

ABSTRACT
Macrophages are mononuclear phagocytes that constitute a first line of defense against pathogens. While lethal to many microbes, they are the primary host cells of Leishmania spp. parasites, the obligate intracellular pathogens that cause leishmaniasis. We conducted transcriptomic profiling of two Leishmania species and the human macrophage over the course of intracellular infection by using high-throughput RNA sequencing to characterize the global gene expression changes and reprogramming events that underlie the interactions between the pathogen and its host. A systematic exclusion of the generic effects of large-particle phagocytosis revealed a vigorous, parasite-specific response of the human macrophage early in the infection that was greatly tempered at later time points. An analogous temporal expression pattern was observed with the parasite, suggesting that much of the reprogramming that occurs as parasites transform into intracellular forms generally stabilizes shortly after entry. Following that, the parasite establishes an intracellular niche within macrophages, with minimal communication between the parasite and the host cell later during the infection. No significant difference was observed between parasite species transcriptomes or in the transcriptional response of macrophages infected with each species. Our comparative analysis of gene expression changes that occur as mouse and human macrophages are infected by Leishmania spp. points toward a general signature of the Leishmania-macrophage infectome.
IMPORTANCE
Little is known about the transcriptional changes that occur within mammalian cells harboring intracellular pathogens. This study characterizes the gene expression signatures of Leishmania spp. parasites and the coordinated response of infected human macrophages as the pathogen enters and persists within them. After accounting for the generic effects of large-particle phagocytosis, we observed a parasite-specific response of the human macrophages early in infection that was reduced at later time points. A similar expression pattern was observed in the parasites. Our analyses provide specific insights into the interplay between human macrophages and Leishmania parasites and constitute an important general resource for the study of how pathogens evade host defenses and modulate the functions of the cell to survive intracellularly.
INTRODUCTION
Leishmaniasis is a vector-borne disease caused by a digenetic protozoan parasite from the Leishmania genus. The yearly incidence is currently estimated to be about 1.8 million cases in regions where leishmaniasasis is endemic (1). The parasites replicate as extracellular promastigotes inside the midgut of their sand fly vector and differentiate into infective, nondividing, metacyclic promastigotes, which are regurgitated when the sand fly takes a blood meal (2). Once inside the mammalian host, metacyclic promastigotes are taken up by professional phagocytes and subsequently differentiate and replicate intracellularly as amastigotes within the phagolysosomes (3).
A wide range of clinical outcomes result from infection with Leishmania spp., with some species causing cutaneous, mucocutaneous, or diffuse cutaneous leishmaniasis, where symptoms remain localized to the skin or mucosal surfaces. Other Leishmania species cause visceral leishmaniasis after migration to internal organs, such as the liver, spleen, and bone marrow. The propensities for rapid self-cure, dissemination, persistence, latency, and reactivation are influenced by factors related to the species of the parasite and also to the host’s acquired and innate immunities (4, 5). Despite the striking variability in pathogenicity and tissue tropism of different Leishmania species, their genomes are remarkably similar and display a high degree of conservation in gene content and synteny (6–9).
Macrophages are widely regarded as primary host cells of Leishmania parasites, although recent studies have demonstrated that neutrophils (10, 11) and dendritic cells (12–14) can also be infected. Nonetheless, the most documented evidence for parasite replication and long-term survival lies within the mononuclear phagocytes, which are a hostile environment that is lethal to other microbes (15).
A limited number of studies have been performed to determine transcriptional changes that occur within macrophages harboring different pathogens, including Mycobacterium tuberculosis, Mycobacterium bovis, Mycobacterium avium, Yersinia enterocolitica, Escherichia coli, Legionella pneumophila, and Leptospira interrogans (16–24). An inflammatory response occurs with all of these pathogens; however, the availability of only partial lists of differentially expressed (DE) genes hinders comparisons of global profiles and the identification of specific responses to the pathogens.
Early studies reported that Leishmania-infected macrophages were impaired in their response to gamma interferon (IFN-γ), and those studies attributed chronic infections to macrophage deactivation (25–30). Overall results from recent transcriptome analyses of human or murine macrophages infected by different species of Leishmania and generated mostly using microarray platforms have indicated that infected cells downmodulate the expression of many proinflammatory genes and upregulate the expression of several genes implicated in anti-inflammatory responses (31–38). Whether a general suppression of gene expression in infected human macrophages occurs is yet to be elucidated, and the current reports remain contradictory (31–33, 37–40). Since data collected to date represent a miscellany of experiments carried out in different macrophage types, host backgrounds, and parasite species, these data have been collected at different postinfection time points, and because the interpretations of most results have been focused on a limited set of markers, it is not possible to integrate these findings to comprehensively describe the state of Leishmania-infected macrophages.
The characterization of the global gene expression profiles and the transcriptomic reprogramming events in an intracellular pathogen and its host cell is a fundamental step toward better comprehending the complexity of the host-pathogen interplay. High-throughput RNA sequencing (RNA-seq) has been used as a novel tool for the global identification of changes in RNA levels in infected cells, yet few studies have applied the technology to interrogate changes in the transcriptomes of the two biological systems simultaneously to identify host-pathogen interactions within the same biological samples (41–45).
Here, we used RNA-seq to simultaneously and comprehensively interrogate the transcriptome profiles of both human macrophages and Leishmania parasites in the context of a dual biological system. We utilized a well-defined time course to measure the effects over time, collected multiple biological replicates, employed careful statistical analyses to account for batch effects, and discerned pathogen-specific responses from large-particle phagocytosis effects. Finally, we compared the profiles of infection by two Leishmania species that cause cutaneous leishmaniasis with potentially different clinical manifestations: L. major (cutaneous self-healing leishmaniasis) and L. amazonensis (cutaneous self-healing/cutaneous diffuse leishmaniasis).
RESULTS AND DISCUSSION
Study design.
In order to capture the global transcriptional response during the initiation and maintenance of intramacrophage infection by 2 different species of Leishmania, the transcriptomes of the parasite and the infected human macrophage were simultaneously profiled using RNA-seq at 4, 24, 48, and 72 h postinfection (hpi). CD14+ monocytes were obtained from 4 donors and allowed to differentiate into macrophages after an 8-day incubation period in macrophage colony-stimulating factor (M-CSF) (Fig. 1). In accordance with the recently proposed standards for macrophage activation nomenclature (46), we consider these cells to be M-CSF monocyte-derived macrophages, and we refer to them simply as macrophages throughout the manuscript. These macrophages were neither polarized nor activated with additional cytokine stimulation and were chosen due to their potential for a broad range of responses.
FIG 1 .

Simultaneous interrogation of the parasite and host transcriptomes. The figure illustrates the study design, donors, and time points. Monocyte-derived human macrophages were infected with the metacyclic promastigote form of L. major (dark blue) or L. amazonensis (light blue) parasites. Following phagocytosis, metacyclic promastigotes transform into the amastigote form (3 to 10 hpi) and reside in membrane-bound compartments called phagosomes. As amastigotes divide, starting at ~24 hpi, each phagosome matures into a membrane-bound singular (L. major) or communal (L. amazonensis) vacuole inside a macrophage. Samples were collected from infected macrophages, macrophages that were allowed to phagocytose latex beads (red), and uninfected controls over 4 time points spanning from 4 to 72 hpi, as pictured. Total RNA was isolated from each sample and analyzed by RNA-seq. A total of 4 biological replicates were collected from 4 different human donors (H2 to H5; represented in different shades of green). A second collection from two of the donors was used as a technical replicate within the L. major/uninfected data set (H2′ and H3′) and for an L. amazonensis and latex bead experiment that constituted a later addition to the study design. The experimental design is further detailed in Data Set S1 in the supplemental material.
Macrophages from each donor were infected with purified metacyclic promastigotes from Leishmania major or Leishmania amazonensis in the presence of human serum. In parallel, macrophages were allowed to phagocytose latex beads to evaluate the effect of inert particle phagocytosis on global gene expression (here referred to as the phagocytosis effect). Uninfected macrophages were used as controls. The dynamics of the infection were monitored at each time point by determining the number of parasites per 100 macrophages and the percentage of infected cells (Fig. 2; see also Data Set S1 in the supplemental material). The two Leishmania species infected macrophages at similar percentages (~90% for L. major and ~80% for L. amazonensis) (Fig. 2). The overall parasite loads (number of parasites per infected cell) were slightly higher for L. major-infected macrophages (ranging from 6.3 to 18) than for L. amazonensis-infected cells (6.1 to 8). The intracellular growth rate of each species was evaluated by determining the number of parasites per 100 cells. L. major showed a significant increase in growth between 4 hpi and the remaining time points, and L. amazonensis growth remained somewhat constant.
FIG 2 .
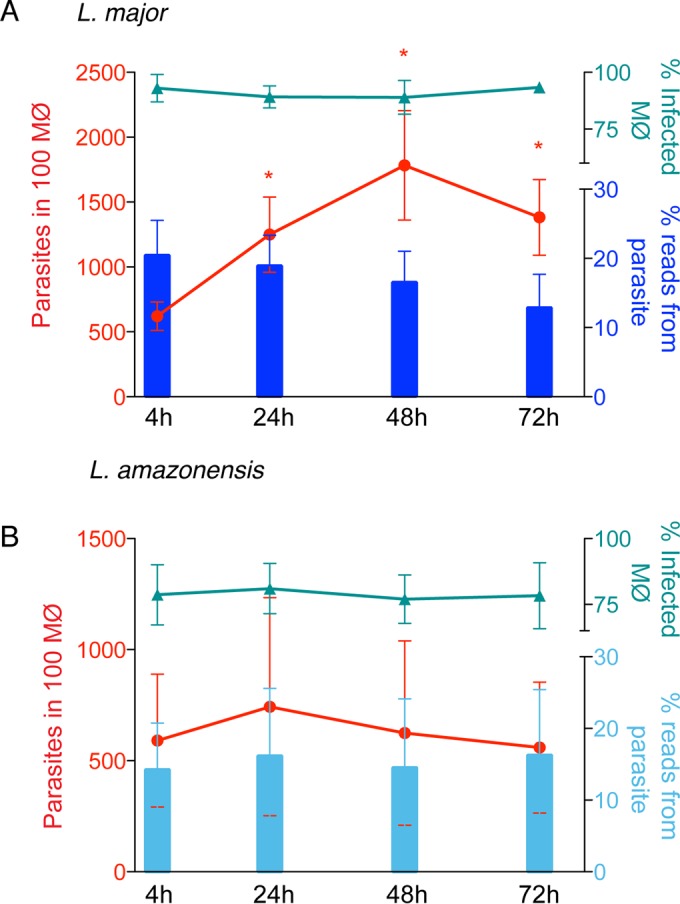
Dynamics and characteristics of M-CSF-induced human macrophage (MΦ) infection with Leishmania parasites. M-CSF-induced human macrophages were infected with either L. major (A) or L. amazonensis (B) for 4 h, washed, and further incubated until 24 hpi, 48 hpi, or 72 hpi. Samples collected at 4, 24, 48, and 72 hpi were subjected to transcriptional profiling by RNA-seq. The number of internalized parasites and percentage of infected cells were determined microscopically. Bars indicate the percentages of trimmed RNA-seq reads that mapped to the respective parasite reference genome. Red lines indicate the number of parasites per 100 macrophages, and cyan lines indicate the percent infected macrophages. The graphs incorporate data from all experiments, ± standard deviations. *, P < 0.05 (Student’s t test to compare each time point to 4-hpi results).
