

Abstract
Cyclodextrin glycosyltransferases (CGTases) (EC 2.4.1.19) catalyze the conversion of starch or starch derivates into mixtures of α-, β-, and γ-cyclodextrins. Because time-consuming and expensive purification procedures hinder the widespread application of single-ingredient cyclodextrins, enzymes with enhanced specificity are needed. In this study, we tested the hypothesis that the α-cyclodextrin selectivity of Paenibacillus macerans α-CGTase could be augmented by masking subsite −7 of the active site, blocking the formation of larger cyclodextrins, particularly β-cyclodextrin. Five single mutants and three double mutants designed to remove hydrogen-bonding interactions between the enzyme and substrate at subsite −7 were constructed and characterized in detail. Although the rates of α-cyclodextrin formation varied only modestly, the rate of β-cyclodextrin formation decreased dramatically in these mutants. The increase in α-cyclodextrin selectivity was directly proportional to the increase in the ratio of their kcat values for α- and β-cyclodextrin formation. The R146A/D147P and R146P/D147A double mutants exhibited ratios of α-cyclodextrin to total cyclodextrin production of 75.1% and 76.1%, approximately one-fifth greater than that of the wild-type enzyme (63.2%), without loss of thermostability. Thus, these double mutants may be more suitable for the industrial production of α-cyclodextrin than the wild-type enzyme. The production of β-cyclodextrin by these mutants was almost identical to their production of γ-cyclodextrin, which was unaffected by the mutations in subsite −7, suggesting that subsite −7 was effectively blocked by these mutations. Further increases in α-cyclodextrin selectivity will require identification of the mechanism or mechanisms by which these small quantities of larger cyclodextrins are formed.
INTRODUCTION
Cyclodextrin glycosyltransferase (EC 2.4.1.19, CGTase), which belongs to glycohydrolase (GH) family 13, catalyzes the cyclization, coupling, disproportionation, and hydrolysis of starch or linear α-(1,4)-linked glucans (1–3). Industrial interest in these enzymes arises from their preference for transglycosylation over hydrolysis (2, 4, 5) and from their ability to catalyze the synthesis of circular α-(1,4)-linked oligosaccharides, which are known as cyclodextrins (3, 6). All known CGTases catalyze the formation of mixtures of cyclodextrins containing 6 (α-cyclodextrin), 7 (β-cyclodextrin), or 8 (γ-cyclodextrin) glucose units. If these reactions were allowed to reach equilibrium, the α-cyclodextrin/β-cyclodextrin/γ-cyclodextrin ratio in the mixture would become 27:58:15 (7). In practice, the ratio of these products is determined by the kinetics of their production, which varies with each individual enzyme (2, 8). The CGTases are classified as α-, β-, or γ-CGTases or mixed CTGases on the basis of the cyclodextrin that they produce in the main proportion (1–3).
Cyclodextrins are unique natural materials. They adopt the structure of a truncated cone with a hydrophilic external surface and a hydrophobic internal cavity (9) that allow them to bind hydrophobic molecules within their internal cavities and carry them into aqueous solution. These advantageous properties have led to their widespread use in industries related to food, pharmaceuticals, and related products (10–12). Single-ingredient cyclodextrins are preferred over cyclodextrin mixtures in almost all industrial applications that require the formation of a complex with a specific hydrophobic molecule.
Since natural CGTases produce mixtures of α-, β-, and γ-cyclodextrins, a time-consuming and expensive procedure is needed to purify the desired product. This added cost prevents the widespread application of single-ingredient cyclodextrins. One potential solution to this problem that has received recent attention is the construction of engineered CGTases with substantially enhanced product specificity. Reviews of recent progress in the artificial modifications of CGTases and their impact on biotechnological processes have been published (2, 3). While some progress has been made, particularly through modifications at subsite −3 or −6 (13, 14), improvements in the α-cyclodextrin specificity of α-CGTases, particularly via modifications at subsite −7, remain underexplored.
In this study, site-directed mutagenesis was performed on Paenibacillus macerans α-CGTase (15) in an effort to enhance the specificity of α-cyclodextrin production under industrial conditions. The hypothesis behind this work is diagrammed in Fig. 1. The synthesis of cyclodextrins begins when a linear maltooligosaccharide chain binds to the subsites of the active site (16). Binding is followed by bond cleavage to yield a covalent glycosyl-enzyme intermediate (17). Cyclodextrin synthesis then proceeds via an intramolecular transglycosylation reaction (cyclization) (2). Structures of maltohexaose and maltoheptaose bound at the donor site have provided insight into the mechanisms governing cyclodextrin size specificity (18). Panels A and B of Fig. 1 present a general diagram representing the CGTase enzyme with covalently bound maltooligosaccharide intermediates that can produce β-cyclodextrin or α-cyclodextrin through subsequent cyclization. Competition between these two patterns is hypothesized to be a key feature in the mechanism that determines the product distribution. The remote end of the donor-binding site, corresponding to subsites −7 and −6, forms numerous hydrogen-bonding interactions with the glucose unit (16, 18). These interactions could play a role in determining cyclodextrin size specificity. The S146P mutant, designed to disturb the hydrogen-bonding network at subsite −7 of Bacillus circulans 251 β-CGTase, showed a decrease in kcat for β-cyclodextrin synthesis and an increase in α-cyclodextrin production (19). From this result, we formed the hypothesis that removing hydrogen-bonding interactions and inducing steric hindrance between subsite −7 and the nonreducing end of the maltooligosaccharide would decrease the rate of β-cyclodextrin synthesis, without having a negative effect on the rate of α-cyclodextrin synthesis by P. macerans α-CGTase. Panel C of Fig. 1 displays a general diagram of the CGTase structure that we are trying to achieve through mutagenesis.
