

Abstract
Multipotent adult progenitor cells are a recently described population of stem cells derived from the bone marrow stroma. Research has demonstrated the potential of multipotent adult progenitor cells for treating ischemic injury and cardiovascular repair; however, understanding of multipotent adult progenitor cells in orthopedic applications remains limited. In this study, we evaluate the osteogenic and angiogenic capacity of multipotent adult progenitor cells, both in vitro and loaded onto demineralized bone matrix in vivo, with comparison to mesenchymal stem cells, as the current standard. When compared to mesenchymal stem cells, multipotent adult progenitor cells exhibited a more robust angiogenic protein release profile in vitro and developed more extensive vasculature within 2 weeks in vivo. The establishment of this vascular network is critical to the ossification process, as it allows nutrient exchange and provides an influx of osteoprogenitor cells to the wound site. In vitro assays confirmed the multipotency of multipotent adult progenitor cells along mesodermal lineages and demonstrated the enhanced expression of alkaline phosphatase and production of calcium-containing mineral deposits by multipotent adult progenitor cells, necessary precursors for osteogenesis. In combination with a demineralized bone matrix scaffold, multipotent adult progenitor cells demonstrated enhanced revascularization and new bone formation in vivo in an orthotopic defect model when compared to mesenchymal stem cells on demineralized bone matrix or demineralized bone matrix–only control groups. The potent combination of angiogenic and osteogenic properties provided by multipotent adult progenitor cells appears to create a synergistic amplification of the bone healing process. Our results indicate that multipotent adult progenitor cells have the potential to better promote tissue regeneration and healing and to be a functional cell source for use in orthopedic applications.
Keywords: Multipotent adult progenitor cell, demineralized bone matrix, tissue engineering, angiogenesis, osteogenesis, bone
Introduction
Regenerative medicine emphasizes the use of stem cells in conjunction with biologic and synthetic scaffolds. The application of stem cells to a wound site can substantially improve the time, quality, and overall extent of healing.1–6 It is widely known that production and maintenance of bone tissue are mediated largely by a cascade of molecular signals that are released by and acted upon lineage specific stem cells. These cells can either differentiate or participate in further signal conduction with growth factors and hormones to facilitate bone remodeling.7 Resident bone cells can provide signals to osteoprogenitor cells through pathways including, but not limited to, Wnt, transforming growth factor-beta (TGF-β), and bone morphogenic protein (BMP).7–12
Osteogenesis is traditionally considered the most important element for new bone formation; however, it is widely known that angiogenesis plays an important role in bone repair as well. New blood vessel formation allows for the migration of cells and necessary nutrients to the site of injury. Studies have shown that promotion of vessel formation in an injury model can influence bone healing and that health factors that negatively impact neovascularization (i.e. smoking or diabetes) can cause delayed fracture healing or result in nonunions.13 Furthermore, the literature has demonstrated that vascular endothelial growth factor (VEGF) is crucial to the bone remodeling process. Studies have shown that both VEGF and BMPs increase the differentiation of pre-osteoblasts and that they elicit a synergistic effect on bone formation.13–18
Many orthopedic injuries involve major trauma to the skeletal system and require surgical intervention. Therapeutic approaches to treating damaged or diseased tissue can include a combination of hardware, bioactive large molecules (i.e. growth factors and cytokines), viable cells, and/or natural or synthetic scaffolds.19–22 While hardware and scaffolds assist in maintaining structural support and load-bearing integrity, bioactive large molecules and viable cells provide signals for the host to upregulate new tissue formation and stimulate the healing process. One of the most common cell sources for orthopedic applications is bone marrow–derived mesenchymal stem cells (MSC). MSC are operationally defined as plastic adherent fibroblastic-like cells capable of differentiating along mesodermal lineages, including bone.23,24 While MSC are established, and their applications are well published in the orthopedic field, there may be alternative cell types possessing greater therapeutic implications. One such population, termed multipotent adult progenitor cells (MAPC), was first described in 2002 by Jiang et al.25 MAPC are nonhematopoietic stem cells derived from the bone marrow stroma. MAPC have a broader lineage differentiation capacity than MSC, generating cells of the mesenchymal lineage, as well as endothelium, hematopoietic cells, hepatocyte-like cells, and neuroectoderm-like cells.24–35 In addition to their broader differentiation potential, MAPC can proliferate without obvious signs of senescence and can be expanded to over 70 passage doublings while remaining cytogenetically normal.26,36
To date, research has focused predominantly on the immunomodulatory properties of MAPC as well as their therapeutic efficacy in models of myocardial infarction and hypoxic ischemia.37–40 MAPC have been reported to release proangiogenic factors including VEGF, interleukin 8 (IL-8), and CXCL-5, all of which are required for in vitro vascular tube formation.41–44 The therapeutic potential of MAPC has been studied in numerous in vivo ischemia models, which indicate that grafted MAPC significantly increase angiogenesis, as well as endogenous stem cell proliferation.45–48
MAPC have been shown to differentiate along mesodermal lineages, including undergoing osteogenic differentiation in vitro; however, MAPC have not been fully investigated for their use in orthopedic applications. A previous study performed in a heterotopic model demonstrated increased osteoinductivity of a MAPC-loaded demineralized bone matrix (DBM) scaffold.49 While the authors presented the ability of MAPC to undergo and/or promote ossification in vivo, the study did not utilize an injury model in an orthotopic site. Another limitation of the study was the absence of an injury-induced inflammatory response, which is significant in the bone healing process. Studies have shown that stem cells may require an inflammatory stimulus to initiate the healing response.50,51 While an initial inflammatory response is normal and may trigger advantageous cellular responses, prolonged or persistent inflammation can negatively impact the healing process.51 MAPC possess demonstrated immunomodulatory properties and the ability to attenuate a local host immune response upon implantation.38,39,52–56 Lehman et al.50 established that endothelial cells exhibit a reduced production of vascular cell adhesion molecule (VCAM), E-selectin, and intercellular adhesion molecule (ICAM) when co-cultured with MAPC; the attenuated presence of these proteins reduces neutrophil binding to endothelial cells. This phenomenon leads to decreased endothelial activation that may reduce inflammation and neutrophil infiltration.
This study further investigates the role of MAPC in the orthopedic milieu by applying them in an orthotopic, segmental defect model, while comparing their performance to the current MSC standard. In this study, MAPC isolated from human bone marrow demonstrated a select angiogenic protein release profile which surpassed that of MSC in vitro. In vivo, MAPC seeded onto DBM and implanted into a fibular defect contributed to wound closure and promoted enhanced vascularization. In conjunction with improved neovascularization, treatment groups with MAPC demonstrated increased bone healing. These results suggest a synergistic relationship between angiogenic and osteogenic elements that may accelerate the bone healing process.
