

Abstract
We have previously identified prohibitin (PHB) and annexin A2 (ANX2) as proteins interacting on the surface of vascular endothelial cells in white adipose tissue (WAT) of humans and mice. Here, we demonstrate that ANX2 and PHB also interact in adipocytes. Mice lacking ANX2 have normal WAT vascularization, adipogenesis, and glucose metabolism but display WAT hypotrophy due to reduced fatty acid uptake by WAT endothelium and adipocytes. By using cell culture systems in which ANX2/PHB binding is disrupted either genetically or through treatment with a blocking peptide, we show that fatty acid transport efficiency relies on this protein complex. We also provide evidence that the interaction between ANX2 and PHB mediates fatty acid transport from the endothelium into adipocytes. Moreover, we demonstrate that ANX2 and PHB form a complex with the fatty acid transporter CD36. Finally, we show that the colocalization of PHB and CD36 on adipocyte surface is induced by extracellular fatty acids. Together, our results suggest that an unrecognized biochemical interaction between ANX2 and PHB regulates CD36-mediated fatty acid transport in WAT, thus revealing a new potential pathway for intervention in metabolic diseases.
Prohibitin and annexin A2, in a complex with CD36, facilitate fatty acid uptake by the endothelium and their transport into adipocytes in white adipose tissue.
Introduction
Human obesity, a medical condition associated with a number of life-threatening diseases, is causing escalating social concern (1). Obesity results from excessive expansion of white adipose tissue (WAT). Hypertrophy of mature adipocytes differentiating from proliferative progenitor cells in the process of adipogenesis is ultimately responsible for obesity and metabolic syndrome, along with its pathological consequences (2). WAT remodeling leading to obesity is mediated by adipocyte interactions with stromal and vascular endothelial cells and is controlled by concerted actions of a number of extracellular signals that together form a highly integrated network designed to maintain energy balance (3). Lipid accumulation in the adipocyte is a result of de novo lipogenesis, as well as the uptake of circulating nutrients that are converted into triglycerides packaged into cytosolic lipid droplets (4).
Glucose and fatty acids (FAs) are the major energy sources assimilated by adipocytes (5). Glucose uptake by adipocytes is regulated by integral membrane carrier GLUT4 (6). Uptake of FAs by adipocytes depends on a high-affinity, low-capacity carrier–facilitated transport system (7, 8). The FA transport protein (FATP) and the membrane FA-binding protein (FABPpm) families (9), as well as caveolins (10), participate in the multiple steps of FA trafficking. The receptor stimulating FA entry into the cell is the FA translocase (FAT), also known as CD36 (11, 12). The CD36-dependent FA transporter operates in the context of lipid rafts, the highly ordered lipid microdomains distinguished by specific interaction between sterols and sphingolipids (13, 14). Despite the progress in characterization of FA uptake by cells of the liver and skeletal muscle, many questions remain regarding the molecular control of FA transport in WAT (11, 15). While defined distinct GLUT transporters control glucose uptake in different organs (16), the molecular machinery regulating FA uptake by WAT endothelium and their transfer into adipocytes is incompletely understood. Like adipocytes, WAT vascular endothelial cells display highly active endocytosis, which regulates internalization of macromolecules and particles into transport vesicles derived from the plasma membrane (17). However, it is not clear whether the molecular uptake by adipose endothelium depends on the same transport machinery as those operating in adipocytes, and it is also unclear which transporters regulate nutrient transfer between adipose endothelial cells and adipocytes.
We have previously proposed that WAT endothelium could be used as a target of obesity treatment aimed at cutting off the supply of nutrients and oxygen essential for adipocyte survival (18). In a screen of a combinatorial library for peptides that bind to cell surface receptors expressed in a tissue-specific manner (19–21), we previously identified a peptide (sequence KGGRAKD) that homes to WAT vasculature (22). Further, we demonstrated that KGGRAKD binds to prohibitin-1 (PHB), a protein present on the surface of endothelial cells selectively in WAT. Based on the capacity of KGGRAKD to undergo PHB-mediated endocytosis, we used this peptide to direct an apoptosis-inducing moiety D(KLAKLAK)2 to mouse WAT in an experimental approach to obesity reversal (22). Preclinically, the WAT vascular-targeting capacity of the KGGRAKD-GG-D(KLAKLAK)2 peptide (now known as adipotide) has been validated in mouse, rat, and nonhuman primate models of obesity and has shown to have antidiabetic effects (23–25).
Prohibitins display similarity to several proteins (SPFH, stomatin, flotillin, and HflK/C) containing a conserved transmembrane domain (26). PHB is a multifunctional protein found in various cellular compartments and is also secreted (27). It has been reported to serve as a cell surface receptor for infectious microorganisms (28). Identification of PHB as a component of lipid rafts in association with other integral membrane receptor complexes and its isolation from the macrophage phagosome proteome have suggested a possible role for PHB in endocytosis (29). However, the tissue-specific functions of PHB are still not fully understood. To gain mechanistic insight into PHB as a prospective obesity therapy target, we have begun to characterize its currently undefined function on the cell surface of WAT endothelium. We have identified annexin A2 (ANX2), also known as annexin II, as a PHB-binding protein containing the amino acid sequence KGRRAED mimicked by the WAT-homing peptide KGGRAKD. ANX2 and PHB colocalize in the vasculature of human WAT and directly interact in vitro in the context of lipid rafts (30). The extreme evolutionary conservation between human and mouse orthologs of both PHB and ANX2 (100% amino acid sequence identity in each case) suggests the physiological importance of their interaction.
ANX2 belongs to a family of homologous membrane-bound proteins (annexins) that are known to associate with anionic phospholipids in the presence of Ca2+ and contribute to the formation of ion channels, thus providing a link between calcium signaling and membrane functions (31). As other annexin family members, ANX2 regulates various functions within distinct cellular compartments and is also secreted (32, 33). We have shown that cell surface ANX2, in complex with other proteins, is implicated in fibrinolysis, coagulation, and inflammation (34). PHB and ANX2 have been previously shown to interact (35) and regulate internalization of polypeptides and pathogens by cells through clathrin-independent endocytosis (27, 36). However, the role of a molecular interaction between PHB and ANX2 in the context of adipose tissue physiology has not been interrogated.
Here, we show that mice lacking ANX2 have defective FA uptake by WAT endothelium and adipocytes. By integrating molecular genetics with cell culture assays, we demonstrate the importance of the PHB/ANX2 interaction for FA uptake and show that FAs stimulate cell surface assembly of a complex formed by PHB, ANX2, and CD36. Our data indicate that FA-induced assembly of the complex between PHB, ANX2, and CD36 on the cell surface regulates transendothelial FA transport into adipocytes.
Results
ANX2 is required for PHB localization on the surface of WAT endothelium and adipocytes.
Immunofluorescence (IF) analysis of WAT from WT mice revealed that PHB and ANX2 protein expression patterns are similar (Figure 1A). As in humans (30), PHB and ANX2 were coexpressed in WAT capillaries identified by isolectin B4 (IB4) binding (Figure 1A). Colocalization of PHB and ANX2 in the endothelium was confirmed in WAT whole mounts, paraffin sections, and isolated stromal/vascular fraction (SVF) cells (Supplemental Figure 1, A–C; supplemental material available online with this article; doi:10.1172/jci.insight.86351DS1). Endothelial PHB expression was observed in WAT but not in liver, lung, kidney, or other organs used as negative controls (Figure 1B). Consistent with the established constitutive activity of PHB in the mitochondrial membrane (37, 38), intracellular PHB IF was also observed in some cell types (Figure 1B). Interestingly, although mitochondrial content is low in adipocytes, they were observed to have high expression of both PHB and ANX2 (Figure 1A). To test for protein exposure on adipocyte surface, we used 3T3-L1 cells induced to undergo adipogenesis that were fixed but not rendered membrane permeable. Confocal IF demonstrated plasmalemmal colocalization of PHB and ANX2 in lipid-laden adipocytes. Adipocytes (39.3% ± 7%) were positive for both PHB and ANX2. In contrast, PHB and ANX2 expression on the surface of surrounding nondifferentiated fibroblasts was undetectable (Figure 1C). These data suggested that PHB and ANX2 might interact on adipocyte surface.
