropriate secondary antibody, either horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (MP Biomedicals) or HRP-conjugated goat anti-mouse IgG (MP Biomedicals), using an Amersham ECL Prime Western blotting detection reagent kit (GE Healthcare). Protein band densities were analyzed using the spot density analysis software ImageJ. For native gels, blots were blocked in Odyssey blocking buffer (Li-Cor) for an hour at room temperature before being probed with antibody overnight at 4°C. The signal was developed with an appropriate secondary antibody, either goat anti-rabbit IgG (Li-Cor) or goat anti-mouse IgG (Li-Cor), with IRDye 800CW, at a 1:5,000 dilution. The protein band pattern was analyzed using an Odyssey CLx infrared imaging system (Autoscan s800; Li-Cor Biosciences).
Size exclusion chromatography.
A Superdex HiLoad 26/60 75 preparation-grade size exclusion column (Äkta pure FPLC) was calibrated using proteins with known sizes and elution profiles, including thyroglobulin (670 kDa), γ-globulin (158 kDa), albumin (66 kDa), ovalbumin (44 kDa), carbonic anhydrase (29 kDa), RNase A (13.7 kDa), and vitamin B12 (1.4 kDa). The Stx2a supernatant was concentrated 25-fold and fractionated by size exclusion chromatography. Holotoxin activity in the chromatography fractions was assessed by determining the dilution causing 50% inhibition of protein synthesis (typically about 18 ng ml−1 for purified Stx2a), using Luc2P Vero cells expressing destabilized luciferase as previously described (20). Recovery of Stx2a A and B subunits was quantified by Western blotting.
Stx partitioning assay.
Culture supernatants of Stx1 and Stx2a, purified Stx1 (5 μg ml−1) (BEI Resources) containing an A:B subunit ratio of 1:5 (as demonstrated by staining with Sypro Orange protein dye), and purified cholera toxin (5 μg ml−1) (List Biological) were partitioned via centrifugal filtration (Amicon Ultracel 50-kDa cutoff; Millipore) into two fractions: >50 kDa (corresponding to holotoxin) and <50 kDa (corresponding to unassembled A and B subunits). Cellular toxicities of the Stx1 supernatant, the Stx2 supernatant, and purified Stx1 (1 ng μl−1) were assayed by incubating Luc2P Vero cells with serial dilutions of the unfiltered and filtered fractions. Antigenic recovery of the Stx A and B subunits and of cholera toxin was determined by Western blotting. A rabbit polyclonal antibody (C3062; Sigma-Aldrich, St. Louis, MO) was used for detection of cholera toxin.
Native gel IEF.
Stx1 holotoxin and purified Stx1 A and B subunits loaded in the indicated amounts were resolved using a pH 3 to 10 isoelectric focusing (IEF) gel (Life Technologies). A Serva liquid mix IEF marker 3-10 was used as the pH standard.
In vivo toxicity of Stx.
Briefly, male CD-1 mice of 13 to 15 g, obtained from Charles River Laboratories (Wilmington, MA), were housed in filter-top cages with access to food and water ad libitum and challenged by intraperitoneal injection 3 days after arrival, as previously described (23). For partitioning studies, Stx2a was adjusted to deliver twice the mouse lethal dose (15 ng per mouse) in 0.5 ml phosphate-buffered saline (PBS) and then fractionated as described above. To assess toxicity in the presence of A2 peptides, Stx2a (7 ng) and A2 peptides (1 μg) were incubated alone or together at 37°C for an hour before injection into mice as described above.
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Cincinnati and were conducted in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (24). Mice appearing moribund or losing more than 20% of initial body weight were sacrificed in compliance with IACUC regulations. Kaplan-Meier survival curves were plotted using GraphPad Prism 5.0. Statistical analysis (unpaired Student's t test) was performed with GraphPad Prism 5.0.
Glycolipids and lipids.
Gb3, phosphatidylcholine (PC), and cholesterol (Ch) were purchased from Matreya Inc. (Pleasant Gap, PA).
A2 subunit peptides.
Peptides corresponding to the A2 subunits of wild-type Stx2a (A2-WT), a stop mutant (A2-Stop), and a W277R mutant (A2-W277R) were purchased from Peptide 2.0 (Chantilly, VA). Stock solutions were prepared by suspending the peptides in distilled water. Kaplan-Meier survival curves were plotted using GraphPad Prism 5.0.
Binding of B subunits to A2 peptides.
Peptides were biotinylated on the cysteine residue (C261) by use of maleimide-PEG2-biotin (Pierce) and then immobilized on streptavidin biosensors (ForteBio). Tips were incubated with unconjugated maleimide-PEG2-biotin (0.6 μg ml−1) to block unbound sites and prepare negative-control tips. Binding to the Stx2a B subunit was assessed at 37°C in PBS. In association studies, biosensors were immersed in 10 μM Stx2a B subunits and shaken at 100 rpm. In dissociation studies, biosensors were transferred to PBS and shaken at 500 rpm. In all studies, final binding curves were calculated using ForteBio Data Analysis 7.0 software after subtracting the response units for the negative control.
Glycolipid binding of A2-peptide and B-subunit mixtures.
Gb3 (final concentration, 4 μg ml−1), phosphatidylcholine, and cholesterol were mixed at a molar ratio of 1:3:3 in methanol and immobilized on hydrophobic amino propylsilane biosensors (ForteBio) by using a Sidekick apparatus (ForteBio) at room temperature. Unbound glycolipids were removed by washing in PBS. Association and dissociation studies monitored binding of unbiotinylated A2 peptides, B subunits, 1:1 mixtures of A2 peptides and B monomers, and a toxoided form of Stx2a holotoxin. Binding was assessed at 37°C in PBS containing 100 μM dithiothreitol to avoid nonspecific inter-A2-peptide interactions. Since saturation was not always achieved, KD (equilibrium dissociation constant) values were approximated by plotting the response units (RU) corresponding to the maximum amount of binding during association against protein concentrations, and pseudo-KD values were obtained using the 1:1 binding model in GraphPad Prism 5.0.