Materials and methods
Cell isolation and culture
MAPC used in this study were isolated as previously described by Yasuhara et al.57 and cultured as described by Boozer et al.58 MAPC were characterized according to the methods of Sohni and Verfaillie.37 MAPC were cultured and expanded at 37°C and 3% O2. Passage numbers ranging from 2 to 4 were used for all experiments. MAPC isolated from 10 donors were validated for consistency in morphology, growth rates, surface markers, and cytokine expression (data not shown). Bone marrow–derived MSC (Normal, Human, ATCC®PCS-500-012) were purchased from ATCC (Manassas, VA) and cultured according to manufacturer instructions. MSC used in this study were selected as a model system and have been verified by ATCC to expand to 15 population doublings while still maintaining characteristics of primary MSC. These characteristics include morphology, growth curves, differentiation potential, surface marker expression, immunosuppression, and tube formation.59 These data are comparable to findings published on isolated primary bone marrow–derived MSC.60–63
Osteogenic differentiation
One million MAPC were seeded onto fibronectin (5 ng/mL)-coated flasks and cultured in MAPC maintenance medium (Lonza, Basel, Switzerland) for 3 days at 37°C and 3% O2. After 3 days, the medium was replaced by osteogenic differentiation medium.64 Osteogenic differentiation medium consisted of high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Carlsbad, CA), dexamethasone (Sigma–Aldrich, St Louis, MO), ascorbic acid 2-phosphate (Sigma–Aldrich), β-glycerophosphate (Sigma–Aldrich), fetal bovine serum (Life Technologies), and penicillin–streptomycin (Life Technologies). Cells were cultured at 37°C and 21% O2 for an additional 14, 21, and 28 days, with medium changes every 3–4 days.
Adipogenic differentiation
MAPC were prepared as described in the “Osteogenic differentiation” section with the substitutions of adipogenic differentiation medium65 and Oil Red O staining (Sigma–Aldrich). Adipogenic differentiation medium contained α-minimum essential medium (MEM; Life Technologies), fetal bovine serum, hydrocortisone (Sigma–Aldrich), isobutylmethylxanthine (Sigma–Aldrich), indomethacin (Sigma–Aldrich), penicillin–streptomycin, and dexamethasone.
Chondrogenic differentiation
One million MAPC were seeded onto fibronectin (5 ng/mL)-coated flasks and cultured in maintenance medium for 3 days at 37°C and 3% O2. After 3 days, cells were transferred onto a round bottom 96-well tissue culture plate at a density of 4.5 × 105 cells/well and cultured in chondrogenic differentiation medium64 (high-glucose DMEM, dexamethasone, ascorbic acid 2-phosphate, proline (Sigma–Aldrich), sodium pyruvate (Sigma–Aldrich), penicillin–streptomycin, Insulin transferrin selenium (ITS) + premix (Corning, Corning, NY), and TGF-β1 (Sigma–Aldrich)). MAPC were aggregated by centrifugation at 500g for 5 min and then returned to the hypoxic incubator. After 24 h, the medium was changed and aggregates were gently released from the sides and bottom of the wells by pipetting. Aggregates were cultured for 14, 21, and 28 days, with media changes every 3–4 days. At the end of the culture period, the aggregates were fixed in formalin, dehydrated, embedded in paraffin, and stained with toluidine blue (Sigma–Aldrich).
Qualitative alkaline phosphatase staining
MAPC or MSC were cultured in control medium or osteogenic medium for 8 days. Alkaline phosphatase (ALP) staining was performed using a napthol AS-MX phosphate and fast red violet B salt-based kit (Sigma–Aldrich). Briefly, cells were fixed in citrate-buffered acetone and rinsed with water. Cells were exposed to the alkaline dye mixture for 30 min, rinsed with water, visualized using light microscopy, and imaged. Images were taken at 10× magnification.
Quantitative ALP staining
MAPC or MSC were cultured in osteogenic medium for 8 days. Supernatant media were collected and analyzed for the presence of ALP using QuantiChrom™ ALP Assay Kit (BioAssay Systems, Hayward, CA). Briefly, media were exposed to the supplied working solution, and the optical density was read at 405 nm at t = 0 min and t = 4 min.
Qualitative alizarin red staining
MAPC or MSC were cultured in control or osteogenic medium for 21 days. Alizarin red staining was performed according to standard protocol.66 Briefly, cells were rinsed with phosphate-buffered saline (PBS) and fixed in 10% formalin. Cells were stained with alizarin red (Sigma–Aldrich) for 20 min, rinsed with water, and visualized using light microscopy. Images were taken at 10× magnification.
Quantitative calcium assay
MAPC or MSC were cultured in osteogenic medium for 21 days. Calcium production was analyzed using a calcium reagent set (Pointe Scientific, Canton, MI). Briefly, cells were lysed with 0.5 N HCl to expose the mineral to the acid, and the volume was collected. The samples were incubated with calcium reagents for 10 minutes at room temperature, and the absorbance was read at 570 nm.
Angiogenic protein analysis
MAPC or MSC were plated at a density of 1 × 105 cells/well in a 24-well tissue culture plate. After 24 h, the medium was removed and replaced with fresh serum-free medium. Following an additional 24 h at 37°C, 3% O2 (MAPC), or 21% O2 (MSC), the media were harvested for use in an enzyme-linked immunosorbent assay (ELISA). IL-8, CXCL-5, VEGF, and GRO-α ELISAs (R&D Systems, Minneapolis, MN) were performed according to the manufacturers’ instructions and normalized to total protein levels using the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA).
Matrigel tube formation assay
Human umbilical vein endothelial cells (HUVEC; Lifeline Technology, Frederick, MD) were cultured in standard medium until they reached 70%–80% confluence. Matrigel (Becton Dickinson, Franklin Lakes, NJ) was added to the wells of a µ-angiogenesis slide (ibidi, Verona, WI) and allowed to polymerize for 30 min at 37°C. A total of 1 × 104 HUVEC were added to each well in 25 µL of medium, along with 25 µL of positive control medium (basal maintenance medium supplied by the manufacturer with growth factors for vessel formation), negative control medium (media devoid of growth factors), or conditioned medium from MAPC or MSC. Wells were imaged at 2, 4, and 6 h.
Surgical methodology
Male athymic rats (7–8 weeks old) purchased from Harlan Laboratories (Indianapolis, IN) were used for this study. The animals were acclimated for 48 h prior to surgery. On the day of surgery, animals were anesthetized and surgically prepared. Surgical procedure and postoperative care were conducted according to the established protocol approved by the Institutional Animal Care and Use Committee of the University of Florida. Briefly, the fibula was accessed through a lateral skin incision and blunt dissection of the musculature. A 4-mm segmental defect in the fibula was created unilaterally. Defects in experimental groups were treated with a DBM scaffold seeded with MAPC (MAPC + DBM scaffold) or MSC (MSC + DBM scaffold) at a concentration of 175,000 cells/cm3 of DBM, while defects in the control group received DBM scaffold only. All DBM scaffolds originated from the same donor lot to ensure consistency. Musculature was sutured, and skin wounds were closed using skin clips. Animals were sacrificed at 14 and 28 days post-implantation for histological evaluation of new vessel and bone formation.
