

ABSTRACT
Ingestion of the obligate intracellular protozoan parasite Toxoplasma gondii causes an acute infection that leads to chronic infection of the host. To facilitate the acute phase of the infection, T. gondii manipulates the host response by secreting rhoptry organelle proteins (ROPs) into host cells during its invasion. A few key ROP proteins with signatures of kinases or pseudokinases (ROPKs) act as virulence factors that enhance parasite survival against host gamma interferon-stimulated innate immunity. However, the roles of these and other ROPK proteins in establishing chronic infection have not been tested. Here, we deleted 26 ROPK gene loci encoding 31 unique ROPK proteins of type II T. gondii and show that numerous ROPK proteins influence the development of chronic infection. Cyst burdens were increased in the Δrop16 knockout strain or moderately reduced in 11 ROPK knockout strains. In contrast, deletion of ROP5, ROP17, ROP18, ROP35, or ROP38/29/19 (ROP38, ROP29, and ROP19) severely reduced cyst burdens. Δrop5 and Δrop18 knockout strains were less resistant to host immunity-related GTPases (IRGs) and exhibited >100-fold-reduced virulence. ROP18 kinase activity and association with the parasitophorous vacuole membrane were necessary for resistance to host IRGs. The Δrop17 strain exhibited a >12-fold defect in virulence; however, virulence was not affected in the Δrop35 or Δrop38/29/19 strain. Resistance to host IRGs was not affected in the Δrop17, Δrop35, or Δrop38/29/19 strain. Collectively, these findings provide the first definitive evidence that the type II T. gondii ROPK proteome functions as virulence factors and facilitates additional mechanisms of host manipulation that are essential for chronic infection and transmission of T. gondii.
IMPORTANCE
Reactivation of chronic Toxoplasma gondii infection in individuals with weakened immune systems causes severe toxoplasmosis. Existing treatments for toxoplasmosis are complicated by adverse reactions to chemotherapy. Understanding key parasite molecules required for chronic infection provides new insights into potential mechanisms that can interrupt parasite survival or persistence in the host. This study reveals that key secreted rhoptry molecules are used by the parasite to establish chronic infection of the host. Certain rhoptry proteins were found to be critical virulence factors that resist innate immunity, while other rhoptry proteins were found to influence chronic infection without affecting virulence. This study reveals that rhoptry proteins utilize multiple mechanisms of host manipulation to establish chronic infection of the host. Targeted disruption of parasite rhoptry proteins involved in these biological processes opens new avenues to interfere with chronic infection with the goal to either eliminate chronic infection or to prevent recrudescent infections.
INTRODUCTION
Toxoplasma gondii chronically infects many warm-blooded vertebrates. Infection with T. gondii begins after oral ingestion of tissue cysts or sporocysts. The ensuing acute infection is characterized by rapidly replicating tachyzoite stage parasites that disseminate widely into host tissues (1) until the infection is controlled by host T cell responses and gamma interferon (IFN-γ) (2). While most human infections go unnoticed in immunocompetent individuals, a primary infection during pregnancy can spread through the transplacental route to the fetus, resulting in fetal death or significant congenital disease (1). Prior to elimination of the acute stage infection, tachyzoites access vascular endothelial cells as a replicative niche to breach the blood-brain barrier and enter the central nervous system (3). The replicating tachyzoite stage differentiates into a chronic bradyzoite stage encased in cyst structures that persist to establish chronic infection of the host (4). Neurons are the primary target cell type in which cysts develop in the brain (5, 6). Reactivation of chronic T. gondii cysts when host immunity wanes due to HIV/AIDS, chemotherapy, or transplantation therapy causes toxoplasmosis, a severe opportunistic infection of humans (7). Though bradyzoite stage differentiation and the development and maintenance of cyst stages are biologically crucial events for successful transmission of T. gondii infection, the biology controlling these stages is not yet well understood. While secreted parasite molecules have been shown to enhance parasite survival by manipulation of host cells and innate immunity during acute infection (8, 9), few secreted parasite molecules have been identified as playing crucial roles in successful chronic infection in vivo (10–12).
Comparative genome analysis reveals amplification and diversification of secretory pathogenesis determinants, including rhoptry organelle proteins (ROPs), are key features that distinguish genomes of biologically diverse coccidian parasites (13). ROP molecules play important roles in invasion, in establishing the replicative niche of the parasitophorous vacuole, in host cell manipulation, and in resisting host IFN-γ-stimulated innate immunity (8, 9, 14). Genetic investigations of T. gondii strain types that differ in virulence in mice identified ROP5 (15–18) and ROP18 (15, 18–21) as key virulence factors.
ROPs are secreted from T. gondii rhoptry organelles directly into host cells during host cell invasion. After their secretion into host cells, ROP5 and ROP18 tether to the cytosolic face of parasitophorous vacuole (PV) membrane (PVM) through N-terminal arginine-rich amphipathic helix (RAH) domains (19, 22–24). PVM-associated ROP18 phosphorylates host immunity-related GTPases (IRGs) to prevent their accumulation on the PVM and thereby preserve the integrity of the PV from destruction (19, 25). Association of the ROP5 pseudokinase with ROP18 increases ROP18 kinase activity (26), and most likely the availability of host IRG substrates for their subsequent inactivation by the ROP18 kinase (27–29). ROP5 also associates in macromolecular complexes with ROP17, another PVM-associated kinase that phosphorylates and inactivates IRGs (22).
In laboratory strains of mice, South American and type I strains are virulent, type II strains exhibit reduced virulence, and type III strains are avirulent (30). While type I PVs avoid IRG accumulation (19), type II and type III PVs accumulate IRGs and are efficiently destroyed (31, 32). To explain these virulence differences in archetypal clonal lineages, virulent ROP18 gene alleles were proposed to inhibit accumulation of IRG proteins on the PVs of strain types that also express a virulent ROP5 locus (27, 28). This hypothesis explains increased virulence in type I strains that possess virulent ROP18 and ROP5 gene alleles. This hypothesis also explains avirulent type III strains that possess a virulent ROP5 locus and an avirulent and nonexpressed ROP18 gene allele. However, it is currently unclear how less virulent type II strains that carry a virulent ROP18 gene allele and an avirulent ROP5 gene locus resist host IFN-γ to establish chronic infection.
In addition to ROP5, ROP17, and ROP18, many ROP proteins are active kinases or pseudokinases (ROPKs), and the ROPKs collectively are characterized as the rhoptry kinome. The T. gondii rhoptry kinome was originally identified to consist of a set of 34 unique ROPK proteins and 10 additional degenerate ROPK genes that contain large DNA insertions or deletions within their kinase domains (33). Subsequently, four additional T. gondii ROPK genes were identified as ROP47, ROP48, ROP49, and ROP50 (34). Targeted deletion of ROP47 or ROP48 in a type II strain does not affect cyst burdens or chronic infection in vivo (35). However, the roles of most ROPKs in virulence, cyst development, and chronic infection in less virulent type II strains have not been directly tested. Here, we developed gene deletions of 26 ROPK gene loci encoding 31 unique type II ROPK proteins and examined the ability of these mutants to establish chronic infection in mice. Our results show that while several type II ROPK molecules moderately affect cyst burdens, ROP5, ROP17, ROP18, ROP35, and ROP38/29/19 (ROP38, ROP29, and ROP19) are essential for chronic infection.
RESULTS
Type II rhoptry kinome molecules are essential for chronic infection.
To broadly assess the roles of type II ROPK proteins in chronic infection, we targeted the deletion of 27 previously defined unique ROPK gene loci that encode 32 identified members of the ROPK gene family (ROP2/8, ROP5, ROP11, ROP16, ROP17, ROP18, ROP20, ROP21, ROP22, ROP23, ROP24, ROP25, ROP26, ROP27, ROP28, ROP30, ROP31, ROP32, ROP35, ROP36, ROP37, ROP38/29/19, ROP39, ROP40, ROP41, ROP42/43/44, and ROP45) (33). Additionally, we previously reported the deletion of the type II ROP4/7 locus (11). ROPK gene locus deletions were targeted in the Δku80 genetic model in the Toxoplasma gondii type II Prugniaud (Pru) background (11), and knockout genotypes were validated by PCR to measure targeted deletion of the ROPK gene locus and the correct integration of the HXGPRT selectable marker into the deleted ROPK gene locus (see Fig. S1A in the supplemental material) (36, 37). We validated the isolation of 26 ROPK gene locus knockouts (see Table S1 in the supplemental material). We assessed the roles of specific ROPK proteins in establishing chronic infection in vivo by measuring brain cyst burdens in C57BL/6 mice infected with ROPK knockout strains (Fig. 1). One ROPK knockout (Δrop16) exhibited a significant increase in cyst burdens, and cyst burdens were not significantly modified in 10 other ROPK knockout strains (Δrop11, Δrop20, Δrop23, Δrop24, Δrop26, Δrop28, Δrop32, Δrop39, Δrop42/43/44, and Δrop45 strains). In contrast, cyst burdens were moderately decreased by 46 to 68% in 11 other ROPK knockout strains (Δrop2/8, Δrop4/7, Δrop21, Δrop22, Δrop25, Δrop27, Δrop31, Δrop36, Δrop37, Δrop40, and Δrop41 strains). Importantly, cyst burdens were severely decreased by 75 to 80% in Δrop35 and Δrop38/29/19 strains and by >99.5% in Δrop5, Δrop17, and Δrop18 strains compared to the Δku80 parent (Fig. 1).
FIG 1 .