FIG 1.

Schematic diagrams of the CGTase active site. (A) A maltoheptaose intermediate covalently attached at the cleavage site (arrow), preceding β-cyclodextrin formation. (B) A maltohexaose intermediate covalently attached at the cleavage site, preceding α-cyclodextrin formation. (C) CGTase mutant represented as having a “masked” subsite −7.
MATERIALS AND METHODS
Bacterial strains, plasmids, and materials.
Escherichia coli JM109 and E. coli BL21(DE3) (13) were used as hosts for recombinant DNA manipulations and protein expression, respectively. Plasmid cgt/pET-20b(+) (13), which contains a gene encoding the mature wild-type CGTase from P. macerans strain JFB05-01 (Canadian Clinical Trials Coordinating Centre [CCTCC]: M203062), was used for site-directed mutagenesis and expression of CGTase proteins.
Primer Star HS DNA polymerase and the restriction endonuclease DpnI were purchased from TaKaRa Biotechnology (Dalian, China) Co., Ltd. A plasmid extraction kit, ampicillin sodium salt, and isopropyl-β-d-1-thiogalactopyranoside were purchased from Sangon Biotech (Shanghai, China) Co., Ltd. Yeast extract and tryptone were purchased from Oxoid Co., Ltd. Bovine serum albumin and chromatographically pure α-cyclodextrin, β-cyclodextrin, and γ-cyclodextrin were purchased from Sigma-Aldrich Co., LLC. Soluble starch, methyl orange, and phenolphthalein were purchased from Sinopharm Chemical Reagent (Shanghai, China) Co., Ltd. All other chemicals and reagents were of analytical grade.
Selection of mutation sites based on sequence and structural analysis.
ClustalX was utilized for multiple-sequence alignment (20). Theoretical structures of the mutant CGTases were obtained by using homology modeling performed by the SwissModel protein-modeling server (21). The crystal structure of wild-type CGTase (PDB identifier [ID] 4JCL) was used as the basis for the homology modeling. The PyMol molecular graphics system (DeLano Scientific, San Carlos, CA) was used to visualize and analyze the generated model structures, as well as to construct graphical presentations and illustrative figures.
DNA manipulations and sequencing.
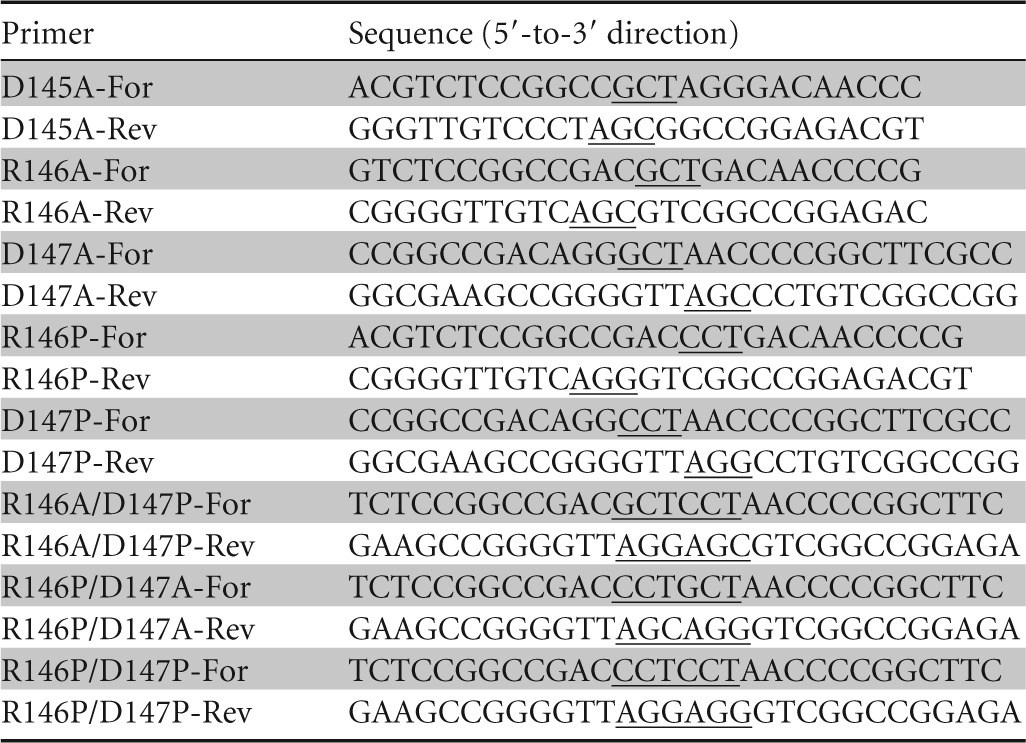
Site-directed mutagenesis was performed using overlap extension PCR (22) with plasmid cgt/pET-20b(+) as the template and a pair of complementary primers. The sequences of the primers carrying the respective mutations are shown in Table 1. DNA manipulations and calcium chloride transformation of E. coli strains were performed as previously described (23). DNA sequencing was performed by Sangon Biotech (Shanghai, China) Co., Ltd. The resulting expression vectors containing the correct mutations were used to transform chemically competent E. coli BL21(DE3) for expression.
TABLE 1.