Global transcriptome profiles of Leishmania and its human macrophage host cell.
The global transcriptome profiles of the parasites and host cells were characterized using RNA-seq. Poly(A)-enriched cDNA libraries were generated for each macrophage sample (uninfected, infected with L. major or L. amazonensis, or those that ingested latex beads) at all time points as well as promastigotes from both species that were used for the infection. Paired-end sequence reads of 100 nucleotides were generated for a total of 74 samples, representing an average of 4 independent biological replicates for each condition and yielding a total of 6.3 billion high-quality reads (see Data Set S1 in the supplemental material for details). Each sample from infected cells consisted of a pool of mixed RNAs from the Leishmania species and human macrophages. To resolve these mixed RNAs, the RNA-seq reads generated from these samples were mapped against the corresponding reference genomes. The fraction of reads mapping to the parasite versus human reference genomes yielded an estimate of the proportion of RNA molecules from each source. While the proportion of L. major reads in parasite-infected macrophages did not vary significantly across time points, the number of intracellular parasites increased over the course of infection (Fig. 2A). This may reflect an increase in global transcriptional activity in the human macrophages or a similar decrease in the parasites. The proportion of parasite-attributable reads also remained constant during L. amazonensis infection, consistent with the attenuated growth rate for the species.
To investigate general trends in the data, principal component analyses (PCA) were carried out both prior to and after accounting for batch effects (Fig. 3; see also Fig. S1 and S2 in the supplemental material). A high degree of similarity between biological replicates was evident in the PCA plots for both of the Leishmania species and for macrophages; similar samples clustered together by infection status per time point. We noted that the spread of samples along the first principal component (PC1; x axis) seemed to reflect the global transcriptional changes in the cells over time (Fig. 3A). Uninfected control macrophages showed notable variance, denoting a drift in the transcriptome as the cells were maintained in culture, thus validating the use of a matched control for each time point. Interestingly, cells that had ingested latex beads clustered tightly with uninfected cells at the 4-h time point, revealing that macrophages are equipped with the cellular components necessary for phagocytosis and experience no marked perturbation in their steady-state transcriptome upon ingestion of particles. Thus, phagocytosis per se is not a trigger for changes in gene expression in macrophages.
FIG 3 .

Global expression profiles of human macrophages and Leishmania parasites during infection. A comprehensive PCA was conducted to evaluate the relationships between samples across time points and to visualize sample-sample distances for human macrophages (uninfected controls, Leishmania-infected cells, and bead-containing cells) (A) and the intracellular forms of Leishmania (B and C). Each sphere represents an experimental sample, with increasing size indicating the progression of time points from 4 to 72 hpi. Sphere colors indicate sample type (shown in color key). In panel A, the clouds highlight the separation of the Leishmania-infected cluster (blue) from the uninfected/bead-containing cluster (orange). All analyses were performed after filtering out nonexpressed or lowly expressed genes, performing quantile normalization, and including experimental batch as a covariate in the statistical model. PCA plots showing the effects of batch adjustment in macrophage and parasite samples are shown in Fig. S1 and Fig. S2 in the supplemental material.
These findings with uninfected macrophages or those that had ingested latex beads contrast with those for macrophages infected with either L. major or L. amazonensis; the infected macrophages clustered together and distinctly away from other samples, suggesting an apparently unique Leishmania-specific human macrophage response early in the infection (4 hpi). As the infection progressed, the separation between Leishmania-infected and bead-containing macrophages became less prominent, and by 24 hpi, macrophages with persisting parasites resembled those harboring inert particles. In addition, both populations displayed a clear trend toward uninfected macrophages at the later time points (48 and 72 hpi). The global transcriptional patterns for intracellular Leishmania parasites of both species were similar among 4-hpi samples, as they were partitioned away from those observed at later time points (Fig. 3B and C). This was consistent with the pronounced and distinct host response to the parasite, which also displayed a clear separation between responses at early and later times postinfection.
Discriminating between parasite- and phagocytosis-driven changes in human macrophages.
We examined the differential expression of individual human macrophage genes following Leishmania infection. In macrophages infected with L. major, the largest host response was observed at 4 hpi, with 5,713 DE genes between uninfected and infected macrophages at an adjusted P value cutoff of <0.05. Later time points showed decreasing numbers of DE genes: 4,846, 4,188, and 2,142 genes at 24, 48, and 72 hpi, respectively (Fig. 4A, top). A similar trend was observed in the L. amazonensis infection group (Fig. 4B, top), with smaller numbers of DE genes detected, which was reflected in a slightly reduced statistical power associated with the fewer biological replicates. Most of the genes that were DE during L. amazonensis infection were also contained within the L. major-infected macrophage data sets (see Data Set S2 in the supplemental material and the comparative DE analyses below). Among the DE genes in macrophages infected with either parasite species, the average proportions of up- and downregulated genes were ~40% and 60% at 4 hpi, respectively, and ~50% and 50% at the remaining time points. While this points to a slightly higher fraction of downregulated genes, we did not observe the general suppression of gene expression in murine macrophages reported in earlier studies (33, 39). Rather, our results are consistent with studies that have reported similar numbers of genes up- and downregulated upon Leishmania infection (31, 32, 37, 38, 40) and challenge the notion that phagocytosis of Leishmania by macrophages induces an overall state of dormancy after uptake.
FIG 4 .

Differentially expressed genes in macrophages infected with Leishmania species and between parasite developmental stages. (A and B) The numbers of DE genes in L. major-infected macrophages (A) and L. amazonensis-infected macrophages (B) relative to uninfected controls both before and after accounting for the phagocytosis effect (top panels and bottom panels, respectively), depicted as horizontal bar plots for 4, 24, 48, and 72 hpi. (C and D) The numbers of parasite genes that were DE between stages/time points were also determined for macrophages infected with L. major (C) or L. amazonensis (D). The box width depicts the number of DE genes downregulated (left; purple) and upregulated (right; blue) at an adjusted P value of <0.05, with the total number of down- and upregulated genes shown. The color shading indicates the proportion of genes with >4-fold differential expression (dark), between 2- and 4-fold differential expression (medium), or 2-fold differential expression (light). The complete lists of DE genes are provided in Data Sets S2 and S5 in the supplemental material.
In order to evaluate the extent of the phagocytosis effect, differential expression analysis was carried out to compare uninfected macrophages to bead-containing macrophages at each time point. This analysis revealed no DE genes at 4 hpi, consistent with the apparent lack of transcriptional response observed at 4 hpi following phagocytosis of beads (Fig. 3A). A well-pronounced phagocytosis effect was observed at later time points, with 3,787, 6,045, and 2,659 DE genes at 24, 48, and 72 hpi, respectively (see Data Set S2 in the supplemental material). In a previous study of the response of murine bone marrow-derived macrophages to L. major and L. donovani, assessed using microarrays, Gregory et al. (33) found that macrophages that had ingested beads were nearly identical to control macrophages at the one time point used in the study (24 hpi), and therefore they did not use bead-containing samples to further account for the effects of phagocytosis. This finding more closely matches our results at 4 hpi and may indicate a time offset in their murine system relative to our human system. Additionally, the observed differences may be reflective of differences in macrophage type and/or incubation conditions.
Subsequent analyses were aimed at distinguishing between changes that constitute a specific response to infection and those attributable to the macrophage response to phagocytosis. Pairwise analyses were conducted at each time point (4, 24, 48, and 72 hpi) for macrophages infected with either L. major or L. amazonensis, evaluated against both matched uninfected macrophages and macrophages that had ingested latex beads. Using a novel approach based on a dual statistical test to identify genes that were differentially regulated not only in infected macrophages relative to uninfected cells but also relative to macrophages that had ingested inert particles, we were able to filter out genes that were differentially regulated due to the phagocytosis effect; thus, we could select genes that were specific to the response of the macrophages to Leishmania infection (see Data Set S2 in the supplemental material). This resulted in significantly reduced numbers of DE genes for each time point (Fig. 4A and B, bottom panels). Remarkably, the parasite-specific response was most pronounced in macrophages at 4 hpi (3,273 and 2,161 DE genes in L. major- and L. amazonensis-infected macrophages, respectively) and greatly attenuated at later time points due to the fact that most of the response was attributable to phagocytosis. These data are consistent with the idea that Leishmania spp. establish a unique niche within macrophages shortly after phagocytosis, with minimal communication between the parasite and the host cell later during infection.
Leishmania-induced remodeling of gene expression in human macrophages.
In examining the DE genes after accounting for the phagocytosis effect, we detected previously identified elements of the macrophage response to Leishmania at 4 hpi. Most recognizable was the upregulation of genes encoding inflammatory cytokines, including interleukin-1β (IL-1β), tumor necrosis factor (TNF), TNF superfamily members, and IL-6, as well as a number of immunomodulators, such as prostaglandin-endoperoxide synthase 2 (PTGS2), colony-stimulating factors 1 and 2 (CSF1 and CSF2), and superoxide dismutase 2 (SOD2 [L. major only]). Some of these gene products have been previously implicated during infection by Leishmania major (31) or other species of Leishmania in both human and murine macrophage systems (32, 37, 38, 40).
Also among the top upregulated genes (up to 136-fold increase with L. major infection and 196-fold increase with L. amazonensis infection) at 4 hpi were multiple metallothionein 1 family members (G, M, H, E, and A). Metallothioneins are known to have an immunomodulatory role (47) and to be induced by a wide range of conditions, including exposure to reactive oxygen species (48), and were recently shown to affect the host response to Listeria spp. infection (49). Although metallothioneins have previously been shown to be highly upregulated in macrophages infected with Leishmania (31, 32), their potential role in the establishment of infection is poorly understood.
L. major and L. amazonensis show different behavioral characteristics, most notably varied clinical outcomes and distinct organization of intracellular amastigotes within macrophages (one vacuole per parasite for L. major, and communal spacious vacuoles that house multiple parasites for L. amazonensis) (3, 50). We explored whether the two species elicited significantly different responses by macrophages across various time points postinfection by using a direct statistical comparison of the gene-level macrophage response to each parasite. This revealed that they did not trigger a significantly different response in human macrophages, with only 4 genes surfacing as differentially expressed at 4 hpi and none at any of the subsequent time points (see Data Set S3 in the supplemental material). Thus, despite differences in clinical presentations and host immune responses to the two parasite species, macrophages do not appear to discriminate between L. major and L. amazonensis.