Association and dissociation binding of B subunits to glycolipids.
Association was assessed by incubating half-log dilutions of Stx B subunits with glycolipid-immobilized biosensors (prepared as described above) without shaking until binding approached saturation. For dissociation, the biosensors were transferred to PBS and shaken at 1,000 rpm. KD values were determined as described above. For multivalency analysis, association and dissociation binding results were plotted using the methods described by Winzor et al. (25).
RESULTS
Stx is not released as an assembled toxin.
The unusual dependence on phage-mediated lysis for toxin release prompted us to investigate Stx assembly. Cultures of E. coli C600 lysogenized with the Stx2a phage 933W or the Stx1 phage H19B were induced to enter the lytic cycle by use of the antibiotic ciprofloxacin. The amounts of A and B subunits in the bacterial lysates were determined by quantitative Western blotting (Fig. 1B to D). The amount of the Stx1 A subunit (32,053 kDa) was determined to be 1.9 ± 1.2 μg ml−1 (59 nM), and that of the Stx1 B subunit (7,690 kDa) was 1.3 ± 1.6 μg ml−1 (170 nM), for an A/B subunit ratio of about 1 to 3. The amount of the Stx2a A subunit (33,194 kDa) was determined to be 4.6 ± 1.4 μg ml−1 (140 nM), and that of the Stx2a B subunit (7,817 kDa) was 9.1 ± 0.82 μg ml−1 (1,160 nM), for an A/B subunit ratio of about 1 to 8.
The assembly state of Stx2a was examined by size exclusion chromatography (Fig. 2). A supernatant with the Stx2a A subunit at 5 μg ml−1 and the Stx2a B subunit at 9 μg ml−1 was concentrated 25-fold to allow for detection by Western blotting, and 1 ml was loaded on a Superdex 75 size exclusion column. Only about 1% of the toxin activity eluted in fractions corresponding to the 72-kDa AB5 holotoxin (124 to 140 ml). About 54% of the toxin activity was recovered in the fractions from 150 to 180 ml, corresponding to AB heterodimers (41 kDa), B pentamers (39 kDa), and A monomers (33 kDa), and about 62% of A-subunit antigenicity and about 78% of B-subunit antigenicity were recovered in these fractions. Another 20% of the B-subunit antigenicity eluted in the fractions from 206 to 230 ml, corresponding to B monomers (7.8 kDa). Similar results were seen for independent repeats. These results demonstrate that the A and B subunits are not released at a 1:5 ratio and that most of the material released by phage lysis is not assembled into holotoxin. Furthermore, the observation that toxin activity also partitioned at a molecular mass lower than that of the 72-kDa holotoxin was unanticipated and suggests that preformed AB5 toxin is not necessary for toxicity.
FIG 2.

Stx2a is not released as a fully assembled AB5 toxin. A C600::933W supernatant concentrated 25-fold was fractionated by size exclusion chromatography on a Superdex 75 column. Calculated elution volumes for Stx2a subunits (arrows) were based on size standards (light gray shading). Toxicity (solid line), plotted as percent activity loaded on the column by use of a logarithmic scale, was assessed by determining the dilution causing 50% inhibition of protein synthesis. Recovery of Stx2a A (circles) and B (squares) subunits was quantified by Western blotting, and the results are expressed as percentages of protein loaded.
Preassembled AB5 holotoxin is not required for toxicity.
The assembly state of the purified toxin was examined (Fig. 3A). Purified Stx holotoxin, containing A and B subunits at a ratio of 1:5, was centrifuged using spin filters with a 50-kDa exclusion cutoff, until about 90% of the volume was in the lower chamber. The fractions were adjusted to the original volume, and antigenic recovery was assessed by Western blotting. Stx was not lost during filtration, and about equal amounts of antigenic Stx1 A and B subunits were recovered from the top (>50 kDa) and bottom (<50 kDa) fractions (Fig. 3B). In contrast, both subunits of the AB5 toxin cholera toxin (molecular mass, 85 kDa) were retained in the top (>50 kDa) fraction (Fig. 3C). Toxin activities of both Stx1 and Stx2a were also recovered in both the top (>50 kDa) and bottom (<50 kDa) fractions (Fig. 3D and E). The top fraction of Stx2a was centrifuged a second time, and toxin activity again partitioned in both the top and bottom chambers. To determine whether unassembled toxin is active in vivo, Stx2a was adjusted to twice the lethal dose (15 ng), filtered, and injected into mice. Mice injected with unfiltered or filtered Stx2a displayed significant weight loss compared to those injected with PBS (Fig. 3F). All of the mice injected with the unfiltered toxin (11 of 11 mice) or the bottom (<50 kDa) fraction (12 of 12 mice) died, and 6 of 12 mice injected with the top (>50 kDa) fraction died (Fig. 3G), demonstrating that toxin activity was distributed in both fractions. These results suggest that Stx in the top fraction was not assembled as stable AB5 and likely existed in an equilibrium between assembled and unassembled states.
FIG 3.

