

Abstract
The Myo1c motor functions as a cargo transporter supporting various cellular events, including vesicular trafficking, cell migration, and stereociliary movements of hair cells. Although its partial crystal structures were recently described, the structural details of its interaction with cargo proteins remain unknown. This study presents the first structural demonstration of a cargo protein, Neph1, attached to Myo1c, providing novel insights into the role of Myo1c in intracellular movements of this critical slit diaphragm protein. Using small angle X-ray scattering studies, models of predominant solution conformation of unliganded full-length Myo1c and Myo1c bound to Neph1 were constructed. The resulting structures show an extended S-shaped Myo1c with Neph1 attached to its C-terminal tail. Importantly, binding of Neph1 did not induce a significant shape change in Myo1c, indicating this as a spontaneous process or event. Analysis of interaction surfaces led to the identification of a critical residue in Neph1 involved in binding to Myo1c. Indeed, a point mutant from this site abolished interaction between Neph1 and Myo1c when tested in the in vitro and in live-cell binding assays. Live-cell imaging, including fluorescence recovery after photobleaching, provided further support for the role of Myo1c in intracellular vesicular movement of Neph1 and its turnover at the membrane.
INTRODUCTION
Podocytes are the critical components of the glomerular filtration assembly whose dysfunction leads to progressive loss of renal function (1, 2). The morphological and structural analysis of podocytes suggests that the podocyte foot processes which surround glomerular capillaries are membrane protrusions supported by parallel F-actin bundles (3, 4). This actin network enables the podocytes to perform a host of biological functions, including motility and maintaining the permeability barrier “slit diaphragm.” Study of various glomerular disease models shows a severely disorganized actin network that is associated with loss of podocyte function and alterations in the podocyte morphology, a process commonly known as podocyte effacement (5–7). Podocyte effacement and the loss of slit diaphragm have been the subjects of intense investigation in recent years, which has led to the identification of several proteins that play a role in maintaining podocyte structure and function (1, 8). Proteins, including Neph1 and nephrin, have been shown to contribute toward the backbone of this structure, thus maintaining the integrity of the slit diaphragm (2, 9, 10). Localization of these proteins at the podocyte cell membrane is significantly altered in many glomerular disorders, which is commonly associated with the loss of glomerular filtration function (11–14). Recent evidence suggests that the trafficking of slit diaphragm proteins, including nephrin and Neph1, might be a critical determinant in the reorganization of podocyte cell membrane that restores the glomerular filtration function during recovery from various glomerular diseases (12–15). However, little is known about the mechanisms that mediate their intracellular movement in podocytes. Our previous studies suggest that Myo1c may regulate the dynamic movement of Neph1, which is essential for podocyte health and defines Neph1 as a cargo for Myo1c.
A recent study by Nabet et al. identified a subset of mRNA transcripts encoding various myosins that were remarkably enriched in glomerular podocytes (16). This report described the existence of both conventional nonmuscle (Myh10 and Myh9) and unconventional (Myo6, Myo1e, Myo18a, Myo5a, Myo1b, and Myo1d) myosins in podocytes (16). Analysis of the slit diaphragm proteome further confirmed the presence of these and additional myosins, including Myo1c (17). The presence of all these various myosins in podocytes suggests that their motor functions play a significant role in the maintenance of podocyte morphology and the slit diaphragm. Among all these myosins, only the mutations in Myh9 and Myo1e genes have been shown to associate with genetic forms of glomerular diseases (18–21). However, we are now beginning to understand the significance of myosins in podocyte biology, and models are currently being developed in various laboratories (including ours) to further establish the role of myosins in maintaining the glomerular filtration function.
Myo1c belongs to the myosin I family of the motor proteins that are ubiquitously expressed in eukaryotic cells (22). The myosin I family motor proteins, including Myo1c, are enriched in membrane structures and localize to filopodia, lamellipodia, ruffles, and the leading edges of migrating cells, in addition to their cytoplasmic distribution (22, 23), and are therefore involved in a variety of cellular functions, including maintenance of cortical tension and related functions, such as cell motility and trafficking (16, 24–26). In podocytes, we demonstrated a similar pattern for Myo1c with partial Neph1 colocalization at the cell periphery. Our results have also shown that Myo1c colocalizes with nephrin and Neph1 in the lipid rafts (15), and a recent report demonstrates an active role for Myo1c in the recycling of lipid rafts that controls cell spreading and migration (27). This further suggests that Myo1c may participate in the recycling events of nephrin and Neph1 trafficking. Podocytes in cell culture do not recapitulate the in vivo foot processes; instead, they form dynamic structures such as lamellipodia, membrane ruffles, and long filopodium-like structures that extend from one cell to the other (15, 23). All these structures are sites of active actin dynamics involving multiple cytoskeletal elements that together help regulate cell migration and/or differentiation (2, 28–30). Since Myo1c is localized in these structures, it is likely that it is involved in the development of podocytes. Indeed, our recent findings suggesting that loss of Myo1c in a zebrafish model system induces proteinuria and podocyte effacement (31) further support an active role for Myo1c in podocyte biology.
Structurally, all the known myosin I family motors are composed of an N-terminal head or motor domain that contains ATP and actin binding sites, a neck domain that consists of one or more calmodulin binding IQ motifs, and the C-terminal variable tail that binds its cargo and provides specificity to myosins (24). Recently, crystal structures of the Myo1c head domain bound to calmodulin (32, 33) and the Myo1c tail bound to calmodulin (34) were reported. Although these studies provided significant insight into the conformation of Myo1c, they analyzed only partial Myo1c structures. Although the C-terminal domain of Myo1c is the designated cargo binding domain, it is interesting that not all proteins that bind Myo1c interact with this domain. The GTPase RalA, calcium-binding protein, and phosphoprotein regulator 14-3-3 have been shown to interact with the regulatory neck region of Myo1c (35, 36). In this report, we describe solution scattering-based structures of the full-length Myo1c and its complex with the cytoplasmic domain (CD) of Neph1. We used structural and biochemical methods to analyze the structural complex of Myo1c and Neph1. We further utilized live-cell imaging to highlight the physiological role of this interaction in regulating the intracellular dynamic movements of Neph1.
MATERIALS AND METHODS
Antibodies and reagents.
Myo1c monoclonal purified antibody (Ab) and polyclonal purified antibody to Neph1 have been previously described (15). Other antibodies, including green fluorescent protein-horseradish peroxidase (GFP-HRP) antibody (Miltenyi Biotec, USA), monoclonal His antibody (Santa Cruz; catalogue no. 8036), and monoclonal Flag antibody (Sigma; catalogue no. F3165), were commercially procured. The cell transfection reagent Lipofectamine 2000 was purchased from Invitrogen (catalogue no. 11668-019). All chemical reagents were commercially obtained from Sigma and Calbiochem.
Plasmids and cell light markers.
Mammalian expression plasmids encoding GFP-tagged Myo1c-FL (full length), the Myo1c tail without the IQ domain, and the Myo1c tail with the 3IQ domain were obtained as described previously (14, 15). The Myo1c head construct was generated by PCR-based cloning at EcoRI and SalI sites in the pEGFP-N1 vector (Clontech). Cerulean fluorescence protein (CFP)-tagged mammalian constructs of Neph1, such as CFP-Neph1-wt (wild type), CFP-Neph1-(−)PDZ, CFP-Neph1-Y637/638F, CFP-Neph1-Y638F, CFP-Neph1-Y716/719F, CFP-Neph1-K761A, and CFP-Neph1-Y762A, were constructed commercially (by Top Gene Technologies, Inc.). Constructs mCherry Neph1-wt and the K761A mutant were constructed in pBABE Puro retroviral vector using standard PCR cloning strategies as described previously (37). Cell light markers for early endosome-GFP (catalogue no. C10586), late endosome-GFP (catalogue no. C10588), Golgi complex-GFP (catalogue no. C10592), and lysosome (catalogue no. C10507) were purchased from Thermo Fisher.
Cell culture.
COS7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 200 U/ml of penicillin and streptomycin (Invitrogen). RPMI 1640-based medium supplemented with 10% FBS (Invitrogen), 2 g/liter of sodium bicarbonate (NaHCO3), insulin-transferrin-selenium (38) supplement (Sigma-Aldrich), and 200 U/ml of penicillin and streptomycin (Invitrogen) was used to culture the human podocytes (14, 15). Lipofectamine 2000 (Invitrogen) was used to perform transfection according to the manufacturer's protocol. Transfected podocytes were grown in 2.5 μg/ml of puromycin-containing RPMI medium for the selection of stable transfectants. Stable mCherry Neph1-wt- and K761A mutant-expressing podocytes were generated using the retroviruses produced by Phoenix cells. Details for the procedure of producing retrovirus and transfecting podocytes to generate a stable cell line have been described previously (15).
Recombinant proteins and peptides.
Proteins, including glutathion S-transferase (GST)—Neph1 cytoplasmic domain (CD), His-Neph1-CD-wt, His-Neph1-CD-R750E, His-Neph1-CD-(−)PDZ, His-Neph1-CD-K761A, and His-Neph1-CD-Y762A, were expressed and purified from Escherichia coli BL21 cells (Stratagene). Details of the expression and purification protocol have been described previously (39). Expression and purification of His-ZO1-PDZ1 protein have also been described previously (14, 39). Recombinant proteins Flag-Myo1c-FL and GFP-His-Myo1c-tail were produced in a baculovirus expression system as described previously (40, 41). Two peptides, ERTPYEAYDPIGKYATATRF and YEKFNSHPFPGAAGYPTYRL, derived from the K761 region of Neph1 and the region of Neph1 not involved in Myo1c binding, respectively, were synthesized chemically using standard 9-fluorenylmethoxy carbonyl (Fmoc) solid-phase chemistry, and 2-chloro trityl resin was used as a solid support (PTI peptide synthesizer). Postsynthesis, the peptides were cleaved using a cocktail of trifluoroacetic acid (TFA) and scavengers and were purified to homogeneity using high-performance liquid chromatography (HPLC) (C18 semiprep and analytical columns attached to a Waters system). Identities of the purified peptides were confirmed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) (Voyager).