Two of the genes that were DE between L. major-infected and L. amazonensis-infected macrophages encode synaptotagmin family members 2 and 8 (SYT2 and SYT8). Synaptotagmins are membrane proteins that regulate vesicle docking and fusion in processes such as exocytosis (51, 52) and phagocytosis (51, 53, 54). While some synaptotagmin family members (SYT5 and SYT11) have been implicated in Leishmania infection (55–57), the involvement of SYT2 and SYT8 has not yet been investigated. Given the general role of synaptotagmins as regulators of membrane trafficking and fusion, it is possible that the higher levels of SYT2 and SYT8 observed during L. major infection compared to L. amazonensis infection may be linked to differences in parasitophorous vacuole maintenance throughout the infection—L. major divides in membrane-bound compartments, with each parasite division maintaining singular parasites in a vacuole; conversely, L. amazonensis may possibly require fewer fission events to maintain its communal vacuoles. Additionally, synaptotagmins are also known to play a role in SNARE activity regulation by mediating membrane fusion in a Ca2+-dependent manner (58–60). Since Leishmania have been shown to target SNARES (VAMP8 in particular) in order to modulate antigen cross-presentation (61), it is possible that transcriptional upregulation of synaptotagmins upon infection by Leishmania may be related to their role in the regulation of SNAREs. CMIP, which encodes c-Maf-inducing protein, and GABRE, which encodes the gamma-aminobutyric acid (GABA) A receptor epsilon, were also expressed at higher levels in L. major-infected macrophages, though the mechanism by which these genes may differentially interact with or be influenced by each parasite species is unclear.
Toward defining the signature of mammalian macrophage infection by Leishmania.
An important aim of this study was to characterize the human macrophage response to infection by Leishmania parasites, with the fundamental goal of defining shared features regarding how intracellular pathogens modulate their host environments. We integrated our findings with data we collected from the same time points in a previous study in L. major-infected murine macrophages (40) as an initial step toward uncovering a common signature in mammalian macrophages infected with Leishmania. Since the differential expression data set collected from murine macrophages did not account for the phagocytosis effect, we sought to restrict this analysis to orthologous genes that were DE in both human and murine macrophages as the result of infection and not phagocytosis. Thus, we identified the mouse orthologs of the human genes that constituted the L. major-specific response and then compared the expression profiles for these genes to corresponding DE gene profiles generated for murine macrophages from the earlier report of Dillon et al. (40). We identified mouse orthologs for 3,017 of the 3,273 genes that were DE in L. major-infected human macrophages at 4 hpi, after accounting for the phagocytosis effect. The relationship between genes in each host system was best visualized using a scatterplot that showed the magnitude and direction of differential expression for each gene and its ortholog (Fig. 5). Of the 1,735 genes that were DE in both host systems (see Data Set S4 in the supplemental material), 1,340 were differentially expressed in the same direction in both systems and thus constitute a unique signature of L. major infection of mammalian macrophages. Those genes localize in quadrants I (567 genes upregulated in both systems) and III (773 genes downregulated in both systems) and contain the most highly differentially expressed genes.
FIG 5 .
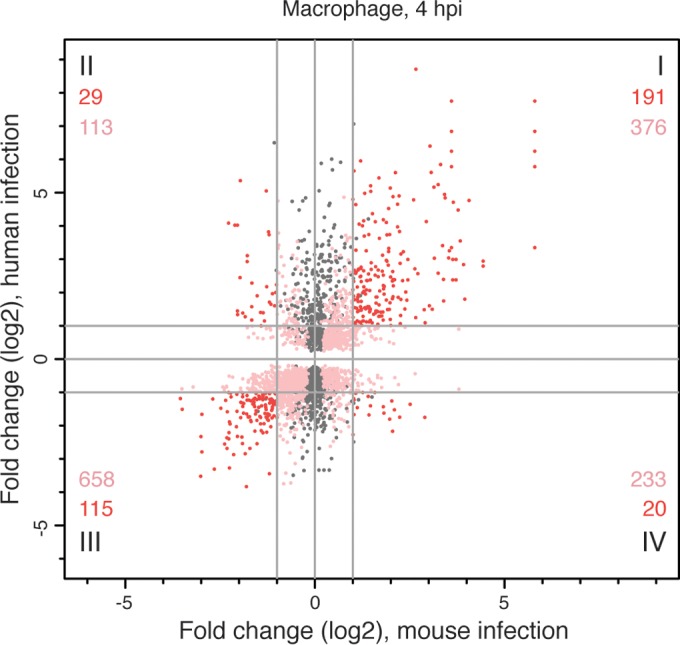
Responses of human and murine macrophages to L. major infection at 4 hpi after accounting for the phagocytosis effect. The differential expression profile of L. major-infected human macrophages was compared to that of L. major-infected murine macrophages collected at the same time point (40). Orthology mapping to mouse was done for the 3,273 human genes identified as differentially expressed at 4 hpi, after accounting for the phagocytosis effect, and the results were compared to the murine expression data set. A scatterplot showing the relationship between fold changes (log2 transformed) in mice (x axis) and humans (y axis) is shown, with each human-mouse gene pair represented by a single point. Some genes duplicates were introduced by the orthology mapping process. Points in gray represent genes in the human data set with an ortholog that was not significantly DE in the murine data set. Points in shades of red represent genes that were significantly DE in both data sets, with those showing <2-fold difference (log2 fold change, <1) in either/both host system(s) shown in pink and those with >2-fold difference (log2 fold change, >1) in both host systems shown in red. The numbers of unique genes represented by the red and pink points are indicated for each quadrant. The complete list of genes that were DE in both systems is provided in Data Set S4 in the supplemental material.
In order to identify known cellular processes within this signature, we used a KEGG pathway enrichment analysis of genes commonly up- or downregulated in both host systems (Table 1; see also Data Set S4 in the supplemental material). Many of the KEGG pathways that were most enriched in the upregulated set of genes are related to immune activation and signaling. Signaling pathways related to the recognition of pathogen-associated molecular patterns (PAMPs; e.g., NOD-like receptor, RIG-I-like receptor, and Toll-like receptor signaling) were implicated, as were many other immune system signaling pathways (cytokine-cytokine receptor interaction, Fc epsilon RI, T cell receptor, Jak-STAT, mitogen-activated protein kinase [MAPK], NF-κB, TNF, vascular endothelial growth factor [VEGF], hypoxia-inducible factor 1 [HIF-1], ErbB, FoxO, phosphatidylinositol 3-kinase–Akt [PI3K-Akt], and transforming growth factor beta [TGF-beta] signaling). Given the effects of many of these pathways on cell growth and metabolism, it is not surprising that many have been previously implicated in cancer and that a number of cancer-related pathways appear in the enriched KEGG pathway lists. KEGG pathways enriched among genes that were downregulated in both the human and murine systems are associated with energy metabolism (amino acid and glycan degradation), lysosome structure and processes, and apoptosis. Interestingly, the FoxO signaling pathway, which is involved in the regulation of cell growth, gluconeogenesis, and adipogenesis, was enriched among both up- and downregulated genes, with 12 members of the pathway upregulated and 14 others downregulated.
TABLE 1 .
KEGG pathways enriched in DE genes common to human and murine macrophages infected with L. major at 4 hpia
| Direction of regulation and KEGG pathway | No. of DE genes | Pathway size | P value |
|---|---|---|---|
| Upregulated KEGG pathways | |||
| TNF signaling pathway | 24 | 109 | 2.07e−13 |
| NF-κB signaling pathway | 14 | 88 | 1.62e−06 |
| Nod-like receptor signaling pathway | 11 | 57 | 3.07e−06 |
| Epstein-Barr virus infection | 21 | 198 | 4.43e−06 |
| Hematopoietic cell lineage | 12 | 82 | 2.17e−05 |
| Shigellosis | 10 | 61 | 3.88e−05 |
| Chagas disease (American trypanosomiasis) | 13 | 103 | 4.98e−05 |
| Proteoglycans in cancer | 19 | 198 | 5.17e−05 |
| Ribosome biogenesis in eukaryotes | 11 | 81 | 9.64e−05 |
| ErbB signaling pathway | 11 | 85 | 1.50e−04 |
| HIF-1 signaling pathway | 12 | 104 | 2.30e−04 |
| Toll-like receptor signaling pathway | 12 | 105 | 2.52e−04 |
| Osteoclast differentiation | 13 | 121 | 2.61e−04 |
| Legionellosis | 8 | 54 | 4.75e−04 |
| Cytokine-cytokine receptor interaction | 19 | 248 | 9.33e−04 |
| Pertussis | 9 | 75 | 1.04e−03 |
| VEGF signaling pathway | 8 | 61 | 1.09e−03 |
| PI3K-Akt signaling pathway | 23 | 335 | 1.27e−03 |
| Herpes simplex infection | 15 | 179 | 1.30e−03 |
| FoxO signaling pathway | 12 | 127 | 1.41e−03 |
| Mineral absorption | 7 | 50 | 1.51e−03 |
| Hepatitis C | 12 | 129 | 1.61e−03 |
| Influenza A virus | 14 | 169 | 2.08e−03 |
| Small cell lung cancer | 9 | 84 | 2.32e−03 |
| MAPK signaling pathway | 18 | 249 | 2.41e−03 |
| Rheumatoid arthritis | 9 | 87 | 2.95e−03 |
| Hepatitis B | 12 | 143 | 3.82e−03 |
| Jak-STAT signaling pathway | 12 | 147 | 4.77e−03 |
| Measles | 11 | 130 | 5.17e−03 |
| TGF-beta signaling pathway | 8 | 79 | 5.66e−03 |
| Pathways in cancer | 23 | 381 | 6.36e−03 |
| HTLV-1 infection | 17 | 254 | 6.79e−03 |
| Central carbon metabolism in cancer | 7 | 66 | 7.35e−03 |
| Prion diseases | 5 | 36 | 7.36e−03 |
| Tight junction | 11 | 137 | 7.62e−03 |
| T cell receptor signaling pathway | 9 | 101 | 7.91e−03 |
| Fc epsilon RI signaling pathway | 7 | 68 | 8.63e−03 |
| Salmonella infection | 8 | 85 | 8.74e−03 |
| RIG-I-like receptor signaling pathway | 7 | 69 | 9.33e−03 |
| Downregulated KEGG pathways | |||
| Valine, leucine, and isoleucine degradation | 10 | 46 | 2.52e−05 |
| Lysine degradation | 9 | 51 | 3.46e−04 |
| Progesterone-mediated oocyte maturation | 12 | 89 | 5.19e−04 |
| Other glycan degradation | 5 | 18 | 8.77e−04 |
| Osteoclast differentiation | 14 | 121 | 8.88e−04 |
| FoxO signaling pathway | 14 | 127 | 1.43e−03 |
| Lysosome | 13 | 120 | 2.44e−03 |
| Glycosphingolipid biosynthesis (globo series) | 4 | 14 | 2.65e−03 |
| Fatty acid elongation | 5 | 23 | 2.87e−03 |
| Apoptosis | 10 | 81 | 2.91e−03 |
| Chronic myeloid leukemia | 9 | 72 | 4.27e−03 |
| Platelet activation | 13 | 128 | 4.29e−03 |
| Chagas disease (American trypanosomiasis) | 11 | 103 | 5.70e−03 |
| Fc gamma R-mediated phagocytosis | 10 | 90 | 6.24e−03 |
| Propanoate metabolism | 5 | 28 | 6.99e−03 |
| Glycosaminoglycan degradation | 4 | 18 | 7.04e−03 |
| Estrogen signaling pathway | 10 | 95 | 9.07e−03 |
KEGG pathway analysis was carried out to identify signaling and metabolic pathways overrepresented in genes that constitute the mammalian response to L. major infection at 4 hpi. Genes that were commonly regulated in both the human and mouse systems were used as input, with up- and downregulated genes considered separately. A P value cutoff of 0.01 was used to define enriched pathways. For each enriched KEGG pathway, the number of DE genes assigned to that pathway, the total number of genes in the pathway, and the P value for the enrichment are reported. The differentially expressed genes corresponding to each enriched KEGG pathway are provided in Data Set S4 in the supplemental material.