Stx A and B subunits can dissociate without a loss of activity. (A) Purified holotoxin was separated into fractions of <50 kDa and >50 kDa by use of 0.5-ml concentrating filters until less than 10% of the volume remained, and the fractions above and below the filter were adjusted to the original volume with PBS. (B and C) Partitioning of purified Stx1 (70 kDa; 5 μg ml−1) (B) and cholera toxin (84 kDa; 5 μg ml−1) (C) was examined by Western blotting. (D and E) Protein synthesis inhibition of the unfiltered and filtered fractions of Stx1 (D) and Stx2a (E) was assessed in vitro using Luc2P Vero cells. (F and G) In vivo toxicity of fractionated Stx2a. (F) Mouse weights (circles, trial 1; squares, trial 2; and triangles, trial 3) 48 h after injection. Data are means ± standard deviations. (G) Kaplan-Meier survival curves demonstrating toxicity of Stx2a in all fractions. The data represent the results of three independent trials. Statistical analysis was performed using unpaired Student's t test in GraphPad Prism 5.0. (H) Native gel isoelectric focusing. Purified Stx1 holotoxin (HT) and Stx1 A and B subunits were loaded at 200, 100, 20, or 10 ng, as indicated, into pH 3 to 10 isoelectric focusing gels, followed by characterization by Western blotting. The first three Western blot panels show replicate samples loaded into the same gel, which was cut into strips and probed with a rabbit polyclonal antibody to Stx1 (α HT), a monoclonal antibody to the Stx1 A subunit (α A-subunit), or a monoclonal antibody to the Stx1 B subunit (α B-subunit). The purified A subunit migrated at pH 8.8, and the purified B subunit migrated at pH 5.7. When samples were loaded at 100 ng (140 nM), a single band was seen for the holotoxin, at pH 7.6. When samples were loaded at 10 ng (14 nM), only the B subunit was detected in the holotoxin lane, and it comigrated with the purified B subunit.
To further validate the assembly state, purified holotoxin and the Stx1 A and B subunits were separated by native gel isoelectric focusing and detected by Western blotting with polyclonal and monoclonal antibodies. The Stx1 subunits were clearly resolved by isoelectric focusing (Fig. 3H). The basic Stx1 A subunit (predicted pI = 9.4) migrated at pH 8.8, and the acidic Stx1 B subunit (predicted pI = 5.6) migrated at pH 5.7. Holotoxin migration was concentration dependent. Stx1 loaded at 100 ng (140 nM) migrated as a single band at about pH 7.6. In contrast, at 10 ng (14 nM), a band was detected which comigrated with the acidic Stx1 B subunit. The other antibodies were unable to detect Stx1 at this concentration. Previous studies using analytical ultracentrifugation determined the half-maximal effective concentration (EC50) for Stx1 B-pentamer formation in solution to be 43 nM (16), a value consistent with the observed fully assembled holotoxin at 140 nM and fully disassembled toxin at 14 nM.
Purified A and B subunits can associate and promote toxicity in vivo.
To test the toxicity of unassembled A and B subunits introduced at the same time, purified A subunits were injected into one side of the peritoneum and purified B subunits were injected into the other side by use of separate syringes. Injection of 1 ml of purified Stx1 A subunits and Stx1 B subunits at 2 to 10 μg each (corresponding to 56 to 280 nM if assembled as an AB5 holotoxin) resulted in statistically significant weight loss (Fig. 4A) and death (Fig. 4B), regardless of the source of A subunits. These results demonstrate that the Stx A and B subunits do not need to be preassembled into AB5 holotoxin to mediate toxicity in vivo.
FIG 4.
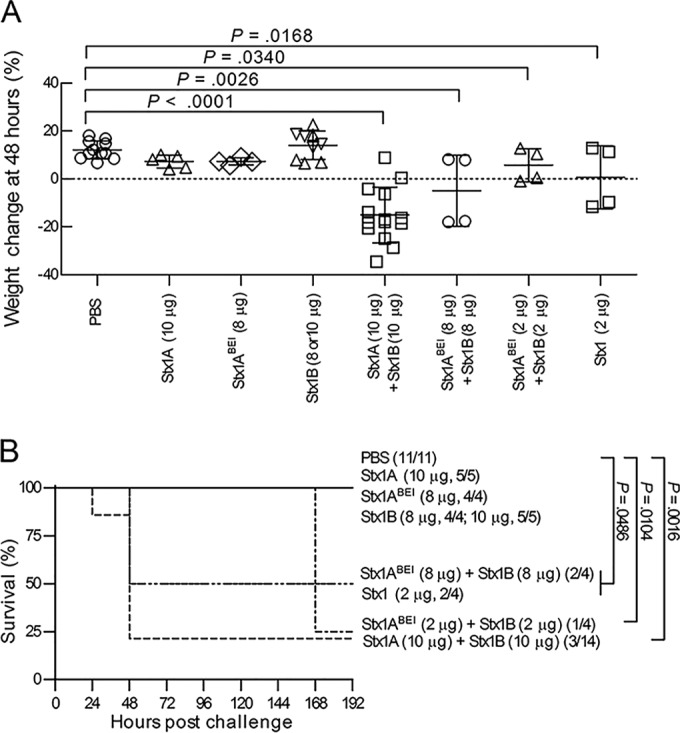
Purified A and B subunits delivered separately are toxic. Mice were injected at the indicated concentrations with the Stx1 A subunit, the Stx1 B subunit, or PBS, alone or in combination. (A) Mouse weights 48 h after injection. Data are means ± standard deviations. (B) Kaplan-Meier survival curves.
Role for receptor binding in pentamer assembly.