Biacore surface plasmon resonance (SPR) assay.
A real-time binding experiment was performed with a Biacore 3000 instrument (Biacore AB). Flag-Myo1c-FL was immobilized on a CM5 chip cell, and the instrument was programmed to perform a series of binding assays with increasing concentrations of His-Neph1-CD-wt. Surface regeneration and complete dissociation between two proteins were achieved by using 2 M NaCl. BIAevaluation software (version 3.2; GE Healthcare) was used for the analysis of sensorgrams and the evaluation of kinetic constants (association constant [Ka], dissociation constant [Kd], and equilibrium dissociation constant [KD]) between Myo1c and Neph1.
SAXS data acquisition and analyses.
The SAXS intensity profiles were collected at the X9 beam line (National Synchrotron Light Source, Brookhaven National Laboratory, NY). Images were acquired on Pilatus detectors and processed as described earlier (39). For each experiment, 120 μl of protein solutions (unliganded Myo1c, its fragments, Neph1-CD, and their mixtures) and their matched buffers were exposed to X rays in a quartz capillary flow cell. Using programmable liquid handling robotics at the X9 beam line, the collection of entire SAXS data described in this report was carried out in triplicate and averaged during processing. To calibrate the beam intensity at zero angles and estimate the actual concentration of protein samples, the SAXS data sets were collected under identical conditions on hen egg lysozyme and recombinant human gelsolin with predetermined concentrations. Data processing provided the scattering intensity (I) as a function of momentum transfer vector, Q (Q = [4πsinθ]/λ), where λ and θ are the wavelengths of the X ray and the scattering angle, respectively. Kratky plots [I(Q)Q2 versus Q] were made from the measured SAXS data to interpret the globular nature of protein molecules and their complexes in solution. The Guinier approximation of the low-Q region was carried out using PRIMUSQT software from the ATSAS 2.6 suite of programs (42) and presuming globular and rod-like scattering shape and provided the radius of gyration (RG) and radius of cross-section (RC) (17) of the predominant shape of protein molecules, respectively. Using the relationship between RG and RC (equation 1), we estimated the persistence length (L) of the molecules:
| (1) |
Additionally, using GNOM45 program (43), we carried out indirect Fourier transformation of the SAXS data (Q range: 0.008 to 0.25 Å−1) to obtain probability of finding various pairwise vectors arising from the scattering shape of the protein molecules in solution. The pair distribution function [P(r)] curve estimation was done presuming zero probability of finding a pairwise vector of length equal to 0 Å and equal to the maximum linear dimension (Dmax) of the molecule. This analysis also provided an estimate of the RG and scattering intensity at zero scattering angles (I0). I0 values are directly proportional to square of the mass of the scattering species in solution, and thus, with the use of standard samples, we could estimate the concentration of unliganded proteins and the extent of formation of their complexes in mixtures.
Shape reconstruction.
To visualize the three-dimensional shape of unliganded Myo1c, the head and tail regions of Myo1c, and Myo1c/Neph1-CD complex, using measured SAXS data profiles and computed P(r) profiles, 10 independent models of each were generated using the DAMMINIF program, aligned using the SUPREF program, averaged using the DAMAVER program, and, finally, minimized using the DAMMIN program (42). This procedure provided a dummy atom model of the predominant scattering shape of the proteins and their complexes in solution. To compare the shape of the SAXS-based model with other complementary information, a structural model of full-length Myo1c was generated bound to three calmodulins on its IQ domain. In brief, the SWISS-MODEL server was used to stitch together the individual crystal structures of the Myo1c head (Protein Data Bank [PDB] code 4BYF) and tail (PDB code 4R8G) regions, both in complex with calmodulin (32, 34, 44). The two structures had common IQ domains which were used for relative referencing of the other domains' coordinates. The coordinates of calmodulin bound to IQ domain was used from one of the structures. The structural model of Myo1c bound to three calmodulins was overlaid with the SAXS-based model of same protein by computationally aligning their inertial axes using the SUPCOMB program (45). The open-source product Pymol version 1.1 was used for graphical analysis and figure generation. The structure of His-Neph1-CD (39) was employed for docking on the composite model of Myo1c (39). Docking of His-Neph1-CD on Myo1c was done using the ZDOCK server, and the model of the 1:1 complex was considered based on agreement with the shape profile of the SAXS data-based envelope shape of the complex (46).
Immunoprecipitation and immunoblotting.
Detailed procedures for immunoprecipitation and immunoblotting experiments have been explained previously (10, 32, 47). Briefly, wild-type Neph1 and its mutants were cotransfected with wild-type Myo1c and its mutants in COS7 cells using Lipofectamine 2000 transfection reagent. Forty-eight hours posttransfection, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (phosphate-buffered 0.9% NaCl [PBS] containing 0.1% SDS, 1% Nonidet P-40, and 0.5% sodium deoxycholate). The lysate was subjected to immunoprecipitation using Neph1 or Flag antibodies. The immunoprecipitated complexes were separated by SDS-PAGE and analyzed by Western blotting using the desired antibodies.
Pulldown.
The recombinant proteins—His-Neph1-CD, the Neph1-CD mutants (including His-Neph1-CD-R750A, His-Neph1-CD-K761A, His-Neph1-CD-Y762A, and His-Neph1-CD-PDZ), and His-ZO1-PDZ1—were expressed and purified in Escherichia coli BL21 cells (Stratagene) as described previously (39). Recombinant proteins Flag-Myo1c-FL and GFP-His-Myo1c-tail were separately mixed with 5 μg of each Neph1 fusion protein, and the His-Neph1-CD/Myo1c complexes were pulled down using Neph1 antibody bound to protein G-agarose. After washing of the agarose beads with PBS containing 0.1% Tween 20, the protein complexes were eluted with SDS sample buffer and resolved by SDS-PAGE prior to immunoblotting with the GFP-HRP, Myo1c, Flag, and His antibodies. Competitive binding of Myo1c and ZO1 with Neph1 was investigated by mixing His-Neph1-CD protein with either Myo1c protein (Flag-Myo1c-FL or GFP-His-Myo1c-tail) in the presence or absence of His-ZO1-PDZ protein. The complexes were pulled down with Neph1 antibody and immunoblotted with Flag or Myo1c antibodies to determine whether Myo1c competes with ZO1 for binding Neph1.
Dot blot experiment.
A peptide representing the K761 region of Neph1 with the sequence ERTPYEAYDPIGKYATATRF (748 to 768) was synthesized. Another peptide derived from a different Neph1 region (718 to 738) that was not involved in binding was used a negative control. The test and control peptides were spot blotted onto a polyvinylidene difluoride (PVDF) membrane along with purified GST-Neph1-CD and His-Neph1-CD proteins as positive controls. The membrane was incubated with 5 μg of either Flag-Myo1c-FL or GFP-His-Myo1c-tail purified proteins for about 5 h in 2% skim milk (in 1× PBS) and then immunoblotted with Myo1c antibody.
FRAP.
Podocyte cells stably expressing mCherry-Neph1-wt or the K761A mutant were mixed with GFP alone in a ratio of 1:3 and plated on 8-chamber dishes (Nunc; catalogue no. 155409). The primary reason for mixing the two cell lines was to obtain a heterogenous population of cells in which mCherry-Neph1-expressing cells are adjacent to GFP-expressing cells. For fluorescence recovery after photobleaching (FRAP) analysis, it is necessary to determine which cell contributes toward the protein dynamics that is being studied. Therefore, the cells grown in this fashion ensured that only the mCherry-Neph1-expressing cell contributed to the junctional Neph1. Thus, we drew the region of interest (ROI) at the junction of mCherry-Neph1- and GFP-expressing cells to ensure that only one mCherry-Neph1-expressing cell contributed to the ROI. The ROI was photobleached and the fluorescence recovery within the photobleached region was monitored at subsequent time intervals of 10 s. A Zeiss confocal microscope equipped with Volocity acquisition software was used. Eight different images were collected, and quantitative analysis was performed.
Live-cell microscopy and cellular distribution of Neph1-containing vesicles.
mCherry-Neph1-wt and the K761A mutant were plated on 35-mm glass-bottom cell culture plates, and the movement of Neph1-containing vesicles was analyzed using live-imaging microscopy. Images were captured for 2 min with 10-s intervals using Zeiss confocal microscopy with Volocity acquisition software. The displacement of Neph1-containing vesicles was plotted against time. The distribution of Neph1 containing vesicles into various subcellular compartments was analyzed using cell light reagent markers of early endosome, late endosome, Golgi complex, and lysosome. Confocal microscopy was performed, and colocalization was analyzed using ImageJ software from single-plane images. Each experiment was repeated at least three times. In each experiment, 2 million cells were plated on each coverslip and 8 or more cells were analyzed for quantification and statistical analysis The values are expressed in terms of Pearson's correlation coefficient (r).
RESULTS
Our previous findings (15) and results from our Biacore study (Fig. 1) demonstrated that Myo1c forms a high-affinity complex with Neph1. To further understand the mechanism through which Myo1c interacts with Neph1 and participates in podocyte maintenance, we investigated the molecular details of this interaction using structural and biochemical approaches.