Primers used for site-directed mutagenesis
Production and purification of CGTase proteins.
LB medium containing 100 μg/ml ampicillin sodium salt was inoculated with a single colony of E. coli BL21(DE3) cells harboring recombinant plasmid and was incubated at 37°C overnight. An aliquot of the overnight culture was then transferred into TB medium containing 100 μg/ml ampicillin sodium salt and 7.5 g/liter glycine and was shaken at 37°C until the optical density at 600 nm reached 1.0. Isopropyl-β-d-1-thiogalactopyranoside was added to give a final concentration of 0.05 mM to induce expression of the target protein. Protein induction was performed at 25°C and continued for 48 h. The culture supernatant, obtained by centrifugation at 10,000 × g for 15 min at 4°C, was subjected to salt fractionation using ammonium sulfate (25% [wt/vol]) and dialyzed against buffer A (20 mM sodium phosphate buffer, pH 6.5). This sample was applied to a DEAE Sepharose column (Pharmacia Biotech) (160 mm by 10 mm) that had been preequilibrated with buffer A. Using a fast protein liquid chromatography (FPLC) system (AKTA FPLC system; GE Healthcare), a linear gradient of 0 to 1 M NaCl in buffer A was used to elute the adsorbed proteins. The fractions containing CGTase activity were pooled and dialyzed against buffer A overnight. Protein concentrations were determined using the Bradford assay (24), with bovine serum albumin as the standard.
CGTase activity assay.
All enzyme assays were performed by incubating 0.1 ml of diluted enzyme (0.35 to 1.0 U/ml) with 2 ml of 2% (wt/vol) soluble starch in 50 mM citric acid/disodium hydrogen phosphate buffer (pH 5.5) at 45°C for 10 min. The α-cyclodextrin-forming activity was determined using the methyl orange method (25), with some modifications. The reaction described above was terminated by the addition of 3.0 M HCl (0.2 ml), and then 0.2 ml of 0.44 mM methyl orange was added. After the reaction mixture had incubated at 16°C for 20 min, the amount of α-cyclodextrin in the mixture was determined spectrophotometrically by measuring the absorbance at 505 nm. One unit of α-cyclodextrin-forming activity was defined as the amount of enzyme that produces 1 μmol of α-cyclodextrin per min. The β-cyclodextrin-forming activity was determined using the phenolphthalein method (26). One unit of each activity was defined as the amount of enzyme that produces 1 μmol of β-cyclodextrin per min.
Effect of pH and temperature on enzyme activity.
The optimum pH of the recombinant enzyme was determined by measuring its activity in citric acid/disodium hydrogen phosphate buffers at pHs ranging from 3.5 to 8.0. The temperature dependence of the recombinant enzyme activity was measured at between 30°C and 65°C in 50 mM citric acid/disodium hydrogen phosphate buffer at pH 5.5. The buffer and soluble starch were preincubated at each temperature for 10 min before initiation of the reaction. The reaction was initiated by the addition of the enzyme and was allowed to proceed for 10 min.
Stability of wild-type and mutant enzymes.
The half-lives of the enzymes at 45°C were investigated to assess the thermostability. Samples diluted in 50 mM citric acid/disodium hydrogen phosphate buffer (pH 5.5) to a protein concentration of 0.3 mg/ml were incubated at 45°C. After heat treatment, the residual activity was measured under the same conditions. The initial activity before heat treatment was taken as 100%. All of the values presented in graphs and tables represent the means of triplicate measurements.
Cyclodextrin production and HPLC analysis.
The formation of cyclodextrins under conditions resembling the industrial no-solvent production process was measured by incubating 5% (wt/vol) soluble starch in 50 mM citric acid/disodium hydrogen phosphate buffer (pH 5.5) with 4.0 U (total cyclization activity) of CGTase per gram of starch at 45°C for 22 h. In order to estimate the performance of the mutant enzymes, the wild-type CGTase served as a control. Reactions were sampled at regular intervals, and the samples were boiled for 10 min to inactivate the enzyme. Acetonitrile was added to give a final concentration of 50% (vol/vol), and then the mixtures were incubated at room temperature for 2 h to precipitate the unconverted starch and dextrin in the boiled samples. Supernatants were obtained by centrifugation at 10,000 × g for 10 min at room temperature. The concentrations of α-, β-, and γ-cyclodextrins were determined by high-performance liquid chromatography (HPLC) using an Aps-2 Hypersil (NH2) column (4.6 mm by 250 mm, 5 μm pore size) eluted with acetonitrile/water (70:30) at a flow rate of 0.8 ml/min and a temperature of 40°C. Products were detected using an Agilent 2410 refractive index detector. All of the values presented in the graphs and tables are the means of the results of three independent replicates.
RESULTS AND DISCUSSION
Selection of mutation sites based on sequence and structural analysis.
To select the mutation sites, the amino acid sequence of P. macerans α-CGTase was aligned with those of a series of 13 CGTases with different product specificities by using ClustalX. As shown in Fig. 2, all of the sequences constituting subsite −7, which is located between conserved region I (conserved in GH family 13) and Glu153 (conserved only in CGTases) (1, 2), are conserved in length among the α/β- and β-CGTases. In addition, alignment of the parsed three-dimensional (3-D) structures of 6 α-, α/β-, and β-CGTases, including that of P. macerans α-CGTase, showed that the extended directions of the loops constituting subsite −7 in the various enzymes are coincident (see Fig. S1A and B in the supplemental material). However, there is no loop that constitutes a subsite similar to subsite −7 in γ-CGTase (see Fig. S1C), and the sequences of γ-CGTases in this region are the shortest among all the CGTases (Fig. 2). The possible explanation is that subsite −7 serves as a steric hindrance in the formation of larger cyclodextrin. And this makes for the production of β- or α-cyclodextrin, as deleting subsite −7 of Bacillus circulans 8 CGTase enhanced the amount of γ-cyclodextrin, with reduced amounts of β- and α-cyclodextrin (27). Thus, we hypothesized that an expansion of this steric hindrance effect by the introduction of mutations that serve as additional steric hindrances would increase the rate of α-cyclodextrin production to the level seen with β-cyclodextrin production.