Since subunit assembly in solution is not favored at concentrations that would be lethal in vivo, we examined the hypothesis that binding to receptors arrayed on a solid surface can template toxin assembly. Analytical ultracentrifugation (16) studies revealed that B-pentamer formation is much more efficient for Stx1 than for Stx2a. Furthermore, the polar amino acid glutamine at position 40 in the hydrophobic interface of the Stx2a B pentamer destabilizes pentamer assembly, since mutation to leucine (the corresponding amino acid in the stable Stx1 B pentamer) resulted in greatly improved pentamer assembly for the Stx2a B-Q40L mutant. Since Stx2a binds best to Gb3 mixed with the membrane components cholesterol (Ch) and phosphatidylcholine (PC) (23, 26), we examined the binding of three variants of Stx B subunits to sensor tips coated with Gb3-PC-Ch in real time by using biolayer interferometry (BLI). Association was assessed in the presence of the B subunit, and protein dissociation was assessed after transfer to buffer without B subunits (Fig. 5). The affinity of Stx B subunits for a single Gb3 glycan is extremely weak, at around 4 mM (27), and stable binding is achieved by engaging multiple binding sites. Stx1 has three glycan binding sites per B subunit (15 per pentamer) (28), and Stx2a has two glycan binding sites per B subunit (10 per pentamer) (26). Traditional kinetic binding equations cannot accurately model the multiple affinity states and partial engagement typical of AB5 toxins (29), and as expected, the binding curves did not display good fits to kinetic kon/koff equations (data not shown). Instead, KD values were calculated by plotting the Bmax values (Fig. 5D; Table 1) at different B-subunit concentrations. A Hill slope value of 1.0 describes monomeric binding to one site with no cooperativity, and as expected for a complex system, Hill values of <1.0 were obtained, reflecting the sum of multiple binding states with different affinities for the ligand (Table 1). The KD for glycolipid binding (Fig. 5) was similar to the EC50 for pentamer formation for Stx1 B-WT and the Stx2a B-Q40L mutant (Table 1), suggesting that pentamer formation is required for binding. However, the KD and EC50 values differed dramatically for Stx2a B-WT, as the KD for glycolipid binding was 14-fold lower than the EC50 for pentamer formation.
FIG 5.
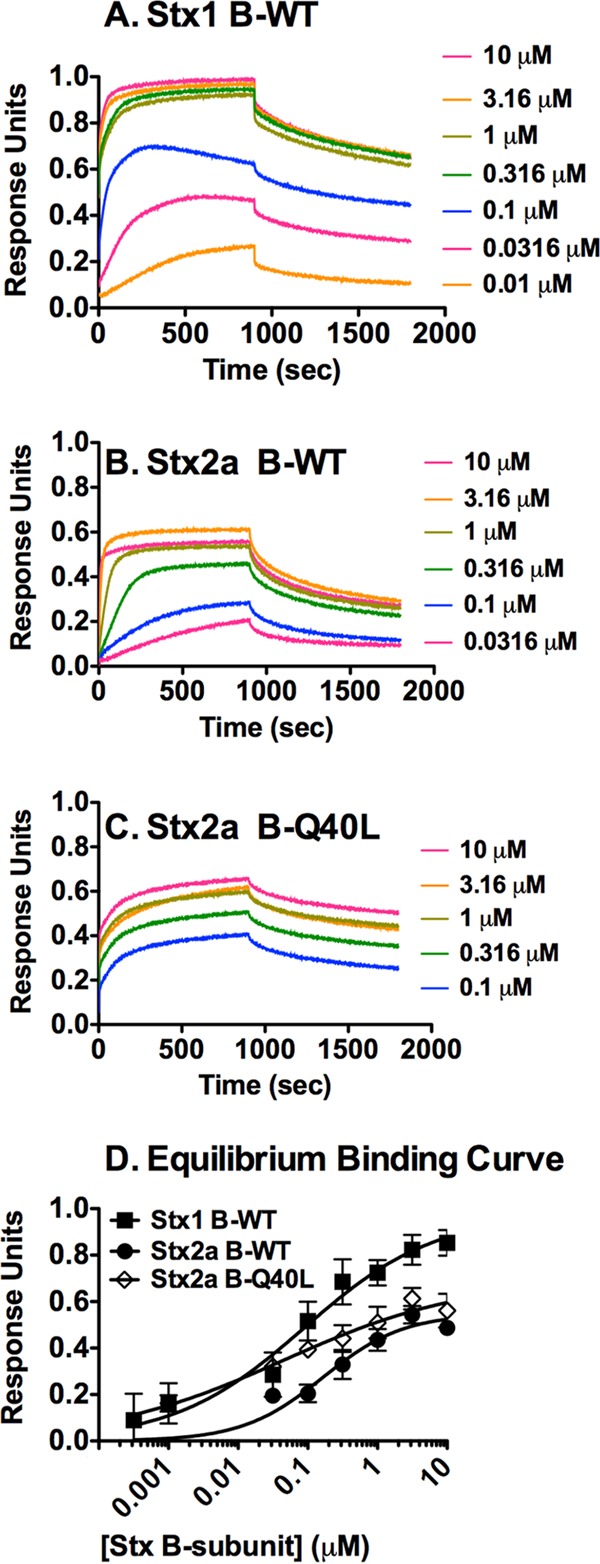
Binding of Stx B subunits in real time as determined by BLI. Gb3-PC-Ch was immobilized on a hydrophobic sensor tip. Association was assessed in wells containing various concentrations of B subunits of Stx1 (A), Stx2a WT (B), and Stx2a Q40L (C) for 900 s, and then the tips were transferred to wells containing only buffer and dissociation was monitored. Means for three independent repeats were plotted. (D) Equilibrium binding at 900 s of association.
TABLE 1.
Properties of subunit associations with glycolipid receptors
| Stx B subunit (EC50 [μM] for pentamer formationa) | KD (μM) for binding of Gb3-PC-Ch | Hill coefficient | Bmax |
|---|---|---|---|
| Stx1 B-WT (0.043) | 0.089 | 0.46 | 0.69 |
| Stx2a B-Q40L (0.11) | 0.045 | 0.33 | 0.47 |
| Stx2a B-WT (2.3) | 0.16 | 0.75 | 0.33 |
| Stx2a B subunits with A2 peptide | |||
| Stx2a B-WT + A2 peptide | 0.038 | 0.98 | 0.89 |
| Stx2a B-WT + A2-Stop | 0.11 | 1.2 | 0.69 |
| Stx2a B-WT + A2-W227R | 0.14 | 0.41 | 1.4 |
Determined by the area under the concentration-time curve.