Histological methodology
After the explants and surrounding tissue were removed, the specimens were fixed, decalcified, and embedded in paraffin using standard protocols. Specimens from the 14-day time point were sectioned transversely, while those from the 28-day time point were sectioned longitudinally to better expose the defect areas for evaluation. Four sections representing different depths into the defect were collected and stained with hematoxylin and eosin (H&E). Blood vessel quantification was performed on the 14-day specimens, and bone repair was evaluated on the 14- and 28-day specimens.
Histological evaluation
All histological evaluation, analysis, and reporting were performed by an outside contract laboratory. The magnitude of vascular network formation and bone healing at the fracture site was evaluated using a semiquantitative severity scoring system with a 0- to 5-point scale width, in which higher scores correlated with normal tissue architecture or improved healing. Tables 1–3 illustrate the comprehensive scoring criteria.
Table 1.
Woven bone scoring key.
| Score | Description |
|---|---|
| 5 (good) | Past woven bone stage |
| 4 | Abundant woven bone |
| 3 | Woven bone becoming organized into trabeculae |
| 2 | Moderate osteoid ECM between osteoblasts |
| 1 | Scant osteoid ECM deposition between mesenchymal cells |
| 0 (bad) | No bone formation |
ECM: extracellular matrix.
Table 2.
Lamellar bone scoring key.
| Score | Description |
|---|---|
| 5 (good) | Lamellar bone completely reformed |
| 4 | Regeneration of lamellar bone |
| 3 | Large regions of lamellar bone |
| 2 | Regional lamellar bone formation |
| 1 | Scant lamellar bone |
| 0 (bad) | No lamellar bone present |
Table 3.
Marrow space scoring key.
| Score | Description |
|---|---|
| 5 (good) | Mixture of lineages filling marrow space |
| 4 | Marrow space between trabeculae with hematopoiesis |
| 3 | Marrow space between trabeculae with scant hematopoiesis |
| 2 | Scant fibrillary stroma with thin-walled vessels |
| 1 | Loose mesenchymal tissue in marrow space |
| 0 (bad) | No marrow space present |
Immunohistochemistry
Immunohistochemical detection of von Willebrand factor (vWF) protein was performed on sections of formalin fixed, paraffin-embedded rat fibulas. Sections were deparaffinized in xylene and then rehydrated through a graded alcohol series. Antigen retrieval was performed for 10 min at 37°C using a Proteinase K solution (Life Technologies). Nonspecific binding was blocked with 10% goat serum (Life Technologies) and 1% bovine serum albumin (BSA; Sigma–Aldrich) in tris-buffered saline and Tween 20 (TBSt) for 1 h at room temperature. Sections were then incubated overnight with anti-vWF antibody (Abcam, Cambridge, England) at a dilution of 1:200 in the blocking buffer. Alexa Fluor 488 goat anti-rabbit IgG (ThermoFisher Scientific, Waltham, MA) was added for 1 h at room temperature, protected from exposure to light. Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies), and coverslips were mounted with Permount Mounting Medium (Thermo Fisher Scientific). Protein-antibody complexes were detected using an Olympus DSU-IX81 spinning disc confocal microscope (Olympus, Shinjuku, Tokyo, Japan). A total of 6–10 images were taken per stack, and images were deconvolved using the nearest-neighbor method. Maximum intensity z-projection was used to obtain two-dimensional (2D) images. Signal intensity and exposure duration were consistent across all groups.
Statistics
All quantitative assays were performed in at least triplicate, and the mean value was used. Data are presented as mean ± standard deviation (SD) with p < 0.05 indicative of significance, unless otherwise specified. If normality passed, a Student’s t test (for comparison of two groups) or a standard analysis of variance (ANOVA; for comparison of three or more groups) was used, followed by Tukey’s post hoc test. If normality failed, a nonparametric Kruskal–Wallis ANOVA on ranks was used followed by a Mann–Whitney U post hoc test. Data were analyzed using SigmaStat software (Systat Software, Inc., Chicago, IL).
Results
MAPC differentiate along the mesoderm lineages
To complement surface marker characterization data as previously described,37 MAPC were cultured in specific medium that promotes differentiation along osteogenic, chondrogenic, and adipogenic lineages. Osteogenic potential was demonstrated by visualization of calcium deposition in the extracellular matrix using alizarin red (Figure 1(b)). Adipogenesis was confirmed by cytoplasmic lipid droplets and intracellular lipid vesicle formation via Oil Red O staining (Figure 1(c)). Finally, chondrogenesis was evidenced by positive staining for glycosaminoglycans using toluidine blue staining (Figure 1(d)). Biphasic organization, indicative of a chondrogenic phenotype, was observed as well. Positive staining was not observed in any of the control conditions (Figure 1(a)). MSC differentiation was performed by ATCC.59
Figure 1.
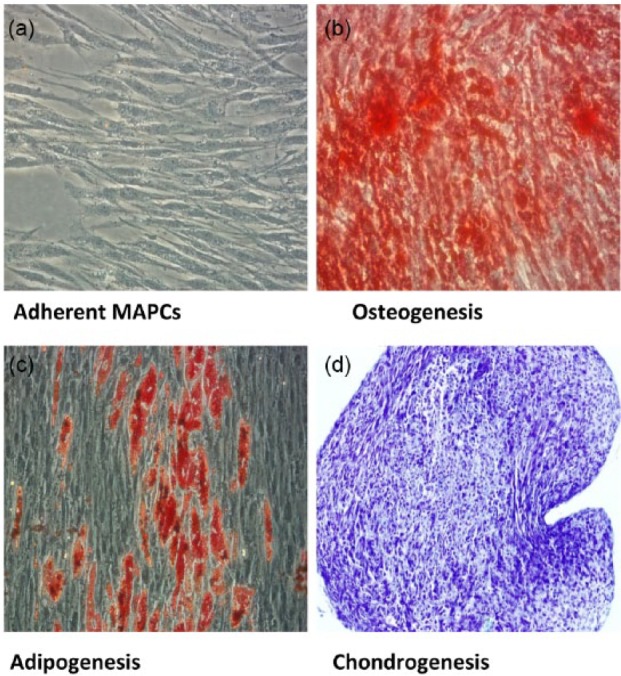
Multipotent differentiation potential of MAPC: (a) MAPC were cultured in control medium and (b–d) selected differentiation media for 21 days: (b) differentiated MAPC displaying calcium deposition with alizarin red staining, (c) differentiated MAPC exhibiting positive staining for lipids using Oil Red O stain, and (d) differentiated MAPC with positive staining for proteoglycans with toluidine blue. Images a–c are at 20× magnification. Image d is at 10× magnification.
MAPC qualitatively express more ALP when compared to MSC
MAPC and MSC were cultured in control and osteogenic media for 8 days. Qualitative staining indicated low levels of ALP expression (dark purple staining) by MAPC in control medium, which became abundant when the cells were exposed to osteogenic conditions (Figure 2(a) and (b)). MSC cultured in control medium did not express ALP (Figure 2(c)), with only mild expression following culture in osteogenic medium, mostly restricted to sparse and random areas (Figure 2(d)).
Figure 2.

Qualitative alkaline phosphatase (ALP—violet color) expression by MAPC and MSC: (a) MAPC in control medium, (b) osteogenic medium, (c) MSC in control medium, and (d) osteogenic medium. All images were visualized using light microscopy; 10× magnification. (e) Quantitative ALP assay indicated enhanced ALP expression in MAPC groups, n = 3. *p < 0.01 when compared to MSC group.