Figure 1. PHB and ANX2 coexpression on WAT endothelium and adipocytes.

(A) Immunofluorescence (IF) on serial mouse white adipose tissue (WAT) paraffin sections with PHB and ANX2 antibodies or a non–immune IgG (control) and green fluorophore–conjugated secondary antibodies. Endothelium (arrows) is counterstained with isolectin B4 (IB4, red). Green/red channel merging indicates expression of PHB and ANX2 in WAT endothelium (yellow). PHB/ANX2 coexpression is also observed in IB4-negative adipocytes (a). (B) IF analysis of mouse paraffin sections with anti-PHB antibodies/IB4 shows that liver, lung, and kidney PHB expression is not detectable in the endothelium (arrows). (C) 3T3-L1 cells after adipogenesis induction were subjected (without permeabilization) to IF with PHB and ANX2 antibodies. Arrows indicate localization of both PHB and ANX2 in differentiated adipocytes (a), for which lipid droplets are shown in the bright field image. Arrows represent nucleated nondifferentiated cells, in which PHB and ANX2 are not detectable. (D) ANX2 requirement for PHB localization to adipocyte surface. WAT-derived cells from of Anxa2+/+ (WT) and Anxa2–/– (ANX2-null) mice adherent to plastic in tissue culture were subjected to PHB (green)/ANX2 (red) IF (without permeabilization). Lipid droplets in differentiated adipocytes (a) are shown in the bright field images. Note the lack of both ANX2 and PHB signal in ANX2-null adipocytes. Arrows indicate cells not differentiated into adipocytes. Nuclei are blue (B–D). (E) Homing of the CKGGRAKDC peptide to WAT is inefficient in the absence of ANX2, as revealed by phage recovery (TU/g of WAT) from visceral WAT extracted from WT and ANX2-null littermates i.v.-injected with 1 × 1010 of CKGGRAKDC-phage transforming units (6 hour circulation). Shown are mean ± SD from n = 4 mice (2 males and 2 females). *P < 0.05 (Student’s t test, WT vs. ANX2-null). Antiphage HRP immunohistochemistry (brown) in sections of WAT demonstrates phage in WAT vasculature in WT mice (arrows). Hematoxylin counterstaining is blue. Scale bars: 50 μm.
We next set out to test whether PHB and ANX2 form a protein complex that operates on adipocyte cell membrane. Confocal IF on nonpermeabilized primary adipocytes in cell culture revealed the presence of both PHB and ANX2 on the surface of Anxa2+/+ (WT) adipocytes (Figure 1D). Interestingly, primary adipocytes from Anxa2–/– (ANX2-null)a mice (39) lacked not only ANX2, but also PHB expression on the cell surface (Figure 1D). This result suggested that ANX2 mediates PHB exposure on the adipocyte membrane. To evaluate whether ANX2 serves a similar role on the vascular endothelial surface, we used the KGGRAKD, the peptide mimicking a PHB-binding domain of ANX2 and homing to WAT endothelium (22, 30). Upon i.v. administration, accumulation of phage particles displaying the KGGRAKD peptide was reduced in WAT of Anxa2–/– mice compared with WT mice (Figure 1E). Combined, these data indicate that ANX2 is required for the assembly of a PHB-containing protein-protein complex on the surface of both WAT endothelial cells and adipocytes.
Anxa2–/– mice have hypotrophic adipocytes despite normal WAT vascularization and adipogenesis.
As reported in our previous studies, Anxa2–/– mice display no apparent developmental abnormalities (39). When raised on a low-fat diet, Anxa2–/– and Anxa2+/+ (WT) mice displayed indistinguishable dynamics of fat body mass accumulation (data not shown). To test whether lack of ANX2 affects induction of experimental obesity, we compared Anxa2–/– and Anxa2+/+ males placed on a high-fat diet (HFD) at weaning. Diet-induced obesity (DIO) onset was delayed in the absence of ANX2; in 3-month old mice, analysis of body composition by Echo MRI demonstrated a comparatively low fat mass in Anxa2–/– DIO males (Figure 2A). Gross anatomical inspection of mouse internal organs revealed less i.p. and s.c. WAT in Anxa2–/– mice (Figure 2B). Histological analysis revealed a notably smaller white adipocyte size in Anxa2–/– animals compared with WT (Figure 2C).
Figure 2. ANX2 is required for adipocyte hypertrophy but not for adipogenesis.
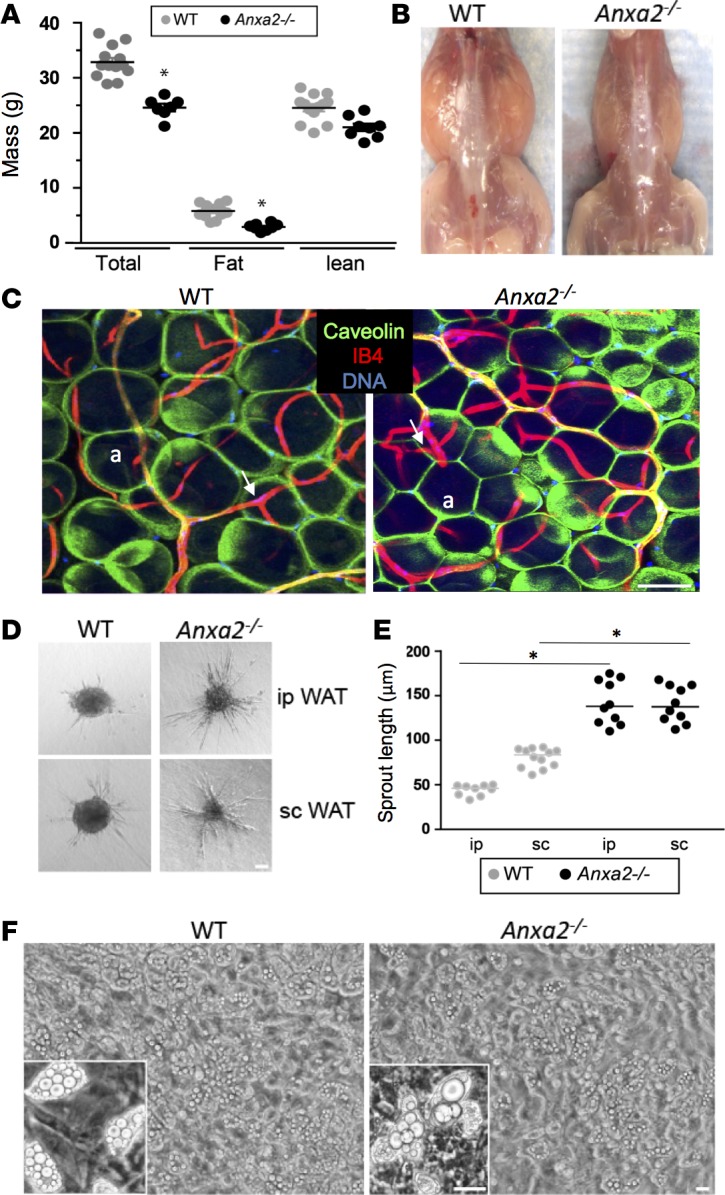
(A) Total fat and lean body mass of Anxa2+/+ (WT) males (n = 13) and Anxa2–/– (ANX2-null) males (n = 8) after feeding HFD for 7 weeks. (B) Representative mice with skin removed revealing lower amount of WAT in ANX2-null mice. (C) Immunofluorescence analysis of whole mounts of WAT from WT and ANX2-null mice with caveolin-1 (green) antibodies revealing reduced adipocyte (a) size in ANX2-null mice. Isolectin B4 (red) counterstaining the endothelium (arrows) reveals vasculature. (D) Angiogenesis in an ex vivo sprouting assay. When embedded into collagen-I/methylcellulose matrix, the stromal/vascular fraction from i.p. or s.c. WAT of ANX2-null mice display increased endothelial sprout formation, compared with WT mice. (E) Data in D quantified based on the analysis of n = 10 view fields. (F) ANX2 is not required for adipocyte differentiation; upon adipogenesis induction (5 days), the timing of lipid droplet formation is comparable for WT and ANX2-null SVF. In A and E, error bars ± SEM; * P<0.05 (Student’s t test, WT vs. ANX2-null). Scale bars: 50 μm.
