

Abstract
Recent innovations in DNA nanofabrication allow the creation of intricately shaped nanostructures ideally suited for many biological applications. To advance the use of DNA nanotechnology for the controlled release of bioactive molecules, we report a general strategy that uses light to liberate encapsulated cargoes from DNA nanostructures with high spatiotemporal precision. Through the incorporation of a custom, photolabile cross-linker, we encapsulated cargoes ranging in size from small molecules to full-sized proteins within DNA nanocages and then released such cargoes upon brief exposure to light. This novel molecular uncaging technique offers a general approach for precisely releasing a large variety of bioactive molecules, allowing investigation into their mechanism of action, or finely tuned delivery with high temporal precision for broad biomedical and materials applications.
Keywords: nanotechnology, DNA origami, controlled release, uncaging, photolabile cross-linker, bioactive cargo
Graphical Abstract
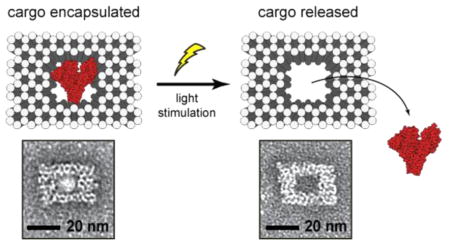
Rapid advances in structural DNA nanotechnology allow the creation of intricately shaped nanostructures that can be functionalized with a high degree of control at precise locations.1–4 For example, DNA origami can be reliably and efficiently self-assembled by folding large, single stranded DNA with a set of specifically designed short oligonucleotide strands.5 This technique affords a tremendous amount of control over the size and shape of the nanostructure whose designs can now be assisted by well-developed software tools.6–8 Molecularly programmed, static,9–11 or dynamic12–14 DNA architectures hold promise for applications in areas such as cell biology,15 NMR spectroscopy,16 super resolution microscopy,17 and nanotherapeutics,18 many of which would be advanced if DNA nanostructures were capable of releasing bound cargos at precise times.
Attempts to obtain controlled release from DNA origami nanostructures thus far have utilized two approaches through either noncovalent or covalent attachment of the cargo to origami. For example, the chemotherapy drug doxorubicin has been found to be able to noncovalently bind to DNA nano-structures through interactions with the DNA helices.19,20 By controlling the DNA origami structure configuration, it was shown that doxorubicin release from the nanostructures could elicit a cytotoxic response in regular and drug-resistant cancer cells. Noncovalent attachment strategies however critically depend on a chemical’s ability to intercalate into DNA helices. This binding mechanism cannot be generalized to most chemicals, and the binding sites within an origami cannot be easily controlled spatially. Direct covalent attachment of cargo to DNA origami nanostructures can overcome most of these limitations. To covalently attach a cargo to DNA helices, short DNA strands can be designed to protrude at specific locations on the surface of the nanostructures, which can then bind to a variety of different chemical moieties including inorganic nanoparticles,21,22 proteins,23 antibodies,18 and fluorophores.24 Placement of cleavable linkages within these DNA strands permits the release of the bound cargo in a highly controllable fashion. However, such strategies often leave a chemical remnant, the chemical group that connects the cargo to DNA strands25 on the molecules being released, which may compromise their native biological function, limiting this approach to applications where the bio-activity of the cargo is important. Here, we demonstrate a novel and general method that releases chemically unaltered cargoes using brief pulses of light that can be broadly applied to a large variety of molecules.
We designed a novel, photolabile linker to append cargo molecules into the cavities of DNA nanostructures, so that light irradiation-induced breakage of the linker would allow the molecules to diffuse away from the protective cavity (Figure 1). This photolabile cross-linker possesses an o-nitrobenzyl (o-NB) motif for photocleavage, an azido group for attachment to alkyne functionalized oligonucleotides, and an activated carbonate group for attachment to cargo molecules possessing a free amino functional group (Figure 1A). The linker is designed to release cargo upon photo cleavage in its original state with no chemical remnants remaining attached. Given the fact that most peptides, proteins, and bioactive compounds contain exposed amino residues, the cross-linker design is broadly applicable to attach many molecules to DNA nanostructures, beyond the examples described here.
Figure 1.

Design and creation of light-triggered, cargo-releasing nanocages. (A) Scheme of the chemical activation of a cargo molecule with the photolabile cross-linker and an oligonucleotide. (B) Depiction of the DNA nanostructure formation. The solid cylinders represent DNA helices as shown by the inset. (C) Agarose gel electrophoresis showing the high folding yield of the crude DNA nanocage sample. Lane L contains the 1-kb ladder, lane m13 contains the single stranded DNA starting material, and lane cage contains the crude reaction mixture. (D) TEM images of DNA nanocages. Scale bars are 200 and 25 nm, respectively. (E) Schematic depiction of the encapsulation of cargo, the photocleavage reaction, and subsequent cargo release.