FIG 1.
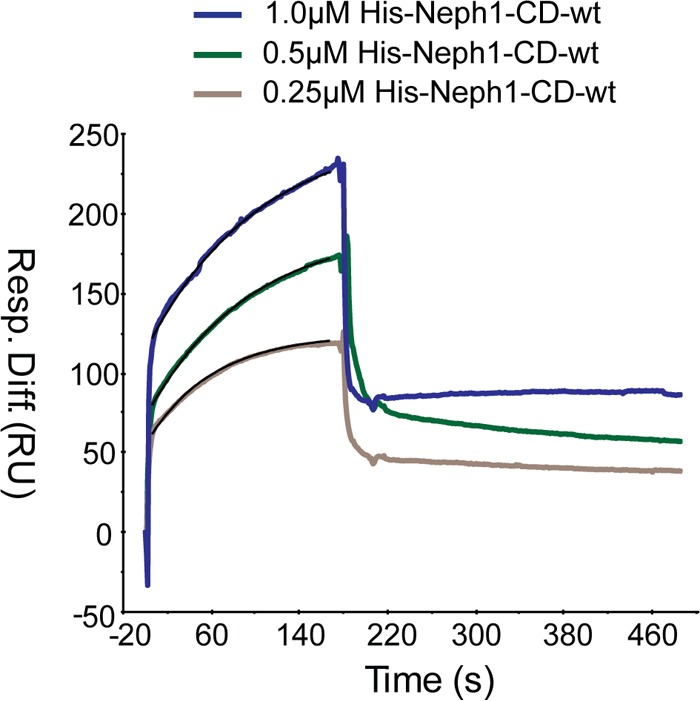
SPR analysis. Sensorgrams for various concentrations of His-Neph1-CD binding to immobilized Flag-Myo1c-FL are shown. Results are expressed as a differential response (in response units [RU]) versus time. Flag-Myo1c-FL was immobilized on a CM5 chip at 826 RU, and His-Neph1-CD protein at concentrations of 1.0, 0.5, and 0.25 μM was passed over the chip for 3 min and allowed to dissociate for about 4 min. Analysis of the difference between binding sensorgrams of the test and control (without protein) was performed using Biacore BIAevaluation software using a 1:1 Langmuir model. The calculated affinity constant (KD) for Myo1c-Neph1 interaction was 1.1 × 10−8 M (11 nM).
SPR analysis of Neph1 and Myo1c interaction.
Flag-Myo1c-FL was immobilized on a CM5 chip cell 2 at 826 RU. Cell 1 was used as a negative control with no protein bound to the chip. His-Neph1-CD protein at concentrations of 1.0, 0.5, and 0.25 μM was passed over the chip for 3 min and allowed to dissociate for about 4 min, and then complete dissociation of His-Neph1-CD was achieved by injection of 2 M NaCl. Analysis of the difference between binding sensorgrams of cells 1 and 2 was performed using Biacore BIAevaluation software and the 1:1 Langmuir model. The analyte binding capacity of the surface (Rmax) for the analysis was 110 RU, and the X2 was 1.25. The calculated binding parameters for Myo1c and Neph1 binding were an affinity constant (KD) of 1.1 × 10−8 M (11 nM) and rate constants of 5.59 × 104 (1/Ms) (Ka [association]) and 6.11 × 10−4 (1/s) (Kd [dissociation]) (Fig. 1).
SAXS-based solution shape analysis of the unliganded Myo1c (and Neph1).
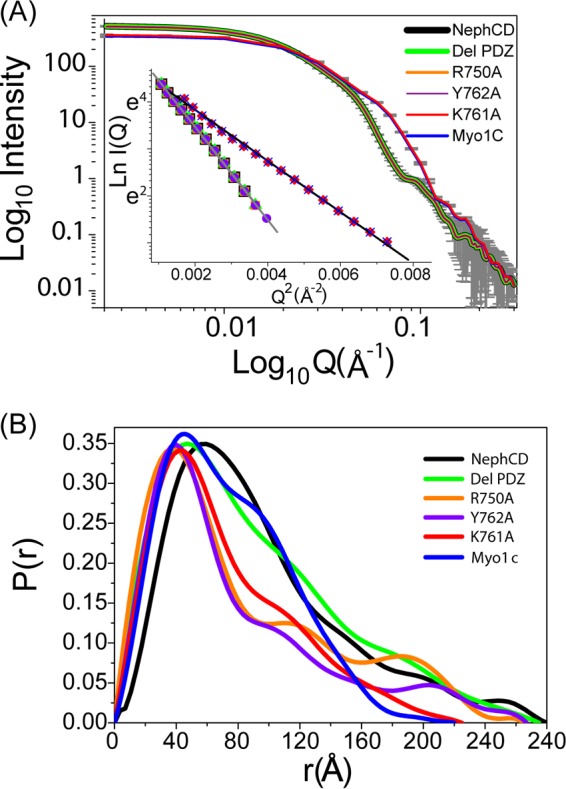
In order to gain structural insight into Myo1c and its interaction with Neph1-CD, we acquired SAXS data for unliganded full-length Myo1c (Myo1c-FL; residues 1 to 1028), the head (residues 1 to 766, with the IQ domain), and the tail (residues 698 to 1028, with the IQ domain) (Fig. 2A). It is pertinent to mention here that the protein Myo1c-FL and its head and tail domains used in this study were purified in the presence of three calmodulin molecules that remained bound to their IQ regions during our SAXS data collection (47). The scattering intensity profiles from the protein components were plotted in log-versus-log mode and indicated lack of aggregation in the samples (Fig. 2A, left). Also, linearity of the data in the Guinier plot for globular particles confirmed the monodisperse nature of the samples during data collection (Fig. 2A, left inset). The single peak-like profiles of Kratky plots of the Myo1c head and tail portions and the two peaks for the full-length protein indicated single-domain and two-domain globular scattering shapes for these proteins in solution (Fig. 2A, right inset). Further, indirect Fourier transformation of the SAXS data (Q range: 0.008 to 0.25 Å−1) provided the Dmax and RG for Myo1c-FL and its head and tail domains to be 220, 152, and 157 Å and 76.9 ± 1.1, 49.1 ± 0.7, and 49.7 ± 0.5 Å, respectively (Fig. 2A, right; Table 1). The two peak profile of the computed P(r) curve for full-length Myo1c supported the idea that the unliganded protein adopts a solution shape which can be characterized as two lobes or domains connected by a nonflexible linker. Importantly, the two-peak profiles were distinctly absent in the P(r) curves calculated for the head and tail domains, indicating that these two domains represent the two lobes in full-length Myo1c. I0 values from SAXS data on standard protein solutions acquired under identical conditions were estimated to calibrate beam intensity, and comparison of estimated I0 values from the solutions of (calmodulin-bound) full-length Myo1c and its head and tail domains suggested that the actual concentrations of full-length Myo1c and its head and tail domains were 3, 1.7, and 2.1 mg/ml, respectively. In addition to these Myo1c constructs, SAXS data were also acquired from the solutions of unliganded Neph1-CD-wt and its mutants [Neph1-CD-K761A, -Y762A, -R750A, and -(−)PDZ] under identical conditions. The data analyses revealed that the shape parameters for Neph1-CD protein and its mutants were similar to those in our previously published report (39). Briefly, we reconfirmed that Neph1-CD and its mutants were globular and monodispersed in solution, with an RG and Dmax of 21.1 ± 0.2 and 70°, respectively. Further analysis of the data confirmed that as reported previously (39), these point mutants did not induce substantial alterations in the global shape of mutant Neph1 compared to the wild type.
FIG 2.

SAXS data of unliganded protein samples. (A) (Left) SAXS intensity profiles acquired from the solutions of Myo1c-FL and its head and tail domains are presented. The inset shows linear fit to the Guinier region of the measured data sets. (Right) The P(r) curves computed for Myo1c-FL and its head and tail domains demonstrate the frequency distribution of interatomic vectors in the predominant scattering species. The inset shows the Kratky plots of the data sets. (B) Scattering shape of the proteins restored from dummy atom modeling using the SAXS data as a reference. The envelope shape (represented as mesh) of the predominant shape computed for full-length Myo1c and its head and tail domains is shown. Crystal structures of the head, tail, and full-length Myo1c (reconstructed by joining head and tail regions) with bound calmodulins (represented as surface) were overlaid by automated alignment of inertial axes with SAXS models. The structure of Myo1c is represented in red, while three calmodulins are represented in green, blue, and cyan.
TABLE 1.
Shape parameters deduced from the SAXS data analysis of proteins and their complexes
| Protein | Mass (kDa) | Indirect Fourier transformation |
Actual concn (mg/ml) | ||
|---|---|---|---|---|---|
| Dmax (Å) | RG (Å) | I0 (AU) | |||
| Standard proteins | |||||
| Lysozyme | 14.2 | 44 | 14.2 | 20 | 1a |
| Gelsolin | 82 | 100 | 30.2 | 115 | 1a |
| Unliganded Myo1c | |||||
| Full length | 168.6 | 220 | 59 | 712 | 3 |
| Myo1c-head | 88.8 | 152 | 49.05 | 215 | 1.7 |
| Myo1c-tail | 37.7 | 157 | 49.7 | 112 | 2.1 |
| Unliganded Neph1-CD and its mutants | |||||
| Neph1-CD | 35.0 | 70 | 21.5 | 120 | 2.4 |
| Neph1-(−)PDZ1 | 34.7 | 70 | 21.1 | 74 | 1.5 |
| Neph1-R750A | 35.0 | 70 | 21.2 | 101 | 2.1 |
| Neph1-K761A | 35.0 | 70 | 21.3 | 96 | 1.9 |
| Neph1-Y762A | 35.0 | 70 | 21.4 | 62 | 1.2 |
The values for lysozyme and gelsolin were estimated based on a concentration series of these proteins with predetermined concentrations (for lysozyme, 3 to 5.7 mg/ml, and for gelsolin, 2 to 6 mg/ml) and extrapolated for 1 mg/ml. Based on these standards, the I0 value for 1 kDa of protein at a concentration of 1 mg/ml would be 1.4 AU, and this was used to estimate protein concentrations.