WAT development and maintenance rely on the blood vessels that deliver oxygen and nutrients to adipocytes (40). Hajjar and collaborators have previously reported that postnatal angiogenesis is reduced in Anxa2–/– mice (39). Therefore, we tested the possibility that altered WAT expansion in Anxa2–/– DIO mice might be due to a defect in vascularization. However, inspection of WAT whole mounts stained with IB4 to mark the endothelium did not display obviously detectable differences in either blood vessel size or branching between Anxa2+/+ and Anxa2–/– DIO animals. In fact, the density of microvessels in both i.p. and s.c. WAT of Anxa2–/– mice was significantly higher than in WT mice (Figure 2C and Supplemental Figure 1D). Moreover, in ex vivo explant assays, cells of the SVF from both i.p. and s.c. WAT of Anxa2–/– mice displayed increased sprouting, compared with Anxa2+/+ mice (Figure 2, D and E). We therefore reasoned that an angiogenesis-independent ANX2 function might be responsible for the observed WAT phenotype.
The lack of obvious developmental abnormalities in WAT populated with typical unilocular adipocytes in Anxa2+/+ mice (Figure 2C) indicates that the PHB/ANX2 complex is not essential for adipogenesis. We, nevertheless, tested whether the WAT phenotype in Anxa2+/+ DIO animals could be due to a subtle defect in adipocyte differentiation. Upon adipogenesis induction, comparable timing of lipid droplet formation was observed in the SVF derived from Anxa2+/+ and Anxa2–/– mice (Figure 2F). These data indicated that the adipocyte hypotrophy observed in the absence of ANX2 is due to its function required after adipocyte differentiation.
Anxa2–/– mice have increased metabolic rate.
We analyzed Anxa2–/– mice for indications of WAT dysfunction. There was no detectable difference in HFD consumption between Anxa2+/+ and Anxa2–/– mice (Supplemental Figure 2A). Stool analysis did not reveal a significant difference in fecal triglyceride content (Supplemental Figure 2B), indicating normal intestinal lipid absorption in Anxa2–/– mice. Spontaneous locomotor activity was also comparable in Anxa2+/+ and Anxa2–/– mice (Supplemental Figure 2C). However, indirect calorimetry revealed that both oxygen consumption and carbon dioxide production were elevated in Anxa2–/– mice during both dark and light cycles (Supplemental Figure 2D). No significant differences in the plasma levels of free FAs, glycerol, triglycerides, cholesterol, β-hydroxybutyrate, or insulin were detected between Anxa2–/– and Anxa2+/+ mice (Supplemental Figure 2E). Steady-state serum glucose levels were reduced in Anxa2–/– mice (Supplemental Figure 2E). However, no significant difference in glucose tolerance was observed between Anxa2–/– and Anxa2+/+ mice, either (Supplemental Figure 2F). We also assessed glucose uptake by injecting mice with a near-infrared fluorophore-labeled glucose analog. Whole body imaging and resected organ analysis did not reveal a significant difference in WAT glucose accumulation between Anxa2–/– and Anxa2+/+ mice (Supplemental Figure 2G).
Deficient FA uptake in WAT of Anxa2–/– mice.
Based on the evidence for altered lipid metabolism and energy expenditure in Anxa2–/– mice, we reasoned that the PHB/ANX2 complex might regulate the processing of dietary FAs. To test this hypothesis, we performed a lipid tolerance test. Indeed, Anxa2–/– mice showed a delayed triglyceride clearance upon i.v. FAs (Intralipid) infusion (Figure 3A). To determine whether this results from reduced FA uptake in WAT, we i.v. administered a BSA-emulsified FAs conjugated to a lipophilic fluorophore 4,4-difluoro-3a,4a-diaza-s-indacene (BODIPY). Whole body imaging 15 minutes after administration revealed medium-chain (C12) lauric acid BODIPY conjugate accumulation throughout the abdominal area of Anxa2+/+ mice, consistent with the probe taken up by WAT (Figure 3B). Anxa2–/– mice displayed a markedly reduced signal in the abdomen, suggesting a WAT uptake inefficiency (Figure 3C). These experiments were performed with a fluorophore-conjugated long-chain (C16) palmitic acid with similar results (Supplemental Figure 2H). To assess FA biodistribution upon i.v. administration, we measured FA fluorescence in individual organs. While nonconjugated fluorophore did not accumulate in WAT and was mainly observed in the liver for both Anxa2+/+ and Anxa2–/– mice, high fluorescence was observed in both s.c. (inguinal) and i.p. (mesenteric and epididymal) WAT depots of Anxa2+/+ mice. This was observed for both C12 and C16 fluorescent FAs (Figure 3, D and E, Supplemental Figure 2I). In contrast, Anxa2–/– mice displayed a reduced accumulation of lauric and palmitic acid in both s.c. and i.p. WAT, while signals observed for kidneys, spleen, intestines, and the skeletal muscle were not significantly different between Anxa2–/– and Anxa2+/+ mice (Figure 3, D and E). An increased FA signal in the liver of Anxa2–/– mice (Figure 3, D and E) suggests that hepatocytes clear FAs that cannot be efficiently absorbed in WAT without ANX2.
Figure 3. Deficient WAT lipid uptake in ANX2-null mice.

(A) Triglyceride blood concentration analysis upon i.v. Intralipid infusion into prestarved mice shows a delay in clearance for ANX2-null compared with WT mice (n = 4). (B) Abdominal fluorescence of lauric acid boron-dipyrromethene conjugate (BODIPY-FL-C12) measured 15 min. after injection. As quantified for 4 abdominal locations (C), a significantly lower signal is observed for Anxa2–/– mice compared with WT mice. (D) Analysis of individual organs from WT and Anxa2–/– mice 15 min. after injection of palmitic acid (C16) labeled with IRDye-680CW (left) or an equimolar amount of uncoupled IRDye-680 (right). (E) Quantification of D showing a significant reduction of FA localization to s.c. and i.p. WAT of Anxa2–/– mice compared with WT mice (n = 4). FA (but not free fluorophore) accumulation in the liver is higher in Anxa2–/– than in WT mice, while kidney and skeletal muscle accumulation is comparable. (F) SVF from i.p. WAT of WT and Anxa2–/– mice with vascular structures (arrows) were incubated with 0.3 μM BODIPY-FL-C12 (red) and then washed. Note FA uptake (arrows) in WT but not Anxa2–/– endothelium. (G) One hour after red BODIPY-FL-C12 i.v injection into WT and Anxa2–/– mice, adipocytes were isolated after WAT disaggregation using collagenase. Note FA accumulation (arrows) inside WT but not Anxa2–/– nucleated adipocytes. Insets: bright field (BF) images. In A, C, and E, error bars ± SEM; *P<0.05 (Student’s t test, WT vs. Anxa2–/–). Nuclei are blue. Scale bars: 50 μm.
