

Abstract
Understanding of the mechanistic progess of Amyloid-β peptide (Aβ) aggregation is critical for elucidating the underlying pathogenesis of Alzheimer’s disease (AD). Herein, we report for the first time the effects of two cholesterol derivatives, negatively charged cholesterol sulfate (cholesterol-SO4) and positively charged 3β-[N-(dimethylaminoethane)carbamoyl]-cholesterol (DC-cholesterol), on the fibrillization of Aβ40. Our results demonstrate that both of the nonvesicle forms of cholesterol-SO4 and DC-cholesterol moderately accelerate the aggregation rate of Aβ40. This effect is similar to that observed for unmodified cholesterol, indicating the importance of hydrophobic interactions in binding of Aβ40 to these steroid molecules. Furthermore, we show that the vesicles formed at higher concentrations of anionic cholesterol-SO4 facilitate Aβ40 aggregation rate markedly. In contrast, the cationic DC-cholesterol vesicles show the ability to inhibit Aβ40 fibril formation under appropriate experimental conditions. The results suggest that the electrostatic interactions between Aβ40 and the charged vesicles can be of great importance in regulating Aβ40-vesicle interaction. Our results also indicate that the structural properties of the aggregates of the cholesterol derivatives, including the surface charge and the size of the vesicles, are critical in regulating the effects of these vesicles on Aβ40 aggregation kinetics.

INTRODUCTION
Anomalous protein aggregation and fibril formation is one of the dominant characteristics in the pathogenesis of a number of neurodegenerative diseases, such as Alzheimer’s, Parkinson’s and Creutzfeldt–Jakob diseases.1-3 In Alzheimer’s disease (AD), extensive genetic, biochemical and pathological evidence links accumulation and amyloid fibril formation of amyloid-β (Aβ) peptides (e.g., the major components Aβ40 and Aβ42), produced by the β- and γ-secretase cleavage of the parental amyloid precursor protein (APP), to the AD phenotype.4, 5 Aβ amyloid fibril contains a typical cross-β-sheet architecture extending in a direction parallel to the fibril axis, identified by high resolution techniques such as solid-state NMR at the molecular level.6-8 Moreover, recent evidences suggest that the oligomeric, diffusible assemblies of Aβ peptides formed in the early stages of aggregation, appear to be highly toxic species in AD.9-11
Although it has been reconciled that both Aβ oligomers and fibrillar plaques may play roles in the progressive degeneration of neurons,12 the fundamental mechanism by which the assembly process causes the toxicity leading to cell death is still unclear. A growing body of recent research highlights the importance of cellular membranes in mediating Aβ self-assembly and the consequent cellular toxicity.13-15 Cholesterol is an essential component of the eukaryotic plasma membrane, necessary for membrane fluidity, permeability and receptor function. Elevated levels of cholesterol have been recgonized as one important risk factor for AD, and the role of cholesterol in APP processing and Aβ generation has been supported by recent studies.16-18 Sparks et al. reported a dose-dependent Aβ amyloid accumulation in the brain of rabbits fed with a high-cholesterol diet.19 Cerebral Aβ generation was reported to be cholesterol dependent,20 and guinea pigs treated with high doses of simvastatin, a widely used cholesterol-lowering drug, showed a strong and reversible reduction of cerebral Aβ levels in the cerebrospinal fluid and brain homogenate.21 Although the mechanism by which cholesterol modulate Aβ generation is unclear, lipid rafts, the cholesterol-rich membrane microdomains, appear to promote β- and γ-secretase processing function.22, 23 Furthermore, increased free cholesterol in the cytoplasm has also been found to affect the aggregation of Aβ peptides into fibrils.24, 25 These suggest that one of the possible roles for cholesterol in AD may be to directly interact with Aβ and consequently modulate the amyloidogenic process of Aβ. However, most of the reports available so far have mainly focused on cholesterol as the component in cellular membranes or lipid bilayer or monolayer model membranes,26-29 leaving the direct investigation of the effects of the pure form of cholesterol on Aβ amyloid formation largely neglected. Although increasing efforts have been provided to put insight into the interactions between cholesterol and Aβ peptides,30-32 a detailed mechanistic view of cholesterol-mediated Aβ fibrillogenesis is unclear.
Cholesterol, as a neutral and hydrophobic steroid molecule, can be decorated to form a series of derivatives, such as the oxidation metabolite 27-hydroxycholesterol and 24S-hydroxycholesterol. The effects of these derivatives in the pathology of AD have been suggested in recent studies.33, 34 Cholesterol sulfate (cholesterol-SO4, Fig. 1) is one of the most important known sterol sulfates, and has emerged as a significant lipid constituent in a variety of human tissues,35 with a concentration of ~110-170 μg/mL in human plasma.36, 37 While its definite functions in human physiology remain poorly understood, considering the potential use of the level of Aβ including the aggregated Aβ species in plasma as a biomarker for early diagnosis of AD,38 the effect of this cholesterol derivative on Aβ amyloidogenesis is of physiological interest. The sulfate moiety of cholesterol-SO4 has a pKa of ~3.3, indicating that it is normally ionized under physiological conditions. The presence of this negatively charged moiety makes cholesterol-SO4 an amphiphilic compound in comparison to a rather rigid hydrophobic cholesterol analog. On the other hand, a cationic cholesterol derivative, 3β-[N-(dimethylaminoethane)carbamoyl]-cholesterol, or DC-cholesterol (Fig. 1), has been commonly utilized for preparation of lipid membranes with cationic charged groups. DC-cholesterol contains a cholesterol moiety attached by an ester bond to a positively charged dimethylethylenediamine group. Although rarely studied to date, recent simulation results suggested the crucial role of hydrophobic force in regulating cholesterol–Aβ interaction.32 Owing to the additional charges in cholesterol-SO4 and DC-cholesterol molecules, these cholesterol derivatives may exhibit unique characteristics in mediating Aβ amyloid formation. Elucidating the effects of these derivatives on Aβ fibrillization will provide new mechanistic and structural insights into the underlying function of steroid-based molecules in modulating Aβ aggregation pathway, complementing the current efforts towards a deep understanding of the roles of cholesterol and the mutants in AD.
Figure 1.

Chemical structures of cholesterol and the two derivatives.