We first synthesized this photolabile cross-linker using conventional organic synthesis techniques. Gram scale product was easily produced from inexpensive, commercially available starting materials (Scheme S1). This photolabile cross-linker was then reacted with cargo molecules including glutamate, bovine serum albumin (BSA), and biotin amine, and subsequently conjugated to oligonucleotides allowing the cargo to be incorporated into preassembled DNA origami through DNA base pairing (Scheme S2).
In parallel, we computationally designed a multilayered, brick-like nanocage structure with a well-defined cavity in its center, similar to those previously reported.6,20,26 The nanocage contains 14 addressable, single-stranded DNA extensions in its cavity, which are complementary to those presented on the activated cargo (Figure 1B–D). Nanostructures were then self-assembled in a single step by slowly cooling a heated mixture of the DNA components. Analysis of the assembly by agarose gel electrophoresis showed a single, dominant product band that migrated faster relative to the single stranded DNA starting material (m13 DNA), consistent with that generally observed for multilayered DNA origami structures (Figure 1C).9,21 Further examination with transmission electron microscopy (TEM) revealed properly assembled structures with the desired shape and a clearly visible central cavity (Figure 1D). The short single-stranded DNA extensions, however, were too small to be resolved using TEM. Purification of fully formed nano-structures from excess oligonucleotides or subsequent cargo molecules was accomplished using poly(ethylene glycol) precipitation.27
Fully assembled and purified DNA nanocages were then incubated with the activated cargo to attach them to the interior of the nanocage cavity. When positioned inside of the nano-structure, the cargo is protected from the exterior environment and unable to bind to its native sites of action. Release from the cage was then achieved with light irradiation, which cleaved the photolabile bonds within the cross-linker (Figure 1E).
To first validate the photocleavage of our cross-linkers, we used it to conjugate an oligonucleotide to the small fluorescent molecule Oregon Green cadaverine (OG). We irradiated the compound with a low-power light source over time and quantified the degree of separation of OG from the oligonucleotide using HPLC (Figure 2A). We found that an increasing duration of light exposure led to a larger fraction of free OG dye. After 11 s of low-powered light irradiation, 50% of OG was released. Nearly complete cleavage was achieved after 40 s of exposure, consistent with the time course for the cleavage of the o-NB motif within the cross-linker.28
Figure 2.
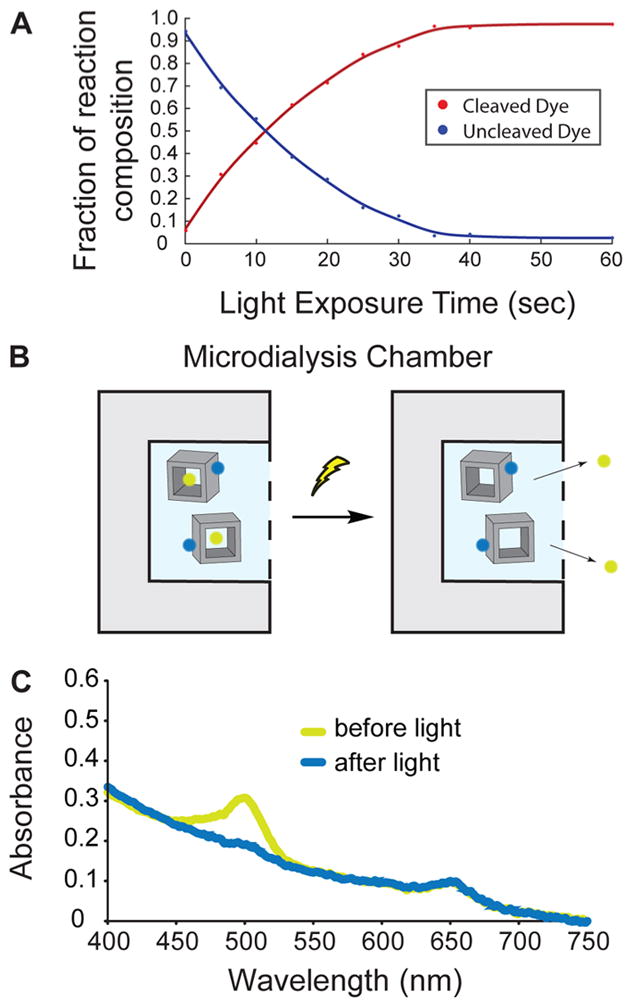
Light-triggered release of small molecules from nanocages. (A) Photolysis data showing increased irradiation duration results in an increase in the cleavage of Oregon Green/oligonucleotides conjugate. (B) Schematic depiction of the dye uncaging experiment. DNA nanostructures remain in the microdialysis chamber, whereas small dyes are able to diffuse out. (C) Absorption spectra of a duel dye tagged nanocage before (yellow curve) and after light irradiation (blue curve).