To identify WAT cell populations that fail to efficiently uptake FAs in the absence of ANX2, we injected BODIPY-FL-C12 into mice and subjected SVF to flow cytometric analysis. Gating on CD31 revealed a 5-fold lower frequency of FA-positive endothelial cells in Anxa2–/– mice compared with WT mice (Supplemental Figure 3A). Ex vivo experiments with cultured SVF treated with BODIPY-FL-C12 (300 nM) confirmed efficient FA uptake by WT but not Anxa2–/– vascular endothelium (Figure 3F). To determine whether reduced endothelial FA uptake results in their lack of deposition into adipocytes, we analyzed enzymatically disaggregating WAT cells 1 hour after FA injection. FA-BODIPY accumulation was observed for adipocytes recovered from WT mice but not from Anxa2–/– mice (Figure 3G). We also measured FA-BODIPY uptake in SVF induced to differentiate into adipocytes. A statistically significant defect in FA uptake by Anxa2–/– adipocytes was detected by quantitative real-time assay (Supplemental Figure 3B). Together, these data suggested that ANX2 expression in both endothelium and adipocytes facilitates lipid transport into adipocytes.
FA uptake in adipocytes is mediated by PHB/ANX2 interaction.
The presence of PHB/ANX2 complex on WAT cell surface (Figure 1) suggested FA uptake, disrupted in the absence of ANX2, is regulated by this protein-protein interaction. We, therefore, tested whether a similar phenotype would be observed in the absence of PHB. Previously, a role of PHB in adipogenesis has been assessed by its overexpression and the knockdown of its expression in 3T3-L1 preadipocytes prior to adipogenesis induction (37, 41). In order to focus on the role of PHB in differentiated adipocytes, we have instead transfected preadipocytes with the same siRNA after adipogenesis induction. A reduction in PHB expression was detected for 2 clones after 72 hours by immunoblotting (Figure 4A). Upon treatment with FA-BODIPY, approximately 60% of control adipocytes displayed FA accumulation in lipid droplets, while fewer than 20% of knock-down cells displayed FA uptake (Figure 4B). IF analysis revealed FAs only in adipocytes that still expressed PHB, while there was no FAs in adipocytes lacking PHB due to RNA interference (Figure 4C). In a reciprocal gain-of-function experiment, we used bEnd.3, a cell line derived from the brain endothelium (42). Consistent with the lack of surface PHB expression, bEnd.3 cells did not take up FA-BODIPY efficiently. Ectopic expression of PHB in these cells using a lentiviral vector promoted FA uptake by 4-fold, indicating that PHB is a limiting factor in this process (Supplemental Figure 3C).
Figure 4. PHB regulates FA uptake in adipocytes through interaction with ANX2.

(A) 3T3-L1 adipocytes after differentiation were transfected with control untargeted siRNA or siRNA targeting PHB. As assessed by PHB and β-actin (loading control) immunoblotting 72 hours after transfection, a substantial reduction of PHB protein expression is observed for siRNA clones 12 and 13. (B) Upon treatment with 0.3 μM boron-dipyrromethene–conjugated (BODIPY-conjugated) C16 fatty acid (FA) (green), anti-PHB immunofluorescence (red) reveals FA uptake by the majority of control PHB-expressing adipocytes (arrows), while for clone 12, FA uptake is not observed for adipocytes (a) that lost PHB expression. Nuclei are blue. The graph quantifies percentage of adipocytes with detectable FA fluorescence in n = 5 view fields. Error bars ± SEM; *P < 0.05 (Student’s t test, WT vs. PHB knockdown). Scale bar: 50 μm. (C) 3T3-L1 adipocytes expressing GFP-tagged WT ANX2 or ANX2 mutant engineered to lack PHB binding were treated with 1 μM BODIPY-FL-C12 for 30 min. Note the abundant FA uptake (red lipid droplets) by adipocytes (a) expressing WT ANX2 on the cell membrane (arrows) and lack of FA uptake in adipocytes expressing ANX2 mutant, which fails to localize to cell membrane. (D) Anxa2–/– cells were differentiated into adipocytes and then transduced with GFP-tagged WT ANX2 or GFP-tagged ANX2 mutant engineered to lack PHB binding or with empty GFP vector. Note GFP signal in transduced adipocytes, which is absent in nontransduced (N.T.) cells. Arrows indicate cell membrane WT ANX2 localization. Nuclei are blue. Scale bar: 50 μm. (E) Fluorescent and bright-field microscopy on cells from D treated with 0.3 μM BODIPY-FL-C12 for 30 min. Note abundant FA uptake (red lipid droplets) by adipocytes (a) expressing WT ANX2 but not ANX2 mutant. Nuclei are blue. Scale bar: 50 μm. (F) Real-time measurement of FA uptake. Cells from D were plated in triplicate, prestarved in 1% FBS/DMEM-low glucose for 2 hours, and then subjected to the QBT assay. Intracellular long-chain FA accumulation is monitored by measuring fluorescence (excitation 485 nm/emission 515 nm). Plotted are mean values; error bars ± SEM; *P < 0.01 (Student’s t test, WT ANX2 vs. ANX2 mutant).
To directly interrogate the function of interaction between PHB and ANX2, we analyzed 3T3-L1 adipocytes transduced with lentivirus that expresses GFP that has either been fused with WT ANX2 or with a ANX2 mutant in which the PHB-binding sequence (KGRRAED) was replaced with a sequence AAAAAAA. While WT ANX2-GFP displayed clear cell membrane expression, the ANX2 mutant localization was largely cytoplasmic (Figure 4C). Importantly, adipocytes expressing ectopic WT ANX2 appeared to have higher BODIPY-FL-C12 accumulation in lipid droplets than adipocytes expressing mutant ANX2. Because endogenous ANX2 expression by 3T3-L1 masks the phenotype of ectopic ANX2 mutant, we introduced the same expression vectors into primary adipocytes from Anxa2–/– mice (Figure 4D). Uptake of both BODIPY-FL-C12 was markedly lower in adipocytes expressing mutant ANX2 compared with WT ANX2 (Figure 4E). Quantitative real-time FA-BODIPY uptake analysis confirmed that cells ectopically expressing WT ANX2 had higher FA uptake compared with cells ectopically expressing mutant ANX2 (Figure 4F). Collectively, these results indicate that a functional protein interaction between PHB and ANX2 promotes FA uptake.
Cell surface PHB/ANX2 complex regulates FA intercellular transport.
Because both PHB and ANX2 have been reported to function in multiple cell compartments, we next designed experiments to test whether FA uptake is mediated by their interaction at the cell surface. We predicted that a fragment of ANX2 spanning the PHB binding site KGRRAED (30) would act as a dominant negative competitor of the binding between PHB and ANX2. To evaluate that hypothesis, a targeted “blocking” peptide encompassing the native motif (sequence AKGRRAEDGSVIDYELI) was chemically synthesized. A scrambled peptide (sequence RDAGRSDALVIYEIGKE) was synthesized in parallel to serve as a negative control. To test whether the PHB/ANX2 interaction regulates FA uptake by the vasculature, we used the bEnd.3 cell model. FA uptake in endothelial cells ectopically expressing PHB was inhibited by AKGRRAEDGSVIDYELI but not by the scrambled peptide (Supplemental Figure 3C). We also analyzed FA uptake into primary endothelial cells treated with these peptides. Uptake of both BODIPY-FL-C12 and BODIPY-FL-C16 was abrogated by the blocking peptide but not by the scrambled peptide (Supplemental Figure 3D). Treatment of differentiated 3T3-L1 cells with AKGRRAEDGSVIDYELI, but not with the scrambled peptide, also reduced FA-BODIPY uptake by adipocytes (Figure 5A). These data demonstrate that the cell surface ANX2/PHB interaction indeed mediates FA uptake by both endothelial cells and adipocytes.
Figure 5. PHB/ANX2 complex mediates lipid transport in WAT.