In the present work, we study for the first time the mechanistic effect of pure cholesterol-SO4 and DC-cholesterol on aggregation of Aβ40 peptide. We employ a combination of fluorescent spectroscopy and atomic force microscopy (AFM) imaging to assess the roles of specific intermolecular forces, e.g., hydrophobic and electrostatic interactions in lipid molecules and Aβ40 peptide association and their impact on Aβ fibrillogenesis. Our results demonstrate that low concentration of non-vesicle cholesterol-SO4 and DC-cholesterol both moderately accelerate Aβ40 fibril formation, similar to that of cholesterol. This implicates the dominating role of hydrophobic interactions between Aβ40 and cholesterol derivatives in facilitating the aggregation rate. Furthermore, we show that the vesicle structures formed by high concentrations of anionic cholesterol-SO4 facilitate Aβ40 aggregation rate markedly. In contrast, the vesicles of cationic DC-cholesterol have the ability to inhibit Aβ40 fibril formation under appropriate experimental conditions, distinct from those of the cholesterol-SO4. Our results suggest the critical role of the structural properties of the vesicles of the cholesterol derivatives, including the surface charge properties and the size, in mediating their effects on Aβ40 amyloid formation.
EXPERIMENTAL METHODS
Materials
All chemical reagents were obtained from commercial suppliers and used without further purification unless otherwise mentioned. Lipid molecules cholesterol, DC-cholesterol and cholesterol-SO4 were purchased from Sigma-Aldrich. Solutions of DC-cholesterol and cholesterol-SO4 were prepared by thoroughly mixing appropriate amounts of compound powder in dimethyl sulfoxide (DMSO), while cholesterol was dissolved in a mixture of hexane/chloroform/ethanol 11:5:4 v/v/v according to a previously reported protocol by Fantini et al.39
Synthesis and preparation of Aβ40 peptide
Aβ40 peptide was synthesized on a PS3 solid phase peptide synthesizer (Protein Technologies) using standard Fmoc strategy. The resulting crude peptide was purified by reversed phase high-performance liquid chromatography (RP-HPLC) using a C18 reverse phase column and characterized by matrix-assisted laser desorption ionization mass spectrometry (MALDI). The Aβ40 peptide utilized in the kinetic aggregation assay was monomerized as described previously.40 Briefly, lyophilized Aβ40 powder was dissolved in aqueous NaOH solution (2 mM) and the pH was adjusted to 11 by using 100 mM NaOH solution. The solution was sonicated for 1 h in ice-water bath, then filtered through 0.22-μm filter (Millipore) and kept on ice before use. The concentration of Aβ40 was determined by using the tyrosine UV absorbance at 280 nm (ε = 1,280 M−1cm−1).
Kinetic aggregation assay of Aβ40 using thioflavin T (ThT)
The aggregation kinetics followed by ThT fluorescence were conducted as described previously.41 In brief, the monomerized Aβ40 peptide solution was diluted to a final concentration of 12 μM in pH 7.4 phosphate buffer (50 mM Na phosphate, 150 mM NaCl) containing 20 μM ThT. 100 μL solution was transferred into wells of a 96-well microplate (Costar black, clear bottom). The plate was sealed with a microplate cover and loaded into a Gemini SpectraMax EM fluorescence plate reader (Molecular Devices, Sunnyvale, CA), where it was incubated at 37 °C. The fluorescence of ThT was measured every 10 min after shaking for 5 s with excitation wavelength of 440 nm and emission wavelength of 480 nm.
For the assays with cholesterol derivatives, a stock solution of DC-cholesterol or cholesterol-SO4 was prepared by dissolving 5.0 mg of the samples in 1.0 mL DMSO and then different stock solutions were prepared by further dilution. The stock solution of cholesterol was prepared by dissolving 5.0 mg of the sample in a mixed solvent composed of hexane:chloroform:ethanol 11:5:4 v/v/v, and further dilutions were applied using the same mixed solvent to obtain stock solutions with different concentration of cholesterol. A particular amount of stock solutions was added to Aβ40 solution (final concentration of 12 μM) in pH 7.4 phosphate buffer (50 mM Na phosphate, 150 mM NaCl) containing 20 μM ThT. The solution was then pipetted into the plate reader (100 μL/well) for the kinetic assay. In consideration of the high sensitivity of Aβ40 aggregation rate on the experimental conditions and therefore the day-to-day assay variance of lag time in Aβ40 aggregation kinetics, an Aβ40-alone sample was always measured in each assay as the control.
The kinetic parameter lag time (tlag) was extracted by fitting the kinetic data using the following equation42:
where F is the fluorescence intensity at time t, Fmax is the maximum steady-state fluorescence, k is the apparent rate constant of amyloid accumulation, tm is the point of the maximum elongation rate, and v represents the asymmetry of the aggregation kinetics curve. The lag time (tlag) is the time at which the tangent at tm crosses the initial baseline and is given by tm–(1 + v)/k.
Atomic force microscopy (AFM)
Aliquots (20 μL) of the Aβ40 solution were adsorbed onto the surface of freshly cleaved mica (5×5 mm) for 5 min at room temperature. The liquid was wicked off by absorption into filter paper. Salts and unbound materials were removed by three washes with 30 μL of water. The samples were dried overnight, and AFM images were acquired in tapping mode utilizing an Asylum Research MFP 3D AFM system with MikroMasch NSC15/Al BS cantilevers.
Critical micelle concentration (CMC) determination
The CMC values of the cholesterol derivatives used in this study were measured following a previously reported protocol.43 Briefly, a series of solutions for each lipid derivative were prepared that differ by an order of concentration. The cholesterol derivatives were dissolved in pH 7.4 phosphate buffer (50 mM Na phosphate, 150 mM NaCl) containing 5% DMSO. The final concentration range of the cholesterol derivatives was from 0.50 ng/mL to 0.25 mg/mL (~ 9 × 10−10 M to 4 × 10−4 M). Using a FluoroMax-4 spectrofluorometer (HORIBA Jobin Yvon) at slit width of 10 nm and excitation and emission wavelength of 460 nm, light intensity for each solution was measured 3 times and then was averaged. Data were plotted as logarithm of sample concentration versus average light scattering intensity, and CMC values were calculated as the intercept with x-axis of a linear regression line for the concentrations where a rapid increase in light scattering intensity was observed.
Vesicle size measurement using dynamic light scattering
100 μL aliquots of different concentrations of each cholesterol derivative in pH 7.4 phosphate buffer (50 mM Na phosphate, 150 mM NaCl) containing 5% DMSO were incubated at 37 °C for 24 h. Then samples were diluted to 200 μL with Millipore H2O and stirred for 30 s. Size measurements were performed using a model Zetasizer Nano ZS instrument (Malvern Instruments, Worcestershire, UK) with disposable cuvettes. The average diameters of vesicles were calculated as the mean of three measurements, with 12-15 runs per measurement.