We then loaded the activated OG into the cavities of the nanostructures by incubating the OG/DNA conjugate with preassembled nanocages. To quantify loading efficiency, we incorporated a nonlabile dye (Alexa Fluor 647N, AF647) for comparison by attaching it to a region on the nanostructure distal to the cavity (Figure 2B). UV absorbance spectra analysis of the product showed two distinct absorption peaks centered around 500 and 647 nm, corresponding to the two dyes used (Figure 2C, yellow trace). The ratio of the dye concentrations for OG versus AF647 was 7.4 to 1, suggesting that about half of the 14 DNA extensions on each cage designed to bind OG were bound, which is likely a representative loading capacity for small molecules of similar size.
To measure the efficiency of the light-induced release of OG from the nanocages, we irradiated the structures with a low powered lamp for 60 s, and then analyzed the absorbance spectra of the reaction solution after extensive sample dialysis of released free OG (Figure 2B). We observed that the peak absorption at 500 nm corresponding to the photolabile OG dye was completely absent after irradiation, whereas the 647 nm absorption peak corresponding to the nonlabile AF647 remained (Figure 2C, blue trace). Together, these results demonstrate that our uncaging strategy can successfully release small molecular cargo from the DNA nanostructure upon brief low energy light irradiation.
We then explored the possibility of releasing large proteins from the nanocages, using bovine serum albumin (BSA) and streptavidin as examples that can be easily observed and analyzed using TEM. BSA was directly caged through the reaction of our cross-linker with the surface amino groups on the protein. Streptavidin was indirectly caged by attaching biotin-amine to the nanocage cavity and then subsequently mixing with the protein. TEM analysis of nanostructures at different orientations revealed clearly visible BSA and streptavidin proteins within the cavity of the DNA cage (Figure 3). None were seen tethered to the cage exterior. The number of DNA nanostructures with and without proteins was determined via particle counting of TEM images, and a loading efficiency of 93% for BSA and 71% for streptavidin was observed. After low power light irradiation for 60 s, we found only 19% of nanocages contained BSA, and 9% cages contained streptavidin, which corresponds to uncaging efficiencies of 79% for BSA and 87% for streptavidin. Together, these results demonstrate that full sized proteins can be effiectively encapsulated and uncaged with high efficiency.
Figure 3.

Light-triggered release of proteins, (A) streptavidin and (B) bovine serum albumin from nanocages. (i) Schematic depictions of the DNA nanocages with and without proteins. (ii) TEM images of nanocaged proteins before (left) and after (middle) irradiation with light. Scale bars are 25 nm. (iii) Graphs showing percentage of nanocages containing protein as determined by TEM image counting before and after light are shown on the right. Numbers in parentheses indicate the number of particles counted per condition.
To demonstrate that molecules released from the DNA nanocages retain their bioactivity, we tested uncaging of the small molecule glutamic acid, an excitatory neurotransmitter that has been shown to be successfully uncaged in numerous instances (Figure 4A).29–31 The bioactivity of the released glutamate from the nanocages was measured by glutamate mediated calcium changes in cultured neurons using real-time fluorescence imaging. Primary hippocampal neuron cultures were incubated with the intracellular calcium dye Fluo-4 and the glutamate-containing DNA nanocages. Before light illumination, little basal calcium activity was observed in the nine-day-old cultures, consistent with the general activity patterns observed in neuron cultures of this age (Figure 4B and C).32 Immediately following a 1 ms light pulse illumination (240–400 nm), we observed an increase in intracellular calcium levels in 16.22% (N = 185 neurons, analyzed in two tests) (Figure 4C, Video S1). Activated cells exhibited heterogeneity in response amplitude with activation onsets ranged from 509 ms to 18.19 s after the light pulse, which could be due to difference in difiusion time from the releasing site to the cell surface, the concentration of released glutamic acid on a given cell, and intrinsic variability of cellular calcium responses (Figure 4D). The fact that light irradiation was delivered for 1 ms suggests that uncaging can be performed with millisecond temporal resolution. In the absence of the DNA nanocages, no cells exhibited a change in calcium levels upon light illumination (N = 124 neurons, analyzed in two tests) (Figure 4B, Video S2). Together, these results demonstrate that DNA nanocages can be used to release functional bioactive molecules with millisecond temporal precision.