(A) 3T3-L1 adipocytes were incubated for 30 min. in medium containing 0.1 mM blocking or a scrambled peptide and then treated with 0.3 μM BODIPY-conjugated C16 fatty acid (FA) (green). Note reduced FA uptake upon PHB/ANX2 binding blockade. Graph: data quantified based on the analysis of n = 5 view fields. Error bars ± SEM. *P<0.05 (Student’s t test, blocking vs. scrambled). (B) A schematic of the intercellular transfer assay measuring endothelium-adipocyte FA transfer: (1) bEnd.3 endothelial cells (EC) are plated as a monolayer; (2) EC are incubated with BODIPY-FL-C16 (green fluorescence) fatty acid (FA) and washed, after which a part of the plate is scraped off; and (3) 3T3-L1 adipocytes coated with magnetic nanoparticles are added and forced to the bottom using magnetic field in the presence or absence of the blocking peptide. After washing, CD31 immunofluorescence (red) is performed, and adipocytes that received FA from interacting endothelial cells are observed (green). (C) The intercellular transfer assay performed in the presence of 0.1 mM RDAGRSDALVIYEIGKE scrambled peptide control (no blocking) or AKGRRAEDGSVIDYELI (blocking) peptide. Note that peptide blocking the PHB/ANX2 binding, but not the scramble peptide, prevents FA (green) transfer from the endothelium (red) to adipocytes (arrows). Arrowheads indicate nondifferentiated 3T3-L1 cells. Shown below are bright-field images demonstrating adipocytes attached to plastic and to EC monolayer below the scraped area. Scale bar: 50 μm.
Based on our data, we hypothesized that the ANX2/PHB complex regulates FA transport from the endothelium into adipocytes. To test this, we designed an intercellular transfer assay (Figure 5B). Confluent bEnd.3 cells (42) were treated with a high dose of green-fluorescent FA-BODIPY, leading to its internalization (Supplemental Figure 4A). After unbound FA-BODIPY was washed off, cells were removed from a part of the plate. Then, a suspension of differentiated 3T3-L1 adipocytes pretreated with either the blocking AKGRRAEDGSVIDYELI or the scrambled control peptide was added. Based on a published approach (42, 43), adipocytes in this experiment were also precoated with magnetic nanoparticles, which were used to force their quick sedimentation onto the endothelial feeder layer with the magnetic field (Supplemental Figure 4A). After adherent adipocytes were incubated to allow FA transfer, cells were washed, fixed, and subjected to red CD31 IF. Numerous FA-BODIPY–positive (22.4% ± 5%) adipocytes were observed in the areas where they were adherent to the endothelium, while only 8% ± 2.5% of plastic-adherent adipocytes were FA-positive (green) (Figure 5C). This result indicated that the assay specifically reads out the contact-mediated transfer of FA-BODIPY from the endothelium to adipocytes. Notably, FA transfer was competitively inhibited by the blocking AKGRRAEDGSVIDYELI peptide (Figure 5C), again indicating specificity.
In another intercellular transfer experiment, we tested whether the ANX2/PHB complex on the endothelium regulates FA transport by comparing cells derived from WAT of WT and Anxa2–/– mice. Upon incubation of SVF monolayers with a high concentration (4 μM) of FA-BODIPY, FA signals were comparable in WT and Anxa2–/– endothelial cells (Supplemental Figure 4B). After extracellular FAs were washed off, 3T3-L1 adipocytes were adhered to the SVF monolayers using the magnetic field. Analysis of FA accumulation in adipocytes indicated that FA transfer was more efficient from WT than from Anxa2–/– endothelium (Supplemental Figure 4C). Combined, these results suggest that the PHB/ANX2 interaction in both endothelial cells and adipocytes regulates FA transport.
ANX2 and PHB are components of the FAT complex.
Because the FAT CD36 expressed on the surface of endothelium and adipocytes is known to stimulate FA uptake (44), we tested a hypothesis that ANX2 and PHB interact with CD36. IF on SVF stimulated to undergo adipogenesis revealed CD36 colocalization with both PHB and ANX2 in 32% ± 7% adipocytes (Figure 6A). CD36 colocalization with both PHB and ANX2 was also observed in 21% ± 4% of vascular structures formed by the endothelial cells, while expression of CD36, PHB, and ANX2 on the surrounding stromal cells was undetectable (Figure 6A).
Figure 6. FA treatment induces plasmalemmal interaction of ANX2, PHB, and CD36.

(A) SVF stimulated to undergo adipogenesis were subjected (without permeabilization) to immunofluorescence with CD36 (red) and PHB (green) or ANX2 (green) antibodies. Both ANX2 and PHB are colocalized (yellow) with CD36 in adipocytes (a) and in vascular structures (arrows). Scale bar: 50 μm. (B) Membrane proteins immunoprecipitated (IP) with CD36 antibodies from WT or ANX2-null mouse–derived adipocytes were subjected to Western blotting (WB) with antibodies against CD36, ANX2, or PHB. Note that CD36 antibodies coimmunoprecipitate PHB and ANX2 from WT but not from ANX2-null extracts. (C) Membrane proteins immunoprecipitated with non–immune IgG or ANX2 antibodies from WT or ANX2-null mouse–derived adipocytes were subjected to immunoblotting with antibodies against CD36, ANX2, or PHB. Note that ANX2 antibodies coimmunoprecipitate PHB and CD36 from WT but not from ANX2-null extracts. Expression of CD36 in ANX2-null cells is confirmed by whole extract (input) immunoblotting. (D) Proteins precipitated with PHB or ANX2 antibodies (or control IgG) from surface-biotinylated 3T3-L1 adipocytes either untreated (0) or pretreated with 1% Intralipid fatty acids (FA) for the indicated time were subjected to anti-PHB or anti-ANX2 WB (Total) or streptavidin-conjugated IRDye-800CW (Surface). Note the increase in biotinylated PHB recovery upon FA treatment. (E) Cell surface proteins precipitated with streptavidin beads from surface-biotinylated 3T3-L1 adipocytes either untreated (-) or treated with FA (1% Intralipid) for 1 hour (+) were subjected to immunoblotting with antibodies against CD36 or PHB. Note that FA treatment increases the amount of CD36 and PHB (arrows) but not of other proteins (nonspecific bands) at the surface. (F) 3T3-L1 adipocytes either untreated or treated with 1 μM lauric acid for 10 minutes were fixed and subjected to immunofluorescence with ANX2 (red), PHB (green), and CD36 (blue) antibodies. Note plasmalemmal colocalization of the 3 proteins (white) in adipocytes (a) upon FA treatment (arrows). Scale bar: 50 μm. (G) A working model: ANX2/PHB/CD36 complex assembly on the cell surface is induced by the postprandial extracellular FA influx and contributes to the uptake of FA from the circulation by the endothelium and FA transfer into adipocytes.
To determine whether ANX2 and PHB form a complex with CD36, we biochemically analyzed membrane proteins purified from mouse adipocytes. Anti-CD36 antibodies coimmunoprecipitated PHB and ANX2 with CD36 from Anxa2+/+ — but not from Anxa2–/– — membrane protein extracts (Figure 6B). PHB and CD36 were not coimmunoprecipitated with ANX2 antibodies from similarly processed Anxa2–/– membrane protein extracts (Figure 6B). We confirmed that the 3 proteins form a complex in a reciprocal experiment. Anti-ANX2 antibodies, but not control IgG, coimmunoprecipitated PHB and CD36 with ANX2 (Figure 6C). Identical observations were made for experiments performed with extracts from mouse SVF (Supplemental Figure 5A). To test the dependence of ANX2/PHB-mediated FA uptake on CD36, we used 3T3-L1 adipocytes in which CD36 was blocked by an inhibitory antibody (Supplemental Figure 5B). FA uptake was decreased by either CD36 blockade or ANX2/PHB binding blockade with the dominant-negative peptide. Importantly, these agents did not have an additive effect, which was also quantified by the plate-reader–based assay (Supplemental Figure 5B). These data suggest that the ANX2/PHB interaction and CD36 function are parts of the same process.