Zeta potential measurement
A 100 μL aliquot of the samples in pH 7.4 phosphate buffer (50 mM Na phosphate, 150 mM NaCl) containing 5% DMSO was incubated at 37 °C for 24 h. Then the sample was 10-fold diluted to 1 mL with Millipore H2O and stirred for few seconds. Zeta potential measurements were performed using a model Zetasizer Nano ZS instrument (Malvern Instruments, Worcestershire, UK) with a disposable polycarbonate folded capillary cell (model DTS1070) with a path length of 4 mm. The values of zeta potential are reported as the mean of three measurements.
RESULTS AND DISCUSSION
Effects of low concentrations of cholesterol derivatives on Aβ40 aggregation
Cholesterol and its derivatives, like other lipid molecules, can self-associate and form aggregated structures in aqueous solution above the concentration of the CMC value. In order to study the effect of pure non-aggregating cholesterol derivatives on aggregation of Aβ40, we first measured the CMC values of cholesterol-SO4 and DC-cholesterol following a protocol reported by Avdulov et al.43 As shown in Table 1, the CMC value of cholesterol-SO4 in pH 7.4 phosphate buffer (50 mM Na-phosphate, 150 mM NaCl) is approximately 12.6 nM (equivalent to 6.2 ng/mL), and the CMC value of DC-cholesterol is approximately 14 nM (equivalent to 7.5 ng/mL). The CMC values for these two derivatives are slightly lower than that of the unmodified cholesterol, which is ~31 nM (equivalent to ~12.1 ng/mL), as reported previously.43, 44
Table 1.
CMC value of the cholesterol derivatives.
| Lipid molecule | CMC value (ng/mL) |
|---|---|
| Cholesterol-SO4 | 6.2 ± 1.6 |
| DC-cholesterol | 7.5 ± 2.6 |
| Cholesterol* | 12.1 |
Data from ref. 43.
The kinetics of fibrillization of monomeric Aβ40 peptide (12 μM) in phosphate buffer (50 mM Na-phosphate, 150 mM NaCl, pH 7.4) was monitored via a ThT fluorescence experiment. ThT molecule can selectively bind to the fibrillar structure, leading to a significant increase of its fluorescence quantum yield.45 As shown in Fig. 2a, the aggregation kinetics of Aβ40 peptide had a sigmoidal appearance containing a lag phase associated with nucleation, a fast growth phase related to the propagation of fibrils, and a final stationary phase. This is consistent with previous studies which indicated that Aβ amyloidogenesis proceeds by a nucleated polymerization mechanism.46-49.
Figure 2.

Effect of low concentration of DC-cholesterol (below the CMC value) on the aggregation of Aβ40. (a) The aggregation kinetics of Aβ40 (12 μM) in the absence or the presence of different amount of DC-cholesterol measured by ThT fluorescence at 37 °C. (b) Growth phase half time (t50) of the aggregation kinetics of Aβ40 in the absence or the presence of DC-cholesterol. Data are reported as means ± the standard deviation of triplicate results.
The effect of DC-cholesterol on amyloidogenesis of Aβ40 was examined using the same ThT fluorescence assay. DC-cholesterol was used at concentrations below its CMC value in this case, allowing to assess the mechanistic effect of non-aggregating DC-cholesterol structures on Aβ aggregation. Here we reasonably assume that DC-cholesterol mainly exists as monomeric molecules under the current experimental conditions, although one could not exclude the possibility of the formation of oligomeric structures of DC-cholesterol. As shown in Fig. 2a, the presence of a low concentration of DC-cholesterol (0.5 to 2.5 ng/mL) slightly but noticeably facilitates the aggregation rate of Aβ40, with a shorter lag phase as compared to that measured without DC-cholesterol. This suggests that DC-cholesterol likely facilitates the early nucleation reactions to form crucial oligomeric intermediates toward the formation of fibrillar structures. The aggregation half time (t50), which is defined as the time at which the ThT fluorescence intensity reaches half of the maximum, is decreased approximately 21% from ~ 10.5 h to ~ 8.3 h in the presence of DC-cholesterol at 2.5 ng/mL (Fig. 2b). This result is comparable to that observed for cholesterol (Fig. S2), wherein the addition of 2.5 ng/mL cholesterol also moderately accelerates the aggregation rate of Aβ40, as revealed by an approximately 27% decrease of t50 from ~10 h to ~ 7.3 h. These results implicate that the additional moiety attached on the hydrophobic cyclic sterol ring structure, including the presence of the positively charged dimethylethylenediamine residue, does not dramatically change the regulating function of cholesterol on Aβ fibril formation. This is also consistent with recent simulation studies,30, 32 which reported that the hydrophobic interactions between cholesterol and the Aβ hydrophobic region play a critical role in association between these molecules.
The effect of low concentration of cholesterol-SO4 on aggregation of Aβ40 was also studied. Similarly, the concentrations below the CMC value of cholesterol-SO4 were used in order to provide insight into the non-aggregating cholesterol-SO4 mediated Aβ40 amyloidogenesis. As shown in Fig. 3, additional cholesterol-SO4 (0.5 to 2.5 ng/mL) also moderately facilitated the aggregation rate of Aβ40, with the aggregation t50 value decreased from ~ 11.2 h to ~ 9.8 h. Again, the small difference between the t50 values (Fig. 3b) suggests that under the current condition, the kinetic effect of cholesterol-SO4 on Aβ40 aggregation is not significant. This is similar to that of the unmodified cholesterol and DC-cholesterol, suggesting that the negatively charged sulfate moiety of cholesterol-SO4 does not dramatically influence its function in mediating Aβ aggregation. Taken together, if non-aggregating cholesterol and the derivatives modify the aggregation of Aβ via binding with the peptide, as suggested by recent simulation studies,30, 32 they presumably interact with Aβ peptides mainly through hydrophobic interactions. The positively or negatively charged groups in the cholesterol derivatives studied here likely are not involved in critical interactions in this process.
Figure 3.

Effect of low concentration of cholesterol-SO4 (below the CMC value) on the aggregation of Aβ40. (a) The aggregation kinetics of Aβ40 (12 μM) in the absence or the presence of different amount of DC-cholesterol measured by ThT fluorescence at 37 °C. (b) Growth phase half time (t50) of the aggregation kinetics of Aβ40 in the absence or the presence of cholesterol-SO4. Data are reported as means ± the standard deviation of triplicate results.