To visualize the predominant solution shape of the unliganded Myo1c-FL and its head and tail domains, 10 independent dummy residue models were generated presuming uniform scattering density and were averaged as described in Materials and Methods (Fig. 2B). As expected from the P(r) curve, the model of the full-length Myo1c resembled a bilobal extended S-shaped curve, while the models for the head and tail domains indicated bilobal and curved shapes, respectively. For each system, 10 models were generated and compared with each other by calculating normalized spatial disposition (NSD) relative to one reference model. NSD values for the Myo1c head, tail, and full-length protein were 0.494 ± 0.009, 0.523 ± 0.014, and 0.637 ± 0.031, respectively. Rejecting any model which had NSD values greater than twice the standard deviation (SD), 10, 9, and 9 models were considered for the head, tail, and full-length protein, respectively. Their inertial axes were aligned and averaged, followed by one round of further minimization using the DAMMIN program. The resultant dummy residue models for the head, tail, and full-length protein showed χ2 values of 2.436, 2.191, and 1.751, with their respective experimental SAXS data profile. Recently described crystal structures of the Myo1c head group with the IQ domain bound to calmodulins (PDB code 4BYF) and the tail group with the IQ group bound to calmodulins (PDB code 4R8G) were used for comparison with the SAXS data-based models of Myo1c (32, 34). Further, as described in Materials and Methods, a composite structural model of Myo1c bound to three calmodulins was generated using available crystal structures of the head and tail groups. Interestingly, automated alignment of the inertial axes of the crystal structures of head and tail groups and the model of full-length Myo1c with their respective global shape models solved using SAXS data showed similarity in three dimensions (Fig. 2B).
SAXS data-based insight into Myo1c/Neph1-CD complex.
Following the confirmation of the actual concentration of proteins, 1:1 molar mixtures of Myo1c-FL and Neph1-CD (and its mutants) were prepared for SAXS data collection (Fig. 3A). An increase in the scattered intensity units (as Q→0) compared to that observed for unliganded Myo1c supported the binding of Myo1c with Neph1-CD available in the mixture (Fig. 3A). The peak-like profile of the Kratky plot supported a globular shape for the complexes of Myo1c and Neph1-CD (data not shown). A linear fit to the Guinier region indicated that the predominant shape of the Myo1c/Neph1-CD complex could be characterized by an RG of 84.3 Å. In close correlation, the P(r) curve computed an RG of 76.9 ± 1.1 Å and a Dmax of 280 ± 0.1 Å for predominant scattering species in the mixture. Importantly, the I0 value computed from P(r) analysis suggested almost complete (∼96%) binding between the Myo1c and Neph1-CD molecules in the mixture (Fig. 3B; Table 2). Since I0 values are proportional to the square of the mass of predominant scattering species, and knowing the precise concentrations of protein used for preparing mixtures, we estimated that for the complexes of Myo1c and Neph1 (and its point mutants), I0 values close to 530 AU and 370 AU will represent scattering from a system with 100% and no binding between available proteins. (Below, we demonstrate a correlation between SAXS-based observations and pulldown experiments.).
FIG 3.
SAXS data of the Myo1c-Neph1 complex. (A) SAXS intensity profiles from unliganded Myo1c (diluted to same molar concentration) and an ∼1:1 molar mixture of Myo1c with different Neph1-CD. The Guinier analysis of the data sets presuming globular scattering shape is shown in the inset. (B) P(r) computed from the indirect Fourier transformation of the data sets.
TABLE 2.
Shape parameters and extent of binding deduced from the SAXS data analysis of Myo1c (9.1 μM) with Neph1 and Neph1 mutants
| Protein in complex with Myo1c | Molar ratio of Myo1c to protein | Dmax (Å) | RG (Å) | I0 | Calculated I0 | Extent of binding (%) |
|---|---|---|---|---|---|---|
| Neph1-CD | 1:1.13 | 280 | 76.9 | 523 | 532 | 96 |
| Neph1-(−)PDZ1 | 1:1.14 | 275 | 75 | 525 | 533 | 97 |
| Neph1-R750A | 1:1.12 | 265 | 71.7 | 520 | 529 | 94 |
| Neph1-K761A | 1:1.12 | 225 | 59.7 | 371 | 529 | ∼0 |
| Neph1-Y762A | 1:1.12 | 267 | 68.1 | 522 | 529 | 95 |
| None (buffer) | 183 | 62.8 | 355 | 356 |
The three-dimensional shape reflecting predominant solution shape of the Myo1c and Neph1-CD binary complex was obtained by averaging 10 independent dummy atom models restored using measured SAXS data as reference (Fig. 4). The NSD value for the 10 models was 0.615 ± 0.118 and 9 models could be averaged, with a final χ2 value of 1.364 between the resultant model of complex versus the experimental SAXS data. By manually aligning the larger head group of the Myo1c in the unliganded and Neph1-bound shape, we clearly observed additional volume at one end of the complex which could easily accommodate one Neph1CD molecule. On the basis of the two rotated views presented in Fig. 4A, and considering the shape profile of the unliganded Myo1c, it appeared that this additional volume was very likely near the tail group. The SAXS data-based envelope shape of the complex was used to perceive which epitopes of Neph1CD interact with the tail group, and therefore, the structural model of Neph1CD (reported earlier) was docked on the structural model of Myo1c containing the atomic details. Of the lowest 10 docking poses, 4 poses showed that Neph1-CD interacts with the tail region, while the other 2 interacted close to the IQ domain and the remaining 4 preferred a position close to the head region of Myo1c. In Fig. 4B, the six poses (four with the tail and two with the IQ domain) are shown. The SAXS data-based model of the complex clearly ruled out interactions with the IQ domain. Considering the increased length of the complex and the shape profile, we concluded that poses shown in the magenta box in Fig. 4B (right side of panel) agreed most with the SAXS-based modeling results. Between the two poses which closely resembled each other, we considered the one which had lower energy in the docking experiment (Fig. 5). This selected model of the Myo1c/Neph1-CD complex suggested that the Neph1-CD molecule interacts primarily with the tail region close to the C-terminal end of Myo1c, thus reducing the probability of interaction at alternate sites (Fig. 5). The detailed analysis of our model suggested that the C-terminal THV motif of Neph1-CD may not be in direct contact with Myo1c, but its orientation in the complex should occlude its binding with ZO1-PDZ1 (39). Further analysis revealed that residue K761 in Neph1-CD was in direct contact with the tail region of Myo1c; if true, this would suggest that mutating the K761 residue may lead to loss of Neph1-CD binding with Myo1c. To validate the structural model of the Myo1c/Neph1-CD complex, various Neph1 mutants were constructed [His-Neph1-CD-K761A, His-Neph1-CD-Y762A, His-Neph1-CD-R750A, and His-Neph1-CD-(−)PDZ], and their ability to interact with Myo1c-FL was investigated by mixing them in the desired molar ratios and subjecting them to SAXS analysis (Table 2). Data analyses indicated that point mutation of Lys-761 to alanine completely abrogated the formation of complex (an I0 value close to 370 AU clearly supported 0% binding of the mixed proteins under experimental conditions). In contrast, mutating Tyr-762 and Arg-750 to alanine or deletion of the THV motif provided I0 values close to 530 AU, suggesting the molecular mass of the scattering species to be close to be 200 kDa for these mixtures. In other words, data supported almost complete binding of these mutants with Myo1c-FL (Table 2). Overall, our SAXS data analysis showed that Lys-761 in Neph1-CD is critical for Myo1c interaction with Neph1-CD.
FIG 4.

SAXS data-based shape of the Myo1c-Neph1 complex. (A) Scattering shape of Myo1c-Neph1 complex restored from dummy atom modeling using the SAXS data (I); manual alignment of full-length Myo1c (salmon) on the complex shows an additional volume at the tail region (II and III). (B) The six lowest-energy docking poses of Myo1c-Neph1 complex (I and II); alignment of docking models on SAXS models clearly ruled out two possible docking models (X), and the remaining four could be the more probable structural models of the Myo1c-Neph1 complex (III). The lowest-energy model (√) was used for further interaction analysis (IV).
FIG 5.

Structural model of the Myo1c/Neph1-CD complex. (A) Myo1c and Neph1-CD proteins are depicted in red and magenta surface representation. The three calmodulins bound to the IQ domain of Myo1c are shown in green, blue, and cyan surface representation mode. The residues K761 and THV (PDZ-binding residues) in Neph1-CD are present in interaction interface. (B) The binding site for ZO1-PDZ1 (yellow) in Neph1-CD (magenta ribbon) is occluded when Neph1-CD interacts with Myo1c.
Neph1 interacts with the C-terminal region of Myo1c.