MAPC quantitatively express more ALP when compared to MSC
MAPC and MSC were cultured in control media for 8 days. The quantitative assay indicated significantly higher expression of ALP by MAPC when compared to MSC (Figure 2(e)).
MAPC produce mineral deposits
No evidence of mineral deposition was observed in MAPC, and MSC cultured under control conditions for 21 days (Figure 3(a) and (c)). However, calcium-containing mineral deposits were present in the extracellular matrix of both cell types under osteogenic conditions after 21 days (Figure 3(b) and (d)); both cell cultures exhibited similar levels of expression qualitatively; however, quantitative data demonstrated a significant increase in expression in the MAPC group (Figure 3(e)).
Figure 3.
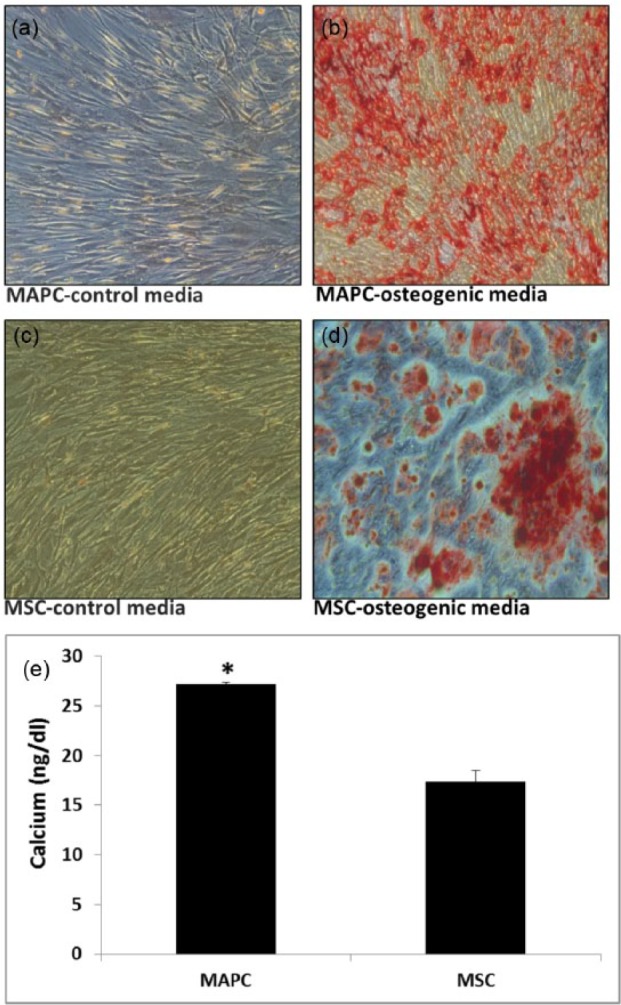
Presence of mineral deposits in MAPC and MSC extracellular matrix: (a) MAPC and (c) MSC cultured in respective control media and (b) MAPC and (d) MSC cultured in osteogenic medium. Alizarin red staining performed after 21 days. All images were visualized using light microscopy; 10× magnification. (e) Quantitative calcium assay indicated elevated levels of calcium in MAPC groups, n = 3. *p < 0.01 when compared to MSC group.
MAPC exhibit increased angiogenic protein expression compared to MSC
An angiogenic cytokine array was performed to qualitatively evaluate the secretion of angiogenic signaling molecules in MAPC and MSC (data not shown). We subsequently quantitatively measured the expression of four commonly known angiogenic markers: VEGF, GRO-α, IL-8, and CXCL-5,41–44,50 using ELISAs. MAPC produced IL-8 (Figure 4(a)) and GRO-α (Figure 4(b)) at significantly higher levels than MSC, while CXCL-5 (Figure 4(d)) was highly produced by MAPC, but undetectable for MSC. VEGF expression was comparable between MAPC and MSC (Figure 4(c)). A matrigel tube formation assay was performed to determine the impact of these significant differences in protein secretion on endothelial cells. The endothelial cells treated with MAPC conditioned medium exhibited a dense tube-like formation within 6 h, while those in the MSC conditioned medium group had only very sporadic tube formation at the same time point (Figure 4(e)).
Figure 4.

In vitro angiogenic potential of MAPC and MSC. (a–d) ELISA analysis using conditioned media from MAPC and MSC in control conditions; (a) ELISA analysis of interleukin 8 (IL-8) secretion in MAPC versus MSC, (b) GRO-α secretion in MAPC versus MSC, and (c) VEGF secretion in MAPC versus MSC. (d) CXCL-5 secretion in MAPC versus MSC. CXCL-5 was below detectable limits of the assay for MSC (n = 3 for each, *p < 0.01). (e) Matrigel tube formation assay with human umbilical vein endothelial cells (HUVEC) exposed to MAPC and MSC conditioned media, n = 3, 6-h time point.
MAPC induce new blood vessel formation in a fibular defect after 14 days
MAPC + DBM scaffold or MSC + DBM scaffold were implanted into a bone void, and the resultant blood vessel formation was examined after 14 days (Figure 5). Histological evaluation revealed that MAPC + DBM scaffold groups developed significantly more blood vessels when compared to the DBM scaffold control and MSC + DBM scaffold groups (Figure 5(a)–(c) and (g)). To highlight these differences in blood vessel formation, slides were stained with vWF (Figure 5(d)–(f)). An increased number of blood vessels were evident in defects treated with MAPC + DBM scaffold (Figure 5(b)and (e)) compared to those treated with MSC + DBM scaffold and the DBM scaffold alone (Figure 5(a), (c), (d), and (f)). In addition, vessels in the MAPC + DBM scaffold group were larger in size and exhibited more mature morphology (Figure 5(b) and (e)).
Figure 5.

In vivo angiogenic potential of MAPC and MSC seeded onto demineralized bone matrix (DBM) scaffolds. Representative photomicrographs of formation of new blood vessels in 2-week-old fractures treated with (a) scaffold only, (b) MAPC + DBM scaffold, or (c) MSC + DBM scaffold. Capillaries (arrows) within the developing fracture calluses illustrate more vascularization in fractures treated with MAPC or MSC in addition to the DBM scaffold (*). Hematoxylin and eosin stain, 40× magnification. (d)–(f) von Willebrand Factor (vWF) staining to illustrate vasculature, at 20× magnification. Scaffold-only, MAPC + DBM scaffold, or MSC + DBM scaffold group was implanted into a bone void, and the resultant blood vessel formation was examined after 14 days. Statistically significant blood vessels were noticeable in defects receiving (e) MAPC + DBM scaffold, compared to either (f) MSC + DBM scaffold or (d) scaffold only. (b and e) The blood vessels in the MAPC + DBM scaffold treatment group appeared larger in size and more fully formed or functional (circular). (g) Histological evaluation revealed a significant increase in blood vessel ingrowth in defects treated with MAPC + DBM scaffold compared to MSC + DBM scaffold and scaffold only. Blood vessels were quantified via H&E stained sections (n = 16 sections analyzed for each, *p < 0.05 when compared to scaffold-only control, #p < 0.05 when compared to MAPC + DBM scaffold).