Time and concentration dependence of receptor binding.
To analyze these data further, we used a mathematical approach developed by Winzor and colleagues to distinguish simple monovalent interactions from complex, multivalent interactions (25). Association data are plotted as ln(1 − Rt/Re) versus time, where Rt is the amount bound at time t and Re is the value at equilibrium. Dissociation data are plotted as ln(Rt/R0) versus time, where R0 is the value at the beginning of the dissociation phase. By using log-transformed values, simple 1:1 binding interactions display first-order kinetics, yielding a straight line as a function of time. However, more complex interactions change as a function of time, as weak initial interactions involving engagement of a few ligands are converted to stronger associations by engagement of additional binding sites. Furthermore, multimeric proteins, such as Stx, may display concentration dependence if monomer-to-pentamer conversion is necessary for binding.
The Winzor dissociation plots were not linear with time (Fig. 6B, D, and F), with initial dissociation occurring more rapidly than that at later times. These results are consistent with heterogeneous binding interactions, where molecules bound by a few sites dissociate more rapidly than molecules with all binding sites engaged. Similar to previously made observations (29), the association binding (Fig. 6A, C, and D) appeared to be somewhat less time dependent than dissociation, likely because BLI cannot distinguish weak binding mediated by engagement of a few binding sites from strong binding mediated by engagement of many binding sites.
FIG 6.
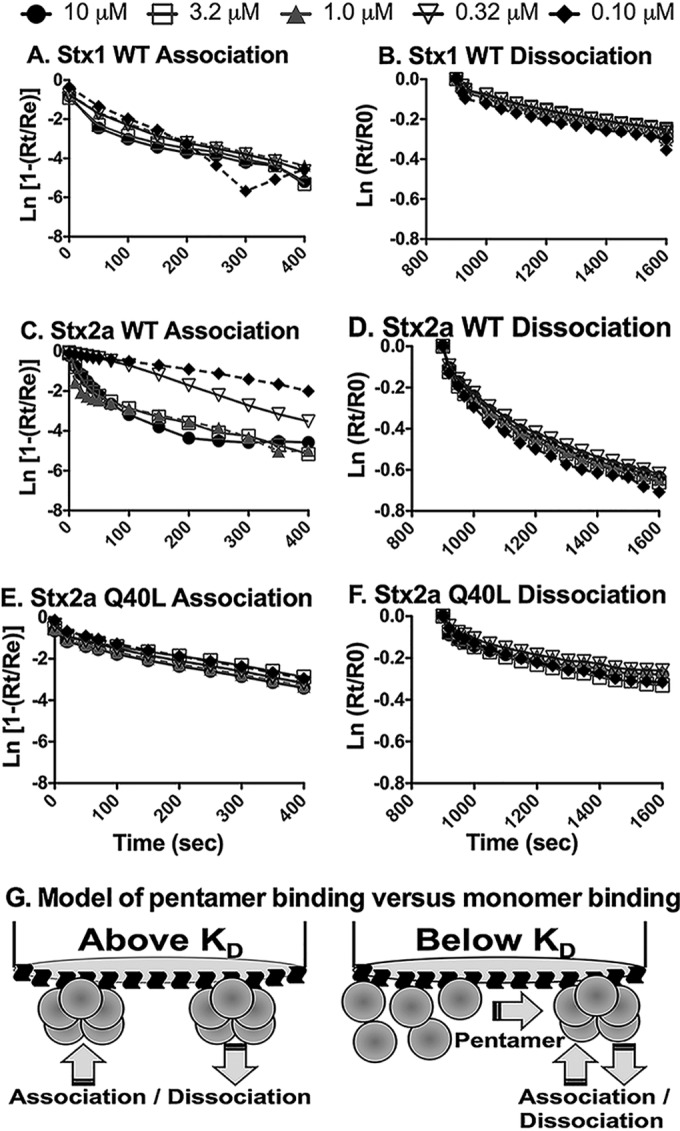
Multivalency analysis of B-subunit binding to Gb3-PC-Ch. The compliance of association and dissociation kinetics of the Stx B subunits from BLI experiments with pseudo-first-order kinetics of the association phase was determined by plotting ln(1 − Rt/Re) versus time, whereas the dissociation phase was analyzed by plotting ln(Rt/R0) versus time. Rt is the BLI response at time t, Re is the BLI response at equilibrium during the association phase, and R0 is the BLI response at the commencement of the dissociation phase. (A) Association data for Stx1; (B) dissociation data for Stx1; (C) association data for Stx2a wild type; (D) dissociation data for Stx2a wild type; (E) association data for Stx2a Q40L mutant; (F) dissociation data for Stx2a Q40L mutant. (G) (Left) Model of pentamer binding at concentrations greater than the KD for pentamer formation, for example, with Stx1 and Stx2a Q40L. (Right) For binding at concentrations below the KD for pentamer formation, for example, with the Stx2a wild type, weak binding of B monomers to Gb3 can lead to an increase in the local concentration of B monomers near the surface of the tip, driving pentamer formation, which in turn results in more avid binding.