To further validate the structural models of Myo1c and its complex with Neph1, and better define the interacting regions in Myo1c and Neph1, we evaluated the interaction between Myo1c and Neph1 in live cells and under in vitro conditions. Therefore, various Myo1c constructs, including GFP-Myo1c-FL and its deletion mutants representing the C-terminal tail (GFP-Myo1c-tail) and the head region (GFP-Myo1c-head), were coexpressed with Flag-Neph1-wt in COS7 cells and the immunoprecipitated Neph1 complex was analyzed for the presence of Myo1c. Additionally, based on the structural data (Fig. 6A), site-specific mutant Neph1 (Neph1-K761A) was also constructed and subjected to the binding assay in a similar fashion. As shown in Fig. 6A, Neph1 interacted with GFP-Myo1c-FL and GFP-Myo1c-tail with or without the IQ domain, whereas GFP-Myo1c-head failed to interact with Neph1 (Fig. 6B). A reciprocal experiment was performed in which various Neph1 mutants, including the phosphorylation site mutants (CFP-Neph1-Y637/638 and -Y716/719), the deletion mutant CFP-Neph1-(−)PDZ (without the PDZ binding domain), and the CFP-Neph1-K761 mutant (predicted from the SAXS analysis), were coexpressed with GFP-Myo1c-FL in COS7 cells and tested for binding to Myo1c. As shown in Fig. 6C, all Neph1 mutants except CFP-Neph1-Y761A interacted with Myo1c. Collectively, these results provided biochemical validation for the SAXS-based global shape data suggesting that Neph1 interacts with the C-terminal domain of Myo1c and that residue K761 in Neph1 is involved in this interaction.
FIG 6.

Neph1 interacts with the IQ domains and C-terminal region of Myo1c. (A and B) COS7 cells were cotransfected with Flag-Neph1-wt and various Myo1c constructs, including GFP-Myo1c-FL, GFP-Myo1c-tail (with no IQ domain), GFP-Myo1c (3IQ domain), and GFP-Myo1c (head), and immunoprecipitated (IP) with Neph1 antibody. Immune complexes were evaluated for binding with Myo1c by Western blotting (WB) with GFP-HRP. Neph1 interacted with GFP-Myo1c-FL and GFP-Myo1c tail with or without the IQ domains but not with the GFP-Myo1c head. (C) In a reciprocal experiment, Neph1 mutants, including the phosphorylation site mutants (CFP-Neph1-Y637/638 and -Y716/719) and the deletion mutant CFP-Neph1-(−)PDZ (without the PDZ binding domain) and the CFP-Neph1-K761 mutant, were cotransfected with GFP-Myo1c-FL in COS7 cells and tested for binding to Myo1c. All the Neph1 mutants interacted with Myo1c except CFP-Neph1-Y761A. (D) The recombinant His-tagged Neph1 (His-Neph1 CD-wt) and its mutants, including His-Neph1-CD-R750E, His-Neph1-CD-(−)PDZ, His-Neph1-CD-K761A, and His-Neph1-CD-Y762A, were separately mixed with the purified Flag-Myo1c-FL protein produced in baculovirus, and pulldown was performed using Neph1 antibody. Western blot analysis using Myo1c antibody showed loss of interaction with Myo1c in the His-Neph1-CD-K761A mutant. (E) A peptide binding experiment showed that similar to purified His-Neph1-CD-wt and GST-Neph1-CD-wt, the peptide containing the K761 region had strong affinity toward Flag-Myo1c-FL and GFP-Myo1c-tail. IB, immunoblotting. (F) Recombinant proteins His-Neph1-CD-wt and either Flag-Myo1c-FL or GFP-His-Myo1c-tail were mixed and pulled down with Neph1 antibody. Western blotting using myo1c antibody was performed to evaluate binding of Neph1 with the Myo1c tail region.
To further determine if these residues are directly involved in this interaction, an in vitro pulldown experiment was performed using purified proteins. Recombinantly produced His-tagged Neph1 (His-Neph1-CD-wt) and its mutants, including His-Neph1 CD-R750E, His-Neph1-CD-(−)PDZ, His-Neph1-CD K761A, and His-Neph1-CD-Y762A, were mixed with the purified Flag-Myo1c-FL protein produced in baculovirus, and the pulldown was performed using Neph1 antibody. Consistent with the binding experiments with COS7 cells and SAXS analysis, the interaction between Myo1c and Neph1 was completely abolished when the lysine at position 761 was mutated to alanine in Neph1 (Fig. 6D). Additionally, a peptide derived from this region (Fig. 6E) also displayed affinity toward Flag-Myo1c-FL and GFP-Myo1c-tail in a dot blot assay. Further, direct binding of Myo1c tail region with Neph1 was evaluated in an in vitro binding assay in which purified recombinant Flag-Myo1c-FL or GFP-His-Myo1c tail was mixed with purified His-Neph1-CD-wt and the pulldown was performed using Neph1 antibody (Fig. 6F). The results presented in Fig. 6F provide further evidence that Neph1 interacts directly with the Myo1c tail. Collectively, the live-cell and in vitro results suggest that the interaction between Neph1 and Myo1c is mediated through the C-terminal domain of Myo1c and that residue K761 of Neph1 is directly involved in this interaction. Overall, these results are entirely consistent with our SAXS and molecular modeling analyses, providing a molecular overview of the Neph1 and Myo1c interaction.
Myo1c competes with ZO1 for binding to Neph1.
Molecular modeling analysis of the Neph1-CD and Myo1c complex suggests that when Neph1 docks at the C-terminal region of Myo1c, its PDZ binding site (the last three amino acids, i.e., THV), which is normally exposed, remains buried and thus cannot interact with ZO1 (39). To validate this observation, a competitive-binding experiment was performed in which recombinant Flag-Myo1c-FL was mixed with His-Neph1-CD-wt protein in the presence or absence of purified His-ZO1-PDZ1 protein and Neph1 was immunoprecipitated using Neph1 antibody. Immunoprecipitation analysis of the Neph1 complex suggests that binding between Neph1 and Myo1c was significantly reduced in the presence of ZO1 (Fig. 7A). Similar results were obtained when the Myo1c tail was substituted in this assay (Fig. 7B). Input purified protein samples are shown in Fig. 7C. These competitive-binding experiments further confirm the conclusions derived from our SAXS and molecular modeling analysis indicating that Neph1 interacts with Myo1c in a fashion such that its ZO1 binding site becomes inaccessible.
FIG 7.

Myo1c competes with ZO1 for binding with Neph1. (A and B) Competitive-binding experiment in which recombinant Flag-Myo1c-FL was mixed with His-Neph1-CD-wt protein in the presence or absence of purified His-ZO1-PDZ1 protein and Neph1 was immunoprecipitated using Neph1 antibody. Immunoprecipitation analysis of the Neph1 complex suggests that binding between Neph1 and Myo1c was significantly reduced in the presence of ZO1. Similar results were obtained when GFP-His-Myo1c-tail was substituted in this assay (B). (C) Purified protein samples used for binding experiments in panels A and B.
Loss of Myo1c binding reduces the turnover rate of Neph1 at the podocyte cell membrane.
Myo1c is known to function as an adaptation motor by regulating the dynamic movement of stereociliary tip links—structures analogous to the slit diaphragm (48). This led us to believe that Myo1c may affect the dynamic movement of Neph1 at the membrane. Thus, to investigate whether loss of Myo1c binding influences the turnover of Neph1 at the podocyte cell membrane, cultured podocytes stably transfected with mCherry-Neph1-wt and mCherry-Neph1-K761A were analyzed by fluorescence recovery after photobleaching (FRAP). Since Neph1 is localized at the cell-cell junctions, it is likely that cells on either side of the junction can contribute toward the turnover of Neph1 at the membrane. Therefore, mCherry-Neph1-expressing cells were cocultured with GFP-expressing cells (Fig. 8A, left image) and the cellular density was carefully selected and monitored to ensure that only one particular cell (that is photobleached) contributes toward the Neph1 turnover at the membrane. A region of interest at the cell junction indicated in Fig. 8A was selected and photobleached. The recovery of Neph1 in the photobleached region was recorded over a period of 5 min. The FRAP analysis suggested a high turnover rate for Neph1-wt with >50% fluorescence recovery (n = 3 experiments) at the membrane, within 3 min of recovery time (Fig. 8A, top row; see also Movie S1A in the supplemental material), whereas the Myo1c-binding Neph1-K761A mutant displayed <10% (n = 3 experiments) recovery under similar conditions (Fig. 8A, bottom row; see also Movie S1B in the supplemental material). Notably, the observed mobile fraction for Neph1-wt was close to 80%, whereas for the Neph1 K761A mutant, it was only 20% (Fig. 8B and C). Collectively, these results suggest that interaction with Myo1c plays a role in maintaining the turnover rate for Neph1 at the podocyte cell membrane.
FIG 8.

The turnover rate of Neph1 at the podocyte cell membrane is decreased upon loss of Myo1c binding. (A) The turnover rate of Neph1 at the podocyte cell membrane was analyzed in cultured podocytes stably transfected with mCherry-Neph1-wt and mCherry-Neph1-K761A by fluorescence recovery after photobleaching (FRAP). A region of interest (ROI) at the cell junction was selected and photobleached. The recovery of Neph1 at the photobleached region was recorded over a period of 5 min. Arrows indicate the localization of mCherry-Neph1-wt at the cell-cell junctions. (B) The FRAP analysis suggests a high turnover rate for Neph1-wt with >50% fluorescence recovery (n = 3 experiments) at the membrane within 3 min of recovery time, whereas the Myo1c-binding Neph1-K761A mutant displayed <10% (n = 3 experiments) recovery under similar conditions. (C) The observed mobile fraction for mCherry-Neph1-wt was about 80%, compared to 20% for the Neph1-K761A mutant. Data are presented as means ± SEM.