It is important to note that there is a significant amount of redundancy within the gene candidates that drive the enrichment of known pathways. An analysis of the overlap between the DE genes contained in different pathways revealed that the KEGG enrichment results can be solely attributed to 142 upregulated and 85 downregulated genes. This is explained by the nature of KEGG pathways (and similar databases), which reflect finite groupings that largely represent areas of research emphasis. Genes driving the KEGG results among the 39 upregulated pathways include TNF (22 pathways), PIK3CB (22 pathways), IL-6 (21 pathways), MAPK8 (20 pathways), NFKBIA (19 pathways), MAPK11 (18 pathways), IL-1B (17 pathways), MAP2K1 (17 pathways), MYC (12 pathways), IL-1A (10 pathways), TRAF2 (10 pathways), CDKN1A (10 pathways), NFKB1B (8 pathways), TlCAM1 (8 pathways), and VEGFA (7 pathways), while those driving results among the 17 downregulated pathways include AKT1 (9 pathways), PIK3CG (9 pathways), PIK3R2 (9 pathways), MAPK14 (5 pathways), GNAI1 (4 pathways), HADHA (4 pathways), HEXA (4 pathways), HEXB (4 pathways), PRKACA (4 pathways), PRKACB (4 pathways), TGFBR1 (4 pathways), and TFGBR2 (4 pathways).
Disease-specific KEGG pathways appeared prominently in the enrichment analysis lists (including those for Chagas disease, Epstein-Barr virus infection, hepatitis, herpes simplex infection, human T cell leukemia virus 1 [HTLV-1] infection, influenza, legionellosis, measles, pertussis, salmonella infection, and shigellosis). These enrichments were largely driven by redundant gene entries within those pathways, as described above, and KEGG pathways for individual diseases appeared to represent a comprehensive assemblage of observations from multiple studies of individual genes (rather than a consensus) from different experiments, hosts, cell types, and time points.
To further extend the interpretation of our L. major and L. amazonensis infection data sets, we attempted to identify some of the common elements in the human macrophage response to infection by comparing the data sets reported here and those of previous reports on the infection of human macrophages infected with Leptospira interrogans (24), Legionella pneumophila (22), and Mycobacterium avium (17). Although we were unable to conduct a comprehensive comparison, since expression values were reported only for subsets of genes in the published data sets, we were able to make some useful observations. A number of cytokines, chemokines, and inflammatory mediators upregulated in the L. major and L. amazonensis infections were also upregulated in these other systems and therefore constitute a shared response to infection. Specifically, IL-6 and TNF were upregulated in infections by the other 3 pathogens, IL-1B was also upregulated in M. avium and L. interrogans infections, TNFSF9 was also upregulated in M. avium and L. pneumophila infections, CCL20 and CXCL3 were also upregulated in L. pneumophila and L. interrogans infections, CXCL1 and CXCL5 were also upregulated during L. interrogans infection, and PTGS2 was also upregulated during M. avium infection. All of these genes except CCL20 and CXCL5 were also upregulated in the L. major-infected murine macrophage data set (40) examined above. Most of these proteins are proinflammatory in nature (IL-6 has both pro- and anti-inflammatory properties) and play a role in the recruitment and stimulation of various immune cells. The involvement of this broad repertoire of gene products reflects the complex and at times opposing immune responses that occur as intracellular pathogens attempt to establish an infection and survive intracellularly.
Identification of differentially expressed genes in Leishmania parasites.
In addition to allowing the characterization of the gene expression patterns of human macrophages as they responded to infection by Leishmania parasites, this study provided an opportunity to simultaneously elucidate changes in the parasites’ gene expression programs as intracellular infections were established and progressed. Differential expression analyses were carried out for parasite genes to determine how Leishmania species modify the expression of their individual genes during the transition from metacyclic promastigotes to intracellular amastigotes and over the course of an intracellular infection. The largest numbers of DE genes were observed as metacyclic promastigotes that had infected human macrophages, with 3,224 L. major genes and 3,896 L. amazonensis genes implicated at an adjusted P value cutoff of <0.05 (Fig. 4C; see also Data Set S5 in the supplemental material). Significantly fewer changes in expression were observed in intracellular parasites across time, with 356 DE genes and 577 DE genes in L. major and L. amazonensis, respectively, during the 4- to 24-hpi transition. Only 3 DE genes were observed for each species during the 24- to 48-hpi transition, and 0 genes were DE during the 48- to 72-hpi transition. This pattern of expression (large numbers of DE genes during the metacyclic-to-early amastigote transition and decreasing numbers of DE genes over time) suggests that the large gene expression reprogramming that occurs as extracellular promastigote parasites transform into intracellular amastigote parasites generally stabilizes by around 24 hpi. This mirrors the changes in host transcriptomes, where large changes were observed during early infection and many fewer changes were observed at later time points, once the phagocytosis effect was considered (see above and Fig. 4A and B, lower panels). The parallel expression patterns between parasite and host suggest that host-pathogen interactions are extensive upon Leishmania entering and establishing infection within the cells but are virtually nonexistent by around 24 h after infection.
In an attempt to directly compare gene expression programs between the two Leishmania species as they transition from the metacyclic form into intracellular forms, we focused our analysis on a set of L. major/L. amazonensis (L. mexicana) orthologous gene clusters that were precomputed in EuPathDB and include ~98% of L. major genes. The DE profiles for each L. major/L. amazonensis gene and its ortholog(s) are available in Data Set S6 in the supplemental material, and a graphical representation of the results is shown in Fig. 6A and B (dots correspond to the differential expression levels for orthologous genes). The orthology mapping between the two species included several one-versus-many relationships because of paralogous gene families, and this is manifested in the scatterplot as rows and columns of perfectly aligned dots representing the amastin and SHERP gene products, among a few others. We observed a set of 1,558 unique gene pairs (737 upregulated and 811 downregulated) that were significantly differentially expressed in the same direction in both species during the metacyclic promastigote-to-intracellular transition at 4 hpi. These represent gene products that underlie a response common to both species as they adapt to the human macrophage environment (Fig. 6A). Two other subsets included transcripts that appeared significantly DE in one species but not in the other. Those included 3,947 unique transcripts in total, with only a small proportion (10%) that were differentially expressed more than 2-fold in one species or the other. A closer inspection revealed that many genes that make up these subsets are members of multigene families in the common expression set (e.g., genes for amastin, gp63, kinesin, flagellar attachment zone protein, AAT family members, dynein, cysteine peptidase B), and/or were DE in the other Leishmania species during the 4- to 24-hpi transition, indicating a time offset in what can be considered a common response (e.g., cathepsin L-like protease, ATG8). A similar phenomenon was apparent during the 4- to 24-hpi intracellular transition (Fig. 6B). While there were only 60 unique gene pairs that were commonly expressed in the same direction, most of the transcripts that appeared to be distinctly DE in one of the species were members of multigene families that were part of the common response, or simply displayed a time-shifted expression pattern.
FIG 6 .

Comparison of Leishmania spp. transcriptomes upon infection of human or murine macrophages. Scatterplots show the relationship between fold changes (log2 transformed) for L. amazonensis (x axis) and L. major (y axis) orthologs for the metacyclic promastigote to 4-hpi intracellular transition (A) and the 4- to 24-hpi intracellular transition (B) within human macrophages. The lists of orthologs and their corresponding fold changes are provided in Data Set S6 in the supplemental material Scatterplots were also used to show a comparison of L. major parasite expression patterns in human macrophages with similar data sets produced using a murine system (40). The relationship between parasite gene fold changes (log2) in the mouse system (x axis) and the human system (y axis) are shown for (C) the metacyclic promastigote to 4-hpi intracellular transition (C) and the 4- to 24-hpi intracellular transition (D). The lists of genes in the comparisons are provided in Data Set S7. Points in gray represent genes that were not significantly DE by L. major in either host system; points shown in shades of purple represent genes that were DE in the same direction in both systems; points shown in shades of green represent genes that were DE in different directions between the systems; points shown in shades of orange or blue represent genes significant in only one system. The numbers of DE genes corresponding to each color are included in parentheses below the graphs. NS, not significantly DE.
The general picture that emerged from the comparison of the two species of Leishmania is that their orthologous gene sets display minimal differences at the transcriptional level when infecting human macrophages. This finding closely aligns with the lack of observed differences in the host response to infection by the two Leishmania species (described above). One gene in particular, which was among those not included in this analysis because they lacked orthologs in the EuPathDB database, was both highly differentially expressed and has supporting evidence of its species specificity. This gene, the virulence factor A2, is present in L. amazonensis but exists only as a pseudogene in L. major (62). It was among the most upregulated genes in L. amazonensis during the metacyclic-to-4-hpi transition and was further upregulated through 24 hpi. A2 is required for visceral infection by L. donovani (63) and has been implicated in the parasite’s resistance to heat shock (64). While L. amazonensis generally causes cutaneous disease, it has also been associated with visceral infections (65). The strong upregulation of the A2 gene during early infection detected here is consistent with that observation.
Toward defining the signatures of Leishmania differentiation and intracellular survival.
In order to characterize the overall signature of L. major differentiation from the metacyclic promastigote developmental stage to intracellular amastigotes and during intracellular infection, we compared the patterns of L. major gene expression identified in human macrophages (this study) to those observed for the same time points in L. major-infected murine macrophages (40). In both host systems, large numbers of DE genes were detected in the parasite during the metacyclic-to-early amastigote transition, and decreasing numbers of DE genes were noted between intracellular time points.
When individual genes were compared between the human and mouse analyses, a composite picture emerged (Fig. 6C and D; see also Data Set S7 in the supplemental material). Among the 4,708 genes that were DE in L. major during the metacyclic promastigote-to-4-hpi intracellular form transition in at least one of the hosts, 28% (1,336) represent a common parasite expression pattern in both human and mouse infections. A gene ontology (GO) enrichment analysis was done to glean insights into the functional overlaps in L. major genes that were differentially expressed as metacyclic promastigote-infected mammalian macrophages over the first 4 h. Two enriched GO categories emerged—peroxiredoxin activity and antioxidant activity—among the 598 L. major genes that were commonly upregulated during both human and mouse infections (see Table S1 in the supplemental material). Tellingly, only 7 genes, peroxidoxin and 6 tryparedoxin peroxidase family members, drove these results (see Data Set S7 in the supplemental material). A greater number and range of GO categories were enriched among the 738 L. major genes downregulated in both host systems. These 13 GO categories were largely related to signaling, microtubule dynamics, and fatty acid biosynthesis. Many of the genes driving the GO results for the genes consistently regulated in both systems have been previously implicated in Leishmania virulence (i.e., tryparedoxin peroxidase family members [66], casein kinase [67], and adenylate cyclase [68]) or regulation of flagellar dynamics (i.e., calmodulin [69], dynein heavy chain [70], kinesin [71, 72], paraflagellar rod protein [73], and multiple MAP and NIMA kinases [74–77]).