Effects of aggregated vesicles of cholesterol derivatives on Aβ40 aggregation
The local concentration of cholesterol-SO4 in human tissuese can be much higher than its CMC value. Therefore, besides addressing its regulating role as a non-aggregating form in Aβ aggregation, it would be valuable to examine the effect of the aggregated vesicles formed at concentrations above the CMC value on the aggregation of Aβ. We thus prepared the aggregated vesicles of the pure cholesterol derivatives formed above their CMC values, and examined their kinetic effects on Aβ amyloidogenesis. As shown in Fig. 4a, addition of large amount of cholesterol-SO4 (concentration of 0.25 μg/mL and higher), under which the molecule self-associates to form vesicle structures, facilitates the aggregation rate of Aβ40 (12 μM) in a concentration-dependent manner. The presence of cholesterol-SO4 of 25 μg/mL leads to a significantly shorter t1ag changed from 12.2 to 3.1 h (~ 75% decrease) (Fig. 4a). With the presence of cholesterol-SO4 of 250 μg/mL, the Aβ40 kinetic aggregation curve shows an immediate growth phase, with no lag phase observed. Similar results were also observed using a lower concentration of Aβ40 (5 μM), as shown in Fig. S1. These results suggest that the cholesterol-SO4 vesicles could influence the early oligomerization reaction, facilitating the formation of oligomeric nucleus and/or the propagation of the oligomers into fibrillar structure. The kinetic effect of cholesterol vesicles were also tested for comparison (Fig. S2). Although only a middle concentration (25 μg/mL) of cholesterol was used here, because of the low solubility of cholesterol under the experimental condition, the results nonetheless demonstrate that the appearance of the cholesterol vesicles increases the aggregation rate of Aβ40 (Fig. S2). This is consistent with previously reported results for cholesterols incorporated into the lipid membrane structure.27, 50 Moreover, the results also suggest that the accelerating activity of cholesterol vesicles is weaker in comparison to that of the cholesterol-SO4 (Fig. 4a vs. Fig. S2).
Figure 4.

Effect of high concentration of cholesterol-SO4 (a) or DC-cholesterol (b) on the aggregation of Aβ40. The aggregation kinetics of Aβ40 (12 μM) in the absence or the presence of different amount of choleterol derivatives were measured by ThT fluorescence at 37 °C. The concentrations of the choleterol derivativies used were above their respective CMC values. The inset shows tlag values of the kinetic aggregation traces with data reported as means ± standard deviation of triplicate results.
Interestingly, the DC-cholesterol vesicles show an unusual effect on Aβ aggregation kinetics. In the presence of relatively low concentration of DC-cholesterol while above its CMC value, Aβ aggregates faster with a shortened lag phase (Fig. 4b). However, when high amount of DC-cholesterol, e.g., 25 μg/mL, was added, the aggregation of Aβ was dramatically interfered. The increase of ThT fluorescence is delayed significantly with the tlag value increased from ~12.4 to 23.6 h. The final stationary fluorescence intensity also decreases dramatically. When 250 μg/mL DC-cholesterol was used in the kinetic assay, there is no observable ThT fluorescence at the ending time point of the kinetics experiment, suggesting a strong inhibiting activity on Aβ fibrillization. In comparison to that of the cholesterol-SO4, these results indicate distinct roles of these two vesicles in the aggregation of Aβ40 under high concentration conditions.
The effect of the lipid vesicles on Aβ40 aggregation was further assessed using AFM imaging. AFM images of Aβ40 (30 μM) in the absence or presence of either cholesterol-SO4 or DC-cholesterol were acquired after incubating the samples for 17 h on a speci-mix aliquot mixer at 37 °C. In the absence of cholesterol derivatives, characteristic fibrillar amyloids were produced (Fig. 5a). Fibers were also observed in the presence of 50 ng/mL cholesterol-SO4 (Fig. 5b). Formation of fibers still occurs with a high concentration of cholesterol-SO4 of 500 μg/mL (Fig. 5c), consistent with the non-inhibiting effect of this derivative on Aβ aggregation, as suggested by the aggregation kinetic results. For DC-cholesterol, Aβ fibers were observed in the image of Aβ40 (30 μM) co-incubated with low concentration of DC-cholesterol (50 ng/mL) (Fig. 5d). However, as shown in Fig. 5e, in the presence of 500 μg/mL DC-cholesterol, no fiber structures were found in the image. This is consistent with our observations from the kinetic results, suggesting an inhibitory activity of the DC-cholesterol species under high concentration condition. These results demonstrate that the DC-cholesterol vesicles formed at high concentration of the molecules may play different roles in Aβ aggregation, in comparison to cholesterol-SO4 vesicles. In view that one of the major structural differences between these two molecules is their opposite charge, the results may suggest the potential effects of electrostatic interactions in mediating interactions between the vesicles and Aβ. However, we do not exclude other factors, including hydrophobic interactions and the properties of particle structures, on impacting the functions of cholesterol derivatives in modulating Aβ aggregation.
Figure 5.

Tapping mode atomic force microscopy images of Aβ40 fibrillization reactions. (a) Aβ40 (30 μM) in pH 7.4 phosphate buffer (50 mM Na phosphate, 150 mM NaCl) incubated on a speci-mix aliquot mixer at 37 °C. Images were also taken for Aβ40 (30 μM) in the presence of 50 ng/mL cholesterol-SO4 (b), 500 μg/mL cholesterol-SO4 (c), 50 ng/mL DC-cholesterol (d), and 500 μg/mL DC-cholesterol (e). Samples were incubated for 17 h before imaging.
Interactions between Aβ40 and the cholesterol derivatives
The importance of cholesterol, as a crucial component of cellular membrane, in modulating the aggregation of Aβ peptide has long been recognized.18 However, most of the previous in vitro studies on the cholesterol-mediated Aβ amyloidogenesis were performed using mixed cholesterol-phospholipid model structures. Studies towards clarification of the particular interactions between Aβ peptide and the cholesterol molecule and the mechanistic effect of cholesterol on regulating Aβ amyloidogenesis are scarce. Nonetheless, direct binding of cholesterol to APP and Aβ amyloid fibrils has been indicated in some recent studies.31, 51 Computational approaches have started to be applied recently to shed light on the interaction details between free cholesterol and Aβ peptide. Di Scala et al. studied the binding between cholesterol and Aβ peptide via both docking modelling and surface pressure measurements of Langmuir monolayers, and their results suggested that a hydrophobic fragment of amino acids 22-35 of Aβ can serve as a potential cholesterol-binding domain, primarily forming hydrophobic interactions with the apolar part of cholesterol.32 Using all-atom molecular dynamic simulations, Zhou et al. reported an important role of steroid-benzyl interaction in cholesterol-mediated aggregation.30 More specifically, the amino acid Phe19 was found to be critical for this interaction. Our experimental results are in accord with these simulation results. At concentrations below the CMC values, where cholesterol-SO4 or DC-cholesterol remains as nonvesicle forms, presumably mainly monomeric structures, they moderately accelerate the aggregation rate of Aβ, similar to that of the unmodified cholesterol. This suggests that none of the negatively or positively charged groups in these mutants could dramatically influence the interactions between Aβ and cholesterol derivatives, suggesting that no critical electrostatic interactions were formed between these charged moieties and Aβ molecule. In other words, we could reasonably speculate that the hydrophobic interactions between the non-aggregating cholesterol derivatives and the Aβ peptide, especially its hydrophobic sequence region, are dominant in the regulating mechanisms on Aβ amyloidogenesis. Recently it has been reported that cholesterol can stimulate the formation of oligomeric Aβ channel structures in membrane,52, 53 and a cholesterol-like compound bexarotene can efficaciously occupy the cholesterol-binding domain in Aβ peptide, thus interfering with the formation of amyloid pore channels.54 Given the similarity of the structures of cholesterol and cholesterol-SO4 or DC-cholesterol derivatives, it would not be surprising that these derivative molecules could bind with Aβ peptide at the same cholesterol-binding site and mediate the oligomerization and fibrillization of Aβ via a similar mechanism.