Rhoptry kinome molecules are essential for chronic infection. C57BL/6 mice were infected i.p. with 200 tachyzoites of each strain, and brain cyst burdens were measured 21 days postinfection. Data shown are cumulative results from one to three independent experiments for each strain tested. ROPK knockout strains and the Δku80 parent strain are organized in descending order by cyst number for each strain, with cyst numbers shown as means plus standard errors of the means (SEM) (error bars). P values were calculated by a Student’s t test, and a P < 0.05 (shown in purple or red color) was considered significant. Data on the ROPK knockout strains and the Δku80 parent strain follow: Δrop16 (two experiments; n = 6 mice; P = 0.0242), Δku80 parent (three experiments; n = 12 mice), Δrop42/43/44 (one experiment; n = 4 mice; P = 0.7188), Δrop39 (two experiments; n = 7 mice; P = 0.5691), Δrop23 (one experiment; n = 4 mice; P = 0.5232), Δrop45 (one experiment; n = 4 mice; P = 0.4186), Δrop24 (one experiment; n = 4 mice; P = 0.4259), Δrop32 (one experiment; n = 4 mice; P = 0.1651), Δrop11 (two experiments; n = 7 mice; P = 0.0703), Δrop20 (two experiments; n = 6 mice; P = 0.0534), Δrop28 (one experiment; n = 4 mice; P = 0.0928), Δrop26 (two experiments; n = 5 mice; P = 0.0500), Δrop41 (two experiments; n = 5 mice; P = 0.0384), Δrop37 (two experiments; n = 5 mice; P = 0.0154), Δrop4/7 (three experiments; n = 12 mice; P = 0.0198), Δrop36 (two experiments; n = 6 mice; P = 0.0070), Δrop31 (two experiments; n = 6 mice; P = 0.0058), Δrop21 (two experiments; n = 7 mice; P = 0.0036), Δrop40 (one experiment; n = 4 mice; P = 0.0217), Δrop27 (two experiments; n = 6 mice; P = 0.0073), Δrop25 (two experiments; n = 7 mice; P = 0.0013), Δrop22 (two experiments; n = 5 mice; P = 0.0095), Δrop2/8 (one experiment; n = 4 mice; P = 0.0063), Δrop38/29/19 (two experiments; n = 8 mice; P < 0.0001), Δrop35 (two experiments; n = 8 mice; P < 0.0001), Δrop18 (two experiments; n = 8 mice; P < 0.0001), Δrop17 (two experiments; n = 8 mice; P < 0.0001), and Δrop5 (three experiments; n = 9 mice; P < 0.0001).
ROPK knockout strains with the most severe defects in chronic infection stage differentiate in vitro.
One hypothesis is that ROPK knockout strains severely deficient in their ability to establish chronic infection may be defective in differentiation from the tachyzoite stage to the bradyzoite stage (4). To examine this, we used high pH to induce differentiation in vitro. Parental Δku80 strain tachyzoites switched normally to the bradyzoite cyst stage based on expression of cytosolic green fluorescent protein (GFP) under control of the bradyzoite stage-specific LDH2 gene promoter (11) and on the development of the cyst wall structure that surrounds the bradyzoites as assessed by Dolichos biflorus agglutinin (DBA) lectin staining of the major cyst wall protein CST1 (12) (Fig. 2A). The Δrop5, Δrop17, Δrop18, Δrop35, and Δrop38/29/19 knockout strains exhibited no defect in their ability to differentiate to cyst stages in vitro (Fig. 2A) or in their frequency of differentiation (data not shown). We next examined whether mice infected with high doses (>105 tachyzoites) of Δrop5, Δrop17, or Δrop18 tachyzoites could rescue normal cyst burdens. Cyst burdens remained extremely low even after high-dose infection of mice with Δrop5, Δrop17, or Δrop18 tachyzoites (Fig. 2B). Therefore, it was not a lack of differentiation that hindered cyst development in these knockout strains.
FIG 2 .
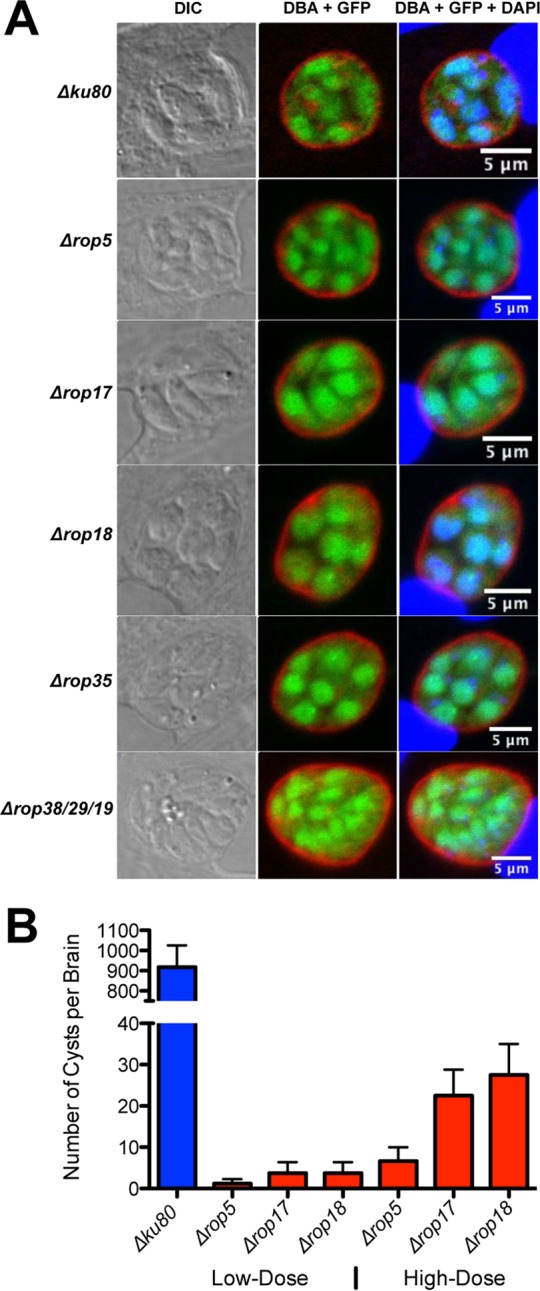
ROPK gene deletions with the most severe defects in cyst burdens differentiate normally in vitro. (A) Infected host cells were treated with bradyzoite inducing conditions (pH 8.1 and CO2 depletion in ambient air) for 3 days. The cyst wall was stained with Dolichos biflorus agglutinin (DBA) (shown in red). Bradyzoites were visualized by expression of GFP (shown in green), which is under control of the bradyzoite stage-specific LDH2 promoter. Host cell and parasite nuclei were stained with DAPI (shown in blue). Samples were imaged by confocal microscopy, and vacuoles were located using differential interference contrast (DIC) microscopy. Representative results are shown for each strain. (B) C57BL/6 mice were infected i.p. with 200 tachyzoites of each strain (low-dose infection) or with 2 × 105 or 2 × 106 tachyzoites of each strain (high-dose infection), and brain cyst burdens were measured. The values shown represent means plus SEM (error bars). Data shown are cumulative results from one to three independent experiments for each strain tested. Data on the ROPK knockout strains and the Δku80 parent strain follow: Δku80 parent low dose (three experiments; n = 12 mice), Δrop5 low dose (three experiments; n = 9 mice), Δrop17 low dose (two experiments; n = 8 mice), Δrop18 low dose (two experiments; n = 8 mice), Δrop5 high dose (three experiments; n = 6 mice), Δrop17 high dose (one experiment; n = 4 mice), and Δrop18 high dose (two experiments; n = 4 mice).
Single wild-type gene alleles rescue the cyst defect in Δrop17 and Δrop18 knockout strains, but not in the Δrop5 knockout strain.
The Δrop5, Δrop17, and Δrop18 knockout strains were complemented with their corresponding wild-type gene alleles targeted into the UPRT gene locus to simultaneously delete UPRT and enable selection of complemented strains in 5-fluorodeoxyuridine (FUDR) (see Fig. S1B in the supplemental material). Complementation of Δrop17 with C-terminal FLAG/hemagglutinin (HA)-tagged ROP17 (ROP17FLHA) (Fig. S2A) rescued cyst burdens (Fig. 3). The Δrop18 knockout strain was complemented with wild-type ROP18, a kinase-dead (KD) ROP18 (ROP18KD) (19), or a ROP18 gene allele [ROP18RAH2(ATF)] lacking the second RAH domain (RAH2) required for PVM association (23, 24). The ROP18 RAH2 domain also associates with ATF6β (ATF), a host endoplasmic reticulum stress sensor (38). Wild-type ROP18 fully rescued cyst burdens. However, expression of mutant ROP18KD or ROP18RAH2(ATF) gene alleles (Fig. S2B) failed to rescue cyst burdens (Fig. 3).
FIG 3 .
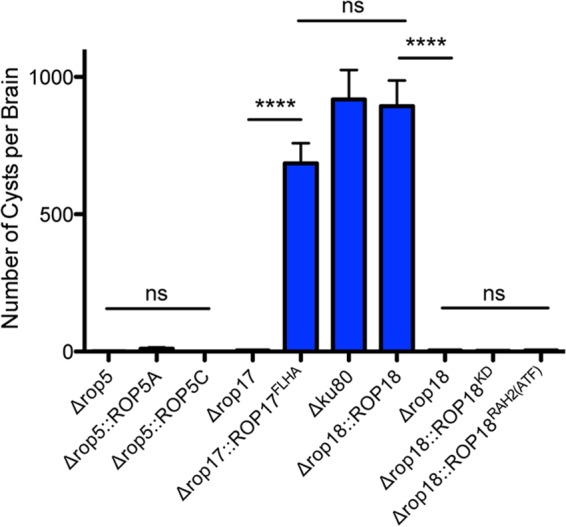
Complementation of Δrop17 and Δrop18 with the corresponding wild-type gene alleles rescues cyst burdens. C57BL/6 mice were infected i.p. with 200 tachyzoites of each strain, and brain cyst burdens were measured 21 days postinfection. Data shown are cumulative results from one to three independent experiments for each strain tested. Data on the Δrop17 and Δrop18 knockout strains and the Δku80 parent strain follow: Δku80 parent (three experiments; n = 12 mice), Δrop5 (three experiments; n = 9 mice), Δrop5::ROP5A (two experiments; n = 8 mice), Δrop5::ROP5C (two experiments; n = 8 mice), Δrop17 (two experiments; n = 8 mice), Δrop17::ROP17FLHA (one experiment; n = 4 mice), Δrop18 (two experiments; n = 8 mice), Δrop18::ROP18 (two experiments; n = 8 mice), Δrop18::ROP18KD (two experiments; n = 8 mice), Δrop18::ROP18RAH2(ATF) (two experiments; n = 8 mice). The values shown represent means ± SEM. The P values were calculated with a Student’s t test. P values that were significantly different are indicated by a bar and four asterisks (P < 0.0001). P values that were not significantly different (ns) are also indicated.