Patterns of parasite gene expression that were unique to either humans or mice were also clearly detectable and likely reflect the biological differences inherent to each host. A total of 1,887 L. major genes were uniquely regulated in human macrophages (1,133 up and 754 down), and 1,626 responded specifically to the infection of murine macrophages (862 up and 764 down). Among the genes that stood out as the most divergent in their behavior in the two host systems were META1, GP63, and surface antigen protein 2, all of which were downregulated in humans and upregulated in mice; conversely, RNA binding protein 5 and cathepsin L-like proteases were upregulated in humans and downregulated in mice. GP63 and META1 are known to play roles in virulence. GP63 is a zinc-dependent metalloprotease found on the surface of the parasite that cleaves C3b to iC3b, thus helping the parasite avoid complement-mediated lysis and promoting parasite entry into the cell via complement receptor 3 (78–80). Entry via this receptor has been found to contribute to parasite survival (81, 82). Additionally, GP63 hydrolyzes host kinase substrates (80, 83) and may thus alter the signaling processes of the host cell. META1 is known to localize to the flagellar pocket of the procyclic form of the parasite, has been implicated in secretory processes, and is upregulated during the procyclic-to-metacyclic promastigote transition (84–86). Cathepsin-L-like proteins have been shown to be key modulators of the host immune response (87, 88) and are regarded as possible molecular targets against leishmaniasis (89, 90). The differential expression of these proteins upon infection of human and mouse macrophages may reflect differences in the intricacies of host-parasite immune response/adaptation and/or the complex signaling pathways triggered by the parasite in the context of each host system.
A GO analysis yielded some additional insights into different parasite behaviors in the two hosts. Several categories related to proton transport were enriched among genes that Leishmania upregulated in the human system but not in the mouse, reflecting an upregulation of the expression of multiple vacuolar ATP synthase genes. This difference in the parasite response to the host system could be indicative of the relative acidity of the phagosomes in the two hosts. A number of categories related to ribosomes and translation were also enriched among genes that parasites upregulated in humans but downregulated in mice. The reason for this difference is unclear. Complete lists of enriched GO categories and the genes associated with each are provided in Table S1 and Data Set S7 in the supplemental material.
As highlighted earlier, the 4- to 24-hpi intracellular transition of the parasite was characterized by considerably smaller numbers of genes that were DE and by lower changes of magnitude. Of the 625 L. major genes that were DE during that transition in at least one of the hosts, only 28 represented a common parasite expression pattern in both human and mouse infections, while 328 and 273 genes were unique to the human and mouse systems, respectively. Despite the large number of differences, almost all were less than 2-fold DE and therefore represented relatively small changes in the context of the host system.
Even though our interpretations of the analyses above focused on previously characterized gene products, it is worth noting that significant proportions of the DE genes across parasite time points postinfeciton encode hypothetical/unspecified proteins: 60% and 67% of genes in the metacyclic to 4-hpi transition and 39% and 55% of genes in the 4- to 24-hpi transition for L. major and L. amazonensis, respectively. While significant numbers of Leishmania hypothetical genes remain uncharacterized to date, they play an integral role in the parasite’s strategy for survival and modulation of functional pathways within the host.
Conclusions.
Leishmania-macrophage interactions have been well studied at the cellular as well as the molecular levels. Most studies thus far have aimed to define the response of either the parasite or the host cell at limited time points and in different host systems and macrophage types. This study was prompted by the lack of a comprehensive picture of transcriptome reprogramming events during infection of human macrophages with Leishmania. We designed our experiments to collect global and simultaneous transcriptome profiles from host and pathogen over a well-defined time course, combined with a careful effort to systematically exclude the effect of large particle phagocytosis from the human response to infection with different Leishmania species.
Perhaps the most intriguing observation is the vigorous, parasite-specific response of the human macrophage (which includes an inflammatory burst) early in the infection. Such a clear picture could only emerge from our systematic exclusion of the “generic” effects of large-particle phagocytosis. The Leishmania-specific response of the macrophage was greatly tempered at later time points, and the transient nature of this response from the host macrophage was rather unexpected and apparently unique to Leishmania, since other pathogens that reside within the macrophage are believed to trigger a prolonged and sustained inflammatory response. Our findings indicate that in the case of Leishmania-infected macrophages, the response at later time points (24 h, 48 h, and 72 h) is mostly indistinguishable from the response to inert particles. While much of this response may be attributable to generic postingestion processes in the macrophage, there may still be underlying components within that response that are relevant to the parasite’s survival. The temporal changes in the global transcriptome of the parasite were similar to the pattern observed in its host cell, suggesting that much of the reprogramming that occurs as the parasites transform into intracellular forms generally stabilizes by around 24 hpi. The analogous expression patterns suggest that the host-pathogen interactions that are active shortly after Leishmania entry into the cell are greatly reduced once the infection is established.
We have collected data from multiple human donors, used time-matched uninfected macrophages as controls, and performed careful statistical analyses to account for batch effects and to discern the phagocytic effect from the pathogen-host modulation effect. This allowed us to detect the reprogramming profiles of the parasite and the host over the course of the infection with high confidence and sensitivity and to establish a robust data set not only for the study of parasite-macrophage interactions but also for the examination of host-pathogen interactions in general. Moreover, the comprehensive data set generated in this study serves as a reference not only to the communities that study host-pathogen interactions but to a broader audience interested in investigating inert particle phagocytosis. It will also serve as a reference for future studies using additional Leishmania strains (or even different pathogens altogether) to examine infection of macrophages from multiple sources and in various states of activation, polarization, or rest. A clearer picture of the signature of intracellular infection will emerge as this data set is examined in light of similar data sets that will be produced for other pathogens, providing additional insights into how pathogens are able to evade host defenses and modulate the biological functions of the cell in order to survive in the mammalian environment.
MATERIALS AND METHODS
Parasites.
Leishmania major (clone VI, MHOM/IL/80/Friedlin) and L. amazonensis (IFLA/BR/67/PH8) promastigotes were cultivated at 26°C in medium 199 (Gibco, Invitrogen) supplemented with 5% penicillin-streptomycin, 0.1% hemin (25 mg/ml in 50% triethanolamine), 20% heat-inactivated fetal bovine serum (FBS), 10 mM adenine (pH 7.5), and 5 mM l-glutamine. Metacyclic forms of L. major were purified by agglutination of stationary-phase promastigote cultures by using peanut agglutinin (Sigma) (91), while enrichment for L. amazonensis metacyclic promastigotes was performed by Ficoll density gradient centrifugation (92).
Human macrophages and infection.
Human macrophages were derived from purified monocytes and purchased fully differentiated from HemaCare (Van Nuys, CA). Briefly, CD14+ monocytes were positively selected via use of immunomagnetic beads and were cultured in the presence of 1,000 IU/ml human recombinant human M-CSF for 10 days. The cells were then harvested and transferred into HypoThermosol (BioLife Solutions) and shipped in suspension. Upon arrival, approximately 2.5 × 106 macrophages were plated per well in a 6-well plate with X-VIVO-15 medium (Lonza) supplemented with 1,000 IU/ml M-CSF (Miltenyi Biotech) and incubated for 24 h at 37°C, 5% CO2.
Macrophages were infected using a ratio of 10 parasites per macrophage for L. amazonensis and 5 parasites per macrophage for L. major for 4 h at 34°C. Infection was performed in X-VIVO-15 medium supplemented with 4% human AB serum (without prior heat inactivation). Macrophages were allowed to ingest 4.35-μm polystyrene beads (Spherotech) at a 1:10 ratio also in the presence of 4% human non-heat-inactivated AB serum (beads were preincubated in AB serum for 30 min). The cells were washed 3 times with phosphate-buffered saline (PBS) and further incubated at 34°C until the 24-, 48-, and 72-h time points. Samples intended for counting were fixed for 5 min at room temperature with Bouin solution (71.4% saturated picric acid, 23.8% formaldehyde, and 4.8% acetic acid), stained with Giemsa, and sequentially dehydrated in acetone, followed by a graded series of acetone-xylol (9:1, 7:3, and 3:7) and, finally, xylol. The number of intracellular parasites was determined by counting the total macrophages, and the total intracellular parasites per microscopic field was determined using a Nikon E200 microscope with a 100× 1.3-numerical aperture oil immersion objective. Counts were performed in triplicate for each period of infection.
RNA isolation and cDNA library preparation.
Total RNA was isolated from macrophages and the metacyclic material used for the L. major and L. amazonensis infections by using a NucleoSpin RNA kit (Macherey-Nagel) according to manufacturer’s protocol. RNA integrity was assessed using an Agilent 2100 bioanalyzer. Poly(A)-enriched cDNA libraries were generated using the Illumina TruSeq sample preparation kit (San Diego, CA) and checked for quality and quantity using the bioanalyzer and quantitative PCR (KAPA Biosystems).
RNA-seq data generation, preprocessing, and quality trimming.
Paired-end reads (100 BP) were obtained using the Illumina HiSeq 1500 platform. Trimmomatic (93) was used to remove any remaining Illumina adapter sequences from reads and to trim bases off the start or the end of a read when the quality score fell below a threshold of 20. Sequence quality metrics were assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Mapping cDNA fragments to the reference genome, abundance estimation, and data normalization.
TopHat (version 2.0.10) (94) was used to align reads to the applicable genome(s), with each genome alignment performed independently. Reads from uninfected, L. major-infected, L. amazonensis-infected, or bead-containing macrophage samples were aligned to the human genome (v. hg19/GRCh37). Reads from L. major-infected or L. amazonensis-infected samples were additionally aligned to the L. major (v. 6.0) or L. mexicana (v. 8.1) genomes, respectively, as were reads from metacyclic promastigote samples. The L. mexicana genome was used for alignment of L. amazonensis-containing samples, since it was the most closely related well-annotated genome that was available. The human genome was obtained from the UCSC Genome Browser (http://genome.ucsc.edu), and the parasite genomes were obtained from the TriTrypDB database (http://www.tritrypdb.org). Two mismatches per read were permitted (default), and reads were allowed to map only to a single locus (-g 1 option in TopHat). The abundance of reads mapping to each gene feature in the aligned genome was determined using HTSeq (95). Each resulting count table was restricted to protein-coding genes (20,956 genes for humans, 8,486 genes for L. major, and 8,336 genes for L. amazonensis/L. mexicana). Nonexpressed and weakly expressed genes, defined as having less than 1 read per million in n samples, where n is the size of the smallest group of replicates (96) (here, n = 3), were removed prior to subsequent analyses, resulting in count tables of 12,666, 8,480, and 8,310 genes for humans, L. major, and L. amazonensis/L. mexicana, respectively.
Global data assessment, visualization, and differential expression analysis.