Our results demonstrate that the negatively charged cholesterol-SO4 vesicles dramatically facilitate the aggregation of Aβ40. Vesicles formed by lipid molecules, such as amphipathic phospholipids, normally contain a liquid crystalline bilayer structure with the hydrophilic head group facing the surrounding aqueous solvent, sequestering the hydrophobic tail regions in the micelle center. Lipid vesicles thus provide an interface that can act as a catalytic or inhibiting surface, and can influence the Aβ aggregation kinetics in similar manner as nanoparticles.55-58 It has been suggested that the head group charge of the phospholipids contributes to the association between the membrane and the Aβ peptide via electrostatic interactions. However, the exact effects of head group charges on Aβ peptide aggregation are somewhat controversial.59 Kremer et al. reported that the affinity of Aβ40 to the zwitterionic1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) is weaker than for the negatively charged 1-palmitoyl-2-oleoyl-snglycero-3-phosphoglycerol (POPG), supporting the view that the charged head groups mediate binding.60 On the contrary, results from Hellstrand et al. suggested that there is no significantly different effect on Aβ aggregation kinetics when increasing amount of negative charge was incorporated into phospholipid vesicles.55 These contradictory results suggest the sensitivity of the kinetic roles of lipid vesicles on their specific structural and surface properties.
The typical steroid structure of cholesterol-SO4 possibly makes it less favourable to form ordered gel- or liquid crystalline-like vesicle structures like other phospholipids. Nonetheless, the size of the vesicles was analysed using dynamic light scattering method assuming a sphere model. The results indicate that the size of the vesicles is dependent on the concentration of cholesterol-SO4 (Fig. 6a and Fig. S3), with larger vesicles formed at higher concentrations. The difference of the width of the size distribution suggests the polydisperse feature of the particles of varied sizes. Considering the amphipathic nature of cholesterol-SO4, it would be conceivable to speculate that considerable amount of negatively charged sulphate ion moieties may still tend to be exposed to face the solvent on the surface of the vesicles. The measured negative zeta potential, i.e., −53 mv for cholesterol-SO4 at a concentration of 250 μg/mL, also suggests the presence of negative charges on the surface of the formed vesicles. Therefore, besides the plausible hydrophobic interactions, it is quite possible that the sulfate group may also play a role in guiding the binding between Aβ and the cholesterol-SO4 vesicles via electrostatic interactions. This is indicated by a stronger facilitating effect of cholesterol-SO4 on the aggregation kinetics of Aβ, in comparison to that of the unmodified cholesterol. One possibility is that the positively charged residues along the Aβ sequence at neutral pH used in this study, including Arg, Lys, and His residues, may interact with the negatively charged sulfate groups exposed on the surface of the vesicles. This will lead to the adsorption of the monomeric Aβ on the surface of vesicles, thus increasing local concentration of Aβ and favouring the formation of critical nucleus for further propagation (Fig 7).
Figure 6.

Size of cholestereol-SO4 (a) and DC-cholesterol (b) vesicles formed at different concentrations of the compounds. The diameters values were determined by dynamic light scattering.
Figure 7.
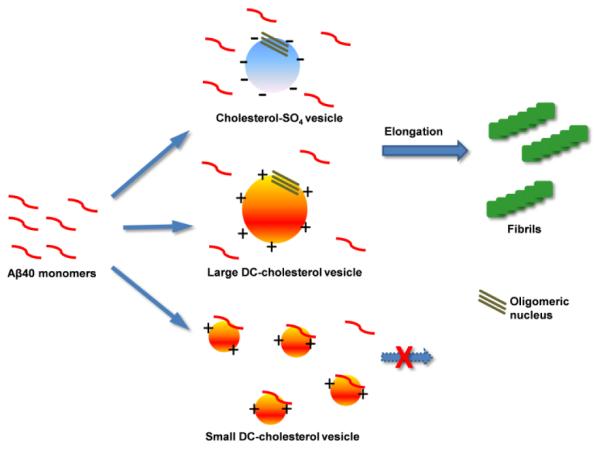
Schematic representation of the possible interactions between Aβ40 and the negatively charged cholesterol-SO4 vesicles and the positively charged DC-cholesterol vesicles with different size.
The surface of DC-cholesterol vesicles is speculated to contain positive charges, opposite to that of cholesterol-SO4. The zeta potential is approximately +32 mV for DC-cholesterol at a concentration of 250 μg/mL, consistent with the proposed surface charge properties of DC-cholesterol vesicles. At low concentrations while above its CMC value, the formed DC-cholesterol vesicles also facilitate the amyloid formation of Aβ. Again, here one would attribute the significantly accelerating effect of DC-cholesterol vesicles partly to the vesicle-aided nucleus formation. The positive charges on the surface of the particles may exhibit as a favourable factor for facilitating the adsorption of Aβ on the vesicles, as Aβ also contains considerable amount of negatively charged residues (i.e., 3 Asp and 3 Glu). With a theoretical isoelectric point pI ~ 5.31, the overall net charge of Aβ40 at pH 7 is approximately −3, and therefore the overall electrostatic attractions between Aβ40 molecule and DC-cholesterol vesicles is expected to be quite favourable. Surprisingly, the vesicles formed at higher concentrations of DC-cholesterol, i.e., 25 μg/mL and above, starts to inhibit the aggregation of the peptide. We found that relatively large vesicle structures with diameters of ~300 nm were formed at low concentrations of DC-cholesterol (Fig. 6b). These vesicles may provide favourable surface area for local binding with Aβ40, thus facilitating the formation of oligomeric nucleus structures for elongation to form fibrils. With increased concentration of DC-cholesterol, the diameter of the formed vesicles becomes surprisingly smaller. At the highest concentration of 250 μg/mL used in the study, the size of the particle is approximately 100 nm (Fig. 6b). It has been previously reported that the size and the curvature properties of the liposome vesicles are important factors in determining the binding between the vesicles and amyloidogenic proteins such as Aβ and α-synuclein.61-63 Considering the small radius and high curvature properties of positively charged DC-cholesterol vesicles formed at high concentrations, and the overall negative net charge of Aβ40, these small vesicles may strongly bind with Aβ monomers, as depicted in Fig. 7. Therefore it would be likely that strong attractive electrostatic interactions, together with hydrophobic interactions, may prevent the diffusion, conformational adjustment, and the self-association of the monomers along the vesicle surface. This will inhibit the formation of critical nucleus on vesicle surface and the further propagation to form fibrillar structures. In addition, because of the large amount of the small vesicles formed under the high concentration condition, such binding will dramatically reduce the concentration of monomeric Aβ40 in solution, thus also possibly leading to retardance or complete inhibition of amyloid fibril formation of Aβ40 peptide.