Finally, we tested whether FAs would act as an extracellular signal inducing assembly of ANX2, PHB, and CD36 into a cell membrane trimolecular complex. After 3T3-L1 adipocytes were treated with FAs, the cell surface proteins were biotinylated and then immunoprecipitated with PHB or ANX2 antibodies (Figure 6D). Presence of both PHB and ANX2 on the cell surface was observed even for untreated adipocytes. PHB cell surface levels were dramatically increased after 5 minutes of FA treatment (Figure 6D). We also tested if FA treatment affects CD36 cell surface localization. Cell surface proteins of FA-treated 3T3-L1 adipocytes were biotinylated and, upon precipitation with streptavidin, PHB and CD36 were analyzed by immunoblotting. This experiment demonstrated that FA stimulation induces localization of both PHB and CD36 to the cell surface (Figure 6E). To confirm this previously unrecognized observation by using IF, we analyzed 3T3-L1 adipocytes upon lauric acid treatment. Increased colocalization of ANX2, PHB, and CD36 at the cell membrane was observed upon stimulation with C12 FAs in 42.2% ± 6% of adipocytes (Figure 6F). Similar observations were made for adipocytes treated with palmitic acid and Intralipid (Supplemental Figure 5, C and D). Taken together, our results show that extracellular FAs trigger the formation of a cell surface complex containing ANX2, PHB, and CD36; this complex mediates medium-chain and long-chain FA transport in WAT.
Discussion
The mechanisms regulating transport of FAs from the circulation into adipocytes remain incompletely understood. We show that a WAT-specific PHB/ANX2 surface complex regulates the uptake of both medium-chain and long-chain FAs by endothelium and adipocytes. Our data also demonstrate that exposure to FAs induces the cell surface presentation of a trimolecular complex containing PHB, ANX2, and CD36. Based on our cell culture experiments showing that this complex regulates FA transfer from endothelium to adipocytes, we propose that the physiological function of a WAT-specific PHB/ANX2 interaction is to mediate FA transport from the circulation into adipocytes (Figure 6G).
Anxa2–/– mice have a phenotype masked by a metabolic adaptation, akin to caveolin-null (10) and to CD36-null (45) mice. Expression of caveolin-1 (Figure 2C) and of CD36 (Figure 6, B and C) are unchanged in Anxa2–/– mice. According to our results, Anxa2–/– mice have a decreased capacity to deposit lipids into adipocytes. The data showing that FA uptake is not reduced in other organs are consistent with the WAT-specificity of the PHB/ANX2 complex. The FA biodistribution data shown here indicate that WAT-evading FAs are shunted to the liver in Anxa2–/– mice. Increased energy expenditure suggests that oxidation of FAs in hepatocytes, and possibly other organs, clears lipids that are not accommodated by WAT. It is unclear to what extent systemic ANX2 deficiency contributes to the phenotype of Anxa2–/– mice. Systemic metabolic changes resulting from the global lack of ANX2, which in many cellular pathways functions independently of PHB, are likely to be quite complex.
ANX2 and PHB are pleiotropic proteins with multiple known functions in various cellular locations (27, 36). This complicates interpretation of phenotypes from a whole cell inactivation of these molecules. Because PHB is an essential protein, PHB KO mice are not viable. Previous knockdown studies have pointed to PHB role in adipocyte physiology (37, 46) and, specifically, adipogenesis (41). However, intracellular functions of PHB and ANX2 could be at least partly responsible for phenotypes previously reported in whole cell inactivation settings. Our approach to specifically block the ANX2/PHB interaction with an extracellular competitor peptide largely circumvents this challenge and demonstrates the role of the complex on the surface of both adipose endothelium and adipocytes. Our studies with ANX2/PHB interaction mutants provide further evidence that this supramolecular complex regulates lipid metabolism in WAT after adipocyte differentiation.
Molecules mediating FA transport into adipocytes are central to mammalian metabolism. There is a clear role for plasma membrane proteins in the cellular uptake of FAs; however, the precise molecular mechanism of their transmembrane transport remains incompletely understood. The evidence for the role of cell membrane–associated CD36 in hepatocytes and myocytes is compelling (8). On the other hand, the occurrence of various types of FA transporters displaying distinct patterns of tissue-specific distribution indicates the sheer complexity of underlying mechanisms. Specifically, the network of proteins regulating FA transport in WAT has remained unclear. It has been proposed that the function of CD36 is likely to be influenced by accessory proteins, which have remained unknown. CD36 has been proposed to act as the receptor of FAs, increasing their concentration at the cell surface and facilitating their diffusion across the phospholipid bilayer (11, 12, 47, 48).
Here, we show that CD36 colocalizes with ANX2 and PHB in both WAT endothelial cells and adipocytes. We also demonstrate ANX2 and PHB to be the components of a CD36 protein complex. Identification of ANX2 and PHB as CD36 interaction partners is an important step in characterization of the mechanism regulating active FA transport in WAT. It has been previously reported that ANX2 translocates to the cell membrane in response to long-chain FAs (49). While CD36, in the context of lipid rafts, stimulates FA uptake by adipocytes (50), ectopic CD36 in certain cell culture models is insufficient to increase FA uptake (8), suggesting that CD36 function relies on accessory proteins. While a basal rate of FA uptake takes place in the absence of CD36, PHB, or ANX2, our data show that FA transport through the receptor-mediated pathway is inefficient when one of these complex components is missing. Our data suggest that the CD36 FAT complex function depends on the PHB/ANX2 interaction at the cell surface. Specifically, we provide evidence that ANX2 is required for cell surface localization of PHB, for the interaction of PHB and CD36, and for efficient FA uptake. We propose that ANX2 and PHB act as membrane chaperones that regulate the recruitment of CD36 to the cell surface and, hence, promote FA uptake in WAT. The possibility that other known CD36-interacting proteins, such as thrombospondin (44); other known FA transporters, such as FATP3/4; or other FATPs, FABPs, and caveolins (9, 10) could be involved in this process remains to be tested.
The action of the PHB/ANX2/CD36 complex in adipocyte FA transport contributes to our understanding of the mechanisms governing WAT expansion. A recent report on obesity promotion resulting from PHB overexpression (51) is consistent with its role in both adipogenesis and FA uptake. The PHB/ANX2 complex is a WAT target of an experimental antiobesity compound adipotide (22). The PHB/ANX2 interaction has been validated in humans (30), and a first-in-human clinical trial of adipotide is currently ongoing in obese prostate cancer patients. Our discovery of PHB expression on adipocytes, in addition to WAT endothelium, increases the possibility that the marked efficacy of adipotide results from it gaining access to adipocytes through transendothelial transport and that adipocyte apoptosis synergizes with endothelial apoptosis caused by this drug candidate. The potential translational relevance of our findings is to be further investigated. In the future, defining the biochemistry of ANX2/PHB/CD36 interaction with FAs and other proteins mediating their trafficking will enable a better understanding of the mechanisms governing lipid metabolism and WAT maintenance.
Methods
Animal experiments.
Mice were anesthetized with Avertin or isofluorane. Anxa2–/– mice have been described previously (39). To avoid strain-specific variations, we performed 10 generations of backcrosses of Anxa2–/– mice into the C57BL/6 background. As control mice, WT littermates or C57BL/6 mice purchased from The Jackson Laboratory were used. For DIO induction, mice were fed 58 kcal% (fat) diet (The Jackson Laboratory, D12331). Food intake was measured as described previously (22, 52). Body composition was measured by EchoMRI-100T (Echo Medical Systems). Glucose tolerance test (6 mice/group) was performed with a glucometer (One Touch Ultra) upon injecting glucose (1 g/kg body weight) into overnight-fasted mice. A lipid tolerance test was performed based on a previously described protocol (53) upon injecting 5.71 μl/g body weight of 20% Intralipid (Sigma-Aldrich) into overnight-fasted mice. Indirect calorimetry and spontaneous locomotor activity studies were performed with OXYMAX (Columbus Instruments) Comprehensive Lab Animal Monitoring System (CLAMS) as described (22). Palmitic acid and lauric acids (Molecular Probes) were conjugated with a fluorophore IRDye 680CW (LI-COR) through the NHS reactive group. BODIPY-FL-C12 (558/568 nm) was purchased from Molecular Probes. For injection, 400 nM of a probe was emulsified in 65 mg/ml BSA. Whole body imaging of FA biodistribution was performed with In-Vivo Multispectral System FX (KODAK); analysis of resected organs was performed with Pearl Impulse system (LI-COR). Steady-state plasma analyses were performed at the Baylor Mouse Metabolism Core (Houston, Texas, USA). Triglyceride quantification was performed using a colorimetric kit from Biovision. Quantification of phage particles homing to WAT was performed by i.v. (tail vein) injection of 1 × 1010 KGGRAKD-phage transforming units, and 6 hours later, recovery of phage from i.p. WAT was measured based on K91 E. coli infection as described (19, 21).