MAPC + DBM scaffold promotes increased bone repair after 14 and 28 days
The extent of repair in an acute long bone defect was evaluated using an osteogenic approach with MAPC + DBM scaffold and MSC + DBM scaffold as treatment groups. Tables 1 and 2 illustrate the scoring criteria for woven and lamellar bone. After 14 days, the MAPC + DBM scaffold treatment groups had significantly more woven and lamellar bone (Figure 6(d)and (e), respectively) when compared to the scaffold-only group. In addition, the average total bone (Figure 6(f)) was increased in the MAPC + DBM scaffold group when compared to either the DBM scaffold-only or the MSC + DBM scaffold group.
Figure 6.

In vivo assessment of bone repair at 14 days. Representative photomicrographs of granulation tissue and new bone formation (arrows) in 14-day fractures treated with (a) scaffold only, (b) MAPC + DBM scaffold, or (c) MSC + DBM scaffold. In fractures treated with (a) scaffold only, a fibroblastic response is present around the cellular scaffold fragments (*). In fractures treated with (b) MAPC or (c) MSC in addition to the scaffold, mesenchymal cells produce osteoid (arrows) or chondroid extracellular matrix with partial incorporation of acellular scaffold fragments. Hematoxylin and eosin stain, 40× magnification. Results indicated that MAPC + DBM scaffold groups had significantly more (d) woven and (e) lamellar bone when compared to the scaffold-only group. (f) MAPC + DBM scaffold groups had significantly more total bone when compared to scaffold-only and MSC + DBM scaffold groups (n = 20 each, *p < 0.001).
Furthermore, at 28 days, mineralized callus containing developing areas of bone marrow was detected in the MAPC + DBM scaffold group with minimal fibrous tissue (Figure 7(b)). The MAPC + DBM scaffold treatment group demonstrated significantly higher deposition of woven bone, when compared to the MSC + DBM scaffold treatment group (Figure 7(d)). While the MSC + DBM scaffold group also contained woven bone, it was to a lesser degree than in the MAPC + DBM scaffold group, with substantial fibrous tissue between proximal and distal ends of the fibular defect in the MSC + DBM scaffold group (Figure 7(a)–(c)).
Figure 7.

In vivo assessment of bone repair at 28 days. Representative photomicrographs of new bone formation (arrows) in 28-day fractures treated with (a) scaffold only, (b) MAPC + DBM scaffold, or (c) MSC + DBM scaffold. In fractures treated with (b) MAPC + DBM scaffold or (c) MSC + DBM scaffold, there is robust incorporation of acellular scaffold fragments (*) by newly formed bone, while in fractures treated with (a) the DBM scaffold only, new bone migrates from the defect margins. Note that the marrow spaces (M) contain hematopoietic cells and thin-walled vascular sinuses. Histological sections were stained with hematoxylin and eosin, and imaged at 20× magnification. Results indicated that MAPC + DBM scaffold groups had significantly more woven bone when compared to (d) MSC + DBM scaffold group. (e) Marrow organization scores among the three groups (n = 20 each, *p < 0.001).
Additionally, higher levels of marrow organization (Table 3) are present in the MAPC + DBM scaffold treatment group (Figure 7(e)), further supporting the accelerated bone healing demonstrated in the 14-day data.
Discussion
Currently, the stem cell landscape has expanded from uncharacterized stromal cells to a variety of identifiable cell types (embryonic cell, MSC, marrow-isolated adult multilineage inducible (MIAMI) cell, very small embryonic-like (VSEL) cell, etc.) intended for a multitude of applications including cardiovascular, neural, and musculoskeletal repair.67–70 One of the more recently studied stem cells, MAPC, has the common ability to differentiate along the osteogenic lineage in vitro.25,27,58 We aimed to confirm the potential role of MAPC in an orthopedic setting by expanding upon recent published data. To this end, we selected an in vivo bone defect model that would evaluate osteogenesis, as well as angiogenesis. The rat fibular defect model was chosen for its associated low levels of inflammation, which can provide an environment conducive to cell signaling.50 Previous studies have demonstrated that 14- and 28-day time points provide sufficient time to evaluate revascularization and bone healing, respectively, in this model.50,67
The osteogenic potential of MAPC was verified through an evaluation of osteogenic markers, specifically ALP and degree of mineralization. ALP, a well-known early marker of the osteogenic phenotype, was measured in vitro. MAPC mineral deposition was demonstrated after 21 days in culture via staining with alizarin red. Both ALP and mineral deposition were expressed at higher levels in MAPC when compared to MSC. In vitro angiogenic results demonstrated that MAPC-secreted proteins promoted neovascularization through their effect on HUVEC in a tube formation assay, to a significant level over MSC. Specific angiogenic proteins, such as GRO, IL-8, and CXCL-5, which may contribute to the increased tube formation in vitro and the upregulated neovasculature observed in vivo,41–44,71 were all expressed at significantly higher levels in MAPC when compared to MSC.
To determine whether in vitro angiogenic and osteogenic potential translated to in vivo efficacy, we performed a 28-day study in a rat fibular defect model with MAPC seeded onto DBM scaffolds (which provided the necessary osteoconductive and osteoinductive elements for bone formation). This constitutes the first time that MAPC have been used in a clinically relevant orthopedic application. In addition, this study compared the overall healing potential of MAPC to that of the more commonly used MSC. The DBM scaffold control allowed us to isolate the contribution of the cellular component to osteogenesis and angiogenesis. Healing was first indicated by the presence of increased vasculature in the MAPC + DBM scaffold treatment group at 14 days in comparison to the scaffold-only or MSC + DBM scaffold controls. This was evaluated quantitatively and confirmed via fluorescent-conjugated antibody staining for vWF. It has previously been demonstrated that MAPC possess angiogenic properties that make them ideal for use in cardiovascular applications.24,26,41,43,44 Although it is rarely emphasized, angiogenesis is an important factor in the bone healing cascade, allowing for delivery of cells and nutrients to the damaged tissue during the healing process. Lack of nutrient transport between damaged tissue and the healthy surrounding tissue can often compound the disruptive effects of a bone injury. Deficient vasculature and subsequent impeded revascularization may slow down the healing process and can lead to partial or incomplete healing such as nonunions.13,15–18 Before osteogenesis can occur, vessel healing and revascularization must begin, making this an essential element in bone repair.
Once in vivo neovascularization was confirmed, repair and bridging in a bone defect model were evaluated. New bone formation after 14 days provided evidence of an osteogenic response at the defect site and defects with implanted MAPC + DBM scaffold demonstrated increased bone repair when compared to MSC + DBM scaffold and scaffold-only controls. We speculate that the increase in angiogenic factors resulted in enhanced neovascularization in lieu of an angio-inductive element, allowing for improved nutrient availability as well as an influx of osteoprogenitor cells. These factors likely contributed to the increased bone healing present at the 14-day time point in the MAPC + DBM scaffold group.