The effect of DC-cholesterol on Aβ40 amyloidogenesis was also investigated with different NaCl salt concentration. The increase of NaCl salt concentration (50-300 mM) did not significantly change the aggregation kinetics of Aβ40 alone (Fig. S4). However, increase of concentration of NaCl led to dramatic decrease of the kinetic difference between Aβ40 alone and Aβ40 with DC-cholesterol (Fig. S4). This suggests the sensitivity of the interaction of Aβ40 and DC-cholesterol on the ionic strength, indicating the critical role of electrostatic force in Aβ40 and DC-cholesterol interactions. On the other hand, increase of NaCl concentration slightly increases the facilitating effect of cholesterol-SO4 on the kinetics of Aβ40 aggregation, possibly because of the screening effect of the salt on repulsive electrostatic interactions between the negatively charged residues in Aβ40 and the sulfate group of cholesterol-SO4.
The specific structural properties of the vesicles could dramatically influence their interactions with Aβ40 peptide. It was reported that small unilamellar gel phase vesicles can show faceted structure,64 where the edges between the facets are highly curved which may be possible that peptides accumulate in such areas.55 Our results show that the vesicles of cholesterol derivatives can affect the aggregation of Aβ peptide, and these effects are highly dependent on the structural properties of the vesicle system. However, it is not trivial to clarify the detailed structural characteristics of these vesicles at molecular level. Some previous studies indicated that cholesterol can form irregular structures under certain conditions.44, 65 Future studies for clarifying the detailed structural characteristics of the vesicles formed by the cholesterol derivatives will be critical for understanding the mechanistic reasons of the distinct regulating roles of the vesicles on Aβ aggregation discovered in this research.
Aβ40 was used as an amyloidogenic model protein in this study, since Aβ40 shows a more moderate aggregation propensity than another aggressive component of the amyloid plaques in AD, Aβ42, which contains two more residues (Ile-Ala) at the C-terminus. This allows the feasibility to carefully investigate the kinetic effects of the cholesterol derivatives on aggregation of Aβ40 peptide. Due to the fast aggregation kinetics of Aβ42 itself, the kinetic effect of the cholesterol derivatives on Aβ42 amyloidogenesis is not as significant as that on Aβ40 (Fig. 4 vs. Fig. S5a). Comparison of the fluorescence intensity of the final stationary phase (Fig. S5b) of the kinetic traces shows that cholesterol-SO4 can facilitate Aβ42 fibrillization, while high concentration of DC-cholesterol interferes with the formation of amyloid fibrils. The results suggest that these cholesterol derivatives may interact with Aβ42 in a mechanism similar to that with Aβ40.
CONCLUSION
In summary, we report for the first time the kinetic effects of both non-aggregating forms and aggregated vesicle structures of cholesterol-SO4 and DC-cholesterol on Aβ40 amyloid formation. Our results demonstrate that the non-aggregating forms of cholesterol-SO4 and DC-cholesterol accelerate the aggregation rate of Aβ40 moderately, similar to that observed for the unmodified cholesterol, indicating the importance of hydrophobic interactions in binding of these steroid molecules and Aβ40. Furthermore, we show that the vesicles formed at high concentrations of pure anionic cholesterol-SO4 facilitate Aβ40 aggregation rate markedly, whereas the cationic DC-cholesterol vesicles have the ability to inhibit Aβ40 fibril formation under appropriate experimental conditions, distinct to that of the cholesterol-SO4. Our results implicate that the characteristics of the protein-vesicle interface, including the surface charge properties and the size of the cholesterol derivative vesicles, are critical in regulating the effects of the vesicles of these cholesterol derivatives on Aβ40 amyloid formation. Our findings not only provide new insights into the mechanistic effect of natural cholesterol derivatives on Aβ amyloidogenesis, but also may illuminate design of specific molecular particles as inhibitors of Aβ fibril formation.
Supplementary Material
ACKNOWLEDGMENT
The authors gratefully thank the startup fund (to D.D.) from Florida Atlantic University. D.D. also gratefully acknowledges the financial support from the National Institutes of Health (R15GM116006) for supporting the research.
Footnotes
Notes
The authors declare no competing financial interests.