Protein expression/fluorescence microscopy.
Paraformaldehyde-fixed cells and formalin-fixed paraffin-embedded tissue sections were analyzed by IF as described (19, 54). WAT whole mounts were prepared and analyzed as described (42). Upon blocking, the following primary antibodies (4°C, 12 hours) and secondary antibodies (room temperature, 1 hours) diluted in phosphate-buffered saline (PBS) with or without 0.05% Tween 20 (for nonpermeabilized cell studies) were used: anti-PHB, rabbit (Abcam, Ab75766, 1:150) and goat (Santa Cruz Biotechnology Inc., sc-18196, 1:50); anti-ANX2, rabbit (Abcam, Ab41803, 1:150) and goat (Santa Cruz Biotechnology Inc., sc-30757, 1:50); anti-CD31 goat (Santa Cruz Biotechnology Inc., sc-1506, 1:75); anti-CD36, rat (Abcam, Ab80080, 1:100), rabbit (Novus Biologicals, NB400-144, 1:200), and biotinylated mouse (Miltenyi Biotec, 130-095-478, 1:50); and anti-GFP, goat (Gene Tex, GTX26673, 1:250). Donkey anti–goat Alexa 488 conjugate (A-11055, 1:150), donkey anti–rabbit Alexa 488 conjugate (A-21206, 1:150), and donkey anti–rabbit Cy3 conjugate (711-165-152, 1:300) from Jackson ImmunoResearch were used. Biotinylated IB4 was from Vector Labs (1:50) and used with Cy3-conjugated streptavidin (Zymax). CD36 antibody was used with Neutravidin DyLight 680 (Pierce). Fluorescein-conjugated peptide KGGRAKD flanked by cyclized cysteines was synthesized by Anaspec. Nuclei were stained with Hoechst 33258 or DRAQ5 (Invitrogen). Images of cultured cells were acquired with Olympus IX70 inverted fluorescence microscope/MagnaFire software. Images of cells fixed on Nunc Lab-Tek II Chamber Slides or tissue preparations were acquired with Carl Zeiss upright Apotome Axio Imager Z1/ZEN2 Core Imaging software or confocal Leica TCS SP5 microscope/LAS AF software.
Recombinant proteins and peptides.
GST-tagged PHB was produced using a vector received from S. Chellappan (Moffitt Cancer Center, Tampa, Florida, USA). To generate ANX2 mutant deficient in PHB binding, PCR-based mutagenesis was performed on ANX2 cDNA as described (20, 55) to replace all amino acids in the sequence KGRRAED with alanines. For inducible expression, cDNA fragments that encoded for either WT ANX2 or ANX2 mutant were cloned in-frame with enhanced GFP sequence into a response pLVX-TREG vector (Clontech). Lentivirus was prepared by using the regulator pLVX-Tet3G plasmid according to the manufacturer’s protocol. After selection in puromycin-containing medium, passage 2 lentivirus transductants were used in functional studies. Expression in transduced cells was induced by addition of 0.1 μg/ml doxycycline to culture medium. For RNA interference experiments, PHB knockdown cells were generated by siRNA molecules reported previously (37). Blocking peptide AKGRRAEDGSVIDYELI and scrambled peptide RDAGRSDALVIYEIGKE were synthesized, purified to > 95% by HPLC, and quality controlled by mass spectroscopy by Celtek.
Cell culture.
Cell lines 3T3-L1 preadipocyte cells (CL-173) and bEnd.3 endothelial cells (CRL-2299) were purchased from American Type Tissue Collection. Cells were maintained in DMEM/10% FBS. Organs were resected, and cell suspensions were made as described (19, 54). Briefly, minced tissues were digested in 0.5 mg/ml collagenase type I (Worthington Biochemical) solution under gentle agitation for 1 hour at 37°C and centrifuged at 200 g for 5 minutes to separate the stromal/vascular pellet from adipocytes. Adipocytes were inspected in suspension under a cover slip within 30 minutes. SVF cells were plated in semiconfluence on uncoated plastic in DMEM/10% FBS. Angiogenesis sprouting assays were performed as previously described (56). Briefly, SVF cells suspended in complete DMEM culture medium containing 0.12% methylcellulose were seeded in nonadherent round-bottom 96-well plates. Single spheroids per well (800 cells per spheroid) were embedded into rat tail collagen I gels (Boehringer Mannheim) by suspending spheroids in DMEM containing 10% FBS and 0.4% methylcellulose and admixing with an equal volume of 0.4% collagen solution. Upon transfer into 48-well plates prewarmed at 37°C, gels were allowed to polymerize and were incubated for 20 hours. Sprouting was quantified by measuring the length of the capillary sprouts (ocular grid at ×100 magnification) that had grown out of each spheroid (5–10 spheroids per experimental group and experiment were measured). For white adipogenesis induction performed as described (19), cells grown to confluence were cultured in medium containing 1.7 μM insulin, 0.5 mM IBMX, 1 μM dexamethasone/0.2 μM indomethacin for 3 days and in medium containing 1.7 μM insulin afterward.
FA uptake and intercellular transfer analysis.
For receptor-mediated FA uptake in cell culture, BODIPY-FL-C12 and BODIPY-FL-C16 (Molecular Probes) were used at 0.3 μM. FA uptake by adipocytes was quantified with the QBT Assay (Molecular Devices), a fluorescent assay that measures real-time kinetics of fluorescent BODIPY-C12 accumulation. QBT stock solutions were dissolved completely in Hank’s balanced salt solution with 20 mM HEPES and 0.2% FA-free BSA. Differentiated adipocytes were seeded in triplicate onto a clear-bottom 96-well plate for 8 hours at 80,000 cells/well. Cells were prestarved with 1% FBS/DMEM low glucose for 2 hours, followed by addition of 100 μl QBT FA uptake solution per well. Fluorescence was monitored at 485 nm excitation/515 nm emission every 30 seconds for 60 minutes. The inhibitory CD36 antibody was described previously (57). For the intercellular transfer assay, bEnd.3 cells plated as a monolayer were incubated with 4 μM BODIPY-FL-C16 for 30 minutes and washed 3 times with culture media containing 5% FA-free BSA, after which a part of the plate is scraped off with a spatula. 3T3-L1 adipocytes predifferentiated for 8 days were coated with magnetic nanoparticles as described (42) and forced to the bottom by placing a magnet underneath the plate. bEnd.3 and 3T3-L1 cells were allowed to interact in for 30 minutes in the presence or absence of 0.1 mM peptide RDAGRSDALVIYEIGKE or AKGRRAEDGSVIDYELI. After washing, cells were fixed with PBS containing 4% paraformaldehyde (PFA), CD31 IF was performed, and the boundary of bEnd.3 monolayer and the endothelium-free area was imaged.
Protein interaction analysis.