Based on the results of this study, it is likely that MAPC + DBM scaffold groups are advancing more rapidly through the stages of bone healing when compared to the MSC + DBM scaffold and scaffold-only groups. At 14 days, there was increased neovascularization, as well as a higher degree of vessel maturity (Figure 5) demonstrated. This is likely a result of the elevated levels of angiogenic factors secreted by MAPC, which subsequently increase the development of blood vessels within the defect. This increase in vasculature resulted in an increase in total bone healing in the MAPC + DBM scaffold group at 14 days (Figure 6). Between 14 and 28 days, the MAPC + DBM scaffold group underwent a significant increase in woven bone, while the MSC + DBM scaffold group maintained a low level, and the DBM scaffold-only group experienced a minor increase (Figure 7). This is a possible indication that MAPC + DBM scaffold groups are progressing more rapidly through the callus ossification stage, in which the vessels that matured in the first 2 weeks provided an influx of osteoprogenitor cells, which then mineralized the cartilage callus and formed woven bone. Additionally, there is early indication of a transition from woven to lamellar bone (Figure 6) in the MAPC + DBM scaffold group, which may suggest that this group is advancing into the bone remodeling stage, the final stage of the ossification process. This is further supported by the early stages of marrow development at 28 days (Figure 7). Figure 8 represents a speculative mechanism of healing of MAPC on DBM scaffold.77
Figure 8.

Proposed Mechanism of MAPC+DBM Scaffold bone healing. Data in this manuscript indicate high degrees of neovascularization and vessel maturity by 2 weeks. This increase in vasculature accelerates formation of the cartilage callus, which is then mineralized to form woven bone within the 2 week period. After 2 weeks, the increase in woven bone begins to plateau, and by 4 weeks there is increased lamellar bone and early marrow development. These results suggest that during the first 2 weeks of healing, defects in the DBM+MAPC scaffold treatment group progress through the callus formation and callus ossification stages, and at 4 weeks the woven bone is being resorbed and replaced with primary lamellar bone, which is characteristic of an advance into the bone remodeling state. Bone illustrations were adapted from Frohlich M (77).
The bone healing mechanism is a complicated process involving multiple steps.72–74 In this study, we investigated two of those steps, angiogenesis and osteogenesis, focusing on the 14-day time point. It is known that MSC condense and undergo chondrogenic differentiation during the process of endochondral ossification.8,74–76 This study demonstrated that MAPC have the capacity to undergo chondrogenesis; however, their involvement in the cartilage stage of endochondral bone formation and their role in callus formation warrant further investigation. Speculatively, MAPC may be responding to the chemical environment at the wound site, recruiting stem cells from the circulation and surrounding tissue, and signaling to host stem cells and osteoprogenitors to stimulate blood vessel formation and bone repair.11 The high base levels of secretion of angiogenic and osteogenic factors observed in vitro may be upregulated in vivo in response to the specific wound environment. The degree of repair demonstrated in this study supports the hypothesis that osteogenic signaling and angiogenic protein release are key mechanisms for the role of MAPC in orthopedic regeneration. This study shows for the first time that MAPC have osteogenic and angiogenic properties in an orthotopic defect model. This study also took the first steps in comparing MAPC + DBM scaffold and MSC + DBM scaffold in terms of angiogenic and osteogenic potential in an orthotopic model. The combination of osteogenic and angiogenic potential from a single cell source, together with the mitigation of a local immune response,39,52–56 can promote enhanced bone healing and result in the more rapid total repair of the defect. The results of this study demonstrate MAPC + DBM scaffold as a promising therapeutic for clinical use in orthopedic applications. Future studies will focus on understanding the signaling mechanisms that drive the strong angiogenic and osteogenic effects of MAPC, the potential early callus formation and chondrogenic effects, cell and scaffold interactions, and the pathways involved.
Conclusion
An in vivo model paired with multiple in vitro assays demonstrated the potential contribution of MAPC at various stages of bone healing. This is the first study to show that MAPC + DBM scaffold exhibit osteogenic and angiogenic properties in an orthotopic, fibular defect model. Furthermore, specific angiogenic factors that may impact revascularization during bone regeneration have been identified, both in vitro and in vivo. Finally, the osteogenic and angiogenic properties of MAPC were compared to those of the more commonly studied MSC. We have demonstrated that MAPC are a promising therapeutic for clinical use in orthopedic applications and have set the foundation for future studies to investigate the mechanisms by which MAPC contribute to the bone healing cascade. This study has implications for the development of synthetic, natural, and composite scaffolds that may benefit from an osteogenic cellular component for the treatment of damaged bone tissue.
Acknowledgments
The authors acknowledge and gratefully thank Dr Peter Supronowicz for his editorial support. Additionally, they thank Dr Elizabeth Whitley for her pathology evaluation and Dr Rasa Zhukauskas for her assistance in the animal studies. Furthermore, they acknowledge Athersys, Inc., for providing guidance regarding multipotent adult progenitor cells.
Footnotes
Declaration of conflicting interests: A.L. and A.H. are employed by RTI Surgical, Inc., as full-time employees.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Machova Urdzikova L, Sedlacek R, Suchy T, et al. Human multipotent mesenchymal stem cells improve healing after collagenase tendon injury in the rat. BioMed Eng Online 2014; 13: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murphy MB, Moncivais K, Caplan AI. Mesenchymal stem cells: environmentally responsive therapeutics for regenerative medicine. Exp Mol Med 2013; 45: e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang L, Burd A. An update review of stem cell applications in burns and wound care. Indian J Plast Surg 2012; 45(2): 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cruz AC, Caon T, Menin A, et al. Adipose-derived stem cells incorporated into platelet-rich plasma improved bone regeneration and maturation in vivo. Dent Traumatol 2015; 31(1): 42–48. [DOI] [PubMed] [Google Scholar]
- 5. Michaeli-Geller G, Zigdon-Giladi H. Bone regeneration induced by stem cells—recent research and future outlook. Refuat Hapeh Vehashinayim 2015; 32(1): 13–20, 59. [PubMed] [Google Scholar]
- 6. Yun JH, Han SH, Choi SH, et al. Effects of bone marrow-derived mesenchymal stem cells and platelet-rich plasma on bone regeneration for osseointegration of dental implants: preliminary study in canine three-wall intrabony defects. J Biomed Mater Res B Appl Biomater 2014; 102(5): 1021–1030. [DOI] [PubMed] [Google Scholar]