Figure 4.

Light-triggered release of glutamate from DNA nanocages. (A) Schematic depiction of glutamate release from DNA nanocage using UV light at 240–400 nm and the subsequent activation of neurons by the freed glutamate. (B,C) Temporal derivative of the normalized fluorescence intensity indicating calcium concentration changes in the control group, neurons illuminated in the absence of nanocages (B, N = 124 neurons), and in the uncaging group, neurons illuminated in the presence of nanocages (C, N = 185 neurons). (D) Normalized fluorescence intensity indicating intracellular calcium activities of responsive cells in the uncaged group, aligned to light onset. Thick blue line indicates the mean, shaded gray indicates standard deviation, and red dots indicate the onset time (N = 30 neurons).
In conclusion, we describe a novel strategy to encapsulate bioactive molecules inside DNA nanostructures and release them using pulses of light. This strategy is realized through tagging DNA origami with a novel photolabile cross-linker that can be broadly used to encapsulate a large variety of molecules. With this cross-linker, a single, general chemical reaction scheme can be used to attach chemicals of interest to DNA origami through reacting with amino groups which are present on many biologically relevant compounds. This technique allows the release of cargo in its unaltered, bioactive state in contrast to existing labile conjugation chemistries, which often leave behind a chemical remnant that may interfere with the natural bioactivity of the cargo. This strategy was shown to be effective for a range of molecular sizes, from small molecules to full-sized proteins. Our nanocage design offers a high degree of addressability and customization, and future versions could be created that accommodate a larger variety of cargo molecules or cocktails of molecules in precise stoichiometries by controlling the shape and dimensions of the nanostructures as well as the sequences of the strands protruding from the cavity. Although light controlled uncaging techniques have been successful in releasing small molecules that rely on small, photochemical blocking chemical groups, our nanocaging platform could be easily designed to release many previously uncagable compounds and accelerate progress in understanding chemical receptor binding or controlled release of therapeutics.
Supplementary Material
Acknowledgments
We thank members of the Han Lab for suggestions related to experimental design and data analysis. We thank James P. Gilbert for providing neuron cultures. The authors would like to thank the W. M. Keck Microscope Facility at the Whitehead Institute for usage of the transmission electron microscope. X.H. acknowledges funding from NIH Director’s new innovator award 1DP2NS082126, Pew Foundation, Alfred P. Sloan Foundation, and Boston University Biomedical Engineering Department. H.Y.M. acknowledges funding from NIH MH079407, S.S.C. acknowledges funding from NIH T32 Training Grant titled Translational Research in Biomaterials.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.6b00530.
- Synthetic schemes, synthetic details, bioconjugation details, experimental details of glutamate uncaging, additional TEM images, and DNA sequences. (PDF)
- Supplemental Video S1: In the presence of DNA nanocages containing glutamate, neurons exhibited an increase in intracellular calcium levels after 1 ms light pulse illumination. (AVI)
- Supplement Video S2: In the absence of the DNA nano-cages, no neurons exhibited a change in calcium levels after 1 ms light pulse illumination. (AVI)
Author Contributions
The research project concept was created by X.H. Molecular design, organic synthesis, nanostructure creation, TEM imaging, and cargo release characterization were performed by R.E.K. Glutamate uncaging experiments were performed by R.E.K. and S.S.C. S.S.C and X.H. analyzed the glutamate uncaging data. H.Y.M. provided neuron cultures and technical support for glutamate uncaging experiments. X.H. supervised the study. R.E.K., S.S.C, H.Y.M., and X. H. wrote the manuscript.