The association curves at different concentrations were superimposable for Stx1 (Fig. 6A) and Stx2a B-Q40L (Fig. 6E), suggesting that binding was not concentration dependent. Binding was assessed at concentrations ranging from 10 down to 0.1 μM, i.e., concentrations at which pentamer formation is favored for these proteins (Stx1 EC50 = 0.043 μM; Stx2a B-Q40L EC50 = 0.11 μM). In contrast, binding of the Stx2a B subunit was concentration dependent (Fig. 6C). The EC50 for the Stx2a B subunit is 2.3 μM, and rapid association was seen at high concentrations (10 to 1 μM), where pentamer formation is more favored, while slow association was seen at 0.32 and 0.1 μM, where pentamer formation is not favored. Concentration dependence was not observed for dissociation, suggesting disengagement of a single species, i.e., bound pentamers.
A2-subunit domains are critical for holotoxin assembly and receptor binding.
Both A and B subunits are necessary for toxicity, and we examined the structural basis for A- and B-subunit association. The Stx A subunit is proteolytically processed, forming two domains joined by a disulfide bond (30). The N-terminal A1 domain possesses the catalytic activity, while the C-terminal A2 domain mediates association with the B pentamer. The crystal structure of Stx2a (Fig. 7A) reveals that A2 associates with the B pentamer via a 19-amino-acid tail that extends through a pore in the B pentamer and via tryptophan 277, which binds a hydrophobic pocket on a single B subunit. We obtained peptides (Fig. 7B) corresponding to the wild-type A2 subunit (A2-WT), a truncated version lacking the C-terminal tail (A2-Stop), and a version with the hydrophobic tryptophan replaced by a bulky, hydrophilic arginine residue (A2-W277R).
FIG 7.

A2-subunit domains are critical for holotoxin assembly and receptor binding. (A) Crystal structure of Stx2a, showing the A1 enzymatic subunit (gray), the B pentamer (red), and the A2 subunit (blue and green). Trp-277 (green) sits in a hydrophobic pocket at the B-subunit interface. The image was generated from PDB entry 1R4P by using PyMOL. (B) Amino acid sequences of A2 peptides. (C) Binding of 10 μM Stx2a B subunits to A2 peptides was assessed by BLI. Means for three independent repeats were plotted. (D) Gb3 binding of A2-peptide and B-subunit mixtures. (E) Equilibrium dissociation constant (KD) values for Gb3 binding of mixtures, calculated from Bmax values for association.
Binding of purified B subunits to immobilized A2 peptides was assessed using BLI (Fig. 7C). The A2-WT peptide mediated rapid association and slow dissociation and displayed the highest binding level compared to the truncated A2-Stop peptide and the A2-W277R peptide. These results suggest that both regions of the A2 peptide are important for optimal B-subunit association.
Binding of the B subunit to Gb3-PC-Ch in the presence of the A2 peptides was also assessed (Fig. 7D). The presence of the A2 peptides resulted in enhanced binding of the B subunit. KD values were calculated (Table 1): the presence of the A2-WT peptide promoted 4-fold more avid binding, as evidenced by a KD of 40 nM in the presence of the A2-WT subunit, compared to the KD of 160 nM for the B subunit alone. A2-Stop and A2-W277R promoted less avid binding. These studies suggest that A- and B-subunit assembly plays a very important role in glycolipid binding of Stx and that both the C-terminal tail and W277 are needed for optimal association.
The A2 peptide rescues mice from lethal doses of Stx2a.
We wanted to determine if the A2 peptides, which lack enzymatic activity, could compete with full-length A subunits and protect mice from a lethal challenge with Stx2a. A lethal dose of purified Stx2a holotoxin (7 ng) was incubated at 37°C for 1 h without or with peptide (1 μg per mouse) prior to injection (about a 2,000-fold molar excess compared to holotoxin). Peptides were not toxic to Vero cells when tested at 100 μg/ml, and no deaths were seen in control groups injected with PBS or peptide alone (Fig. 8). In contrast, only 4 of 22 mice injected with purified Stx2a survived. The A2-WT peptide conferred significant protection (P < 0.03) from Stx2a-mediated mortality, with 15 of 22 mice surviving the challenge. Protection by the A2-Stop and A2-W277R peptides did not reach statistical significance.
FIG 8.
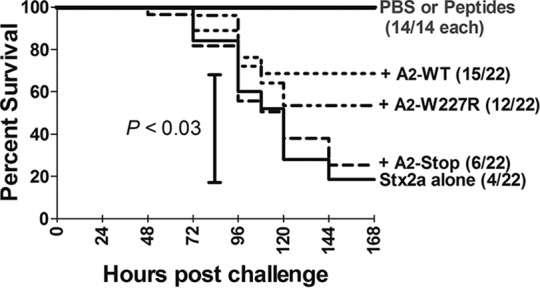
The A2 peptide rescues mice from lethal doses of Stx. Stx2a (7 ng) and peptides (1 μg) were incubated alone or together at 37°C for an hour before injection into mice. Kaplan-Meier survival curves were plotted, with the number of mice per group indicated in parentheses. Statistical analysis was performed using unpaired Student's t test in GraphPad Prism 5.0.
DISCUSSION
Previous studies using electrospray ionization mass spectrometry (17, 18) and analytical ultracentrifugation (16) reported an instability of Stx assembly. Using biochemical techniques, including size exclusion chromatography, centrifugal filtration, and native gel isoelectric focusing, we confirmed that the Stx holotoxin is unstable. We report for the first time that preassembly is not required for toxicity, as unequivocally demonstrated by the death of mice injected separately with purified A and B subunits.
Unlike other AB5 toxins, Stx is phage encoded in E. coli. The toxin is not secreted; rather, it is released by phage-mediated lysis. It is well established that the Stx A subunit needs the B pentamer for cellular entry, and the ability of the individual A and B subunits to retain function compensates for the lack of a presecretion mechanism to ensure holotoxin assembly of subunits released by phage lysis. While holotoxin assembly must occur in vivo, isoelectric focusing studies demonstrate that holotoxin is not the favored assembly state in solution at lethal concentrations. Real-time binding studies using BLI demonstrated that the presence of the A subunit enhances B-subunit binding to Gb3 (Fig. 7D), suggesting that glycolipid receptors arrayed on a surface can template holotoxin assembly.