FIG 2.

Sequence alignment of conserved region I and subsite −7 in CGTases. The CGTases are ordered according to their cyclodextrin product specificity (1–3), with α-CGTases on the top, followed by the α/β-CGTases and β-CGTases and, finally, by the γ-CGTases at the bottom. *, identical; :, conserved substitution; ., semiconserved substitution. Conserved region I of the GH13 family is boxed. The region constituting subsite −7 is rendered with a gray background. The protein sequences utilized are those of Paenibacillus macerans CGTase (GenBank accession number 4JCL_A), Bacillus macerans CGTase (accession number P31835.1), Thermococcus sp. strain B1001 CGTase (accession number BAA88217.1), Klebsiella pneumoniae CGTase (accession number P08704.1), Anaerobranca gottschalkii CGTase (accession number CAH61550.1), Thermoanaerobacterium thermosulfurigenes Em1 CGTase (accession number 1A47_A), Bacillus stearothermophilus N02 CGTase (accession number 1CYG_A), Bacillus circulans 251 CGTase (accession number 1CDG_A), Bacillus circulans 8 CGTase (accession number 1CGT_A), Bacillus sp. strain 1011 CGTase (accession number P05618.1), Bacillus ohbensis CGTase (accession number P27036.2), Bacillus firmus 290-3 CGTase (accession number CAA01436.1) Bacillus sp. strain G-825-6 CGTase (accession number Q25CB6), and Bacillus clarkii CGTase (accession number 4JCM_A).
Previously, it was reported that, in the case of subsite −7 in B. circulans 251 β-CGTase, the side chains of residues S145, S146, and D147, as well as the N-H groups of S146 and D147, were involved in the hydrogen-bonding interactions with the nonreducing end of the substrate oligosaccharide (18). In P. macerans α-CGTase, the analogous residues, D145, R146, and D147, also have polar side chains that have the potential to form hydrogen-bonding interactions with the glucose unit binding at subsite −7. To test the aforementioned hypothesis, the side chains of all three residues were removed, individually, via replacement with alanine to form the D145A, R146A, and D147A mutants. Both the side chain and the backbone N-H bonds were removed from residues R146 and D147, individually, via replacement with proline to form mutants R146P and D147P. In addition, both the R146P and D147P mutants would induce steric hindrance between subsite −7 and the nonreducing end of the maltooligosaccharide. The double mutants R146A/D147P, R146P/D147A, and R146P/D147P were also constructed, with the expectation that these mutants would have a greater effect than the single-site mutations.
Construction, expression, and purification of wild-type and mutant CGTases.
Site-directed mutagenesis was performed using overlap PCR; the resulting sequences were verified and used to transform E. coli BL21(DE3) for expression. Protein concentrations analyzed using the Bradford assay suggested that there were no statistically significant differences (P > 0.05 [Student's t test]) in expression levels among the wild-type and the mutant enzymes. The crude enzymes were purified by ammonium sulfate fractionation and anion-exchange chromatography (see Table S1 and Dataset S1 in the supplemental material) and were demonstrated to be homogeneous by SDS-PAGE. The bands of purified recombinant CGTases were displayed near the 66.2-kDa marker band (see Fig. S2).
Optimum pH and optimum temperature of wild-type and mutant CGTases.
To determine the optimum reaction temperature, α-cyclodextrin-forming activity was measured at pH 5.5 and between 30°C and 65°C. The optimum pH of the enzymes was measured, at 45°C, using citric acid/disodium hydrogen phosphate buffer at pH values ranging from pH 3.5 to pH 8.0. All of the CGTase mutants had pH and temperature activity profiles similar to those of the wild-type CGTase (data not shown). The optimal pH and temperature for the wild-type and mutant CGTases were pH 5.5 and 45°C, respectively.
Cyclization characteristics of wild-type and mutant CGTases.
The cyclization activities of the wild-type and mutant CGTases are summarized in Table 2. None of the mutations in subsite −7 resulted in a substantial (≤11.3%) change in the catalytic constant (kcat) for α-cyclodextrin formation compared with the wild-type enzyme. The D145A and R146A mutants also exhibited only slight (≤10%) decreases in kcat for β-cyclodextrin formation compared with the wild-type enzyme. However, the D147A, R146P, and D147P mutants showed statistical significant (P < 0.01 [Student's t test]), remarkable decreases in kcat for β-cyclodextrin formation, decreasing to 53.4%, 55.3%, and 41.6% of that of the wild-type enzyme, respectively. The decreased kcat values for β-cyclodextrin formation, combined with the slight change in kcat for α-cyclodextrin formation, resulted in a substantial increase in the ratio of these kcat values (α-cyclodextrin formation to β-cyclodextrin formation). Single mutants D147A, R146P, and D147P showed increases of 75.2%, 92.8%, and 119% compared with the wild type, respectively. The kcat values for β-cyclodextrin formation of the R146A/D147P, R146P/D147A, and R146P/D147P double mutants were lower than those for the single mutants, at 42.0%, 42.5%, and 35.2% that of the wild-type enzyme, respectively, which showed statistical significant (P < 0.01 [Student's t test]), remarkable decreases. Their kcat ratios (α-cyclodextrin formation to β-cyclodextrin formation) were much higher than those of the single mutants, with increases of 130%, 145%, and 170% compared with that of the wild-type enzyme, respectively. Values that reflect substrate affinity, such as the substrate Km values, cannot be reported here because the amounts of cyclodextrin formed at the low substrate concentrations needed to determine the Km were too low to be reliably measured. This observation is consistent with those of previous reports (28, 29). In addition, γ-cyclodextrin-forming activity was not analyzed, as it constituted a negligible fraction of the total cyclodextrin-forming activity (13).