The type II ME49 strain ROP5 gene locus is multiallelic and possesses ~10 ROP5 gene alleles (16, 17). Complementation of Δrop5 through expression of the ROP5A or ROP5C gene allele (see Fig. S2C in the supplemental material) failed to rescue cyst burdens (Fig. 3), even after high-dose infection (data not shown). To examine the ROP5 locus in the type II Pru strain, we PCR amplified the full coding region of ROP5A and ROP5C alleles from the Δku80 parent strain and sequenced randomly selected clones. Eight ROP5C alleles were observed for each ROP5A allele (Fig. S3), suggesting that the Pru strain ROP5 locus is highly similar to the ME49 strain ROP5 locus (16, 17).
The Δrop17 knockout strain exhibits mild egress and growth defects in vitro.
ROPK knockout strains deficient in chronic infection may be compromised in their ability to replicate. To globally assess ROPK knockout strains for growth defects, we first examined plaques representing zones of infection where repeated cycles of invasion, intracellular replication, and egress occur. While all other ROPK knockout strains and their complemented strains developed plaques that were similar in size to those of the Δku80 parent (data not shown), the Δrop17 knockout strain developed smaller plaques (Fig. 4A). This small-plaque phenotype was rescued by expression of the ROP17FLHA gene (Fig. 4A). However, the Δrop17 small-plaque phenotype was not associated with any significant defect in the replication rate (see Fig. S4A in the supplemental material) or in the number of parasites per PV at 21 h (data not shown) or 45 h postinfection (Fig. S4B). In addition, the Δrop17, Δrop5, and Δrop18 knockout strains infected host cells as efficiently as the Δku80 parent (Fig. 4B). Consequently, we measured whether the Δrop17 strain exhibited any defect in the egress of replicated parasites from the PV. Tachyzoites egressed from a higher percentage of Δrop17 PVs after 45, 68, and 72 h of infection, and this early egress phenotype was rescued by complementation with ROP17FLHA (Fig. 4C). However, the average numbers of Δrop17 parasites at secondary sites of infection after release from the PVs were not different from those of the Δku80 parent or the ROP17FLHA complemented strain (Fig. S4C), suggesting that this early egress phenotype did not immediately alter invasion or continued replication of Δrop17 parasites.
FIG 4 .
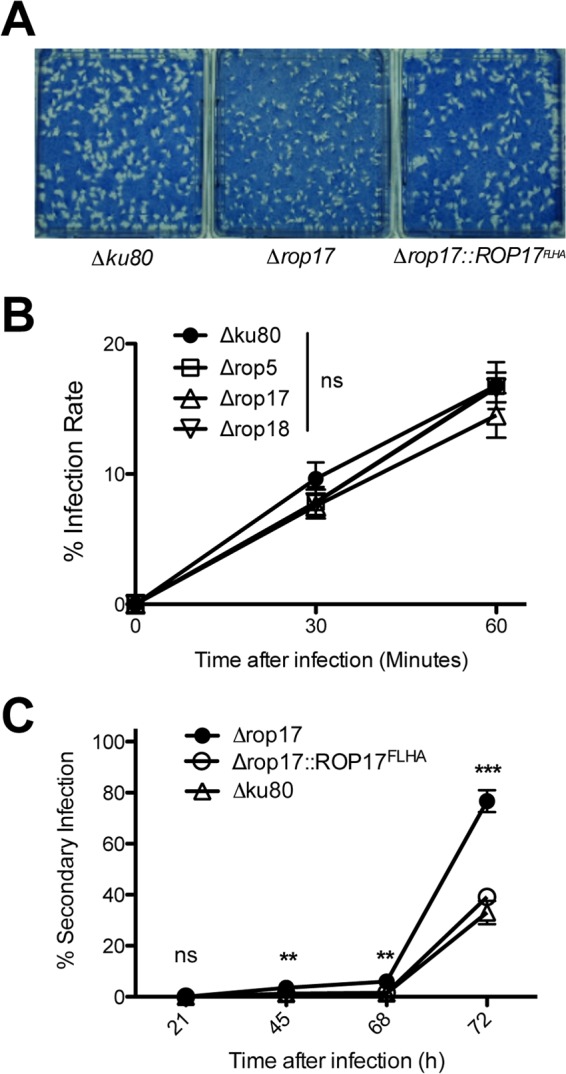
Deletion of ROP17 reduces plaque size and induces early egress. (A) Monolayers of human foreskin fibroblast (HFF) host cells were infected with ~200 tachyzoites of the indicated strains. PFU were visualized 13 days after infection by staining with Coomassie brilliant blue and photographed. The results shown are representative of three independent experiments. (B) Monolayers of HFF cells were infected with the indicated strains, and noninvaded parasites were removed from cultures at 30 or 60 min postinfection and PFU were determined. The infection rate was calculated by comparison to cultures where parasites were not removed (total PFU), and the percent infection rate was determined. Results are from cumulative data obtained in three independent experiments and are shown as means ± SEM (error bars). The values were not significantly different (ns) at 30 min or at 60 min postinfection, calculated with a Student’s t test. (C) Monolayers of HFF host cells were infected at an MOI of ~0.01, and noninvaded parasites were removed 1 h postinfection. At 21, 45, 68, or 72 h after infection, cultures were scored microscopically to measure the percentage of secondary infection sites. Results are from cumulative data obtained in three independent experiments and are shown as means ± SEM. The P values were calculated with a Student’s t test and are indicated as follows: **, P < 0.01; ***, P < 0.001; ns, not significant.
Type II ROP5 and ROP18 molecules actively resist PV killing, but ROP17 does not.
While virulent type I strains effectively resist host IRGs (19), the PVs of type II strains are highly susceptible to coating and killing by host IRGs (31, 32). As expected, only a low percentage of type II Δku80 PVs (~15%) survived in IFN-γ-stimulated mouse embryonic fibroblasts (MEFs) (Fig. 5A). In contrast, survival of the Δrop5 and Δrop18 knockout strains was further reduced by ~10% compared to that of the Δku80 parent. Complementation of Δrop18 with wild-type ROP18 rescued resistance to PV killing (Fig. 5A). Moreover, complementation of Δrop18 with ROP18 gene alleles defective in kinase activity (ROP18KD) or PVM association [ROP18RAH2(ATF)], as expected, failed to rescue Δrop18 resistance to PV killing. Single-copy gene alleles of ROP5A or ROP5C also failed to rescue Δrop5 resistance to PV killing (Fig. 5A). Single-copy gene alleles of virulent type I ROP5A or ROP5C were previously reported to incompletely rescue resistance to IRGs or virulence (16, 17, 28). Surprisingly, the Δrop17, Δrop35, and Δrop38/29/19 knockouts exhibited no defect in their resistance to PV killing in IFN-γ-stimulated MEFs (Fig. 5A).
FIG 5 .

Type II ROP5 and ROP18 resist Irgb6 coating and killing of the PV. (A) MEFs were stimulated with IFN-γ, and parasite survival (measured as PFU) was determined in comparison to nonstimulated MEFs. Results are from at least four independent experiments and are shown as means plus SEM. Significant P values were calculated with a Student’s t test. The values for all strains shown in red were not significantly different from the value for the Δku80 parent. The values for all strains shown in green were significantly different (P < 0.0001) from the value for the Δku80 parent strain. (B) Quantification of Irgb6 coating of PVs 45 min after infection of IFN-γ-stimulated bone marrow-derived macrophages. Representative images are shown of PVs stained with anti-Irgb6 (shown in green) and anti-GRA5 (shown in red). At least 500 PVs were scored to determine significance. Significant P values were calculated with a Student’s t test. The values for all strains shown in red were not significantly different from the value for the Δku80 parent. The values for all strains shown in green were significantly different from the value for the Δku80 parent strain. The P values for the knockout strains shown in green are listed in parentheses after the genotype: Δrop18 (P < 0.0001), Δrop18::ROP18KD (P < 0.0001), Δrop18::ROP18RAH2(ATF) (P < 0.0001), Δrop5 (P < 0.0013), Δrop5::ROP5A (P < 0.0001), and Δrop5::ROP5C (P < 0.0006).
Virulent type I alleles of ROP5, ROP17, and ROP18 are associated with reduced PV coating by Irgb6 in IFN-γ-stimulated macrophages (22). Irgb6 coating was significantly increased on Δrop5 PVs (~55% coating), and a single copy of the ROP5A or ROP5C gene allele failed to rescue resistance to coating (Fig. 5B). Irgb6 coating on Δrop18 PVs was markedly increased (~72% coating), and while wild-type ROP18 rescued this phenotype, the mutant ROP18KD or ROP18RAH2(ATF) gene allele failed to rescue resistance to Irgb6 coating (Fig. 5B). In contrast to Δrop5 and Δrop18 PVs, Δrop38/29/19 and Δrop17 PVs exhibited the same resistance to Irgb6 coating as the Δku80 parental strain (Fig. 5B). Collectively, these results suggest that while type II ROP5 and ROP18 resist IRG coating and PV killing, ROP17, ROP35, and ROP39/29/19, individually, were not required for IRG resistance.