- 7. Bab I, Sela J. Cellular and molecular aspects of bone repair. In: Sela JJ, Bab IA. (eds) Principles of bone regeneration. New York: Springer, 2012, pp. 11–41. [Google Scholar]
- 8. Kronenberg HM. Developmental regulation of the growth plate. Nature 2003; 423(6937): 332–336. [DOI] [PubMed] [Google Scholar]
- 9. Qi H, Aguiar DA, Williams SM, et al. Identification of genes responsible for osteoblast differentiation from human mesodermal progenitor cells. P Natl Acad Sci USA 2003; 100(6): 3305–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ilmer M, Karow M, Geissler C, et al. Human osteoblast-derived factors induce early osteogenic markers in human mesenchymal stem cells. Tissue Eng Part A 2009; 15(9): 2397–2409. [DOI] [PubMed] [Google Scholar]
- 11. Kristensen HB, Anderson TL, Marcussen N, et al. Osteoblast recruitment routes in human cancellous bone remodeling. Am J Pathol 2014; 184(3): 778–789. [DOI] [PubMed] [Google Scholar]
- 12. Gaston MS, Simpson AHRW. Inhibition of fracture healing. J Bone Joint Surg Br 2007; 89(12): 1553–1560. [DOI] [PubMed] [Google Scholar]
- 13. Bahney CS, Hu DP, Miclau T, 3rd, et al. The multifaceted role of the vasculature in endochondral fracture repair. Front Endocrinol 2015; 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sfeir C, Ho L, Doll B, et al. Fracture repair. In: Lieberman J, Friedlaender G. (eds) Bone regeneration and repair. New Jersey: Humana Press, Totowa, 2005, pp. 21–44. [Google Scholar]
- 15. Layliev J, Marchac A, Tanaka R, et al. Endogenous cell therapy improves bone healing. J Craniofac Surg 2015; 26(1): 300–305. [DOI] [PubMed] [Google Scholar]
- 16. Stegen S, van Gastel N, Carmeliet G. Bringing new life to damaged bone: the importance of angiogenesis in bone repair and regeneration. Bone 2015; 70: 19–27. [DOI] [PubMed] [Google Scholar]
- 17. Tabbaa SM, Horton CO, Jeray KJ, et al. Role of vascularity for successful bone formation and repair. Crit Rev Biomed Eng 2014; 42(3–4): 319–348. [DOI] [PubMed] [Google Scholar]
- 18. Matsumoto T, Sato S. Stimulating angiogenesis mitigates the unloading-induced reduction in osteogenesis in early-stage bone repair in rats. Physiol Rep 2015; 3(3): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tam RY, Fuehrmann T, Mitrousis N, et al. Regenerative therapies for central nervous system diseases: a biomaterials approach. Neuropsychopharmacology 2014; 39(1): 169–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amini AR, Laurencin CT, Nukavarapu SP. Bone tissue engineering: recent advances and challenges. Crit Rev Biomed Eng 2012; 40(5): 363–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Correia SI, Pereira H, Silva-Correia J, et al. Current concepts: tissue engineering and regenerative medicine applications in the ankle joint. J R Soc Interface 2014; 11(92): 20130784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perán M, García MA, López-Ruiz E, et al. Functionalized nanostructures with application in regenerative medicine. Int J Mol Sci 2012; 13(3): 3847–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284(5411): 143–147. [DOI] [PubMed] [Google Scholar]
- 24. Roobrouck VD, Clavel C, Jacobs SA, et al. Differentiation potential of human postnatal mesenchymal stem cells, mesoangioblasts, and multipotent adult progenitor cells reflected in their transcriptome and partially influenced by the culture conditions. Stem Cells 2011; 29(5): 871–882. [DOI] [PubMed] [Google Scholar]
- 25. Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002; 418(6893): 41–49. [DOI] [PubMed] [Google Scholar]
- 26. Reyes M, Dudek A, Jahagirdar B, et al. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest 2002; 109(3): 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aranguren XL, Luttun A, Clavel C, et al. In vitro and in vivo arterial differentiation of human multipotent adult progenitor cells. Blood 2007; 109(6): 2634–2642. [DOI] [PubMed] [Google Scholar]
- 28. Beresford JN, Bennett JH, Devlin C, et al. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci 1992; 102(2): 341–351. [DOI] [PubMed] [Google Scholar]
- 29. Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. P Natl Acad Sci USA 1999; 96(19): 10711–10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pereira RF, et al. Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. P Natl Acad Sci USA 1995; 92(11): 4857–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petersen BE, Halford KW, O’Hara MD, et al. Bone marrow as a potential source of hepatic oval cells. Science 1999; 284(5417): 1168–1170. [DOI] [PubMed] [Google Scholar]
- 32. Ulloa-Montoya F, Kidder BL, Pauwelyn KA, et al. Comparative transcriptome analysis of embryonic and adult stem cells with extended and limited differentiation capacity. Genome Biol 2007; 8(8): R163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muguruma Y, Reyes M, Nakamura Y, et al. In vivo and in vitro differentiation of myocytes from human bone marrow-derived multipotent progenitor cells. Exp Hematol 2003; 31(12): 1323–1330. [DOI] [PubMed] [Google Scholar]
- 34. Ross JJ, Hong Z, Willenbring B, et al. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J Clin Invest 2006; 116(12): 3139–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwartz RE, Reyes M, Koodie L, et al. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest 2002; 109(10): 1291–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Verfaillie CM, Crabbe A. Chapter 28. Multipotent adult progenitor cells. In: Lanza R, Gearhart J, Hogan B, et al. (eds) Essentials of stem cell biology. 2nd ed. San Diego, CA: Academic Press, 2009, pp. 233–241. [Google Scholar]
- 37. Sohni A, Verfaillie CM. Multipotent adult progenitor cells. Best Pract Res Clin Haematol 2011; 24(1): 3–11. [DOI] [PubMed] [Google Scholar]
- 38. Woda JM, Hof WV. Use of stem cells to reduce leukocyte extravasation. Patent 20140037596 A1, USA, 2014. [Google Scholar]
- 39. Jacobs SA, Roobrouck VD, Verfaillie CM, et al. Immuno-logical characteristics of human mesenchymal stem cells and multipotent adult progenitor cells. Immunol Cell Biol 2013; 91(1): 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mays RW, van’t Hof W, Ting AE, et al. Development of adult pluripotent stem cell therapies for ischemic injury and disease. Expert Opin Biol Ther 2007; 7(2): 173–184. [DOI] [PubMed] [Google Scholar]
- 41. Lehman N, Cutrone R, Raber A, et al. Development of a surrogate angiogenic potency assay for clinical-grade stem cell production. Cytotherapy 2012; 14(8): 994–1004. [DOI] [PubMed] [Google Scholar]
- 42. Ilan N, Mahooti S, Madri JA. Distinct signal transduction pathways are utilized during the tube formation and survival phases of in vitro angiogenesis. J Cell Sci 1998; 111(24): 3621–3631. [DOI] [PubMed] [Google Scholar]
- 43. Li A, Dubey S, Varney ML, et al. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol 2003; 170(6): 3369–3376. [DOI] [PubMed] [Google Scholar]
- 44. Li A, Varney ML, Valasek J, et al. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis 2005; 8(1): 63–71. [DOI] [PubMed] [Google Scholar]