Loss of Myo1c binding does not affect distribution of Neph1 into endocytic vesicles but affects their movement.
During this study, an interesting observation was made. In a confluent monolayer of podocyte cells, the majority of Neph1 was localized at the cell-cell junctions and at the membrane, as noted previously (Fig. 9 and reference 37), while in a subconfluent state, the majority of Neph1 was present in vesicles that were highly motile and distributed throughout the cytoplasm (Fig. 9). This prompted us to analyze the nature of these vesicles and their intracellular movements. To confirm the identity of these vesicles, podocytes stably expressing either mCherry-Neph1-wt or mCherry-Neph1-K761A were costained with markers of early and late endosomes and the Golgi complex. Live-cell analysis of the stained cells with different markers showed that the majority of Neph1 was present in the late endosomes (Fig. 10). A further quantitative analysis showed that the majority of mCherry-Neph1 vesicles (∼83%) costained with the late endosome marker, while the early endosomes and Golgi markers stained only 23% and 20% mCherry-Neph1 vesicles, respectively (Fig. 10). More importantly, significant motility was noted only in the late endosomes (see Movies S2A and B in the supplemental material), while early endosomes were largely immotile. Further comparison of mCherry-Neph1-wt with mCherry-Neph1-K761A vesicles suggested a similar pattern of distribution (Fig. 10).
FIG 9.

Neph1 intracellular distribution changes with cellular confluency. Untransfected podocytes and mCherry-Neph1-wt-expressing podocytes were cultured at very low confluence (<20%) to full confluence levels (100%) and analyzed by immunofluorescence microscopy for endogenous Neph1 and mCherry Neph1 localization at different stages of confluence. The localization of mCherry Neph1 (top) changed in a fashion similar to endogenous Neph1 (bottom) from mostly intracellular at low confluence to cell-cell junctions at high confluence. Arrowheads indicate the localization of mCherry-Neph1-wt at the cell-cell junctions. Scale bars represent 10 μm.
FIG 10.

Loss of Myo1c binding does not influence distribution of Neph1 into various subcellular compartments. To confirm the identity of Neph1-containing vesicles, podocytes stably expressing either mCherry-Neph1-wt or mCherry-Neph1-K761A were colabeled with markers of the early endosome, late endosome, Golgi complex, and lysosome using cell light reagents. Live-cell imaging was performed using confocal microscopy, and deconvoluted images were constructed and are presented (A). Single-plane images were used for analyzing colocalization of Neph1-wt and Neph1-K761A with the early endosome, late endosome, Golgi complex, and lysosome using ImageJ software. (B) Pearson's correlation coefficients are presented as means ± SEM. Scale bars represent 10 μm.
Since Myo1c has been shown to mediate the intracellular movement of Glut4 vesicles in adipocytes (49), we wanted to know if loss of Myo1c binding affects the movement of Neph1 vesicles in podocytes. Therefore, we recorded the vesicular movements of mCherry-Neph1-wt and mCherry-Neph1-K761A vesicles using time-lapse live-imaging microscopy, and the data were analyzed using Volocity image acquisition software. Representative images of Neph1-containing vesicles were plotted with displacement (micrometers, y axis) versus time (seconds, x axis) (Fig. 11A and B). As shown in Fig. 11, the mCherry-Neph1-wt vesicles were highly motile and showed a displacement of around 10 μm in podocytes, whereas the motility of vesicles containing the Neph1 mutant (K761A) was significantly attenuated (P < 0.001) (Fig. 11C and D; see also Movie S3A and B in the supplemental material). Collectively, these results indicate that Myo1c plays a role in the intracellular movement of Neph1 and may participate in Neph1 trafficking.
FIG 11.

Neph1 vesicular movement requires its interaction with Myo1c. (A and B) mCherry-Neph1-wt and the K761A mutant were plated on a glass-bottom cell culture plate, and the movement of Neph1-containing vesicles was analyzed using time-lapse live imaging. The vesicular movement was plotted as displacement (micrometers, y axis) versus time (seconds, x axis). (C and D) The displacement (micrometers) and velocity (micrometers per second) of mCherry-Neph1-K761A vesicles were significantly decreased compared to those of mCherry-Neph1-wt (P < 0.001). Data are presented as means ± SEM.
DISCUSSION
Although many cargo proteins, including actin, Nemo, Rictor, and the transporter protein Glut4, are known to interact with Myo1c (49–51), none of these interactions have been structurally defined. While this study was being conducted, two reports describing the structures of the Myo1c motor with the first IQ domain and Myo1c tail bound to calmodulin using X-ray crystallography were published (32–34). The Myo1c structure presented in these studies provided insight into the regulation of calmodulin binding through Ca2+ and binding to 14-3-3 (32, 33). However, we were interested in determining the structure of Myo1c that is engaged in transporting a cargo protein. In this report, we describe the full-length solution-based structure of Myo1c and its complex with Neph1 that highlights the molecular mechanism through which Myo1c participates in the intracellular movement of Neph1.
SAXS data-based results presented in this report show an extended S-shaped structure for full-length Myo1c in solution which compares well with the portions of the crystal structures of Myo1c known to date (32, 34). The composite model of Myo1c (with three calmodulins bound) nicely fitted with the SAXS data-based model and was in overall agreement with the head and tail structures that were separately studied by X-ray crystallography (32, 33). To the best of our knowledge, this is the first model of the full-length Myo1c.
Podocytes, along with their specialized junctions, the slit diaphragm, form core components of the glomerular filtration system (1, 29, 52). Over the past decade many proteins, including Neph1, have been described that specifically localize at these junctions and regulate podocyte structure and function and thus glomerular filtration function (1, 10, 12). However, the mechanisms through which Neph1 participates in the maintenance of podocyte function remain unknown. Our previous studies have shown that the activation of Neph1 cytoplasmic domain through an extracellular ligand generates signaling cues that induce reorganization of the actin cytoskeleton (14, 15). Recent analysis of the glomerular diseases and in vitro models of glomerular diseases have shown a dynamic shift in the localization of the slit diaphragm proteins, including nephrin, Neph1, and podocin, from the podocyte cell membrane to cell cytoplasm (11–15). Understanding the mechanisms that regulate these intracellular movements of slit diaphragm proteins is critical in developing therapeutic alternatives for the prevention of glomerular disorders. This study is an attempt to provide mechanistic insights into the intracellular movements of Neph1 that are driven by Myo1c.
We previously defined the solution-based structure of Neph1 cytoplasmic domain and its complex with the first PDZ domain of ZO1 using SWAXS (39). To gain insight into the molecular details of Neph1-Myo1c interaction, a similar approach was used in which solution-based structures of purified Myo1c and its complex with the cytoplasmic domain of Neph1 were studied. The combined approach of SAXS data analysis and molecular modeling revealed a comprehensive three-dimensional overview of this interaction, in which Neph1 is bound as cargo at the extreme C-terminal end of Myo1c. Moreover, the structural analysis suggests that there are no binding-induced changes in the complex, indicating that the interaction between Neph1 and Myo1c is a spontaneous event. At this point it is difficult to speculate what initiates the formation of this motor cargo complex and under what conditions the cargo is released from the motor. Nevertheless, this is the first study that provides structural information about the interaction of a cargo protein with Myo1c. Although many other proteins, including RalA, calcium-binding protein 1, and 14-3-3 (phosphoprotein regulator), are known to interact with the regulatory domain of Myo1c (35, 36), the structural details and the significance of these interactions are not clear.
Unlike X-ray crystallography, SAXS does not provide molecular details of atomic-level resolution of the residues involved in Neph1-Myo1c interaction. However, the recent significant advancements in the field of structure reconstruction from scattering data allowed us to identify interacting residues and other structural details with reliable certainty. Indeed, the residue K761 in Neph1 predicted from molecular modeling analysis was validated through the live-cell and in vitro biochemical experiments performed with mammalian cells and using recombinant purified proteins, respectively. Although it has been described that Neph1 interacts with both ZO1 and Myo1c (14, 15, 39), the structural data presented in this study further highlight the inability of Neph1 to simultaneously interact with ZO1 and Myo1c (Fig. 7B). Indeed, the first PDZ domain of ZO1 competed for binding with Neph1 in an in vitro Neph1-Myo1c binding assay (Fig. 7). This further suggests that either Neph1 interacts with ZO1 and Myo1c in separate cellular compartments or that these complexes exist under different cellular conditions. Since we have previously shown that the Neph1 and ZO1 complex is primarily localized at cell-cell junctions (14), it is likely that Neph1 interacts with Myo1c within the cytoplasmic region, where it may be involved in the intracellular movement of Neph1. While the Neph1 and ZO1 complex was shown to establish the integrity of podocyte cell junctions during recovery from injury (14, 53), this study highlights the role of Neph1 binding to Myo1c in the intracellular dynamic movement of Neph1, which is supported by our live-cell imaging studies. Additional studies may be needed to further delineate the exact functional significance of these associations. Since colocalization of Myo1c and Neph1 was primarily visualized at the podocyte cell membrane (15), we hypothesized that Myo1c plays a role in the membrane dynamics of Neph1. Indeed, the results from FRAP experiments further support this hypothesis. Based on these results, it is tempting to speculate that Myo1c may be involved in multiple cellular events in podocytes, which may include intracellular movement of Neph1 and cross-linking Neph1 with the actin cytoskeleton (Fig. 8). Whether there is a directional movement of Neph1 mediated by Myo1c that translocates Neph1 from cytoplasm to the membrane and either the cargo is released at this point or Myo1c tethers Neph1 at the membrane (in a fashion similar to Glut4) (36, 49) may require further investigation. With the tools developed in this study, we are now in a position to investigate this further.