Type II parasites deficient in ROP5 and ROP18 are less virulent than parasites deficient in ROP17, ROP35, or ROP38/29/19.
Virulence of the Δrop5, Δrop17, and Δrop18 knockout strains was measured in C57BL/6 mice. The Δku80 parent showed dose-dependent survival, and the 50% lethal dose (LD50) was ~1 × 105 tachyzoites (Fig. 6A). In contrast, the Δrop5, Δrop17, and Δrop18 knockout strains failed to exhibit virulence lethality after inoculation of mice with 2 × 105 tachyzoites (Fig. 6A). Furthermore, C57BL/6 mice deficient in the production of IFN-γ (IFN-γ−/− mice) failed to control a low-dose infection of Δrop5, Δrop17, or Δrop18 tachyzoites (Fig. 6B), suggesting that IFN-γ-stimulated mechanisms were necessary to control infection by these mutants. The Δrop17 knockout strain exhibited incomplete virulence lethality after inoculation of 2 × 106 tachyzoites (Fig. 6C). In contrast, all mice survived a 2 × 106 challenge dose of Δrop5 or Δrop18 tachyzoites (Fig. 6C). Mice infected with 2 × 107 Δrop18 tachyzoites also survived infection (Fig. 6D). However, a small fraction of mice infected with 2 × 107 Δrop5 tachyzoites succumbed to infection, whereas all mice infected with 2 × 107 Δrop17 tachyzoites succumbed (Fig. 6D). The Δrop17 LD50 was increased ~12-fold compared to that of the Δku80 parent, and the LD50 of the Δrop5 or Δrop18 strain was increased >100-fold. While single ROP5A and ROP5C gene alleles failed to rescue virulence of the Δrop5 knockout strain, the ROP17FLHA gene allele rescued virulence of the Δrop17 knockout strain, and wild-type ROP18 rescued virulence of the Δrop18 knockout strain (Fig. 6C). The mutant ROP18KD or ROP18RAH2(ATF) gene allele failed to rescue virulence of the Δrop18 knockout strain (Fig. 6C). These findings suggest that ROP5 and ROP18 play significant roles as type II virulence factors that resist host IRGs and parasite clearance, thereby promoting parasite survival and chronic infection. Surprisingly, ROP17 was associated with only a low-virulence phenotype, though this molecule was found to be as essential for chronic infection as ROP5 or ROP18. Moreover, parasites deficient in ROP35 or ROP38/29/19 were not virulence attenuated (Fig. 6E) yet were highly attenuated in cyst burdens (Fig. 1). Collectively, these findings suggest that additional mechanisms of host manipulation beyond direct interference with IRG functions at the PVM are associated with the successful development of chronic infection by type II strains of T. gondii.
FIG 6 .

Type II parasites deficient in ROP5 and ROP18 are less virulent than parasites deficient in ROP17, ROP35, or ROP38/29/19. The Δrop5, Δrop17, Δrop18, Δrop35, and Δrop38/29/19 mutants and complemented strains were evaluated for virulence lethality in mice. (A) C57BL/6 mice were infected i.p. with 2 × 105 tachyzoites of the Δrop5, Δrop17, or Δrop18 knockout strain or the Δku80 parent strain. Data shown are combined from two independent experiments, each with four mice per group. The P values were calculated by a log rank Mantel-Cox test, and a P of <0.05 was considered significant. (B) C57BL/6 IFN-γ−/− knockout mice were infected i.p. with 2 × 102 tachyzoites of the Δrop5, Δrop17, or Δrop18 strain or the Δku80 parental strain. Data shown are from a single experiment with four mice per group. The P values were calculated by a log rank Mantel-Cox test, and a P of <0.05 was considered significant. ns, not significant. (C) C57BL/6 mice were infected i.p. with 2 × 106 tachyzoites of the Δrop5, Δrop17, or Δrop18 strain or the Δku80 parental strain or with 2 × 106 tachyzoites of the Δrop5::ROP5A or Δrop5::ROP5C [shown together as Δrop5::ROP5A/C], Δrop17::ROP17FLHA, Δrop18::ROP18, Δrop18::ROP18KD, or Δrop18::ROP18RAH2(ATF) complemented strain. Data shown are combined from two independent experiments, each with four mice per group. The P values were calculated by a log rank Mantel-Cox test, and a P of <0.05 was considered significant. (D) C57BL/6 mice were infected i.p. with 2 × 107 tachyzoites of the Δrop5, Δrop17, or Δrop18 strain or the Δku80 parental strain. Data shown are combined from two independent experiments, each with four mice per group. The P values were calculated by a log rank Mantel-Cox test, and a P of <0.05 was considered significant. (E) C57BL/6 mice were infected i.p. with 2 × 105 tachyzoites of the Δrop35 or Δrop38/29/19 strain or the Δku80 parent strain. Data shown are combined from two independent experiments, each with four mice per group.
DISCUSSION
Toxoplasma gondii establishes a chronic infection of the host as an obligatory step for its biological transmission to new hosts (1). Herein, using the Δku80 genetic background that enables efficient and reliable genetic dissection of parasite gene families (39, 40), we deleted 26 ROPK gene loci encoding 31 known ROPK proteins and show for the first time that many type II ROPK proteins influence the ability of T. gondii to establish chronic infection. Moreover, several ROPKs, including ROP5, ROP17, ROP18, ROP35, and ROP38/29/19 were found to provide crucial influences on functions that were necessary to establish chronic infection.
The roles of ROP5, ROP17, or ROP18 molecules in establishing chronic infection of the host have not been assessed previously. Type II strains carry a virulent ROP18 gene allele and were previously proposed to behave in an avirulent fashion because type II strains also express avirulent ROP5 gene alleles (16, 17, 26–28). We found that deletion of either type II ROP18 or ROP5 essentially abrogated chronic infection and markedly reduced virulence lethality by >100-fold. These results correlate with the significant defects found in the resistance of Δrop18 and Δrop5 PVs to IRG coating and killing, suggesting that ROP5 and ROP18 cooperate as type II virulence factors despite the absence of a highly virulent ROP5 gene allele. Most likely, it is the absence of ROP5 gene alleles that limits type II virulence in comparison to the more highly virulent type I and South American strain types (15–17). This interpretation is consistent with previous evidence showing that coexpression of the virulent type I ROP5 gene locus in the type II background increased virulence by ~3 to 4 log units (28). Our study is the first to link the importance of type II ROP5 and ROP18 virulence functions to the development of cyst burdens and chronic infection.
Remarkably, nearly complete ablation of chronic infection in the Δrop5 and Δrop18 knockout strains was associated with only an ~10% reduction in parasite survival in IFN-γ-stimulated MEFs in vitro. Therefore, our results do not exclude the possibility that in addition to IRG resistance mechanisms, non-IRG functions of ROP5 and ROP18 complexes could also influence chronic infection. The N-terminal ROP18 RAH2 domain establishes PVM association, which is required for both IRG resistance and virulence but not for intrinsic ROP18 kinase activity (23, 24). The RAH2 domain also mediates ROP18 association with host ATF6β, and this interaction triggers the proteasome-dependent degradation of host ATF6β, and correspondingly, compromises adaptive immune responses mediated by CD8+ T cells (38). However, this specific ROP18-ATF6β interaction has only been previously demonstrated using recombinant ROP18 molecules that were not PVM associated (38). The multifunctional ROP18 molecule also associates with the p65 subunit of NF-κB to mediate its degradation (41). A second ROP18 ligand-binding pocket that influences virulence is also present in the ROP18 protein structure (42). Moreover, additional parasite proteins were recently identified in ROP5 and ROP18 complexes, suggesting there is unexpected biological complexity to their regulation and PVM-associated functions (22). Thus, future studies are necessary to decipher whether ROP5 and ROP18 IRG-independent mechanisms influence chronic infection.
Our findings establish that ROP17 is a key molecule that determines cyst development in type II strains. However, IRG resistance in the Δrop17 knockout strain was unaffected in IFN-γ-stimulated MEFs, and the Δrop17 strain exhibited only a low-virulence defect in comparison to deletion of ROP5 or ROP18. Previously, the deletion of ROP17 in the virulent type I RH strain was shown to reduce virulence, to increase Irgb6 coating of PVs, and to increase clearance of PVs in IFN-γ-stimulated macrophages (22). Together, these findings are consistent with previously reported evidence showing that the PVM-associated ROP17 kinase is polymorphic and under positive selective pressure in the virulent type I strain, which possesses 24-amino-acid differences compared with type II or type III ROP17 (22). Collectively, the data suggest that type II ROP17 is likely to play another role important for establishing chronic infection that is independent of the mild ROP17 virulence defect. The early egress phenotype of the Δrop17 knockout strain was not linked to an immediate defect in parasite invasion or replication, yet 13-day-old Δrop17 plaques were significantly reduced in their size. It is possible that ROP17 may function as a signaling molecule that influences the receptiveness of host cells to parasitism, the ability of the parasite to gain access to the central nervous system, or the development or maintenance of cyst burdens in the central nervous system. Recent elegant work based on rhoptry organelle secretion of Cre recombinase into host cells in contact with or invaded by T. gondii (43, 44) has demonstrated that ROP proteins are primarily injected into neurons within the brain (5). Another possibility is that the early egress phenotype of Δrop17 parasites may reduce the frequency of the overall success rate for tachyzoite differentiation to encysted bradyzoites in vivo. Alternatively, loss of ROP17 or other PVM-associated ROPK proteins may increase the susceptibility of the cyst wall to degradation by host chitinase, which is produced by alternatively activated macrophages in chronically infected mice (45). Future studies are necessary to more clearly identify the mechanisms that determine the key role of ROP17 in establishing chronic infection.