- 45. Aranguren XL, McCue JD, Hendrickx B, et al. Multipotent adult progenitor cells sustain function of ischemic limbs in mice. J Clin Invest 2008; 118(2): 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ryu JC, Davidson BP, Xie A, et al. Molecular imaging of the paracrine proangiogenic effects of progenitor cell therapy in limb ischemia. Circulation 2013; 127(6): 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zeng L, Hu Q, Wang X, et al. Bioenergetic and functional consequences of bone marrow-derived multipotent progenitor cell transplantation in hearts with postinfarction left ventricular remodeling. Circulation 2007; 115(14): 1866–1875. [DOI] [PubMed] [Google Scholar]
- 48. Van’t Hof W, Mal N, Huang Y, et al. Direct delivery of syngeneic and allogeneic large-scale expanded multipotent adult progenitor cells improves cardiac function after myocardial infarct. Cytotherapy 2007; 9(5): 477–487. [DOI] [PubMed] [Google Scholar]
- 49. Supronowicz P, Gill E, Trujillo A, et al. Multipotent adult progenitor cell-loaded demineralized bone matrix for bone tissue engineering. J Tissue Eng Regen Med 2016; 10: 275–283. [DOI] [PubMed] [Google Scholar]
- 50. Lehman N, Woda J, Cutrone R, et al. Multipotent adult progenitor cells modulate endothelial cell adhesion molecule surface expression following activation and reduces inflammation following AMI. Stem Cells 2013. [Google Scholar]
- 51. Imitola J, Comabella M, Chandraker AK, et al. Neural stem/progenitor cells express costimulatory molecules that are differentially regulated by inflammatory and apoptotic stimuli. Am J Pathol 2004; 164(5): 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reading JL, Yang JH, Sabbah S, et al. Clinical-grade multipotent adult progenitor cells durably control pathogenic T cell responses in human models of transplantation and autoimmunity. J Immunol 2013; 190(9): 4542–4552. [DOI] [PubMed] [Google Scholar]
- 53. Jacobs SA, Pinxteren J, Roobrouck VD, et al. Human multipotent adult progenitor cells are nonimmunogenic and exert potent immunomodulatory effects on alloreactive T-cell responses. Cell Transplant 2013; 22(10): 1915–1928. [DOI] [PubMed] [Google Scholar]
- 54. Highfill SL, Kelly RM, O’Shaughnessy MJ, et al. Multi-potent adult progenitor cells can suppress graft-versus-host disease via prostaglandin E2 synthesis and only if localized to sites of allopriming. Blood 2009; 114(3): 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kovacsovics-Bankowski M, Streeter PR, Mauch KA, et al. Clinical scale expanded adult pluripotent stem cells prevent graft-versus-host disease. Cell Immunol 2009; 255(1–2): 55–60. [DOI] [PubMed] [Google Scholar]
- 56. Ringden O, Uzunel M, Rasmusson I, et al. Mesenchymal stem cells for treatment of therapy-resistant graft-versus-host disease. Transplantation 2006; 81(10): 1390–1397. [DOI] [PubMed] [Google Scholar]
- 57. Yasuhara T, Hara K, Maki M, et al. Intravenous grafts recapitulate the neurorestoration afforded by intracerebrally delivered multipotent adult progenitor cells in neonatal hypoxic–ischemic rats. J Cereb Blood Flow Metab 2008; 28(11): 1804–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boozer S, Lehman N, Lakshmipathy U, et al. Global characterization and genomic stability of human multistem, a multipotent adult progenitor cell. J Stem Cells 2009; 4(1): 17–28. [PubMed] [Google Scholar]
- 59. Yin D, Wells J, Clinton J, et al. Comparative analysis of cell proliferation, immunosuppressive action, and multi-lineage differentiation of immortalized MSC and MSC from bone marrow, adipose tissue, and umbilical cord blood. In: Proceedings of the International society for stem cell research, Vancouver, BC, Canada, June 2014. [Google Scholar]
- 60. Boxall SA, Jones E. Markers for characterization of bone marrow multipotential stromal cells. Stem Cells Int 2012; 2012: 975871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brown PT, Squire MW, Li WJ. Characterization and evaluation of mesenchymal stem cells derived from human embryonic stem cells and bone marrow. Cell Tissue Res 2014; 358(1): 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Busser H, Najar M, Raicevic G, et al. Isolation and characterization of human mesenchymal stromal cell subpopulations: comparison of bone marrow and adipose tissue. Stem Cells Dev 2015; 24(18): 2142–2157. [DOI] [PubMed] [Google Scholar]
- 63. Roson-Burgo B, Sanchez-Guijo F, Del Cañizo C, et al. Transcriptomic portrait of human mesenchymal stromal/stem cells isolated from bone marrow and placenta. BMC Genomics 2014; 15(1): 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hwang N, Varghese S, Li H, et al. Regulation of osteogenic and chondrogenic differentiation of mesenchymal stem cells in PEG-ECM hydrogels. Cell Tissue Res 2011; 344(3): 499–509. [DOI] [PubMed] [Google Scholar]
- 65. Hillel AT, Varghese S, Petsche J, et al. Embryonic germ cells are capable of adipogenic differentiation in vitro and in vivo. Tissue Eng Part A 2008; 15(3): 479–486. [DOI] [PubMed] [Google Scholar]
- 66. Gregory CA, Gunn WG, Peister A, et al. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem 2004; 329(1): 77–84. [DOI] [PubMed] [Google Scholar]
- 67. Ilmer M, Vykoukal J, Recio Boiles A, et al. Two sides of the same coin: stem cells in cancer and regenerative medicine. FASEB J 2014; 28(7): 2748–2761. [DOI] [PubMed] [Google Scholar]
- 68. Budniatzky I, Gepstein L. Concise review. Reprogram-ming strategies for cardiovascular regenerative medicine: from induced pluripotent stem cells to direct reprogramming. Stem Cells Transl Med 2014; 3(4): 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Delcroix GJ, Curtis KM, Schiller PC, et al. EGF and bFGF pre-treatment enhances neural specification and the response to neuronal commitment of MIAMI cells. Differentiation 2010; 80(4–5): 213–227. [DOI] [PubMed] [Google Scholar]
- 70. Bianco P, Robey PG. Stem cells in tissue engineering. Nature 2001; 414(6859): 118–121. [DOI] [PubMed] [Google Scholar]
- 71. Ma J, Wang Q, Fei T, et al. MCP-1 mediates TGF-beta-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood 2007; 109(3): 987–994. [DOI] [PubMed] [Google Scholar]
- 72. Chen Z, Yue SX, Zhou G, et al. ERK1 and ERK2 regulate chondrocyte terminal differentiation during endochondral bone formation. J Bone Miner Res 2015; 30: 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ishijima M, Suzuki N, Hozumi K, et al. Perlecan modulates VEGF signaling and is essential for vascularization in endochondral bone formation. Matrix Biol 2012; 31(4): 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol 2013; 5(1): a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zelzer E, Mamluk R, Ferrara N, et al. VEGFA is necessary for chondrocyte survival during bone development. Development 2004; 131(9): 2161–2171. [DOI] [PubMed] [Google Scholar]
- 76. Karsenty G. The complexities of skeletal biology. Nature 2003; 423(6937): 316–318. [DOI] [PubMed] [Google Scholar]
- 77. Frohlich M, Greyson W, Wan L, et al. Tissue engineered bone grafts: biological requirements, tissue culture and clinical relevance. Curr Stem Cell Res Ther 2008; 3(4): 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]