A quantile normalization scheme was applied to all samples (97), and data were log2 transformed. Principal component analysis (PCA) was used to evaluate replicates and to visualize the relationships between samples. The limma program was used to conduct differential expression analyses following application of the voom module to transform the data based on observational-level weights derived from the mean-variance relationship (98). Experimental batch effects were adjusted for by including experimental batch as a covariate in the statistical model (99). Pairwise contrasts were performed within limma to identify DE genes across time points in the parasites and within time points in human macrophages, both before and after accounting for the effect of phagocytosis. Genes with a Benjamini-Hochberg (BH) multiple-testing adjusted P value of <0.05 were defined as differentially expressed. For comparisons of uninfected versus infected samples without accounting for the effects of phagocytosis, P values for infected versus uninfected samples were used as input into the BH multiple-testing adjustment. To determine differential expression while accounting for phagocytosis, infected samples were evaluated relative to both bead-containing macrophages and uninfected control cells. For each gene, the maximum P value from these two contrasts was selected for input into the BH multiple-testing adjustment. Components of the statistical pipeline, named cbcbSEQ, were done in R and can be accessed on GitHub (https://github.com/kokrah/cbcbSEQ/).
Ortholog mapping.
Human-mouse orthologs were defined using the bioMaRt package in R (100). Orthology gene tables from TriTrypDB (v. 8.1) (101) were used to identify orthologs between L. major and L. mexicana.
KEGG pathway analysis.
KEGG pathway analysis using ConsensusPathDB-human (102) was done to identify signaling and metabolic pathways that were overrepresented in the human DE gene lists. For each KEGG pathway, a P value was calculated using a hypergeometric test, and a cutoff of 0.01 was applied to identify enriched KEGG pathways. For each analysis, up- and downregulated genes were considered separately.
Gene ontology analysis.
GO categories enriched among L. major gene sets were identified using the GOseq package in R (103). GOseq was developed specifically to account for transcript length bias in GO analyses based on RNA-seq data. For each comparison, upregulated and downregulated gene sets (no fold change cutoff) were input separately into GOseq. A P value cutoff of 0.05 was used.
Availability of supporting raw data.
Raw sequence data are available at the NCBI Short Read Archive (SRA) under records PRJNA290995, PRJNA252769, and PRJNA292915.
SUPPLEMENTAL MATERIAL
Global gene expression profiles of Leishmania-infected and bead-containing macrophages before and after batch adjustment. RNA-seq was carried out on human macrophages infected with L. major or L. amazonensis at 4, 24, 48, and 72 hpi, as well as on macrophages that had ingested latex beads and on uninfected controls. PCA plots are shown for all human macrophage samples both before (A) and after (B) including experimental batch as a covariate in the statistical model. The first two principal components are shown on the x and y axes, respectively, with the proportion of total variance explained by the PC indicated. Each experimental sample is represented as a single point, with color hue indicating sample type (yellow for L. major-infected cells, blue for L. amazonensis-infected cells, red for bead-containing cells, and gray for uninfected cells), color shade indicating time point (light to dark representing 4 to 72 hpi), and shape indicating experimental batch. Samples are named according to sample type (infLM, L. major-infected cells; infLA, L. amazonensis-infected cells; beads, bead-containing cells; uninf, uninfected cells) and time point (4, 24, 48, or 72 hpi). All analyses were performed after filtering out nonexpressed and lowly expressed genes and quantile normalization. Download
Global gene expression profiles of Leishmania parasites before and after batch adjustment. RNA-seq was carried out on human macrophages infected with L. major or L. amazonensis at 4, 24, 48, and 72 hpi as well as on the metacyclic promastigotes used for the infection. PCA plots are shown for L. major (A and C) and L. amazonensis (B and D) transcriptomes over the course of the experiment both before (A and B) and after (C and D) including experimental batch as a covariate in the statistical model. The first two principal components are shown on the x and y axes, respectively, with the proportion of total variance explained by the PC indicated. Each experimental sample is represented as a single point, with color indicating sample type/time point and shape indicating experimental batch. Samples are named according to sample type (metac, metacyclic promastigotes; amast, intracellular amastigotes) and time point (4, 24, 48, or 72 hpi). All analyses were performed after filtering out nonexpressed and lowly expressed genes and quantile normalization. Download
Enriched GO categories among L. major genes in common or specific to human and/or mouse infections (GOseq was used to identify GO categories enriched among DE genes during the metacyclic promastigote to 4-hpi intracellular transition. Genes that were expressed in the same direction in human and mouse host systems, those specific to infection in the human system, and those specific to infection in the mouse system were evaluated, with up- and downregulated genes considered separately [P < 0.05].)
Experimental design. Download
Differentially expressed gene lists for Leishmania-infected human macrophages before and after accounting for phagocytosis and for bead-containing human macrophages. Download
Differentially expressed gene lists for L. amazonensis-infected human macrophages relative to L. major-infected human macrophages. Download
Comparison of human and murine macrophage responses to L. major infection and genes associated with enriched KEGG pathways. Download
Differentially expressed gene lists for L. major and L. amazonensis parasite stages and time points. Download
Expression profiles for L. major and L. amazonensis orthologs. Download
Comparison of L. major DE profiles during human and murine macrophage infections and genes associated with enriched GO categories. Download
ACKNOWLEDGMENTS
All the sequencing was performed at the University of Maryland Institute for Bioscience and Biotechnology Research (IBBR) sequencing core.
This work was supported by a grant from the National Institutes of Health (grant AI094773 to N.M.E.-S. and D.M.M.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Fernandes MC, Dillon LAL, Belew AT, Bravo HC, Mosser DM, El-Sayed NM. 2016. Dual transcriptome profiling of Leishmania-infected human macrophages reveals distinct reprogramming signatures. mBio 7(3):e00027-16. doi:10.1128/mBio.00027-16.
REFERENCES
- 1.Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M, WHO Leishmaniasis Control Team . 2012. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sacks D, Kamhawi S. 2001. Molecular aspects of parasite-vector and vector-host interactions in leishmaniasis. Annu Rev Microbiol 55:453–483. doi: 10.1146/annurev.micro.55.1.453. [DOI] [PubMed] [Google Scholar]
- 3.Kaye P, Scott P. 2011. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol 9:604–615. doi: 10.1038/nrmicro2608. [DOI] [PubMed] [Google Scholar]
- 4.Herwaldt BL. 1999. Leishmaniasis. Lancet 354:1191–1199. doi: 10.1016/S0140-6736(98)10178-2. [DOI] [PubMed] [Google Scholar]
- 5.Murray HW, Berman JD, Davies CR, Saravia NG. 2005. Advances in leishmaniasis. Lancet 366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- 6.Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream M-A, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G. 2005. The genome of the kinetoplastid parasite, Leishmania major. Science 309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, Kerhornou A, Ivens A, Fraser A, Rajandream M-A, Carver T, Norbertczak H, Chillingworth T, Hance Z, Jagels K, Moule S. 2007. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet 39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Real F, Vidal RO, Carazzolle MF, Mondego JM, Costa GG, Herai RH, Würtele M, de Carvalho LM, Carmona e Ferreira R, Mortara RA, Barbiéri CL, Mieczkowski P, da Silveira JF, Briones MR, Pereira GA, Bahia D. 2013. The genome sequence of Leishmania (Leishmania) amazonensis: functional annotation and extended analysis of gene models. DNA Res 20:567–581. doi: 10.1093/dnares/dst031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, Harris D, Her Y, Herzyk P, Imamura H, Otto TD, Sanders M, Seeger K, Dujardin J-C, Berriman M, Smith DF, Hertz-Fowler C, Mottram JC. 2011. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res 21:2129–2142. doi: 10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laskay T, van Zandbergen G, Solbach W. 2003. Neutrophil granulocytes—Trojan horses for Leishmania major and other intracellular microbes? Trends Microbiol 11:210–214. doi: 10.1016/S0966-842X(03)00075-1. [DOI] [PubMed] [Google Scholar]
- 11.Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. 2008. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321:970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charmoy M, Brunner-Agten S, Aebischer D, Auderset F, Launois P, Milon G, Proudfoot AE, Tacchini-Cottier F. 2010. Neutrophil-derived CCL3 is essential for the rapid recruitment of dendritic cells to the site of Leishmania major inoculation in resistant mice. PLoS Pathog 6:e1000755. doi: 10.1371/journal.ppat.1000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.León B, López-Bravo M, Ardavín C. 2007. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 26:519–531. doi: 10.1016/j.immuni.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 14.Ng LG, Hsu A, Mandell MA, Roediger B, Hoeller C, Mrass P, Iparraguirre A, Cavanagh LL, Triccas JA, Beverley SM, Scott P, Weninger W. 2008. Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS Pathog 4:e1000222. doi: 10.1371/journal.ppat.1000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogdan C, Gessner A, Solbach W, Röllinghoff M. 1996. Invasion, control and persistence of Leishmania parasites. Curr Opin Immunol 8:517–525. doi: 10.1016/S0952-7915(96)80040-9. [DOI] [PubMed] [Google Scholar]
- 16.Bent ZW, Poorey K, Brazel DM, LaBauve AE, Sinha A, Curtis DJ, House SE, Tew KE, Hamblin RY, Williams KP, Branda SS, Young GM, Meagher RJ. 2015. Transcriptomic analysis of Yersinia enterocolitica biovar 1B infecting murine macrophages reveals new mechanisms of extracellular and intracellular survival. Infect Immun 83:2672–2685. doi: 10.1128/IAI.02922-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blumenthal A, Lauber J, Hoffmann R, Ernst M, Keller C, Buer J, Ehlers S, Reiling N. 2005. Common and unique gene expression signatures of human macrophages in response to four strains of Mycobacterium avium that differ in their growth and persistence characteristics. Infect Immun 73:3330–3341. doi: 10.1128/IAI.73.6.3330-3341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kabara E, Kloss CC, Wilson M, Tempelman RJ, Sreevatsan S, Janagama H, Coussens PM. 2010. A large-scale study of differential gene expression in monocyte-derived macrophages infected with several strains of Mycobacterium avium subspecies paratuberculosis. Brief Funct Genomics 9:220–237. doi: 10.1093/bfgp/elq009. [DOI] [PubMed] [Google Scholar]