Severe reductions in cyst burdens were observed in the Δrop35 and Δrop38/29/19 knockout strains in the absence of any virulence defect in vivo or any defect in resistance to IRGs in vitro. These findings are intriguing in view of previous microarray evidence that defines ROP35 and ROP38 as the only ROPK genes that are significantly upregulated in less-virulent strain types (33). In particular, ROP38 transcripts are found to be more abundant in bradyzoite stages (increased >16-fold) compared to tachyzoite stages (33). Consequently, bradyzoite stage-specific expression of ROP38 may be important for the transition to or the maintenance of chronic stages in vivo. Because type II Δrop35 and Δrop39/29/19 knockout strains show severe impairments in cyst formation without changes in virulence, future studies may elucidate novel mechanisms that influence chronic infection.
While ROP38 expression is upregulated during bradyzoite stages, expression of ROP19 and ROP29, also present in the ROP38/29/19 gene locus, is downregulated in the bradyzoite stage (33). Moreover, of 13 ROPK genes showing bradyzoite stage-specific transcriptional regulation in vitro (33), we found that deletion of 9 of these ROPKs (ROP4/7, ROP5, ROP17, ROP18, ROP19, ROP29, ROP38, ROP40, and ROP41) reduced cyst burdens, while deletion of ROP16 increased cyst burdens. Stage regulation of ROPK expression appears to be important for the development or maintenance of cyst burdens in vivo.
ROP35 is expressed in Toxoplasma, Neospora, Eimeria, and Sarcocystis and maps to a basal ROPK cluster designated ROPKL (34). ROP38-related genes are expanded in both Toxoplasma and cyst-forming Neospora coccidian parasites (33). T. gondii ROP38 strongly influences the expression of a large number of host genes involved in mitogen-activated protein kinase (MAPK) signaling cascades, apoptosis, and host cell proliferation (33). ROP16 is another ROPK that strongly influences the expression of a large number of host genes (8, 46, 47), and inhibits the responsiveness of its host cell to IFN-γ signaling (46). Knockdown of the host STAT1 transcriptional coactivator tailless (TLX) was recently shown to repress a subset of IFN-γ-activated genes in the brain, and mice lacking expression of TLX in the brain were found to be impaired in their ability to control chronic infection (48). These observations suggest that ROPK molecules, such as ROP38, ROP35, and ROP16, could influence chronic infection through regulation of host response genes. Our findings provide the first definitive evidence showing that the type II T. gondii rhoptry kinome provides anti-IRG virulence functions in acute infection as well as additional mechanisms of host manipulation necessary to establish chronic infection.
MATERIALS AND METHODS
Ethics statement.
All mouse work was conducted in accordance with the recommendations in the Guide to the Care and Use of Laboratory Animals (49) and Association for the Assessment and Accreditation of Laboratory Animal Care guidelines. The animal protocol was approved by the Dartmouth College Committee on the Use and Care of Animals (Animal Welfare Assurance A3259-01, protocol bzik.dj2). All efforts were made to minimize suffering and pain.
Parasite culture.
Type II Prugniaud (Pru) background Toxoplasma gondii parasites were maintained by serial passage of tachyzoites in human foreskin fibroblast (HFF) monolayers cultured in Eagle’s modified essential medium (EMEM) containing 1% fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin as previously described (50, 51).
Mice and generation of bone marrow-derived macrophages.
Female 7- to 9-week-old C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were maintained at the Center for Comparative Medicine and Research at the Geisel School of Medicine at Dartmouth. Bone marrow-derived macrophages were isolated from bone marrow from C57BL/6 mice by differentiation in Dulbecco’s modified essential medium (DMEM) supplemented with 10% FBS, 1× minimal essential medium nonessential amino acids, 1 mM sodium pyruvate (Life Technologies), antibiotics, and 30% L929 culture supernatant as previously described (52). Bone marrow-derived macrophages were harvested after 5 days of differentiation.
Chronic infection.
Freshly lysed high-viability type II tachyzoites were obtained as previously described (11, 37). Tachyzoites were centrifuged at 900 × g for 7 min, washed, and counted in Dulbecco’s modified phosphate-buffered saline (DPBS). Mice were infected by intraperitoneal (i.p.) injection, and parasite viability was determined in a plaque assay at the time of mouse infection. To induce chronic infection, mice were infected i.p. with 2 × 102 tachyzoites of the tested type II strain. Higher parasite doses (2 × 105 or 2 × 106) were used in certain experiments as indicated.
Tissue cyst burden assay.
The brains from mice infected with type II strains were harvested at 3 weeks postinfection (11). The brains were homogenized in 2-ml volumes of sterile DPBS using a Dounce homogenizer. Cyst counts were performed on a minimum of 10% of each brain. Because Pru background cysts vary greatly in size and the brains of chronically infected mice contain many small, medium, and large cysts (11, 53), cysts were scored using dark-field microscopy with an inverted fluorescence phase-contrast microscope (Olympus CKX41) to reliably identify all cysts. The type II Δku80 strain expresses green fluorescent protein (GFP) under control of the bradyzoite stage-specific LDH2 promoter (11). GFP-positive (GFP+) cysts were scored at a total magnification of ×150 that provided the highest sensitivity for the detection of GFP+ bradyzoites within cysts and to then also verify the presence of a translucent thick cyst wall in bright-field microscopy (11, 37).
Generation of ROPK knockout strains.
Deletion of ROP kinome gene loci was performed using the KU80 knockout strain of the type II PruΔhxgprt strain as previously described (11, 37). Briefly, gene locus knockout targeting plasmids were assembled in yeast shuttle vectors pRS416 or pRS426 using yeast recombinational cloning to fuse three distinct PCR products with 31- to 34-bp crossovers in order; a 5′ GOI (gene of interest) target gene flank, the HXGPRT selectable marker, and a 3′ GOI target flank (see Fig. S1A in the supplemental material) (36). Knockout plasmids were engineered to delete at least 200 nucleotides of the 5′ untranslated region (5′ UTR) and the complete coding region of the ROPK gene locus as defined in the ToxoDB.org database (54). All primers used to construct knockout targeting plasmids and nucleotide definition of ROPK gene loci deletions are listed in Table S2 in the supplemental material. After plasmid validation by DNA sequencing, targeting plasmids were linearized at restriction sites inserted at either the 5′ end of the 5′-targeting flank or at the 3′ end of the 3′-targeting flank. Linearized targeting plasmids were transfected by electroporation into tachyzoites of the PruΔku80Δhxgprt strain and ROPK knockouts were selected in 50 µg/ml mycophenolic acid and 50 µg/ml xanthine, and parasites were cloned by limiting dilution 30 days after transfection. ROPK knockouts were validated by genotype analysis using PCR to measure the following (shown in Fig. S1A): (i) targeted deletion of the coding region of the targeted gene (DF and DR primers) in PCR 1, (ii) correct targeted 5′ integration (CXF and 5′DHFRCXR primers) in PCR 2, and (iii) correct targeted 3′ integration (3′DHFRCXF and CXR primers) in PCR 3 using knockout validation primers shown in Table S3.
Complementation of ROPK knockouts.
Complementation plasmids were designed to complement ROPK knockout strains PruΔku80ΔropGOI through chromosomal integration and expression of wild-type or mutant gene alleles at the uracil phosphoribosyltransferase (UPRT) chromosomal locus (TGME49_312480) as previously described (11) (see Fig. S1B in the supplemental material). Complementation plasmids were developed in the pRS416 or pRS426 yeast shuttle vector using yeast recombination to fuse, in order, a 5′ UPRT target flank, the complementing gene of interest with native 5′ UTR, and the 3′ UPRT target flank (Fig. S1B). Oligonucleotide DNA primers (Table S4) were used to generate the complementing genes, synthesized on one or two PCR products. ROP18 mutant gene alleles possessing a kinase-dead (KD) domain or deleted RAH2 domain [RAH2(ATF)] were engineered as previously described (19, 38). Following plasmid assembly by yeast recombinational cloning, targeting plasmids were validated by DNA sequencing. Prior to transfection, plasmids were linearized via the unique restriction site PmeI. The parasites were cultured for 2 days in normal infection media, and the cultures were then switched to selection medium containing 2 µM 5-fluorodeoxyuridine (FUDR) and cloned 30 days after transfection by limiting dilution. Targeting of complementing genes to the UPRT locus was validated by genotype analysis using PCR assays (strategy shown in Fig. S1B) to measure the following: (i) deletion of UPRT coding region in PCR 4, (ii) correct targeted 5′ integration in PCR 5, and (iii) correct targeted 3′ integration of the complementing transgene at the UPRT locus in PCR 6 using oligonucleotide DNA validation primers (Table S5).
Characterization of ROP5A and ROP5C gene alleles.
Genomic DNA isolated from the Δku80 parent strain was used as the template to PCR amplify the complete coding region of ROP5A and ROP5C gene alleles using primers ATGGCGACGAAGCTCGCTAGAC (forward) and GAGCCGTTTTCTCAAAGCGACTGAG (reverse). PCR products were randomly cloned into the pCR4 Topo vector (Invitrogen), and individual clones were sequenced. All sequences were managed with MacVector and aligned using ClustalW.
Intracellular replication rate assay.
Parasite growth rate was determined using methods described previously employing a direct parasite per vacuole scoring approach (11). Briefly, triplicate monolayers of HFF cells were infected at a multiplicity of infection (MOI) of ~0.2. and parasites were allowed to invade cells for 1 h. Monolayers were then washed three times in DPBS to remove extracellular parasites. At 21 h and 45 h postinfection, tachyzoites per vacuole were scored in at least 50 randomly encountered vacuoles.
Plaque assay.