References
- 1.Seeman NC. Nature. 2003;421:427. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- 2.Seeman NC. Annu Rev Biochem. 2010;79:65. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang F, Nangreave J, Liu Y, Yan H. J Am Chem Soc. 2014;136:11198. doi: 10.1021/ja505101a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones MR, Seeman NC, Mirkin CA. Science. 2015;347:1260901. doi: 10.1126/science.1260901. [DOI] [PubMed] [Google Scholar]
- 5.Rothemund PWK. Nature. 2006;440:297. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 6.Douglas SM, Marblestone AH, Teerapittayanon S, Vazquez A, Church GM, Shih WM. Nucleic Acids Res. 2009;37:5001. doi: 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim DN, Kilchherr F, Dietz H, Bathe M. Nucleic Acids Res. 2012;40:2862. doi: 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan K, Kim DN, Zhang F, Adendorff MR, Yan H, Bathe M. Nat Commun. 2014;5:5578. doi: 10.1038/ncomms6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Douglas SM, Dietz H, Liedl T, Hogberg B, Graf F, Shih WM. Nature. 2009;459:414. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han D, Pal S, Yang Y, Jiang S, Nangreave J, Liu Y, Yan H. Science. 2013;339:1412. doi: 10.1126/science.1232252. [DOI] [PubMed] [Google Scholar]
- 11.Benson E, Mohammed A, Gardell J, Masich S, Czeizler E, Orponen P, Hogberg B. Nature. 2015;523:441. doi: 10.1038/nature14586. [DOI] [PubMed] [Google Scholar]
- 12.Bath J, Turberfield AJ. Nat Nanotechnol. 2007;2:275. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- 13.Kohman RE, Han X. Chem Commun. 2015;51:5747. doi: 10.1039/c5cc00082c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerling T, Wagenbauer KF, Neuner AM, Dietz H. Science. 2015;347:1446. doi: 10.1126/science.aaa5372. [DOI] [PubMed] [Google Scholar]
- 15.Shaw A, Lundin V, Petrova E, Fordos F, Benson E, Al-Amin A, Herland A, Blokzijl A, Hogberg B, Teixeira AI. Nat Methods. 2014;11:841. doi: 10.1038/nmeth.3025. [DOI] [PubMed] [Google Scholar]
- 16.Douglas SM, Chou JJ, Shih WM. Proc Natl Acad Sci U S A. 2007;104:6644. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jungmann R, Avendano MS, Woehrstein JB, Dai M, Shih WM, Yin P. Nat Methods. 2014;11:313. doi: 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Douglas SM, Bachelet I, Church GM. Science. 2012;335:831. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Q, Song C, Nangreave J, Liu X, Lin L, Qiu D, Wang ZG, Zou G, Liang X, Yan H, Ding B. J Am Chem Soc. 2012;134:13396. doi: 10.1021/ja304263n. [DOI] [PubMed] [Google Scholar]
- 20.Zhao YX, Shaw A, Zeng X, Benson E, Nyström AM, Högberg B. ACS Nano. 2012;6:8684. doi: 10.1021/nn3022662. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Z, Jacovetty EL, Liu Y, Yan H. Angew Chem, Int Ed. 2011;50:2041. doi: 10.1002/anie.201006818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreiber R, Do J, Roller EM, Zhang T, Schuller VJ, Nickels PC, Feldmann J, Liedl T. Nat Nanotechnol. 2013;9:74. doi: 10.1038/nnano.2013.253. [DOI] [PubMed] [Google Scholar]
- 23.Rinker S, Ke Y, Liu Y, Chhabra R, Yan H. Nat Nanotechnol. 2008;3:418. doi: 10.1038/nnano.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutta PK, Varghese R, Nangreave J, Lin S, Yan H, Liu Y. J Am Chem Soc. 2011;133:11985. doi: 10.1021/ja1115138. [DOI] [PubMed] [Google Scholar]
- 25.Voigt NV, Torring T, Rotaru A, Jacobsen MF, Ravnsbaek JB, Subramani R, Mamdouh W, Kjems J, Mokhir A, Besenbacher F, Gothelf KV. Nat Nanotechnol. 2010;5:200. doi: 10.1038/nnano.2010.5. [DOI] [PubMed] [Google Scholar]
- 26.Sun W, Boulais E, Hakobyan Y, Wang WL, Guan A, Bathe M, Yin P. Science. 2014;346:1258361. doi: 10.1126/science.1258361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stahl E, Martin TG, Praetorius F, Dietz H. Angew Chem, Int Ed. 2014;53:12735. doi: 10.1002/anie.201405991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmes CP. J Org Chem. 1997;62:2370. doi: 10.1021/jo961602x. [DOI] [PubMed] [Google Scholar]
- 29.Matsuzaki M, Ellis-Davies GCR, Nemoto T, Miyashita Y, Iino M, Kasai H. Nat Neurosci. 2001;4:1086. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fino E, Araya R, Peterka DS, Salierno M, Etchenique R, Yuste R. Front Neural Circuits. 2009;3 doi: 10.3389/neuro.04.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olson JP, Kwon HB, Takasaki KT, Chiu CQ, Higley MJ, Sabatini BL, Ellis-Davies GCR. J Am Chem Soc. 2013;135:5954. doi: 10.1021/ja4019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soriano J, Rodríguez Martínez M, Tlusty T, Moses E. Proc Natl Acad Sci U S A. 2008;105:13758. doi: 10.1073/pnas.0707492105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.