Aggregation kinetics of Aβ peptides with cholesterol and the derivatives, salt effect results, and light scattering data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Selkoe DJ. Folding Proteins in Fatal Ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 2.Kelly JW. Alternative Conformations of Amyloidogenic Proteins Govern Their Behavior. Curr. Opin. Struct. Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Alzheimer's disease: Genotypes, Phenotypes, and Treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 5.Tanzi RE, Bertram L. Twenty Years of the Alzheimer's Disease Amyloid Hypothesis: A Genetic Perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common Core Structure of Amyloid Fibrils by Synchrotron X-Ray Diffraction. J. Mol. Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 7.Petkova AT, Yau WM, Tycko R. Experimental Constraints on Quaternary Structure in Alzheimer's Beta-Amyloid Fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D Structure of Alzheimer's Amyloid-Beta(1-42) Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haass C, Selkoe DJ. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer's Amyloid Beta-Peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 10.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble Pool of Abeta Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer's Disease. Ann. Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 11.Glabe CG. Structural Classification of Toxic Amyloid Oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selkoe DJ. Resolving Controversies on the Path to Alzheimer's Therapeutics. Nat. Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 13.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of Lipid Bilayers Is a Common Conformation-Dependent Activity of Soluble Amyloid Oligomers in Protein Misfolding Diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 14.Yip CM, Darabie AA, McLaurin J. Aβ42-Peptide Assembly on Lipid Bilayers. J. Mol. Biol. 2002;318:97–107. doi: 10.1016/S0022-2836(02)00028-1. [DOI] [PubMed] [Google Scholar]
- 15.Walter J, van Echten-Deckert G. Cross-Talk of Membrane Lipids and Alzheimer-Related Proteins. Mol. Neurodegener. 2013;8:34–45. doi: 10.1186/1750-1326-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bu G. Apolipoprotein E and Its Receptors in Alzheimer's Disease: Pathways, Pathogenesis and Therapy. Nature Rev. Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Basak JM, Holtzman DM. The Role of Apolipoprotein E in Alzheimer's Disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Paolo G, Kim TW. Linking Lipids to Alzheimer's Disease: Cholesterol and Beyond. Nat. Rev. Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sparks DL, Scheff SW, Liu H, Landers TM, Coyne CM, Hunsaker JC., III Increased Incidence of Neurofibrillary Tangles (NFT) in Non-Demented Individuals with Hypertension. J. Neurol. Sci. 1995;131:162–169. doi: 10.1016/0022-510x(95)00105-b. [DOI] [PubMed] [Google Scholar]
- 20.Shobab LA, Hsiung G-YR, Feldman HH. Cholesterol in Alzheimer's Disease. Lancet Neurol. 2005;4:841–852. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- 21.Fassbender K, Simons M, Bergmann C, Stroick M, Lütjohann D, Keller P, Runz H, Kühl S, Bertsch T, Von Bergmann K. Simvastatin Strongly Reduces Levels of Alzheimer's Disease β-Amyloid Peptides Aβ42 and Aβ40 in Vitro and in Vivo. Proc. Natl. Acad. Sci. U. S. A. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanagisawa K, Matsuzaki K. Cholesterol-Dependent Aggregation of Amyloid β-Protein. Ann. N.Y. Acad. Sci. 2002;977:384–386. doi: 10.1111/j.1749-6632.2002.tb04841.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee S-J, Liyanage U, Bickel PE, Xia W, Lansbury PT, Kosik KS. A Detergent-Insoluble Membrance Compartment Contains Aβ in Vivo. Nat. Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 24.Distl R, Meske V, Ohm TG. Tangle-Bearing Neurons Contain More Free Cholesterol Than Adjacent Tangle-Free Neurons. Acta Neuropathol. 2001;101:547–554. doi: 10.1007/s004010000314. [DOI] [PubMed] [Google Scholar]
- 25.Panchal M, Loeper J, Cossec J-C, Perruchini C, Lazar A, Pompon D, Duyckaerts C. Enrichment of Cholesterol in Microdissected Alzheimer's Disease Senile Plaques as Assessed by Mass Spectrometry. J. Lipid Res. 2010;51:598–605. doi: 10.1194/jlr.M001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic Processing of the Alzheimer β-Amyloid Precursor Protein Depends on Lipid Rafts. J. Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X, Zheng J. Cholesterol Promotes the Interaction of Alzheimer β-Amyloid Monomer with Lipid Bilayer. J. Mol. Biol. 2012;421:561–571. doi: 10.1016/j.jmb.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Vestergaard M. d., Hamada T, Takagi M. Using Model Membranes for the Study of Amyloid Beta: Lipid Interactions and Neurotoxicity. Biotechnol. Bioeng. 2008;99:753–763. doi: 10.1002/bit.21731. [DOI] [PubMed] [Google Scholar]
- 29.Yip C, Elton E, Darabie A, Morrison M, McLaurin J. Cholesterol, a Modulator of Membrane-Associated Aβ-Fibrillogenesis and Neurotoxicity. J. Mol. Biol. 2001;311:723–734. doi: 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- 30.Zhou X, Xu J. Free Cholesterol Induces Higher β-Sheet Content in Aβ Peptide Oligomers by Aromatic Interaction with Phe19. PLoS One. 2012;7:e46245. doi: 10.1371/journal.pone.0046245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris JR. Cholesterol Binding to Amyloid-β Fibrils: A Tem Study. Micron. 2008;39:1192–1196. doi: 10.1016/j.micron.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Di Scala C, Yahi N, Leli vre C. m., Garmy N, Chahinian H, Fantini J. Biochemical Identification of a Linear Cholesterol-Binding Domain within Alzheimer’s β Amyloid Peptide. ACS Chem. Neurosci. 2012;4:509–517. doi: 10.1021/cn300203a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gamba P, Guglielmotto M, Testa G, Monteleone D, Zerbinati C, Gargiulo S, Biasi F, Iuliano L, Giaccone G, Mauro A. Up-Regulation of β-Amyloidogenesis in Neuron-Like Human Cells by Both 24- and 27-Hydroxycholesterol: Protective Effect of N-Acetyl-Cysteine. Aging cell. 2014;13:561–572. doi: 10.1111/acel.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schönknecht P, Lütjohann D, Pantel J, Bardenheuer H, Hartmann T, von Bergmann K, Beyreuther K, Schröder J. Cerebrospinal Fluid 24s-Hydroxycholesterol Is Increased in Patients with Alzheimer's Disease Compared to Healthy Controls. Neurosci. Lett. 2002;324:83–85. doi: 10.1016/s0304-3940(02)00164-7. [DOI] [PubMed] [Google Scholar]
- 35.Strott CA, Higashi Y. Cholesterol Sulfate in Human Physiology What's It All About? J. Lipid Res. 2003;44:1268–1278. doi: 10.1194/jlr.R300005-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Eid K, Davignon J. Cholesteryl Sulfate; Measurement with Beta-Sitosteryl Sulfate as an Internal Standard. Can. J. Biochem. 1981;59:602–605. doi: 10.1139/o81-083. [DOI] [PubMed] [Google Scholar]