Confluent monolayers of HFF cells were infected with 200 tachyzoites of a type II strain in infection medium, and cultures were left undisturbed for 13 days to allow localized development of PFU. The medium in the cultures was removed, and the cultures were fixed in 50% methanol and 10% acetic acid and stained with 0.25% Coomassie brilliant blue for 2 days. Cultures were rinsed in water, air dried, and photographed (55).
Infection rate assay.
Parasite infection rate was determined in PFU assays. Briefly, multiple monolayers of HFF cells in 25-cm2 flasks were infected with 500 tachyzoites. At 30 and 60 min postinfection, the monolayers were washed three times in DPBS to remove extracellular parasites, and either infection medium was replaced, or parasites were not removed from continuously incubated control flask. The cultures were incubated continuously for 11 additional days to establish PFU. Percent infection rate was determined as the number of PFU present at each time divided by the number of PFU in the continuously incubated cultures.
Early egress assay.
Tachyzoites in primary type II PruΔku80 PVs egress at ~72 h postinfection (37). HFF cells were infected at an MOI of 0.01, and 1 h after infection, the monolayers were washed three times in DPBS to remove extracellular parasites, and infection medium was replaced. At least 500 infection sites were scored at 21, 45, 68, and 72 h postinfection to measure the percentage of intact primary PVs or secondary sites of infection sites (after parasite egress from the PV). The number of tachyzoites present in secondary infection sites was also measured at 45 h and 72 h postinfection by scoring at least 100 secondary sites of infection.
PV killing assay.
C57BL/6 MEFs (ATCC) were seeded into 24-well trays and were cultured in DMEM in 15% FBS. MEF monolayers were incubated without IFN-γ, or MEF monolayers were stimulated with 200 U/ml IFN-γ (Peprotech) 24 h prior to parasite infection to activate host immunity-related GTPases and cell autonomous killing mechanisms. Triplicate wells of MEFs were infected with 200 or 1,000 tachyzoites, and plaques were allowed to develop for 5 to 7 days. The total number of PFU per well was counted microscopically, and the percentage of PFU survival was calculated as the number of PFU in IFN-γ-stimulated MEFs divided by the number of PFU scored in nonstimulated MEFs (no IFN-γ treatment).
IRG coating assay.
Bone marrow-derived macrophages were harvested, seeded on circular cover glasses (Electron Microscopy Sciences), and incubated overnight, and then macrophages were primed with 100 U/ml IFN-γ and 10 U/ml tumor necrosis factor alpha (TNF-α) (Peprotech) for 6 h. The macrophages on the coverslips were infected with parasites at an MOI of 4 for 45 min, the coverslips were washed in DPBS, and infected cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences), permeabilized with 0.1% saponin (Sigma), and blocked in 10% FBS. For visualization, cultures were incubated with mouse anti-GRA5 (1:2,000) (clone TG 17.113; Biotem) and rabbit anti-Irgb6 (1:1,000) (56), then washed and incubated with secondary antibodies anti-mouse antibodies conjugated to Alexa Fluor 568 and anti-rabbit antibodies conjugated to Alexa Fluor 488 (Invitrogen). Coverslips were mounted in ProLong gold with DAPI (4′,6′-diamidino-2-phenylindole) (Invitrogen) and imaged at 63× with a Nikon A1R SI confocal microscope (Nikon, Inc.). All images were processed with FIJI (57). A minimum of 500 PVs was scored for each strain for quantification of Irgb6 PV coating.
Immunofluorescence assay.
HFF cells were cultured on circular microcover glasses and infected with parasites for ~16 to 30 h. To visualize ROP5, the cultures were fixed in 4% paraformaldehyde and permeabilized with 0.01% Triton X-100. To visualize ROP17 and ROP18, the cultures were fixed with Histochoice (Amresco) and permeabilized in 0.1% saponin (Sigma) for 10 min. All samples were blocked with 10% FBS and incubated with primary antibodies for 1 h at room temperature (RT). The primary antibodies were rabbit anti-ROP5 (MO556; 1:3,000 dilution) (26), rabbit anti-ROP18 (ROP18-His WA525; 1:500 dilution; Sibley Laboratory), and anti-HA to visualize ROP17FLHA (rabbit monoclonal anti-HA tag antibodies; 1:500 dilution; Cell Signaling). Preparations were washed three times with PBS and incubated 1 h at RT with a 1:1,000 dilution of secondary goat anti-rabbit IgG antibodies conjugated to Alexa Fluor 488. Samples were mounted in SlowFade gold with DAPI (Life Technologies).
In vitro cyst differentiation assay.
Tachyzoites were differentiated in vitro into bradyzoites within cysts essentially as previously and elegantly described by Tobin and colleagues (58). Differentiation media contained Roswell Park Memorial Institute medium (RPMI) without bicarbonate supplemented with 2.05 mM l-glutamine (Hyclone), 20 mM HEPES-free acid (IBI Scientific), 1% XL-glutamine (a long-lasting stable form of glutamine; VWR), 1% FBS, and 1% penicillin-streptomycin. The pH of differentiation medium was adjusted to 8.1 with sodium hydroxide and filter sterilized. HFF cells were cultured on circular microcover glass, and confluent monolayers were infected with type II parasites at an MOI of ~0.5. Three hours after infection, the infected cells were washed once in DPBS supplemented with Ca2+ and Mg2+, and cultures were incubated in differentiation media for 3 days at 37°C in ambient air. Infected cells were fixed in 4% paraformaldehyde, and the excess was quenched with 0.1 M glycine. All samples were permeabilized and blocked in 3% FBS plus 0.2% Triton X-100 for 30 min at room temperature, and then they were incubated with a 1:250 dilution of rhodamine-labeled Dolichos biflorus agglutinin (Vector Laboratories) for 1 h at RT. The preparations were washed three times with DPBS, mounted in SlowFade gold antifade with DAPI (Life Technologies), and imaged by confocal microscopy.
SUPPLEMENTAL MATERIAL
Knockout and complementation strategy. (A) Knockout strategy to insert HXGPRT at deleted gene loci. The design of genotype validation PCR is shown. MPA, mycophenolic acid; X, xanthine. B) Complementation strategy at the UPRT locus. The design of genotype validation PCR is shown. FUDR, 5-fluorodeoxyuridine. Download
Complementation of Δrop5, Δrop17, and Δrop18. Wild-type or mutant gene alleles or ROP5, ROP17, or ROP18, as designated, were evaluated for expression and rhoptry localization of protein products. Nuclei were stained with DAPI, and PVs were identified using DIC microscopy. Download
Amino acid sequence of type II ROP5 alleles. Type II ROP5 gene alleles were characterized by gene sequencing and compared with the corresponding ROP5 gene alleles of strain ME49. ROP5A allele-specific amino acid changes are shown in green, and ROP5C allele-specific amino acid changes are shown in red. Download
Intracellular replication rates of ROPK knockouts that influence chronic infection. (A) Replication rate, measured as the average number of tachyzoites per vacuole 45 h postinfection, was determined for ROPK knockout strains that exhibited reductions or increases in cyst burdens. (B) The percentage of parasite vacuoles containing various number of tachyzoites was measured 45 h postinfection for the Δrop17 and Δrop17::ROP17FLHA knockout strains and the parental Δku80 strain. (C) The number of tachyzoites present in secondary sites of infection was determined at 45 h and 72 h postinfection for the Δrop17 and Δrop17::ROP17FLHA strains and the parental Δku80 strain. Download
Genotypes of strains developed or used in this study.
Primers used to construct ROPK knockouts. The sequences of oligonucleotide forward primers (FP) and reverse primers (RP) used for construction of deletion-targeting plasmids pROP2/8P (Δrop2/8), pROP5P (Δrop5), pROP11P (Δrop11), pROP16P (Δrop16), pROP17P (Δrop17), pROP18P (Δrop18), pROP20P (Δrop20), pROP21P (Δrop21), pROP22P (Δrop22), pROP23P (Δrop23), pROP24P (Δrop24), pROP25P (Δrop25), pROP26P (Δrop26), pROP27P (Δrop27), pROP28P (Δrop28), pROP30P (Δrop30), pROP31P (Δrop31), pROP32P (Δrop32), pROP35P (Δrop35), pROP36P (Δrop36), pROP37P (Δrop37), and pROP38P (Δrop38/29/19), pROP39P (Δrop39), pROP40P (Δrop40), pROP41P (Δrop41), pROP42,43,44P (Δrop42/43/44), and pROP45P (Δrop45) are shown. The corresponding T. gondii ME49 (TgME49) gene locus, chromosome, and nucleotides deleted for each knockout were determined from data in toxodb.org.
Primers used to validate ROPK knockouts. The sequences of oligonucleotide primers used for validation of Δrop2/8, Δrop5, Δrop11, Δrop16, Δrop17, Δrop18, Δrop20, Δrop21, Δrop22, Δrop23, Δrop24, Δrop25, Δrop26, Δrop27, Δrop28, Δrop30, Δrop31, Δrop32, Δrop35, Δrop36, Δrop37, Δrop38/29/19, Δrop39, Δrop40, Δrop41, Δrop42/43/44, and Δrop45 knockouts are shown. The primers were designed from TgME49 data in toxodb.org.
Primers used to develop targeting plasmids for complementation. The sequences of oligonucleotide forward primers (FP) and reverse primers (RP) used for construction of Pru complementation vectors pURO5A (::ROP5A), pURO5C (::ROP5C), pURO17FLHA (::ROP17FLHA), pURO18 (::ROP18), pURO18KD (::ROP18KD), and pURO18RAH2(ATF) [::ROP18RAH2(ATF)] are shown. The corresponding TgME49 gene locus, chromosome, and nucleotides deleted for each knockout were determined from data in toxodb.org.