- 19.Magee DA, Taraktsoglou M, Killick KE, Nalpas NC, Browne JA, Park SD, Conlon KM, Lynn DJ, Hokamp K, Gordon SV, Gormley E, MacHugh DE. 2012. Global gene expression and systems biology analysis of bovine monocyte-derived macrophages in response to in vitro challenge with Mycobacterium bovis. PLoS One 7:e32034. doi: 10.1371/journal.pone.0032034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mavromatis CH, Bokil NJ, Totsika M, Kakkanat A, Schaale K, Cannistraci CV, Ryu T, Beatson SA, Ulett GC, Schembri MA, Sweet MJ, Ravasi T. 2015. The co-transcriptome of uropathogenic Escherichia coli-infected mouse macrophages reveals new insights into host-pathogen interactions. Cell Microbiol 17:730–746. doi: 10.1111/cmi.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nalpas NC, Park SD, Magee DA, Taraktsoglou M, Browne JA, Conlon KM, Rue-Albrecht K, Killick KE, Hokamp K, Lohan AJ, Loftus BJ, Gormley E, Gordon SV, MacHugh DE. 2013. Whole-transcriptome, high-throughput RNA sequence analysis of the bovine macrophage response to Mycobacterium bovis infection in vitro. BMC Genomics 14:230. doi: 10.1186/1471-2164-14-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price CT, Abu Kwaik Y. 2014. The transcriptome of Legionella pneumophila-infected human monocyte-derived macrophages. PLoS One 9:e114914. doi: 10.1371/journal.pone.0114914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subbian S, Tsenova L, Kim M-J, Wainwright HC, Visser A, Bandyopadhyay N, Bader JS, Karakousis PC, Murrmann GB, Bekker L-G, Russell DG, Kaplan G. 2015. Lesion-specific immune response in granulomas of patients with pulmonary tuberculosis: a pilot study. PLoS One 10:e0132249. doi: 10.1371/journal.pone.0132249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue F, Zhao X, Yang Y, Zhao J, Yang Y, Cao Y, Hong C, Liu Y, Sun L, Huang M, Gu J. 2013. Responses of murine and human macrophages to leptospiral infection: a study using comparative array analysis. PLoS Negl Trop Dis 7:e2477. doi: 10.1371/journal.pntd.0002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forget G, Siminovitch KA, Brochu S, Rivest S, Radzioch D, Olivier M. 2001. Role of host phosphotyrosine phosphatase SHP-1 in the development of murine leishmaniasis. Eur J Immunol 31:3185–3196. doi: 10.1002/1521-4141(200111)31:11<3185::AID-IMMU3185>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Kwan WC, McMaster WR, Wong N, Reiner NE. 1992. Inhibition of expression of major histocompatibility complex class II molecules in macrophages infected with Leishmania donovani occurs at the level of gene transcription via a cyclic AMP-independent mechanism. Infect Immun 60:2115–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nandan D, Reiner NE. 1995. Attenuation of gamma interferon-induced tyrosine phosphorylation in mononuclear phagocytes infected with Leishmania donovani: selective inhibition of signaling through Janus kinases and Stat1. Infect Immun 63:4495–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nandan D, Lo R, Reiner NE. 1999. Activation of phosphotyrosine phosphatase activity attenuates mitogen-activated protein kinase signaling and inhibits c-fos and nitric oxide synthase expression in macrophages infected with Leishmania donovani. Infect Immun 67:4055–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olivier M, Brownsey RW, Reiner NE. 1992. Defective stimulus-response coupling in human monocytes infected with Leishmania donovani is associated with altered activation and translocation of protein kinase C. Proc Natl Acad Sci U S A 89:7481–7485. doi: 10.1073/pnas.89.16.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiner NE. 1994. Altered cell signaling and mononuclear phagocyte deactivation during intracellular infection. Immunol Today 15:374–381. doi: 10.1016/0167-5699(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 31.Chaussabel D, Semnani RT, McDowell MA, Sacks D, Sher A, Nutman TB. 2003. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 102:672–681. doi: 10.1182/blood-2002-10-3232. [DOI] [PubMed] [Google Scholar]
- 32.Ettinger NA, Wilson ME. 2008. Macrophage and T-cell gene expression in a model of early infection with the protozoan Leishmania chagasi. PLoS Negl Trop Dis 2:e252.doi: 10.1371/journal.pntd.0000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gregory DJ, Sladek R, Olivier M, Matlashewski G. 2008. Comparison of the effects of Leishmania major or Leishmania donovani infection on macrophage gene expression. Infect Immun 76:1186–1192. doi: 10.1128/IAI.01320-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osorio y Fortéa J, de la Llave E, Regnault B, Coppée J-Y, Milon G, Lang T, Prina E. 2009. Transcriptional signatures of BALB/c mouse macrophages housing multiplying Leishmania amazonensis amastigotes. BMC Genomics 10:119. doi: 10.1186/1471-2164-10-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ovalle-Bracho C, Franco-Muñoz C, Londoño-Barbosa D, Restrepo-Montoya D, Clavijo-Ramírez C. 2015. Changes in macrophage gene expression associated with Leishmania (Viannia) braziliensis infection. PLoS One 10:e0128934. doi: 10.1371/journal.pone.0128934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabhi I, Rabhi S, Ben-Othman R, Rasche A, Daskalaki A, Trentin B, Piquemal D, Regnault B, Descoteaux A, Guizani-Tabbane L, Sysco Consortium . 2012. Transcriptomic signature of Leishmania infected mice macrophages: a metabolic point of view. PLoS Negl Trop Dis 6:e1763. doi: 10.1371/journal.pntd.0001763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramírez C, Díaz-Toro Y, Tellez J, Castilho TM, Rojas R, Ettinger NA, Tikhonova I, Alexander ND, Valderrama L, Hager J, Wilson ME, Lin A, Zhao H, Saravia NG, McMahon-Pratt D. 2012. Human macrophage response to L. (Viannia) panamensis: microarray evidence for an early inflammatory response. PLoS Negl Trop Dis 6:e1866. doi: 10.1371/journal.pntd.0001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez NE, Chang HK, Wilson ME. 2004. Novel program of macrophage gene expression induced by phagocytosis of Leishmania chagasi. Infect Immun 72:2111–2122. doi: 10.1128/IAI.72.4.2111-2122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buates S, Matlashewski G. 2001. General suppression of macrophage gene expression during Leishmania donovani infection. J Immunol 166:3416–3422. doi: 10.4049/jimmunol.166.5.3416. [DOI] [PubMed] [Google Scholar]
- 40.Dillon LA, Suresh R, Okrah K, Corrada Bravo H, Mosser DM, El-Sayed NM. 2015. Simultaneous transcriptional profiling of Leishmania major and its murine macrophage host cell reveals insights into host-pathogen interactions. BMC Genomics 16:1108. doi: 10.1186/s12864-015-2237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Humphrys MS, Creasy T, Sun Y, Shetty AC, Chibucos MC, Drabek EF, Fraser CM, Farooq U, Sengamalay N, Ott S, Shou H, Bavoil PM, Mahurkar A, Myers GS. 2013. Simultaneous transcriptional profiling of bacteria and their host cells. PLoS One 8:e80597. doi: 10.1371/journal.pone.0080597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maretti-Mira AC, Bittner J, Oliveira-Neto MP, Liu M, Kang D, Li H, Pirmez C, Craft N. 2012. Transcriptome patterns from primary cutaneous Leishmania braziliensis infections associate with eventual development of mucosal disease in humans. PLoS Negl Trop Dis 6:e1816. doi: 10.1371/journal.pntd.0001816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pittman KJ, Aliota MT, Knoll LJ. 2014. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics 15:806. doi: 10.1186/1471-2164-15-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tierney L, Linde J, Müller S, Brunke S, Molina JC, Hube B, Schöck U, Guthke R, Kuchler K. 2012. An interspecies regulatory network inferred from simultaneous RNA-seq of Candida albicans invading innate immune cells. Front Microbiol 3:85. doi: 10.3389/fmicb.2012.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westermann AJ, Gorski SA, Vogel J. 2012. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10:618–630. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- 46.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege J-L, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lynes MA, Borghesi LA, Youn J, Olson EA. 1993. Immunomodulatory activities of extracellular metallothionein. I. Metallothionein effects on antibody production. Toxicology 85:161–177. doi: 10.1016/0300-483X(93)90040-Y. [DOI] [PubMed] [Google Scholar]
- 48.Ghoshal K, Jacob ST. 2001. Regulation of metallothionein gene expression. Prog Nucleic Acid Res Mol Biol 66:357–384. doi: 10.1016/S0079-6603(00)66034-8. [DOI] [PubMed] [Google Scholar]
- 49.Emeny RT, Kasten-Jolly J, Mondal T, Lynes MA, Lawrence DA. 2015. Metallothionein differentially affects the host response to listeria infection both with and without an additional stress from cold-restraint. Cell Stress Chaperones 20:1013–1022. doi: 10.1007/s12192-015-0630-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Real F, Florentino PT, Reis LC, Ramos-Sanchez EM, Veras PS, Goto H, Mortara RA. 2014. Cell-to-cell transfer of Leishmania amazonensis amastigotes is mediated by immunomodulatory lamp-rich parasitophorous extrusions. Cell Microbiol 16:1549–1564. doi: 10.1111/cmi.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arango Duque G, Fukuda M, Descoteaux A. 2013. Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. J Immunol 190:1737–1745. doi: 10.4049/jimmunol.1202500. [DOI] [PubMed] [Google Scholar]
- 52.Baram D, Adachi R, Medalia O, Tuvim M, Dickey BF, Mekori YA, Sagi-Eisenberg R. 1999. Synaptotagmin II negatively regulates Ca2+-triggered exocytosis of lysosomes in mast cells. J Exp Med 189:1649–1658. doi: 10.1084/jem.189.10.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Czibener C, Sherer NM, Becker SM, Pypaert M, Hui E, Chapman ER, Mothes W, Andrews NW. 2006. Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J Cell Biol 174:997–1007. doi: 10.1083/jcb.200605004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vinet AF, Fukuda M, Descoteaux A. 2008. The exocytosis regulator synaptotagmin V controls phagocytosis in macrophages. J Immunol 181:5289–5295. doi: 10.4049/jimmunol.181.8.5289. [DOI] [PubMed] [Google Scholar]
- 55.Arango Duque G, Fukuda M, Turco SJ, Stäger S, Descoteaux A. 2014. Leishmania promastigotes induce cytokine secretion in macrophages through the degradation of synaptotagmin XI. J Immunol 193:2363–2372. doi: 10.4049/jimmunol.1303043. [DOI] [PubMed] [Google Scholar]
- 56.Vinet AF, Fukuda M, Turco SJ, Descoteaux A. 2009. The Leishmania donovani lipophosphoglycan excludes the vesicular proton-ATPase from phagosomes by impairing the recruitment of synaptotagmin V. PLoS Pathog 5:e1000628. doi: 10.1371/journal.ppat.1000628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vinet AF, Jananji S, Turco SJ, Fukuda M, Descoteaux A. 2011. Exclusion of synaptotagmin V at the phagocytic cup by Leishmania donovani lipophosphoglycan results in decreased promastigote internalization. Microbiology 157:2619–2628. doi: 10.1099/mic.0.050252-0. [DOI] [PubMed] [Google Scholar]
- 58.Andrews NW, Chakrabarti S. 2005. There’s more to life than neurotransmission: the regulation of exocytosis by synaptotagmin VII. Trends Cell Biol 15:626–631. doi: 10.1016/j.tcb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 59.Südhof TC, Rothman JE. 2009. Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tucker WC, Chapman ER. 2002. Role of synaptotagmin in Ca2+-triggered exocytosis. Biochem J 366:1–13. doi: 10.1042/BJ20020776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matheoud D, Moradin N, Bellemare-Pelletier A, Shio MT, Hong WJ, Olivier M, Gagnon E, Desjardins M, Descoteaux A. 2013. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 14:15–25. doi: 10.1016/j.chom.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 62.Zhang W-W, Mendez S, Ghosh A, Myler P, Ivens A, Clos J, Sacks DL, Matlashewski G. 2003. Comparison of the A2 gene locus in Leishmania donovani and Leishmania major and its control over cutaneous infection. J Biol Chem 278:35508–35515. doi: 10.1074/jbc.M305030200. [DOI] [PubMed] [Google Scholar]
- 63.Zhang WW, Matlashewski G. 2001. Characterization of the A2-A2rel gene cluster in Leishmania donovani: involvement of A2 in visceralization during infection. Mol Microbiol 39:935–948. doi: 10.1046/j.1365-2958.2001.02286.x. [DOI] [PubMed] [Google Scholar]