TABLE 2.
Cyclization activities of wild-type and mutant CGTases from P. macerans
| CGTase | Cyclization activity |
||
|---|---|---|---|
| kcat α-CD (s−1)a | kcat β-CD (s−1) | Ratio (kcat α-CD/kcat β-CD) | |
| Wild type | 147.3 ± 1.7 | 21.9 ± 0.8 | 6.7 ± 0.3 |
| D145A | 130.6 ± 3.8 | 19.7 ± 1.0 | 6.6 ± 0.3 |
| R146A | 159.1 ± 4.3 | 19.8 ± 0.9 | 8.0 ± 0.4 |
| D147A | 137.9 ± 5.2 | 11.7 ± 0.4 | 11.8 ± 0.4 |
| R146P | 156.1 ± 1.8 | 12.1 ± 0.4 | 12.9 ± 0.4 |
| D147P | 134.3 ± 2.0 | 9.1 ± 0.3 | 14.8 ± 0.5 |
| R146A/D147P | 142.3 ± 3.9 | 9.2 ± 0.5 | 15.5 ± 0.8 |
| R146P/D147A | 153.2 ± 3.6 | 9.3 ± 0.2 | 16.5 ± 0.4 |
| R146P/D147P | 139.9 ± 4.1 | 7.7 ± 0.3 | 18.2 ± 0.7 |
CD, cyclodextrin.
An alignment of the parsed structures of P. macerans α-CGTase and B. circulans 251 β-CGTase is shown in Fig. 3A. Numerous hydrogen-bonding interactions between subsite −7 and the glucose at the nonreducing end of the oligosaccharide were found within this alignment (Fig. 3B). Moreover, three pairs of hydrogen-bonding interactions were retained in the case of P. macerans α-CGTase (Fig. 3C). Hydrogen-bonding interactions between polar groups are known to play an important role in substrate binding in this enzyme (17, 18). In the present study, replacement of D145 or R146 with alanine did not have a notable influence on the synthesis of β-cyclodextrin. However, replacement of D147 with alanine resulted in a remarkable decrease in the kcat for β-cyclodextrin production. One interpretation is that the latter mutation results in elimination of a hydrogen bond (3.3 Å; Fig. 3C) between side chain Oδ1 of D147 and O4 of the glucose unit in subsite −7. Mutants R146P and D147P, which lack both a polar side chain and a backbone NH, exhibited a further decrease in kcat of β-cyclodextrin formation, compared with R146A and D147A, respectively. In the case of structural modeling, Fig. 3D and E show obvious steric hindrance generated between the Cδ of proline and the glucose unit in subsite −7 (a distance of 1.9Å for both R146P and D147P), which would disrupt the hydrogen-bonding interactions in this region. This result suggests that steric hindrance may also be helpful in decreasing β-cyclodextrin formation.
FIG 3.

Structural modeling of wild-type and mutant CGTases in cartoon format. (A) Alignment of P. macerans CGTase (PDB ID 4JCL) and B. circulans 251 CGTase (PDB ID 1EO5) in complex with maltoheptaose. One of the two structures and the non-reducing-end glucose are shown in panels B, C, D, and E. (B) Wild-type B. circulans 251 CGTase. (C) Wild-type P. macerans CGTase. (D) Model of P. macerans CGTase mutant R146P. (E) Model of P. macerans CGTase mutant D147P. The models of the R146P and D147P mutants were created using wild-type P. macerans CGTase (PDB ID 4JCL) as the template, and the two mutant models have QMEAN4 scores of −0.72 and −0.80, respectively (30). Residues of B. circulans 251 CGTase (C, baby blue; N, blue; O, red) and P. macerans CGTase (C, green; N, blue; O, red), directly involved in binding to the glucose unit (C, pink; O, red) in subsite −7, as well as the residues next to them and the glucose unit, are rendered as sticks. The distances between the polar groups, which represent the hydrogen-bonding interactions, are indicated with red stippled lines and black numbers. The figure was produced using PyMol.
Stability of the wild-type and mutant CGTases.
The wild-type and mutant CGTases were individually incubated at 45°C and pH 5.5 to evaluate their stability. These incubations were sampled at specified intervals, and the α-cyclodextrin-forming activity of the samples was measured. Mutants D145A, D147A, and D147P had shorter half-lives (3.3, 3.7, and 2.9 h, respectively) than their wild-type parent (5.2 h) (see Fig. S3 in the supplemental material). In contrast, the R146A and R146P mutants displayed longer half-lives (9.2 and 8.8 h, respectively). The R146A/D147P, R146P/D147A, and R146P/D147P double mutants displayed half-lives (5.5, 5.8, and 4.4 h, respectively) similar to that of the wild-type enzyme.