It is important that Stx2a is one of the most potent bacterial toxins, causing death in mice at picomolar concentrations (20), and its unusual structural properties are likely to play a role in enhancing potency. Stx crystal structures demonstrate that receptor recognition is mediated primarily by the two terminal sugars on Gb3, Galα1-4Gal (28). This motif is widely expressed on glycoproteins as well as glycolipids, and off-target binding can reduce toxicity. Stx2a is about 100 times more toxic to mice than Stx1. It has been hypothesized that enhanced receptor discrimination of Stx2a limits its association with inappropriate target tissues. In mice injected with radiolabeled toxin (31), Stx2a accumulated primarily in susceptible tissues (the kidneys and the brain), while high levels of Stx1 were detected in the lungs, which are tissues not affected by the toxin. However, in vitro studies comparing levels of binding to receptor mimics typically report stronger binding for the less potent Stx1 than for Stx2a (22), and the weak binding of the more potent Stx2a toxin is likely because we have not yet elucidated the actual structure that mediates high-affinity cellular binding of Stx2a in vivo.
Two unusual features of Stx2 may limit off-target recognition and ensure that binding occurs only to densely arrayed glycans, such as Gb3 in lipid rafts. Sequences of hundreds of Stx2 variants deposited in GenBank reveal that the pentamer-destabilizing amino acid Q40 is highly conserved, suggesting that the destabilized pentamer phenotype confers a strong selective advantage. In addition, the crystal structure of Stx2a (26) reveals that the B subunit has two glycan binding sites. While one site is contained within a single B subunit, the other is formed by amino acids from adjacent subunits, and this glycan binding site would be present only in the pentamer. In our studies, for wild-type Stx2a, the KD for glycolipid binding was 14-fold lower than the EC50 for pentamer formation (Table 1). While this may suggest that pentamers are not required for Stx2a binding, it seems unlikely given that the affinity of the Stx2a B subunit for Gb3 monomers in solution is very low, in the millimolar range (27). It is more likely that receptors displayed on a solid surface enhance pentamer assembly. At concentrations below the EC50 for pentamer assembly, Stx2a binding to Gb3 is time dependent (Fig. 6C) and can be modeled as a multistep process (Fig. 6G), as follows. First, a monomer associates with the receptor. While the weak affinity for glycan is not sufficient to promote stable binding, the weak on-off associations can increase B-subunit accumulation near the surface of the receptor, resulting in an increase in the local concentration of subunit, which can ultimately drive pentamer formation. Once formed, the pentamer creates the second, intersubunit binding site, which can engage ligand and lock the pentamer to the receptor. A similar process likely occurs for Stx1, but our methods are not sensitive enough to detect binding at values below the EC50 (0.043 μM) for pentamer formation of Stx1.
At present, treatment of STEC infection is primarily limited to supportive therapy. Antibiotics are not recommended due to their ability to activate the bacteriophage SOS response, which greatly increases toxin expression (32). The ability of the A2 peptide to protect mice from lethal doses of Stx suggests that agents that interrupt Stx assembly hold promise as therapeutics for treating Stx-related diseases.
ACKNOWLEDGMENTS
The Biodefense and Emerging Infections Research Resources Repository (BEI) provided purified Stx1 and Stx2a and antibodies to Stx. We thank Rhett Kovall for identifying interaction sites in the Stx crystal structures.
This work was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (grants U01AI075498 and R01 AI 064893 to A.A.W.). C.A.P. was supported by an NSF IGERT predoctoral training fellowship.
REFERENCES
- 1.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gyles CL. 2007. Shiga toxin-producing Escherichia coli: an overview. J Anim Sci 85:E45–E62. doi: 10.2527/jas.2006-508. [DOI] [PubMed] [Google Scholar]
- 3.Melton-Celsa A, Mohawk K, Teel L, O'Brien A. 2012. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr Top Microbiol Immunol 357:67–103. doi: 10.1007/82_2011_176. [DOI] [PubMed] [Google Scholar]
- 4.Mayer CL, Leibowitz CS, Kurosawa S, Stearns-Kurosawa DJ. 2012. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins 4:1261–1287. doi: 10.3390/toxins4111261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Proulx F, Siedman EG, Karpman D. 2001. Pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Pediatr Res 50:163–171. doi: 10.1203/00006450-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Zoja C, Buelli S, Morigi M. 2010. Shiga toxin-associated hemolytic uremic syndrome: pathophysiology of endothelial dysfunction. Pediatr Nephrol 25:2231–2240. doi: 10.1007/s00467-010-1522-1. [DOI] [PubMed] [Google Scholar]
- 7.McCluskey AJ, Poon GM, Bolewska-Pedyczak E, Srikumar T, Jeram SM, Raught B, Gariepy J. 2008. The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J Mol Biol 378:375–386. doi: 10.1016/j.jmb.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Obata F, Tohyama K, Bonev AD, Kolling GL, Keepers TR, Gross LK, Nelson MT, Sato S, Obrig TG. 2008. Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. J Infect Dis 198:1398–1406. doi: 10.1086/591911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soltyk AM, MacKenzie CR, Wolski VM, Hirama T, Kitov PI, Bundle DR, Brunton JL. 2002. A mutational analysis of the globotriaosylceramide-binding sites of verotoxin VT1. J Biol Chem 277:5351–5359. doi: 10.1074/jbc.M107472200. [DOI] [PubMed] [Google Scholar]