The slit diaphragm is uniquely situated at the distal end of a podocyte, away from the cell body, where it connects the tertiary foot processes of two different podocytes. This implies that proteins, like Neph1, that are the critical backbone of this structure must employ transport mechanisms that mediate the movement of this protein from the podocyte cell body to tertiary processes (anterograde transport), where it contributes to the formation of slit diaphragm. Additionally, in the event of a glomerular injury the protein complexes at the slit diaphragm are disassembled (14, 15), which may result in the retrograde transport of Neph1 toward the podocyte cell body. Understanding the trafficking mechanisms that mediate such intracellular movements of Neph1 is critical in developing therapeutic alternatives for restoring renal function in various glomerular disorders. In this study, we demonstrated that intracellular dynamic movement of Neph1 requires binding to Myo1c. Since Myo1c tightly associates with actin (23, 54, 55) and Neph1 activation is involved in actin reorganization (14, 56), it is likely that Myo1c serves as a linker between Neph1 and the actin cytoskeleton. How this linkage affects Neph1 dynamics at the membrane, especially since Myo1c also has been shown to function as a tethering protein (57), remains to be investigated.
To further study the cellular dynamics of Neph1, the kinetics of diffusion of Neph1 and its Myo1c binding mutant at the membrane was studied using FRAP in live cultured podocytes. The Neph1 mutant that did not interact with Myo1c demonstrated slower membrane dynamics, and its intracellular mobility was also significantly decreased. The high turnover rate of Neph1 at the membrane compared to that of its mutant suggests that Neph1 interaction with Myo1c may play a key role in the intracellular movement of Neph1. Whether this intracellular movement is related to the trafficking of Neph1 that involves specific cellular processes, such as endocytosis or exocytosis, will require further investigation. Moreover, how this movement affects physiological function of podocytes needs to be explored further. A recent report suggests that the velocity of Glut4 vesicles contributed by Myo1c is 0.3 to 0.4 μm/s (57), which is quite comparable to the velocities reported in this study (∼0.5 μm/s) (Fig. 11) for Neph1 vesicles. Although we demonstrate that Myo1c interaction plays a crucial role in Neph1 vesicular movement, the possibility that other factors contribute to the movements of Neph1-containing vesicles cannot be ruled out. However, our analysis of the Myo1c-binding Neph1 mutant whose mobility was significantly impaired provides sufficient evidence that the intracellular movements of Neph1 in large part are mediated by Myo1c. It is important to note that both Neph1 and Myo1c localize in lipid rafts that house several signaling and adapter proteins that regularly traffic between cell membrane and perinuclear storage compartments. Interestingly, the levels of lipid rafts at the cell surface are regulated in a Myo1c-dependent fashion in which Myo1c facilitates the delivery of raft-enriched cargo to the cell surface (27). Since Myo1c also interacts with another slit diaphragm protein, nephrin, this further implies that Myo1c plays a major role in trafficking of slit diaphragm proteins that are generally localized in lipid rafts.
Although the exact function of Neph1 at the podocyte cell membrane is unclear, its role in the podocyte development is supported from the studies with Drosophila and Caenorhabditis elegans models in which it has been shown to be involved in axonal guidance and synapse development, respectively (58, 59). Since Myo1c also localizes to the actin-rich periphery and plasma membrane and has been shown to be involved in regulating dynamic cellular processes, including vesicle trafficking, cell migration, cell spreading, and membrane tension (15, 23, 27, 55, 60), it is likely that the interaction between Neph1 and Myo1c contributes toward the regulation of Neph1 signaling and trafficking. Although our study using a zebrafish model system demonstrated that Myo1c plays a critical role in maintaining renal function (31), it did not provide a specific function for Myo1c in podocytes. We are currently in the process of developing a podocyte-specific Myo1c knockout mouse model that may provide answers to some of these questions.
Supplementary Material
ACKNOWLEDGMENTS
National Institutes of Health, NIDDK, grants RO1 2R01DK087956-06A1 to D.N. and DK080751 to L.B.H. are duly acknowledged. SAXS experiments were supported by CSIR 12FYP BioDiscovery and Unseen projects to A. Use of the National Synchrotron Light Source (X9 Beamline), Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract DE-AC02-98CH10886. Biacore work was performed in the Department of Regenerative Medicine and Cell Biology Proteomics Core, which is supported by the South Carolina COBRE for Developmentally based Cardiovascular Diseases (5P30GM103342) from NIH.
We declare that no conflict of interest exists.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00020-16.
REFERENCES
- 1.Tryggvason K, Patrakka J, Wartiovaara J. 2006. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med 354:1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 2.Tryggvason K, Pikkarainen T, Patrakka J. 2006. Nck links nephrin to actin in kidney podocytes. Cell 125:221–224. doi: 10.1016/j.cell.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Welsh GI, Saleem MA. 2012. The podocyte cytoskeleton—key to a functioning glomerulus in health and disease. Nat Rev Nephrol 8:14–21. [DOI] [PubMed] [Google Scholar]
- 4.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. 2007. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Buccione R, Orth JD, McNiven MA. 2004. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol 5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 6.Kerjaschki D. 2001. Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J Clin Invest 108:1583–1587. doi: 10.1172/JCI200114629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda T, McQuistan T, Orlando RA, Farquhar MG. 2001. Loss of glomerular foot processes is associated with uncoupling of podocalyxin from the actin cytoskeleton. J Clin Invest 108:289–301. doi: 10.1172/JCI12539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Agati VD, Kaskel FJ, Falk RJ. 2011. Focal segmental glomerulosclerosis. N Engl J Med 365:2398–2411. doi: 10.1056/NEJMra1106556. [DOI] [PubMed] [Google Scholar]
- 9.Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. 2006. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest 116:1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barletta GM, Kovari IA, Verma RK, Kerjaschki D, Holzman LB. 2003. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J Biol Chem 278:19266–19271. doi: 10.1074/jbc.M301279200. [DOI] [PubMed] [Google Scholar]
- 11.Doublier S, Ruotsalainen V, Salvidio G, Lupia E, Biancone L, Conaldi PG, Reponen P, Tryggvason K, Camussi G. 2001. Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am J Pathol 158:1723–1731. doi: 10.1016/S0002-9440(10)64128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otaki Y, Miyauchi N, Higa M, Takada A, Kuroda T, Gejyo F, Shimizu F, Kawachi H. 2008. Dissociation of NEPH1 from nephrin is involved in development of a rat model of focal segmental glomerulosclerosis. Am J Physiol Renal Physiol 295:F1376–F1387. doi: 10.1152/ajprenal.00075.2008. [DOI] [PubMed] [Google Scholar]
- 13.Wernerson A, Duner F, Pettersson E, Widholm SM, Berg U, Ruotsalainen V, Tryggvason K, Hultenby K, Soderberg M. 2003. Altered ultrastructural distribution of nephrin in minimal change nephrotic syndrome. Nephrol Dial Transplant 18:70–76. doi: 10.1093/ndt/18.1.70. [DOI] [PubMed] [Google Scholar]
- 14.Wagner MC, Rhodes G, Wang E, Pruthi V, Arif E, Saleem MA, Wean SE, Garg P, Verma R, Holzman LB, Gattone V, Molitoris BA, Nihalani D. 2008. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem 283:35579–35589. doi: 10.1074/jbc.M805507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arif E, Wagner MC, Johnstone DB, Wong HN, George B, Pruthi PA, Lazzara MJ, Nihalani D. 2011. Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol Cell Biol 31:2134–2150. doi: 10.1128/MCB.05051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nabet B, Tsai A, Tobias JW, Carstens RP. 2009. Identification of a putative network of actin-associated cytoskeletal proteins in glomerular podocytes defined by co-purified mRNAs. PLoS One 4:e6491. doi: 10.1371/journal.pone.0006491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierchala BA, Munoz MR, Tsui CC. 2010. Proteomic analysis of the slit diaphragm complex: CLIC5 is a protein critical for podocyte morphology and function. Kidney Int 78:868–882. doi: 10.1038/ki.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krendel M, Osterweil EK, Mooseker MS. 2007. Myosin 1E interacts with synaptojanin-1 and dynamin and is involved in endocytosis. FEBS Lett 581:644–650. doi: 10.1016/j.febslet.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, Buelli S, Tomasoni S, Piras R, Krendel M, Bettoni S, Morigi M, Delledonne M, Pecoraro C, Abbate I, Capobianchi MR, Hildebrandt F, Otto E, Schaefer F, Macciardi F, Ozaltin F, Emre S, Ibsirlioglu T, Benigni A, Remuzzi G, Noris M. 2011. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 365:295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pecci A, Panza E, Pujol-Moix N, Klersy C, Di Bari F, Bozzi V, Gresele P, Lethagen S, Fabris F, Dufour C, Granata A, Doubek M, Pecoraro C, Koivisto PA, Heller PG, Iolascon A, Alvisi P, Schwabe D, De Candia E, Rocca B, Russo U, Ramenghi U, Noris P, Seri M, Balduini CL, Savoia A. 2008. Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of MYH9-related disease. Hum Mutat 29:409–417. doi: 10.1002/humu.20661. [DOI] [PubMed] [Google Scholar]