- 37.Serizawa S, Nagai T, Ito M, Sato Y. Simplified Determination of Serum Cholesterol Sulfate by Gas-Liquid Chromatography Combined with Cyclohexylsilane-Bonded Phase Column Purification. Arch. Dermatol. Res. 1989;281:411–416. doi: 10.1007/BF00455327. [DOI] [PubMed] [Google Scholar]
- 38.Cedazo-Minguez A, Winblad B. Biomarkers for Alzheimer’s Disease and Other Forms of Dementia: Clinical Needs, Limitations and Future Aspects. Exp. Gerontol. 2010;45:5–14. doi: 10.1016/j.exger.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Fantini J, Yahi N, Garmy N. Cholesterol Accelerates the Binding of Alzheimer's β-Amyloid Peptide to Ganglioside GM1 through a Universal Hydrogen-Bond-Dependent Sterol Tuning of Glycolipid Conformation. Front. Physiol. 2013;4:120. doi: 10.3389/fphys.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ojha B, Liu H, Dutta S, Rao PP, Wojcikiewicz EP, Du D. Poly (4-Styrenesulfonate) as an Inhibitor of Aβ40 Amyloid Fibril Formation. J. Phys. Chem. B. 2013;117:13975–13984. doi: 10.1021/jp4065467. [DOI] [PubMed] [Google Scholar]
- 41.Liu H, Lantz R, Cosme P, Rivera N, Andino C, Gonzalez WG, Terentis AC, Wojcikiewicz EP, Oyola R, Miksovska J, et al. Site-Specific Dynamics of Amyloid Formation and Fibrillar Configuration of Abeta(1-23) Using an Unnatural Amino Acid. Chem. Commun. 2015;51:7000–7003. doi: 10.1039/c5cc00149h. [DOI] [PubMed] [Google Scholar]
- 42.Pagano RS, Medus ML, Gómez GE, Couto PM, Labanda MS, Landolfo L, D’Alessio C, Caramelo JJ. Protein Fibrillation Lag Times During Kinetic Inhibition. Biophys. J. 2014;107:711–720. doi: 10.1016/j.bpj.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG. Lipid Binding to Amyloid β-Peptide Aggregates: Preferential Binding of Cholesterol as Compared with Phosphatidylcholine and Fatty Acids. J. Neurochem. 1997;69:1746–1752. doi: 10.1046/j.1471-4159.1997.69041746.x. [DOI] [PubMed] [Google Scholar]
- 44.Haberland ME, Reynolds JA. Self-Association of Cholesterol in Aqueous Solution. Proc. Natl. Acad. Sci. U. S. A. 1973;70:2313–2316. doi: 10.1073/pnas.70.8.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LeVine H., III Quantification of Beta-Sheet Amyloid Fibril Structures with Thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 46.Teplow DB. Structural and Kinetic Features of Amyloid Beta-Protein Fibrillogenesis. Amyloid. 1998;5:121–142. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 47.Harper JD, Lansbury PT. Models of Amyloid Seeding in Alzheimier's Disease and Scrapie: Mechanistic Truths and Physiological Consequences of the Time-Dependent Solubility of Amyloid Proteins. Annu. Rev. Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 48.Powers ET, Powers DL. Mechanisms of Protein Fibril Formation: Nucleated Polymerization with Competing Off-Pathway Aggregation. Biophys. J. 2008;94:379–391. doi: 10.1529/biophysj.107.117168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du D, Murray AN, Cohen E, Kim HE, Simkovsky R, Dillin A, Kelly JW. A Kinetic Aggregation Assay Allowing Selective and Sensitive Amyloid-Beta Quantification in Cells and Tissues. Biochemistry. 2011;50:1607–1617. doi: 10.1021/bi1013744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider A, Schulz-Schaeffer W, Hartmann T, Schulz JB, Simons M. Cholesterol Depletion Reduces Aggregation of Amyloid-Beta Peptide in Hippocampal Neurons. Neurobiol. Dis. 2006;23:573–577. doi: 10.1016/j.nbd.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 51.Beel AJ, Sakakura M, Barrett PJ, Sanders CR. Direct Binding of Cholesterol to the Amyloid Precursor Protein: An Important Interaction in Lipid–Alzheimer's Disease Relationships? BBA-Mol. Cell Biol. L. 2010;1801:975–982. doi: 10.1016/j.bbalip.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu L, Lewis A, Como J, Vaughn MW, Huang J, Somerharju P, Virtanen J, Cheng KH. Cholesterol Modulates the Interaction of β-Amyloid Peptide with Lipid Bilayers. Biophys. J. 2009;96:4299–4307. doi: 10.1016/j.bpj.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of Alzheimer’s β-Amyloid Peptides with Cholesterol: Mechanistic Insights into Amyloid Pore Formation. Biochemistry. 2014;53:4489–4502. doi: 10.1021/bi500373k. [DOI] [PubMed] [Google Scholar]
- 54.Fantini J, Di Scala C, Yahi N, Troadec J-D, Sadelli K, Chahinian H, Garmy N. Bexarotene Blocks Calcium-Permeable Ion Channels Formed by Neurotoxic Alzheimer’s β-Amyloid Peptides. ACS Chem. Neurosci. 2014;5:216–224. doi: 10.1021/cn400183w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hellstrand E, Sparr E, Linse S. Retardation of Aβ Fibril Formation by Phospholipid Vesicles Depends on Membrane Phase Behavior. Biophys. J. 2010;98:2206–2214. doi: 10.1016/j.bpj.2010.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linse S, Cabaleiro-Lago C, Xue WF, Lynch I, Lindman S, Thulin E, Radford SE, Dawson KA. Nucleation of Protein Fibrillation by Nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2007;104:8691–8696. doi: 10.1073/pnas.0701250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-Beta Forms Fibrils by Nucleated Conformational Conversion of Oligomers. Nat. Chem. Biol. 2011;7:602–609. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hane F, Drolle E, Gaikwad R, Faught E, Leonenko Z. Amyloid-Beta Aggregation on Model Lipid Membranes: An Atomic Force Microscopy Study. J. Alzheimers Dis. 2011;26:485–494. doi: 10.3233/JAD-2011-102112. [DOI] [PubMed] [Google Scholar]
- 59.Matsuzaki K. Physicochemical Interactions of Amyloid Beta-Peptide with Lipid Bilayers. Biochim. Biophys. Acta. 2007;1768:1935–1942. doi: 10.1016/j.bbamem.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 60.Kremer JJ, Murphy RM. Kinetics of Adsorption of β-Amyloid Peptide Aβ (1–40) to Lipid Bilayers. J. Biochem. Bioph. Methods. 2003;57:159–169. doi: 10.1016/s0165-022x(03)00103-9. [DOI] [PubMed] [Google Scholar]
- 61.Terakawa MS, Yagi H, Adachi M, Lee Y-H, Goto Y. Small Liposomes Accelerate the Fibrillation of Amyloid β (1–40) J. Biol. Chem. 2015;290:815–826. doi: 10.1074/jbc.M114.592527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith PE, Brender JR, Ramamoorthy A. Induction of Negative Curvature as a Mechanism of Cell Toxicity by Amyloidogenic Peptides: The Case of Islet Amyloid Polypeptide. J. Am. Chem. Soc. 2009;131:4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang Z, de Messieres M, Lee JC. Membrane Remodeling by α-Synuclein and Effects on Amyloid Formation. J. Am. Chem. Soc. 2013;135:15970–15973. doi: 10.1021/ja405993r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dierksen K, Typke D, Hegerl R, Walz J, Sackmann E, Baumeister W. Three-Dimensional Structure of Lipid Vesicles Embedded in Vitreous Ice and Investigated by Automated Electron Tomography. Biophys. J. 1995;68:1416–1422. doi: 10.1016/S0006-3495(95)80314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castanho M, Brown W, Prieto M. Rod-Like Cholesterol Micelles in Aqueous Solution Studied Using Polarized and Depolarized Dynamic Light Scattering. Biophys. J. 1992;63:1455–1461. doi: 10.1016/S0006-3495(92)81733-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.