Production of cyclodextrins by wild-type and mutant CGTases.
The time courses of cyclodextrin production in reaction mixtures containing 5% (wt/vol) soluble starch and 4.0 U of each CGTase per gram of starch were analyzed by using HPLC. All of the CGTase variants produced a mixture of cyclodextrins at each time point (Fig. 4). After 22 h of incubation, 30.2% to 36.9% of the soluble starch had been converted into cyclodextrins by each of the CGTases. In each case, the ratio of α-cyclodextrin to total cyclodextrins produced tended to decrease with incubation time. Thus, the concentrations of β- and γ-cyclodextrin increased during the entire 22 h of incubation time, while that of α-cyclodextrin reached a plateau at approximately 10 h.
FIG 4.

Production of cyclodextrins from 5% (wt/vol) soluble starch by the action of wild-type and mutant P. macerans CGTases at 45°C and pH 5.5. (A) Wild-type CGTase. (B) Mutant D145A. (C) Mutant R146A. (D) Mutant D147A. (E) Mutant R146P. (F) Mutant D147P. (G) Double mutant R146A/D147P. (H) Double mutant R146P/D147A. (I) Double mutant R146P/D147P. The concentrations of the cyclodextrin products (indicated in grams per liter; left axis) were determined using HPLC. The percentage of the total of cyclodextrin products represented by each of the cyclodextrins (%, right axis) at each time point was calculated from the HPLC data. CD, cyclodextrin.
Despite this overall similarity, there were important differences in cyclodextrin production among the mutants and wild-type CGTases. At the 10-h time point, the wild-type CGTase produced 10.28 g/liter α-cyclodextrin, 4.14 g/liter β-cyclodextrin, and 1.84 g/liter γ-cyclodextrin (Fig. 5), which yields a percentage of α-cyclodextrin in total cyclodextrins of 63.2%. Each of the mutants, except the D145A mutant, produced a smaller amount of β-cyclodextrin, which showed a statistically significant difference (P < 0.02 [for R146A] and P < 0.01 [for the others] [Student's t test]). All these mutants produced a slightly larger amount of α-cyclodextrin than the wild-type CGTase; among these mutants, the R146A, R146P, and D147P single mutants and the double mutants showed a statistically significant difference (P < 0.05 [Student's t test]). These increases in α-cyclodextrin production and decreases in β-cyclodextrin production led to remarkable increases in the percentage of α-cyclodextrin compared to total cyclodextrins for mutants R146A (68.1%), D147A (68.8%), R146P (71.4%), and D147P (73.0%) compared with that of wild-type enzyme (63.2%). The R146A/D147P, R146P/D147A, and R146P/D147P double mutants showed greater increases in α-cyclodextrin production and greater decreases in β-cyclodextrin production than the single-site mutants. Thus, the double mutants had even greater percentages of α-cyclodextrin compared to total cyclodextrins (75.1%, 76.1%, and 76.8%, respectively) than the single-site mutants. For these mutants, β-cyclodextrin production was almost as low as γ-cyclodextrin production. In general, the differences in γ-cyclodextrin production between these mutants and wild-type CGTase were negligible (1.63 to 2.05 g/liter; P > 0.05 [Student's t test]).
FIG 5.

Production of cyclodextrins from 5% (wt/vol) soluble starch by the action of wild-type and mutant P. macerans CGTases after incubation at 45°C and pH 5.5 for 10 h. The concentration of each cyclodextrin (indicated in grams per liter) was determined by HPLC analysis. CD, cyclodextrin.
Mechanistic considerations and future directions.
The results presented above are consistent with our original mechanism-based hypothesis. The percentages of α-cyclodextrin production compared to total cyclodextrin production for the various CGTases were directly proportional to the ratios of their kcat values for α- and β-cyclodextrin production (Table 2 and Fig. 5). Linear regression results in a coefficient of determination (R2) of 0.948 (see Dataset S1 to S4 in the supplemental material), providing convincing evidence that the key to selective formation of a single cyclodextrin is modification of the rates at which the individual cyclodextrins are produced.
Close inspection of the individual kcat values for the production of α- and β-cyclodextrin revealed that the increase in the kcat ratio was not driven by increases in the rate of α-cyclodextrin formation but rather by the decrease in the rate of β-cyclodextrin formation. Since α-cyclodextrin, which is composed of six glucose units, is the smallest cyclodextrin produced by CGTases (1, 2), hindering the formation of larger cyclodextrins seemed an unambiguous way to design CGTase mutants with enhanced α-cyclodextrin specificity. The data presented here demonstrate that masking subsite −7 accomplished this goal. Interestingly, the small amount of γ-cyclodextrin produced and its negligible variation among all the CGTases tested suggest that γ-cyclodextrin production does not involve binding at subsite −7. The fact that the R146P/D147P double mutant reduced the β-cyclodextrin production to essentially the same level as the γ-cyclodextrin production suggests that subsite −7 was fully blocked by this mutant and that further increases in α-cyclodextrin specificity must be achieved by addressing the mechanism or mechanisms by which these low levels of β- and γ-cyclodextrin are being produced.