- 21.Sekine T, Konno M, Sasaki S, Moritani S, Miura T, Wong WS, Nishio H, Nishiguchi T, Ohuchi MY, Tsuchiya S, Matsuyama T, Kanegane H, Ida K, Miura K, Harita Y, Hattori M, Horita S, Igarashi T, Saito H, Kunishima S. 2010. Patients with Epstein-Fechtner syndromes owing to MYH9 R702 mutations develop progressive proteinuric renal disease. Kidney Int 78:207–214. doi: 10.1038/ki.2010.21. [DOI] [PubMed] [Google Scholar]
- 22.Wagner MC, Barylko B, Albanesi JP. 1992. Tissue distribution and subcellular localization of mammalian myosin I. J Cell Biol 119:163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barylko B, Jung G, Albanesi JP. 2005. Structure, function, and regulation of myosin 1C. Acta Biochim Pol 52:373–380. [PubMed] [Google Scholar]
- 24.Sellers JR. 2000. Myosins: a diverse superfamily. Biochim Biophys Acta 1496:3–22. doi: 10.1016/S0167-4889(00)00005-7. [DOI] [PubMed] [Google Scholar]
- 25.Gillespie PG, Cyr JL. 2004. Myosin-1c, the hair cell's adaptation motor. Annu Rev Physiol 66:521–545. doi: 10.1146/annurev.physiol.66.032102.112842. [DOI] [PubMed] [Google Scholar]
- 26.Tang N, Lin T, Ostap EM. 2002. Dynamics of Myo1c (myosin-Ibeta) lipid binding and dissociation. J Biol Chem 277:42763–42768. doi: 10.1074/jbc.M206388200. [DOI] [PubMed] [Google Scholar]
- 27.Brandstaetter H, Kendrick-Jones J, Buss F. 2012. Myo1c regulates lipid raft recycling to control cell spreading, migration and Salmonella invasion. J Cell Sci 125:1991–2003. doi: 10.1242/jcs.097212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnstone DB, Holzman LB. 2006. Clinical impact of research on the podocyte slit diaphragm. Nat Clin Pract Nephrol 2:271–282. doi: 10.1038/ncpneph0180. [DOI] [PubMed] [Google Scholar]
- 29.Patrakka J, Tryggvason K. 2010. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun 396:164–169. doi: 10.1016/j.bbrc.2010.04.069. [DOI] [PubMed] [Google Scholar]
- 30.Saleem MA, Ni L, Witherden I, Tryggvason K, Ruotsalainen V, Mundel P, Mathieson PW. 2002. Co-localization of nephrin, podocin, and the actin cytoskeleton: evidence for a role in podocyte foot process formation. Am J Pathol 161:1459–1466. doi: 10.1016/S0002-9440(10)64421-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arif E, Kumari B, Wagner MC, Zhou W, Holzman LB, Nihalani D. 29 May 2013. Myo1c is an unconventional myosin required for zebrafish glomerular development. Kidney Int doi: 10.1038/ki.2013.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Münnich S, Taft MH, Manstein DJ. 2014. Crystal structure of human myosin 1c–the motor in GLUT4 exocytosis: implications for Ca2+ regulation and 14-3-3 binding. J Mol Biol 426:2070–2081. doi: 10.1016/j.jmb.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Münnich S, Manstein DJ. 2013. Expression, purification, crystallization and preliminary X-ray crystallographic analysis of human myosin 1c in complex with calmodulin. Acta Crystallogr Sect F Struct Biol Cryst Commun 69:1020–1022. doi: 10.1107/S1744309113020988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Q, Li J, Ye F, Zhang M. 2015. Structure of myosin-1c tail bound to calmodulin provides insights into calcium-mediated conformational coupling. Nat Struct Mol Biol 22:81–88. [DOI] [PubMed] [Google Scholar]
- 35.Yip MF, Ramm G, Larance M, Hoehn KL, Wagner MC, Guilhaus M, James DE. 2008. CaMKII-mediated phosphorylation of the myosin motor Myo1c is required for insulin-stimulated GLUT4 translocation in adipocytes. Cell Metab 8:384–398. doi: 10.1016/j.cmet.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. 2007. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell 13:391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Arif E, Rathore YS, Kumari B, Ashish F, Wong HN, Holzman LB, Nihalani D. 2014. Slit diaphragm protein Neph1 and its signaling: a novel therapeutic target for protection of podocytes against glomerular injury. J Biol Chem 289:9502–9518. doi: 10.1074/jbc.M113.505743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin X-S, Tsukaguchi H, Shono A, Yamamoto A, Kurihara H, Doi T. 2009. Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis. J Am Soc Nephrol 20:2534–2545. doi: 10.1681/ASN.2009010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mallik L, Arif E, Sharma P, Rathore YS, Wong HN, Holzman LB, Ashish, Nihalani D. 2012. Solution structure analysis of cytoplasmic domain of podocyte protein Neph1 using small/wide angle x-ray scattering (SWAXS). J Biol Chem 287:9441–9453. doi: 10.1074/jbc.M111.284927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin T, Greenberg MJ, Moore JR, Ostap EM. 2011. A hearing loss-associated myo1c mutation (R156W) decreases the myosin duty ratio and force sensitivity. Biochemistry 50:1831–1838. doi: 10.1021/bi1016777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hokanson DE, Ostap EM. 2006. Myo1c binds tightly and specifically to phosphatidylinositol 4,5-bisphosphate and inositol 1,4,5-trisphosphate. Proc Natl Acad Sci U S A 103:3118–3123. doi: 10.1073/pnas.0505685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 43.Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25:495–503. doi: 10.1107/S0021889892001663. [DOI] [Google Scholar]
- 44.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozin MB, Svergun DI. 2001. Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34:33–41. doi: 10.1107/S0021889800014126. [DOI] [Google Scholar]
- 46.Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. 2014. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30:1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKenna JM, Ostap EM. 2009. Kinetics of the interaction of myo1c with phosphoinositides. J Biol Chem 284:28650–28659. doi: 10.1074/jbc.M109.049791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adamek N, Coluccio LM, Geeves MA. 2008. Calcium sensitivity of the cross-bridge cycle of Myo1c, the adaptation motor in the inner ear. Proc Natl Acad Sci U S A 105:5710–5715. doi: 10.1073/pnas.0710520105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bose A, Guilherme A, Robida SI, Nicoloro SM, Zhou QL, Jiang ZY, Pomerleau DP, Czech MP. 2002. Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature 420:821–824. doi: 10.1038/nature01246. [DOI] [PubMed] [Google Scholar]
- 50.Nakamori Y, Emoto M, Fukuda N, Taguchi A, Okuya S, Tajiri M, Miyagishi M, Taira K, Wada Y, Tanizawa Y. 2006. Myosin motor Myo1c and its receptor NEMO/IKK-gamma promote TNF-alpha-induced serine307 phosphorylation of IRS-1. J Cell Biol 173:665–671. doi: 10.1083/jcb.200601065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hagan GN, Lin Y, Magnuson MA, Avruch J, Czech MP. 2008. A Rictor-Myo1c complex participates in dynamic cortical actin events in 3T3-L1 adipocytes. Mol Cell Biol 28:4215–4226. doi: 10.1128/MCB.00867-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huber TB, Benzing T. 2005. The slit diaphragm: a signaling platform to regulate podocyte function. Curr Opin Nephrol Hypertens 14:211–216. doi: 10.1097/01.mnh.0000165885.85803.a8. [DOI] [PubMed] [Google Scholar]
- 53.Huber TB, Schmidts M, Gerke P, Schermer B, Zahn A, Hartleben B, Sellin L, Walz G, Benzing T. 2003. The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J Biol Chem 278:13417–13421. doi: 10.1074/jbc.C200678200. [DOI] [PubMed] [Google Scholar]
- 54.Pyrpassopoulos S, Feeser EA, Mazerik JN, Tyska MJ, Ostap EM. 2012. Membrane-bound myo1c powers asymmetric motility of actin filaments. Curr Biol 22:1688–1692. doi: 10.1016/j.cub.2012.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fan Y, Eswarappa SM, Hitomi M, Fox PL. 2012. Myo1c facilitates G-actin transport to the leading edge of migrating endothelial cells. J Cell Biol 198:47–55. doi: 10.1083/jcb.201111088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. 2007. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol 27:8698–8712. doi: 10.1128/MCB.00948-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boguslavsky S, Chiu T, Foley KP, Osorio-Fuentealba C, Antonescu CN, Bayer KU, Bilan PJ, Klip A. 2012. Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles. Mol Biol Cell 23:4065–4078. doi: 10.1091/mbc.E12-04-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugie A, Umetsu D, Yasugi T, Fischbach KF, Tabata T. 2010. Recognition of pre- and postsynaptic neurons via nephrin/NEPH1 homologs is a basis for the formation of the Drosophila retinotopic map. Development 137:3303–3313. doi: 10.1242/dev.047332. [DOI] [PubMed] [Google Scholar]
- 59.Neumann-Haefelin E, Kramer-Zucker A, Slanchev K, Hartleben B, Noutsou F, Martin K, Wanner N, Ritter A, Godel M, Pagel P, Fu X, Muller A, Baumeister R, Walz G, Huber TB. 2010. A model organism approach: defining the role of Neph proteins as regulators of neuron and kidney morphogenesis. Hum Mol Genet 19:2347–2359. doi: 10.1093/hmg/ddq108. [DOI] [PubMed] [Google Scholar]
- 60.Greenberg MJ, Lin T, Goldman YE, Shuman H, Ostap EM. 2012. Myosin IC generates power over a range of loads via a new tension-sensing mechanism. Proc Natl Acad Sci U S A 109:E2433–E2440. doi: 10.1073/pnas.1207811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.