Primers used to validate complementation of ROPK knockouts. The sequences of oligonucleotide primers used to validate integration of complementation vectors pURO5A (::ROP5A), pURO5C (::ROP5C), pURO17FLHA (::ROP17FLHA), pURO18 (::ROP18), pURO18KD (::ROP18KD), and pURO18RAH2(ATF) [::ROP18RAH2(ATF)] are shown. Primers were designed from TgME49 data in toxodb.org.
ACKNOWLEDGMENTS
We are grateful to developers of the ToxoDB.org Genome Resource. ToxoDB and EuPathDB are part of the National Institutes of Health/National Institutes of Allergy and Infectious Diseases (NIH/NIAID)-funded Bioinformatics Resource Center. We thank David Sibley for providing antibodies specific to ROP5 and ROP18.
This work was sponsored by grants AI041930, AI075931, AI084570, AI097018, and AI104514 from the National Institutes of Health (NIH). L.M.R. was a trainee on NIH training grants 5T32AI007363 and 2T32AI007519. R.B.G. was a trainee on NIH training grant 2T32AI007519.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Citation Fox BA, Rommereim LM, Guevara RB, Falla A, Hortua Triana MA, Sun Y, Bzik DJ. 2016. The Toxoplasma gondii rhoptry kinome is essential for chronic infection. mBio 7(3):e00193-16. doi:10.1128/mBio.00193-16.
REFERENCES
- 1.Dubey JP. 2007. The history and life cycle of Toxoplasma gondii, p 1–18. In Weiss LM, Kim K (), Toxoplasma gondii: the model apicomplexan parasite: perspectives and methods. Elsevier, London, United Kingdom. [Google Scholar]
- 2.Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, Sher A. 1991. Synergistic role of CD4+ and CD8+ T lymphocytes in IFN-gamma production and protective immunity induced by an attenuated Toxoplasma gondii vaccine. J Immunol 146:286–292. [PubMed] [Google Scholar]
- 3.Konradt C, Ueno N, Christian DA, Delong JH, Pritchard GH, Herz J, Bzik DJ, Koshy AA, McGavern DB, Lodoen MB, Hunter CA. 2016. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat Microbiol 1:16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss LM, Kim K. 2000. The development and biology of bradyzoites of Toxoplasma gondii. Front Biosci 5:D391–D405. doi: 10.2741/Weiss. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabral CM, Tuladhar S, Dietrich HK, Nguyen E, MacDonald WR, Trivedi T, Devineni A, Koshy AA. 2016. Neurons are the primary target cell for the brain-tropic intracellular parasite Toxoplasma gondii. PLoS Pathog 12:e1005447. doi: 10.1371/journal.ppat.1005447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melzer TC, Cranston HJ, Weiss LM, Halonen SK. 2010. Host cell preference of Toxoplasma gondii cysts in murine brain: a confocal study. J Neuroparasitol 1:N100505. doi: 10.4303/jnp/N100505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contini C. 2008. Clinical and diagnostic management of toxoplasmosis in the immunocompromised patient. Parassitologia 50:45–50. [PubMed] [Google Scholar]
- 8.Denkers EY, Bzik DJ, Fox BA, Butcher BA. 2012. An inside job: hacking into Janus kinase/signal transducer and activator of transcription signaling cascades by the intracellular protozoan Toxoplasma gondii. Infect Immun 80:476–482. doi: 10.1128/IAI.05974-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter CA, Sibley LD. 2012. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol 10:766–778. doi: 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchholz KR, Bowyer PW, Boothroyd JC. 2013. Bradyzoite pseudokinase 1 is crucial for efficient oral infectivity of the Toxoplasma gondii tissue cyst. Eukaryot Cell 12:399–410. doi: 10.1128/EC.00343-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox BA, Falla A, Rommereim LM, Tomita T, Gigley JP, Mercier C, Cesbron-Delauw MF, Weiss LM, Bzik DJ. 2011. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot Cell 10:1193–1206. doi: 10.1128/EC.00297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomita T, Bzik DJ, Ma YF, Fox BA, Markillie LM, Taylor RC, Kim K, Weiss LM. 2013. The Toxoplasma gondii cyst wall protein CST1 is critical for cyst wall integrity and promotes bradyzoite persistence. PLoS Pathog 9:e1003823. doi: 10.1371/journal.ppat.1003823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorenzi H, Khan A, Behnke MS, Namasivayam S, Swapna LS, Hadjithomas M, Karamycheva S, Pinney D, Brunk BP, Ajioka JW, Ajzenberg D, Boothroyd JC, Boyle JP, Darde ML, Diaz-Miranda MA, Dubey JP, Fritz HM, Gennari SM, Gregory BD, Kim K, Saeij JP, Su C, White MW, Zhu XQ, Howe DK, Rosenthal BM, Grigg ME, Parkinson J, Liu L, Kissinger JC, Roos DS, Sibley LD. 2016. Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat Commun 7:10147. doi: 10.1038/ncomms10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boothroyd JC, Dubremetz JF. 2008. Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat Rev Microbiol 6:79–88. doi: 10.1038/nrmicro1800. [DOI] [PubMed] [Google Scholar]
- 15.Behnke MS, Khan A, Lauron EJ, Jimah JR, Wang Q, Tolia NH, Sibley LD. 2015. Rhoptry proteins ROP5 and ROP18 are major murine virulence factors in genetically divergent South American strains of Toxoplasma gondii. PLoS Genet 11:e1005434. doi: 10.1371/journal.pgen.1005434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behnke MS, Khan A, Wootton JC, Dubey JP, Tang K, Sibley LD. 2011. Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc Natl Acad Sci U S A 108:9631–9636. doi: 10.1073/pnas.1015338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reese ML, Zeiner GM, Saeij JP, Boothroyd JC, Boyle JP. 2011. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc Natl Acad Sci U S A 108:9625–9630. doi: 10.1073/pnas.1015980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shwab EK, Jiang T, Pena HF, Gennari SM, Dubey JP, Su C. 2016. The ROP18 and ROP5 gene allele types are highly predictive of virulence in mice across globally distributed strains of Toxoplasma gondii. Int J Parasitol 46:141–146. doi: 10.1016/j.ijpara.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Fentress SJ, Behnke MS, Dunay IR, Mashayekhi M, Rommereim LM, Fox BA, Bzik DJ, Taylor GA, Turk BE, Lichti CF, Townsend RR, Qiu W, Hui R, Beatty WL, Sibley LD. 2010. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8:484–495. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saeij JP, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783. doi: 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor S, Barragan A, Su C, Fux B, Fentress SJ, Tang K, Beatty WL, Hajj HE, Jerome M, Behnke MS, White M, Wootton JC, Sibley LD. 2006. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314:1776–1780. doi: 10.1126/science.1133643. [DOI] [PubMed] [Google Scholar]
- 22.Etheridge RD, Alaganan A, Tang K, Lou HJ, Turk BE, Sibley LD. 2014. The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe 15:537–550. doi: 10.1016/j.chom.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fentress SJ, Steinfeldt T, Howard JC, Sibley LD. 2012. The arginine-rich N-terminal domain of ROP18 is necessary for vacuole targeting and virulence of Toxoplasma gondii. Cell Microbiol 14:1921–1933. doi: 10.1111/cmi.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reese ML, Boothroyd JC. 2009. A helical membrane-binding domain targets the Toxoplasma ROP2 family to the parasitophorous vacuole. Traffic 10:1458–1470. doi: 10.1111/j.1600-0854.2009.00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinfeldt T, Könen-Waisman S, Tong L, Pawlowski N, Lamkemeyer T, Sibley LD, Hunn JP, Howard JC. 2010. Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol 8:e1000576. doi: 10.1371/journal.pbio.1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behnke MS, Fentress SJ, Mashayekhi M, Li LX, Taylor GA, Sibley LD. 2012. The polymorphic pseudokinase ROP5 controls virulence in Toxoplasma gondii by regulating the active kinase ROP18. PLoS Pathog 8:e1002992. doi: 10.1371/journal.ppat.1002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleckenstein MC, Reese ML, Könen-Waisman S, Boothroyd JC, Howard JC, Steinfeldt T. 2012. A Toxoplasma gondii pseudokinase inhibits host IRG resistance proteins. PLOS Biol 10:e1001358. doi: 10.1371/journal.pbio.1001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niedelman W, Gold DA, Rosowski EE, Sprokholt JK, Lim D, Farid Arenas A, Melo MB, Spooner E, Yaffe MB, Saeij JP. 2012. The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-gamma response. PLoS Pathog 8:e1002784. doi: 10.1371/journal.ppat.1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reese ML, Shah N, Boothroyd JC. 2014. The Toxoplasma pseudokinase ROP5 is an allosteric inhibitor of the immunity-related GTPases. J Biol Chem 289:27849–27858. doi: 10.1074/jbc.M114.567057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sibley LD, Ajioka JW. 2008. Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu Rev Microbiol 62:329–351. doi: 10.1146/annurev.micro.62.081307.162925. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS. 2009. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J Immunol 182:3775–3781. doi: 10.4049/jimmunol.0804190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao YO, Khaminets A, Hunn JP, Howard JC. 2009. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog 5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peixoto L, Chen F, Harb OS, Davis PH, Beiting DP, Brownback CS, Ouloguem D, Roos DS. 2010. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe 8:208–218. doi: 10.1016/j.chom.