- 64.McCall L-I, Matlashewski G. 2012. Involvement of the Leishmania donovani virulence factor A2 in protection against heat and oxidative stress. Exp Parasitol 132:109–115. doi: 10.1016/j.exppara.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 65.Almeida RP, Barral-Netto M, De Jesus AM, De Freitas LA, Carvalho EM, Barral A. 1996. Biological behavior of Leishmania amazonensis isolated from humans with cutaneous, mucosal, or visceral leishmaniasis in BALB/c mice. Am J Trop Med Hyg 54:178–184. [DOI] [PubMed] [Google Scholar]
- 66.Iyer JP, Kaprakkaden A, Choudhary ML, Shaha C. 2008. Crucial role of cytosolic tryparedoxin peroxidase in Leishmania donovani survival, drug response and virulence. Mol Microbiol 68:372–391. doi: 10.1111/j.1365-2958.2008.06154.x. [DOI] [PubMed] [Google Scholar]
- 67.Dan-Goor M, Nasereddin A, Jaber H, Jaffe CL. 2013. Identification of a secreted casein kinase 1 in Leishmania donovani: effect of protein over expression on parasite growth and virulence. PLoS One 8:e79287. doi: 10.1371/journal.pone.0079287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Biswas A, Bhattacharya A, Das PK. 2011. Role of cAMP signaling in the survival and infectivity of the protozoan parasite, Leishmania donovani. Mol Biol Int 2011:782971. doi: 10.4061/2011/782971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ginger ML, Collingridge PW, Brown RW, Sproat R, Shaw MK, Gull K. 2013. Calmodulin is required for paraflagellar rod assembly and flagellum-cell body attachment in trypanosomes. Protist 164:528–540. doi: 10.1016/j.protis.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 70.Harder S, Thiel M, Clos J, Bruchhaus I. 2010. Characterization of a subunit of the outer dynein arm docking complex necessary for correct flagellar assembly in Leishmania donovani. PLoS Negl Trop Dis 4:e586. doi: 10.1371/journal.pntd.0000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blaineau C, Tessier M, Dubessay P, Tasse L, Crobu L, Pagès M, Bastien P. 2007. A novel microtubule-depolymerizing kinesin involved in length control of a eukaryotic flagellum. Curr Biol 17:778–782. doi: 10.1016/j.cub.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 72.Vicente JJ, Wordeman L. 2015. Mitosis, microtubule dynamics and the evolution of kinesins. Exp Cell Res 334:61–69. doi: 10.1016/j.yexcr.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lander N, Li Z-H, Niyogi S, Docampo R. 2015. CRISPR/Cas9-induced disruption of paraflagellar rod protein 1 and 2 genes in Trypanosoma cruzi reveals their role in flagellar attachment. mBio 6:e01012. doi: 10.1128/mBio.01012-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bengs F, Scholz A, Kuhn D, Wiese M. 2005. LmxMPK9, a mitogen-activated protein kinase homologue affects flagellar length in Leishmania mexicana. Mol Microbiol 55:1606–1615. doi: 10.1111/j.1365-2958.2005.04498.x. [DOI] [PubMed] [Google Scholar]
- 75.Cao M, Li G, Pan J. 2009. Regulation of cilia assembly, disassembly, and length by protein phosphorylation. Methods Cell Biol 94:333–346. doi: 10.1016/S0091-679X(08)94017-6. [DOI] [PubMed] [Google Scholar]
- 76.Erdmann M, Scholz A, Melzer IM, Schmetz C, Wiese M. 2006. Interacting protein kinases involved in the regulation of flagellar length. Mol Biol Cell 17:2035–2045. doi: 10.1091/mbc.E05-10-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wiese M, Kuhn D, Grünfelder CG. 2003. Protein kinase involved in flagellar-length control. Eukaryot Cell 2:769–777. doi: 10.1128/EC.2.4.769-777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brittingham A, Morrison CJ, McMaster WR, McGwire BS, Chang KP, Mosser DM. 1995. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J Immunol 155:3102–3111. doi: 10.1016/0169-4758(95)80054-9. [DOI] [PubMed] [Google Scholar]
- 79.Mosser DM, Edelson PJ. 1985. The mouse macrophage receptor for C3bi (CR3) is a major mechanism in the phagocytosis of Leishmania promastigotes. J Immunol 135:2785–2789. [PubMed] [Google Scholar]
- 80.Olivier M, Atayde VD, Isnard A, Hassani K, Shio MT. 2012. Leishmania virulence factors: focus on the metalloprotease GP63. Microbes Infect 14:1377–1389. doi: 10.1016/j.micinf.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 81.Da Silva RP, Hall BF, Joiner KA, Sacks DL. 1989. CR1, the C3b receptor, mediates binding of infective Leishmania major metacyclic promastigotes to human macrophages. J Immunol 143:617–622. [PubMed] [Google Scholar]
- 82.Mosser DM, Edelson PJ. 1987. The third component of complement (C3) is responsible for the intracellular survival of Leishmania major. Nature 327:329–331. doi: 10.1038/327329b0. [DOI] [PubMed] [Google Scholar]
- 83.Corradin S, Ransijn A, Corradin G, Roggero MA, Schmitz AA, Schneider P, Mauël J, Vergères G. 1999. MARCKS-related protein (MRP) is a substrate for the Leishmania major surface protease leishmanolysin (gp63). J Biol Chem 274:25411–25418. doi: 10.1074/jbc.274.36.25411. [DOI] [PubMed] [Google Scholar]
- 84.Dillon LA, Okrah K, Hughitt VK, Suresh R, Li Y, Fernandes MC, Belew AT, Corrada Bravo H, Mosser DM, El-Sayed NM. 2015. Transcriptomic profiling of gene expression and RNA processing during Leishmania major differentiation. Nucleic Acids Res 43:6799–6813. doi: 10.1093/nar/gkv656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nourbakhsh F, Uliana SR, Smith DF. 1996. Characterisation and expression of a stage-regulated gene of Leishmania major. Mol Biochem Parasitol 76:201–213. doi: 10.1016/0166-6851(95)02559-6. [DOI] [PubMed] [Google Scholar]
- 86.Puri V, Goyal A, Sankaranarayanan R, Enright AJ, Vaidya T. 2011. Evolutionary and functional insights into Leishmania META1: evidence for lateral gene transfer and a role for META1 in secretion. BMC Evol Biol 11:334. doi: 10.1186/1471-2148-11-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alexander J, Coombs GH, Mottram JC. 1998. Leishmania mexicana cysteine proteinase-deficient mutants have attenuated virulence for mice and potentiate a Th1 response. J Immunol 161:6794–6801. [PubMed] [Google Scholar]
- 88.Mottram JC, Coombs GH, Alexander J. 2004. Cysteine peptidases as virulence factors of Leishmania. Curr Opin Microbiol 7:375–381. doi: 10.1016/j.mib.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 89.De Sousa LR, Wu H, Nebo L, Fernandes JB, da Silva MF, Kiefer W, Schirmeister T, Vieira PC. 2015. Natural products as inhibitors of recombinant cathepsin L of Leishmania mexicana. Exp Parasitol 156:42–48. doi: 10.1016/j.exppara.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 90.Desai PV, Patny A, Gut J, Rosenthal PJ, Tekwani B, Srivastava A, Avery M. 2006. Identification of novel parasitic cysteine protease inhibitors by use of virtual screening. 2. The available chemical directory. J Med Chem 49:1576–1584. doi: 10.1021/jm0505765. [DOI] [PubMed] [Google Scholar]
- 91.Sacks DL, Hieny S, Sher A. 1985. Identification of cell surface carbohydrate and antigenic changes between noninfective and infective developmental stages of Leishmania major promastigotes. J Immunol 135:564–569. [PubMed] [Google Scholar]
- 92.Späth GF, Beverley SM. 2001. A lipophosphoglycan-independent method for isolation of infective Leishmania metacyclic promastigotes by density gradient centrifugation. Exp Parasitol 99:97–103. doi: 10.1006/expr.2001.4656. [DOI] [PubMed] [Google Scholar]
- 93.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. 2013. Count-based differential expression analysis of RNA sequencing data using R and bioconductor. Nat Protoc 8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 97.Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 98.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. 2010. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. 2005. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 101.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC. 2010. TriTrypDB: a functional genomic resource for the trypanosomatidae. Nucleic Acids Res 38:D457–D462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. 2011. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res 39:D712–D717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Young MD, Wakefield MJ, Smyth GK, Oshlack A. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11:R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Global gene expression profiles of Leishmania-infected and bead-containing macrophages before and after batch adjustment. RNA-seq was carried out on human macrophages infected with L. major or L. amazonensis at 4, 24, 48, and 72 hpi, as well as on macrophages that had ingested latex beads and on uninfected controls. PCA plots are shown for all human macrophage samples both before (A) and after (B) including experimental batch as a covariate in the statistical model. The first two principal components are shown on the x and y axes, respectively, with the proportion of total variance explained by the PC indicated. Each experimental sample is represented as a single point, with color hue indicating sample type (yellow for L. major-infected cells, blue for L. amazonensis-infected cells, red for bead-containing cells, and gray for uninfected cells), color shade indicating time point (light to dark representing 4 to 72 hpi), and shape indicating experimental batch. Samples are named according to sample type (infLM, L. major-infected cells; infLA, L. amazonensis-infected cells; beads, bead-containing cells; uninf, uninfected cells) and time point (4, 24, 48, or 72 hpi). All analyses were performed after filtering out nonexpressed and lowly expressed genes and quantile normalization. Download
Global gene expression profiles of Leishmania parasites before and after batch adjustment. RNA-seq was carried out on human macrophages infected with L. major or L. amazonensis at 4, 24, 48, and 72 hpi as well as on the metacyclic promastigotes used for the infection. PCA plots are shown for L. major (A and C) and L. amazonensis (B and D) transcriptomes over the course of the experiment both before (A and B) and after (C and D) including experimental batch as a covariate in the statistical model. The first two principal components are shown on the x and y axes, respectively, with the proportion of total variance explained by the PC indicated. Each experimental sample is represented as a single point, with color indicating sample type/time point and shape indicating experimental batch. Samples are named according to sample type (metac, metacyclic promastigotes; amast, intracellular amastigotes) and time point (4, 24, 48, or 72 hpi). All analyses were performed after filtering out nonexpressed and lowly expressed genes and quantile normalization. Download
Enriched GO categories among L. major genes in common or specific to human and/or mouse infections (GOseq was used to identify GO categories enriched among DE genes during the metacyclic promastigote to 4-hpi intracellular transition. Genes that were expressed in the same direction in human and mouse host systems, those specific to infection in the human system, and those specific to infection in the mouse system were evaluated, with up- and downregulated genes considered separately [P < 0.05].)
Experimental design. Download
Differentially expressed gene lists for Leishmania-infected human macrophages before and after accounting for phagocytosis and for bead-containing human macrophages. Download
Differentially expressed gene lists for L. amazonensis-infected human macrophages relative to L. major-infected human macrophages. Download
Comparison of human and murine macrophage responses to L. major infection and genes associated with enriched KEGG pathways. Download
Differentially expressed gene lists for L. major and L. amazonensis parasite stages and time points. Download
Expression profiles for L. major and L. amazonensis orthologs. Download
Comparison of L. major DE profiles during human and murine macrophage infections and genes associated with enriched GO categories. Download