In addition to α-cyclodextrin specificity, the amount of α-cyclodextrin resulting from the activity of the CGTases is an important property for α-cyclodextrin production. The amounts of cyclodextrins are the result of comprehensive effects of cyclodextrin generation and degradation (coupling, disproportionation, and hydrolysis) (7, 8); nevertheless, they were all based on active enzymes. In this study, despite there being a poor R2 value, the △(α-cyclodextrin) value (the difference between the amounts of α-cyclodextrin from the mutant and wild-type enzymes) increased along with the ratios of the kcat values for α- and β-cyclodextrin (see Dataset S2 in the supplemental material). However, a decent R2 value of 0.9178 (see Dataset S3) between the half-lives of the enzymes and the deviation of △(α-cyclodextrin) [i.e., the difference between △(α-cyclodextrin) and its corresponding value calculated from the regression line in Dataset S2] was observed. When considering this regression line (Dataset S3) as the factor of enzyme stability and further amending the △(α-cyclodextrin) value by deducting the calculated value from the regression line in Dataset S3 from △(α-cyclodextrin), a significant R2 of 0.9805 for the amended △(α-cyclodextrin) and the ratios of the kcat values for α- and β-cyclodextrins is observed (see Dataset S4 in the supplemental material). Thus, the poor R2 between △(α-cyclodextrin) and the ratios of the kcat values for α- and β-cyclodextrin (see Dataset S2 in the supplemental material) was closely related to the changes in the stability of the enzymes. The amount of α-cyclodextrin obtained in the product is determined by both the specificity and the stability of the enzyme, and greater α-cyclodextrin production would be achieved by increasing both the stability and the specificity of the mutant.
In conclusion, the ratio of kcat values for α- and β-cyclodextrin of CGTase was crucial for α-cyclodextrin production. The higher the ratio of kcat values, the higher the percentages and amounts of α-cyclodextrin produced by the CGTase mutant. Incidentally, high stability of CGTase would have a positive effect on α-cyclodextrin production. The present report describes the design and characterization of mutants in subsite −7 of the P. macerans α-CGTase for the first time. The data suggest that the double mutants effectively blocked subsite −7 of the active site cleft and boosted the α-cyclodextrin specificity by as much as 22% to 76.8% of the total cyclodextrins produced. Notably, double mutants R146A/D147P and R146P/D147A exhibited both greatly enhanced α-cyclodextrin specificity and greatly enhanced stability, suggesting that these mutants have the potential to provide better performance than the wild-type enzyme in an industrial setting.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Science Foundation for Distinguished Young Scholars (31425020), the National Natural Science Foundation of China (31271813, 31401636, and 31501419), the Natural Science Foundation of Jiangsu Province (BK20140142), the 111 Project (no. 111-2-06), Fundamental Research Funds for the Central Universities (JUSRP11511), and the Independent Research Program of the State Key Laboratory of Food Science and Technology, Jiangnan University (5812060204150170).
Funding Statement
The National Natural Science Foundation of China (31501419), 111 Project (no. 111-2-06), Fundamental Research Funds for the Central Universities (JUSRP11511), and Independent Research Program of the State Key Laboratory of Food Science and Technology, Jiangnan University (5812060204150170) provided funding for this research.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03535-15.
REFERENCES
- 1.van der Veen BA, Uitdehaag JC, Dijkstra BW, Dijkhuizen L. 2000. Engineering of cyclodextrin glycosyltransferase reaction and product specificity. Biochim Biophys Acta 1543:336–360. doi: 10.1016/S0167-4838(00)00233-8. [DOI] [PubMed] [Google Scholar]
- 2.Kelly RM, Dijkhuizen L, Leemhuis H. 2009. The evolution of cyclodextrin glucanotransferase product specificity. Appl Microbiol Biotechnol 84:119–133. doi: 10.1007/s00253-009-1988-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leemhuis H, Kelly RM, Dijkhuizen L. 2010. Engineering of cyclodextrin glucanotransferases and the impact for biotechnological applications. Appl Microbiol Biotechnol 85:823–835. doi: 10.1007/s00253-009-2221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Veen BA, Leemhuis H, Kralj S, Uitdehaag JC, Dijkstra BW, Dijkhuizen L. 2001. Hydrophobic amino acid residues in the acceptor binding site are main determinants for reaction mechanism and specificity of cyclodextrin-glycosyltransferase. J Biol Chem 276:44557–44562. doi: 10.1074/jbc.M107533200. [DOI] [PubMed] [Google Scholar]
- 5.Leemhuis H, Dijkstra BW, Dijkhuizen L. 2002. Mutations converting cyclodextrin glycosyltransferase from a transglycosylase into a starch hydrolase. FEBS Lett 514:189–192. doi: 10.1016/S0014-5793(02)02362-1. [DOI] [PubMed] [Google Scholar]
- 6.van der Maarel MJ, van der Veen B, Uitdehaag JC, Leemhuis H, Dijkhuizen L. 2002. Properties and applications of starch-converting enzymes of the alpha-amylase family. J Biotechnol 94:137–155. doi: 10.1016/S0168-1656(01)00407-2. [DOI] [PubMed] [Google Scholar]