- 10.Müthing J, Schweppe CH, Karch H, Friedrich AW. 2009. Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thromb Haemost 101:252–264. doi: 10.1160/th08-05-0317. [DOI] [PubMed] [Google Scholar]
- 11.Sandkvist M, Michel LO, Hough LP, Morales VM, Bagdasarian M, Koomey M, DiRita VJ. 1997. General secretion pathway (eps) genes required for toxin secretion and outer membrane biogenesis in Vibrio cholerae. J Bacteriol 179:6994–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiss AA, Johnson FD, Burns DL. 1993. Molecular characterization of an operon required for pertussis toxin secretion. Proc Natl Acad Sci U S A 90:2970–2974. doi: 10.1073/pnas.90.7.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Brien AD, Newland JW, Miller SF, Holmes RK, Smith HW, Formal SB. 1984. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 226:694–696. doi: 10.1126/science.6387911. [DOI] [PubMed] [Google Scholar]
- 14.Mauro SA, Koudelka GB. 2011. Shiga toxin: expression, distribution, and its role in the environment. Toxins (Basel) 3:608–625. doi: 10.3390/toxins3060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimmitt PT, Harwood CR, Barer MR. 2000. Toxin gene expression by Shiga toxin-producing Escherichia coli: the role of antibiotics and the bacterial SOS response. Emerg Infect Dis 6:458–465. doi: 10.3201/eid0605.000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrady DG, Flagler MJ, Friedmann DR, Vander Wielen BD, Kovall RA, Weiss AA, Herr AB. 2010. Molecular basis of differential B-pentamer stability of Shiga toxins 1 and 2. PLoS One 5:e15153. doi: 10.1371/journal.pone.0015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitova EN, Daneshfar R, Marcato P, Mulvey GL, Armstrong G, Klassen JS. 2005. Stability of the homopentameric B subunits of Shiga toxins 1 and 2 in solution and the gas phase as revealed by nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry. J Am Soc Mass Spectrom 16:1957–1968. doi: 10.1016/j.jasms.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Kitova EN, Mulvey GL, Dingle T, Sinelnikov I, Wee S, Griener TP, Armstrong GD, Klassen JS. 2009. Assembly and stability of the Shiga toxins investigated by electrospray ionization mass spectrometry. Biochemistry 48:5365–5374. doi: 10.1021/bi9003155. [DOI] [PubMed] [Google Scholar]
- 19.Le Nours J, Paton AW, Byres E, Troy S, Herdman BP, Johnson MD, Paton JC, Rossjohn J, Beddoe T. 2013. Structural basis of subtilase cytotoxin SubAB assembly. J Biol Chem 288:27505–27516. doi: 10.1074/jbc.M113.462622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuller CA, Pellino CA, Flagler MJ, Strasser JE, Weiss AA. 2011. Shiga toxin subtypes display dramatic differences in potency. Infect Immun 79:1329–1337. doi: 10.1128/IAI.01182-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karve SS, Weiss AA. 2014. Glycolipid binding preferences of Shiga toxin variants. PLoS One 9:e101173. doi: 10.1371/journal.pone.0101173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flagler MJ, Mahajan SS, Kulkarni AA, Iyer SS, Weiss AA. 2010. Comparison of binding platforms yields insights into receptor binding differences between Shiga toxins 1 and 2. Biochemistry 49:1649–1657. doi: 10.1021/bi902084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaston MA, Pellino CA, Weiss AA. 2013. Failure of manganese to protect from Shiga toxin. PLoS One 8:e69823. doi: 10.1371/journal.pone.0069823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 25.Kalinin NL, Ward LD, Winzor DJ. 1995. Effects of solute multivalence on the evaluation of binding constants by biosensor technology: studies with concanavalin A and interleukin-6 as partitioning proteins. Anal Biochem 228:238–244. doi: 10.1006/abio.1995.1345. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson JM, Yin J, Kitov PI, Mulvey GL, Griener TP, James MN, Armstrong GD, Bundle DR. 2014. The crystal structure of Shiga toxin type 2 with bound disaccharide guides the design of a heterobifunctional toxin inhibitor. J Biol Chem 289:885–894. doi: 10.1074/jbc.M113.518886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallegos KM, Conrady DG, Karve SS, Gunasekera TS, Herr AB, Weiss AA. 2012. Shiga toxin binding to glycolipids and glycans. PLoS One 7:e30368. doi: 10.1371/journal.pone.0030368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling H, Boodhoo A, Hazes B, Cummings MD, Armstrong GD, Brunton JL, Read RJ. 1998. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 37:1777–1788. doi: 10.1021/bi971806n. [DOI] [PubMed] [Google Scholar]
- 29.Millen SH, Lewallen DM, Herr AB, Iyer SS, Weiss AA. 2010. Identification and characterization of the carbohydrate ligands recognized by pertussis toxin via a glycan microarray and surface plasmon resonance. Biochemistry 49:5954–5967. doi: 10.1021/bi100474z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garred Deurs B, Sandvig K. 1995. Furin-induced cleavage and activation of Shiga toxin. J Biol Chem 270:10817–10821. doi: 10.1074/jbc.270.18.10817. [DOI] [PubMed] [Google Scholar]
- 31.Rutjes NW, Binnington BA, Smith CR, Maloney MD, Lingwood CA. 2002. Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int 62:832–845. doi: 10.1046/j.1523-1755.2002.00502.x. [DOI] [PubMed] [Google Scholar]
- 32.McGannon CM, Fuller CA, Weiss AA. 2010. Different classes of antibiotics differentially influence Shiga toxin production. Antimicrob Agents Chemother 54:3790–3798. doi: 10.1128/AAC.01783-09. [DOI] [PMC free article] [PubMed] [Google Scholar]