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talevich E, Kannan N. 2013. Structural and evolutionary adaptation of rhoptry kinases and pseudokinases, a family of coccidian virulence factors. BMC Evol Biol 13:117. doi: 10.1186/1471-2148-13-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Camejo A, Gold DA, Lu D, McFetridge K, Julien L, Yang N, Jensen KD, Saeij JP. 2014. Identification of three novel Toxoplasma gondii rhoptry proteins. Int J Parasitol 44:147–160. doi: 10.1016/j.ijpara.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. 2009. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot Cell 8:520–529. doi: 10.1128/EC.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rommereim LM, Hortua Triana MA, Falla A, Sanders KL, Guevara RB, Bzik DJ, Fox BA. 2013. Genetic manipulation in Deltaku80 strains for functional genomic analysis of Toxoplasma gondii. J Vis Exp 77:e50598. doi: 10.3791/50598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto M, Ma JS, Mueller C, Kamiyama N, Saiga H, Kubo E, Kimura T, Okamoto T, Okuyama M, Kayama H, Nagamune K, Takashima S, Matsuura Y, Soldati-Favre D, Takeda K. 2011. ATF6beta is a host cellular target of the Toxoplasma gondii virulence factor ROP18. J Exp Med 208:1533–1546. doi: 10.1084/jem.20101660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bzik DJ. 2016. Serial dissection of parasite gene families. Infect Immun 84:1252–1254. doi: 10.1128/IAI.00175-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long S, Wang Q, Sibley LD. 2016. Analysis of non-canonical calcium dependent protein kinases in Toxoplasma gondii by targeted gene deletion using CRISPR/Cas9. Infect Immun 84:1262–1273. doi: 10.1128/IAI.01173-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du J, An R, Chen L, Shen Y, Chen Y, Cheng L, Jiang Z, Zhang A, Yu L, Chu D, Shen Y, Luo Q, Chen H, Wan L, Li M, Xu X, Shen J. 2014. Toxoplasma gondii virulence factor ROP18 inhibits the host NF-kappaB pathway by promoting p65 degradation. J Biol Chem 289:12578–12592. doi: 10.1074/jbc.M113.544718. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Lim D, Gold DA, Julien L, Rosowski EE, Niedelman W, Yaffe MB, Saeij JP. 2013. Structure of the Toxoplasma gondii ROP18 kinase domain reveals a second ligand binding pocket required for acute virulence. J Biol Chem 288:34968–34980. doi: 10.1074/jbc.M113.523266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christian DA, Koshy AA, Reuter MA, Betts MR, Boothroyd JC, Hunter CA. 2014. Use of transgenic parasites and host reporters to dissect events that promote interleukin-12 production during toxoplasmosis. Infect Immun 82:4056–4067. doi: 10.1128/IAI.01643-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koshy AA, Dietrich HK, Christian DA, Melehani JH, Shastri AJ, Hunter CA, Boothroyd JC. 2012. Toxoplasma co-opts host cells it does not invade. PLoS Pathog 8:e1002825. doi: 10.1371/journal.ppat.1002825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nance JP, Vannella KM, Worth D, David C, Carter D, Noor S, Hubeau C, Fitz L, Lane TE, Wynn TA, Wilson EH. 2012. Chitinase dependent control of protozoan cyst burden in the brain. PLoS Pathog 8:e1002990. doi: 10.1371/journal.ppat.1002990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denkers EY. 2011. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog 7:e1002236. doi: 10.1371/journal.ppat.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jensen KD, Wang Y, Wojno ED, Shastri AJ, Hu K, Cornel L, Boedec E, Ong YC, Chien YH, Hunter CA, Boothroyd JC, Saeij JP. 2011. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 9:472–483. doi: 10.1016/j.chom.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beiting DP, Hidano S, Baggs JE, Geskes JM, Fang Q, Wherry EJ, Hunter CA, Roos DS, Cherry S. 2015. The orphan nuclear receptor TLX is an enhancer of STAT1-mediated transcription and immunity to Toxoplasma gondii. PLoS Biol 13:e1002200. doi: 10.1371/journal.pbio.1002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 50.Fox BA, Gigley JP, Bzik DJ. 2004. Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int J Parasitol 34:323–331. doi: 10.1016/j.ijpara.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Gigley JP, Fox BA, Bzik DJ. 2009. Cell-mediated immunity to Toxoplasma gondii develops primarily by local Th1 host immune responses in the absence of parasite replication. J Immunol 182:1069–1078. doi: 10.4049/jimmunol.182.2.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim L, Butcher BA, Denkers EY. 2004. Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J Immunol 172:3003–3010. doi: 10.4049/jimmunol.172.5.3003. [DOI] [PubMed] [Google Scholar]
- 53.Aldebert D, Hypolite M, Cavailles P, Touquet B, Flori P, Loeuillet C, Cesbron-Delauw MF. 2011. Development of high-throughput methods to quantify cysts of Toxoplasma gondii. Cytometry A 79:952–958. doi: 10.1002/cyto.a.21138. [DOI] [PubMed] [Google Scholar]
- 54.Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, Heiges M, Iodice J, Kissinger JC, Mackey AJ, Pinney DF, Roos DS, Stoeckert CJ Jr, Wang H, Brunk BP. 2008. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res 36:D553–D556. doi: 10.1093/nar/gkm981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fox BA, Bzik DJ. 2010. Avirulent uracil auxotrophs based on disruption of orotidine-5′-monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii. Infect Immun 78:3744–3752. doi: 10.1128/IAI.00287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henry SC, Daniell XG, Burroughs AR, Indaram M, Howell DN, Coers J, Starnbach MN, Hunn JP, Howard JC, Feng CG, Sher A, Taylor GA. 2009. Balance of Irgm protein activities determines IFN-gamma-induced host defense. J Leukoc Biol 85:877–885. doi: 10.1189/jlb.1008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tobin C, Pollard A, Knoll L. 2010. Toxoplasma gondii cyst wall formation in activated bone marrow-derived macrophages and bradyzoite conditions. J Vis Exp 2010(42):e2091. doi: 10.3791/2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Knockout and complementation strategy. (A) Knockout strategy to insert HXGPRT at deleted gene loci. The design of genotype validation PCR is shown. MPA, mycophenolic acid; X, xanthine. B) Complementation strategy at the UPRT locus. The design of genotype validation PCR is shown. FUDR, 5-fluorodeoxyuridine. Download
Complementation of Δrop5, Δrop17, and Δrop18. Wild-type or mutant gene alleles or ROP5, ROP17, or ROP18, as designated, were evaluated for expression and rhoptry localization of protein products. Nuclei were stained with DAPI, and PVs were identified using DIC microscopy. Download
Amino acid sequence of type II ROP5 alleles. Type II ROP5 gene alleles were characterized by gene sequencing and compared with the corresponding ROP5 gene alleles of strain ME49. ROP5A allele-specific amino acid changes are shown in green, and ROP5C allele-specific amino acid changes are shown in red. Download
Intracellular replication rates of ROPK knockouts that influence chronic infection. (A) Replication rate, measured as the average number of tachyzoites per vacuole 45 h postinfection, was determined for ROPK knockout strains that exhibited reductions or increases in cyst burdens. (B) The percentage of parasite vacuoles containing various number of tachyzoites was measured 45 h postinfection for the Δrop17 and Δrop17::ROP17FLHA knockout strains and the parental Δku80 strain. (C) The number of tachyzoites present in secondary sites of infection was determined at 45 h and 72 h postinfection for the Δrop17 and Δrop17::ROP17FLHA strains and the parental Δku80 strain. Download
Genotypes of strains developed or used in this study.
Primers used to construct ROPK knockouts. The sequences of oligonucleotide forward primers (FP) and reverse primers (RP) used for construction of deletion-targeting plasmids pROP2/8P (Δrop2/8), pROP5P (Δrop5), pROP11P (Δrop11), pROP16P (Δrop16), pROP17P (Δrop17), pROP18P (Δrop18), pROP20P (Δrop20), pROP21P (Δrop21), pROP22P (Δrop22), pROP23P (Δrop23), pROP24P (Δrop24), pROP25P (Δrop25), pROP26P (Δrop26), pROP27P (Δrop27), pROP28P (Δrop28), pROP30P (Δrop30), pROP31P (Δrop31), pROP32P (Δrop32), pROP35P (Δrop35), pROP36P (Δrop36), pROP37P (Δrop37), and pROP38P (Δrop38/29/19), pROP39P (Δrop39), pROP40P (Δrop40), pROP41P (Δrop41), pROP42,43,44P (Δrop42/43/44), and pROP45P (Δrop45) are shown. The corresponding T. gondii ME49 (TgME49) gene locus, chromosome, and nucleotides deleted for each knockout were determined from data in toxodb.org.
Primers used to validate ROPK knockouts. The sequences of oligonucleotide primers used for validation of Δrop2/8, Δrop5, Δrop11, Δrop16, Δrop17, Δrop18, Δrop20, Δrop21, Δrop22, Δrop23, Δrop24, Δrop25, Δrop26, Δrop27, Δrop28, Δrop30, Δrop31, Δrop32, Δrop35, Δrop36, Δrop37, Δrop38/29/19, Δrop39, Δrop40, Δrop41, Δrop42/43/44, and Δrop45 knockouts are shown. The primers were designed from TgME49 data in toxodb.org.
Primers used to develop targeting plasmids for complementation. The sequences of oligonucleotide forward primers (FP) and reverse primers (RP) used for construction of Pru complementation vectors pURO5A (::ROP5A), pURO5C (::ROP5C), pURO17FLHA (::ROP17FLHA), pURO18 (::ROP18), pURO18KD (::ROP18KD), and pURO18RAH2(ATF) [::ROP18RAH2(ATF)] are shown. The corresponding TgME49 gene locus, chromosome, and nucleotides deleted for each knockout were determined from data in toxodb.org.
Primers used to validate complementation of ROPK knockouts. The sequences of oligonucleotide primers used to validate integration of complementation vectors pURO5A (::ROP5A), pURO5C (::ROP5C), pURO17FLHA (::ROP17FLHA), pURO18 (::ROP18), pURO18KD (::ROP18KD), and pURO18RAH2(ATF) [::ROP18RAH2(ATF)] are shown. Primers were designed from TgME49 data in toxodb.org.