- 7.Tewari YB, Goldberg RN, Sato M. 1997. Thermodynamics of the hydrolysis and cyclization reactions of alpha-, beta-, and gamma-cyclodextrin. Carbohydr Res 301:11–22. doi: 10.1016/S0008-6215(97)00073-6. [DOI] [PubMed] [Google Scholar]
- 8.van der Veen BA, van Alebeek GJ, Uitdehaag JC, Dijkstra BW, Dijkhuizen L. 2000. The three transglycosylation reactions catalyzed by cyclodextrin glycosyltransferase from Bacillus circulans (strain 251) proceed via different kinetic mechanisms. Eur J Biochem 267:658–665. doi: 10.1046/j.1432-1327.2000.01031.x. [DOI] [PubMed] [Google Scholar]
- 9.Szejtli J. 1998. Introduction and general overview of cyclodextrin chemistry. Chem Rev 98:1743–1753. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- 10.Uekama K, Hirayama F, Irie T. 1998. Cyclodextrin drug carrier systems. Chem Rev 98:2045–2076. doi: 10.1021/cr970025p. [DOI] [PubMed] [Google Scholar]
- 11.Astray G, Gonzalez-Barreiro C, Mejuto JC, Rial-Otero R, Simal-Gandara J. 2009. A review on the use of cyclodextrins in foods. Food Hydrocolloid 23:1631–1640. doi: 10.1016/j.foodhyd.2009.01.001. [DOI] [Google Scholar]
- 12.Szente L, Szeman J. 2013. Cyclodextrins in analytical chemistry: host-guest type molecular recognition. Anal Chem 85:8024–8030. doi: 10.1021/ac400639y. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Zhang J, Wang M, Gu Z, Du G, Li J, Wu J, Chen J. 2009. Mutations at subsite -3 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhancing alpha-cyclodextrin specificity. Appl Microbiol Biotechnol 83:483–490. doi: 10.1007/s00253-009-1865-3. [DOI] [PubMed] [Google Scholar]
- 14.Yue Y, Song BH, Xie T, Sun Y, Chao YP, Qian SJ. 2014. Enhancement of alpha-cyclodextrin product specificity by enriching histidines of alpha-cyclodextrin glucanotransferase at remote subsite-6. Process Biochem 49:230–236. doi: 10.1016/j.procbio.2013.11.002. [DOI] [Google Scholar]
- 15.Li Z, Li B, Gu Z, Du G, Wu J, Chen J. 2010. Extracellular expression and biochemical characterization of alpha-cyclodextrin glycosyltransferase from Paenibacillus macerans. Carbohydr Res 345:886–892. doi: 10.1016/j.carres.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Strokopytov B, Knegtel RM, Penninga D, Rozeboom HJ, Kalk KH, Dijkhuizen L, Dijkstra BW. 1996. Structure of cyclodextrin glycosyltransferase complexed with a maltononaose inhibitor at 2.6 angstrom resolution. Implications for product specificity. Biochemistry 35:4241–4249. [DOI] [PubMed] [Google Scholar]
- 17.Uitdehaag JCM, Mosi R, Kalk KH, van der Veen BA, Dijkhuizen L, Withers SG, Dijkstra BW. 1999. X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the alpha-amylase family. Nat Struct Biol 6:432–436. doi: 10.1038/8235. [DOI] [PubMed] [Google Scholar]
- 18.Uitdehaag JC, van Alebeek GJ, van Der Veen BA, Dijkhuizen L, Dijkstra BW. 2000. Structures of maltohexaose and maltoheptaose bound at the donor sites of cyclodextrin glycosyltransferase give insight into the mechanisms of transglycosylation activity and cyclodextrin size specificity. Biochemistry 39:7772–7780. doi: 10.1021/bi000340x. [DOI] [PubMed] [Google Scholar]
- 19.van der Veen BA, Uitdehaag JC, Penninga D, van Alebeek GJ, Smith LM, Dijkstra BW, Dijkhuizen L. 2000. Rational design of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 to increase alpha-cyclodextrin production. J Mol Biol 296:1027–1038. doi: 10.1006/jmbi.2000.3528. [DOI] [PubMed] [Google Scholar]
- 20.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustalversion 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 21.Guex N, Peitsch MC, Schwede T. 2009. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30(Suppl 1):S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 22.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 24.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Lejeune A, Sakaguchi K, Imanaka T. 1989. A spectrophotometric assay for the cyclization activity of cyclomaltohexaose (alpha-cyclodextrin) glucanotransferase. Anal Biochem 181:6–11. doi: 10.1016/0003-2697(89)90385-0. [DOI] [PubMed] [Google Scholar]
- 26.Mäkelä M, Korpela T, Laakso S. 1987. Colorimetric determination of beta-cyclodextrin: two assay modifications based on molecular complexation of phenolphtalein. J Biochem Biophys Methods 14:85–92. doi: 10.1016/0165-022X(87)90043-1. [DOI] [PubMed] [Google Scholar]
- 27.Parsiegla G, Schmidt AK, Schulz GE. 1998. Substrate binding to a cyclodextrin glycosyltransferase and mutations increasing the gamma-cyclodextrin production. Eur J Biochem 255:710–717. doi: 10.1046/j.1432-1327.1998.2550710.x. [DOI] [PubMed] [Google Scholar]
- 28.Leemhuis H, Uitdehaag JC, Rozeboom HJ, Dijkstra BW, Dijkhuizen L. 2002. The remote substrate binding subsite -6 in cyclodextrin-glycosyltransferase controls the transferase activity of the enzyme via an induced-fit mechanism. J Biol Chem 277:1113–1119. doi: 10.1074/jbc.M106667200. [DOI] [PubMed] [Google Scholar]
- 29.Li ZF, Zhang JY, Sun Q, Wang M, Gu ZB, Du GC, Wu J, Chen J. 2009. Mutations of lysine 47 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhance beta-cyclodextrin specificity. J Agric Food Chem 57:8386–8391. doi: 10.1021/jf902312u. [DOI] [PubMed] [Google Scholar]
- 30.Benkert P, Biasini M, Schwede T. 2011. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.