WAT-derived cells were disrupted in PBS containing 0.2 mM phenylmethanesulphonylfluoride (Roche) protease inhibitor (PI) cocktail with a Dounce homogenizer; centrifugation (15,000 g for 30 minutes at 4oC) was performed to separate soluble proteins from membrane pellets. Plasma membrane fraction was isolated using Qproteome PM kit (Qiagen). Solubilization of membrane proteins in PBS containing 1 mM CaCl2, 1 mM MgCl2, 50 mM n-Octyl-β-D-glucopyranoside, and PI cocktail (column buffer) was performed as described (58). Immunoprecipitation was performed by using a total of 2 × 106 confluent cells for preparation of the membrane extract based on previously reported techniques (59). Cells were solubilized in 1 ml of lysis buffer: 50 mM Tris-HCI (pH 7.4), 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EGTA, 1 mM EDTA, 2.5 mM Na3VO4, 1 mM NaF, and 1 mM β-glycerophosphate, containing antiprotease cocktail from Sigma-Aldrich. Cell lysate was centrifuged at 10,000 g for 15 minutes, and the supernatants were precleared for 1 hour at 4°C by incubation with 15 μl protein A/G magnetic beads (Invitrogen). The precleared lysates were used for immunoprecipitation with antibodies, after which the solution was centrifuged at 200 g for 4 minutes and washed 3 times with 0.5 ml lysis buffer and once with ice-cold PBS containing 1 mM Na3VO4. For precipitation of membrane proteins, serum/FA-starved cells were treated with FAs where indicated and then incubated with 5 mg/ml of EZ-Link sulfo-NHS-LC-biotin cell reagent (Thermo Scientific) in PBS, pH 8, at 4°C for 30 minutes, enabling surface biotinylation. Upon membrane extraction, proteins were pulled down with streptavidin-coated beads or indicated antibodies. Immunoprecipitates were separated by denaturing 4%–12% SDS-PAGE, blotted onto Immobilon-FL membrane (Millipore), blocked with Odyssey blocking buffer (LI-COR), and immunoblotted with specified antibodies or streptavidin-IRDye 800CW (1:1000; LI-COR 925-32230) in PBS/0.05% Triton X-100. The following antibodies were used for immunoprecipitation: anti-ANX2 (Abcam, Ab41803), anti-CD36 (Sigma-Aldrich, NB400-144), and anti-PHB (Sigma-Aldrich, HPA003280) at 5 μg IgG/mg protein extract. The following antibodies were used for immunoblotting: anti-ANX2 (Santa Cruz Biotechnology Inc., sc-30757, 1:1,000), anti-CD36 (Novus Biologicals, NB400-144, 1:3,000), and anti-PHB (Santa Cruz Biotechnology Inc., sc-18196, 1:500). Signal was detected by Odyssey CLx imaging system (LI-COR).
Statistics.
Graphpad Prism software v.5.03 and Microsoft Excel was used to graph data as mean ± SD or SEM as indicated and to calculate P values by using homoscedastic (1-tailed) Student’s t tests. P < 0.05 were considered statistically significant.
Study approval.
All experiments conformed to the University of Texas regulatory standards. The present studies in animals (protocol AWC-13-016) followed Animal Research Reporting: Reporting of In Vivo Experiments (ARRIVE) guidelines and were reviewed and approved by the Animal Welfare Committee of the University of Texas, Houston, Texas, USA (A3413-01).
Author contributions
MGK designed research, analyzed data, and wrote the first manuscript draft, to which all coauthors provided edits. ACD, AS, ZA, and DIS designed research, performed experiments, and analyzed data. KAH, RP, and WA provided materials and resources, contributed to the conceptual development of the work, analyzed data, and edited the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the grants R01DK088131 (to M.G. Kolonin), HL042493, and March of Dimes FY12-356 (both to K.A. Hajjar); by awards from the Marcus Foundation and the Gillson-Longenbaugh Foundation (to R. Pasqualini and W. Arap); and by the Mouse Metabolism core at BCM, which is supported by the Diabetes Research Center (P30 DK079638). We thank Chieh Tseng, Felipe Amaya-Manzanares, Violeta Gallardo Montejano, Fernando Florez, and Michael G. Ozawa for technical assistance. We thank Perry Bickel and Randy Seeley for many helpful discussions.
Footnotes
Conflict of interest: R. Pasqualini and W. Arap have equity in Ablaris Therapeutics, which is subject to certain restrictions under university policy. The University of New Mexico Health Sciences Center manages and monitors the terms of these arrangements in accordance with its conflict-of-interest policy.
Reference information:JCI Insight. 2016;1(10):e86351. doi:10.1172/jci.insight.86351.
Contributor Information
Ahmad Salameh, Email: Ahmad.S.Salameh@uth.tmc.edu.
Daniela I. Staquicini, Email: DStaquicini@salud.unm.edu.
Zhiqiang An, Email: zhiqiang.an@uth.tmc.edu.
Katherine A. Hajjar, Email: khajjar@med.cornell.edu.
Renata Pasqualini, Email: RPasqual@salud.unm.edu.
Wadih Arap, Email: warap@salud.unm.edu.
References
- 1.Friedman JM. Obesity: Causes and control of excess body fat. Nature. 2009;459(7245):340–342. doi: 10.1038/459340a. [DOI] [PubMed] [Google Scholar]
- 2.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121(6):2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156(1–2):20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149(3):942–949. doi: 10.1210/en.2007-1713. [DOI] [PubMed] [Google Scholar]
- 5.Bederman IR, Foy S, Chandramouli V, Alexander JC, Previs SF. Triglyceride synthesis in epididymal adipose tissue: contribution of glucose and non-glucose carbon sources. J Biol Chem. 2009;284(10):6101–6108. doi: 10.1074/jbc.M808668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blot V, McGraw TE. Use of quantitative immunofluorescence microscopy to study intracellular trafficking: studies of the GLUT4 glucose transporter. Methods Mol Biol. 2008;457:347–366. doi: 10.1007/978-1-59745-261-8_26. [DOI] [PubMed] [Google Scholar]
- 7.McArthur MJ, Atshaves BP, Frolov A, Foxworth WD, Kier AB, Schroeder F. Cellular uptake and intracellular trafficking of long chain fatty acids. J Lipid Res. 1999;40(8):1371–1383. [PubMed] [Google Scholar]
- 8.Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. 2010;90(1):367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 9.Kazantzis M, Stahl A. Fatty acid transport proteins, implications in physiology and disease. Biochim Biophys Acta. 2012;1821(5):852–857. doi: 10.1016/j.bbalip.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asterholm IW, Mundy DI, Weng J, Anderson RG, Scherer PE. Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab. 2012;15(2):171–185. doi: 10.1016/j.cmet.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su X, Abumrad NA. Cellular fatty acid uptake: a pathway under construction. Trends Endocrinol Metab. 2009;20(2):72–77. doi: 10.1016/j.tem.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest. 2002;109(10):1381–1389. doi: 10.1172/JCI14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387(6633):569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 14.Pohl J, et al. Long-chain fatty acid uptake into adipocytes depends on lipid raft function. Biochemistry. 2004;43(14):4179–4187. doi: 10.1021/bi035743m. [DOI] [PubMed] [Google Scholar]
- 15.Kampf JP, Parmley D, Kleinfeld AM. Free fatty acid transport across adipocytes is mediated by an unknown membrane protein pump. Am J Physiol Endocrinol Metab. 2007;293(5):E1207–E1214. doi: 10.1152/ajpendo.00259.2007. [DOI] [PubMed] [Google Scholar]
- 16.Charron MJ, Kahn BB. Divergent molecular mechanisms for insulin-resistant glucose transport in muscle and adipose cells in vivo. J Biol Chem. 1990;265(14):7994–8000. [PubMed] [Google Scholar]
- 17.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 18.Daquinag AC, Zhang Y, Kolonin MG. Vascular targeting of adipose tissue as an anti-obesity approach. Trends Pharmacol Sci. 2011;32(5):300–307. doi: 10.1016/j.tips.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Daquinag AC, Zhang Y, Amaya-Manzanares F, Simmons PJ, Kolonin MG. An isoform of decorin is a resistin receptor on the surface of adipose progenitor cells. Cell Stem Cell. 2011;9(1):74–86. doi: 10.1016/j.stem.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Kolonin MG, et al. Synchronous selection of homing peptides for multiple tissues by in vivo phage display. FASEB J. 2006;20(7):979–981. doi: 10.1096/fj.05-5186fje. [DOI] [PubMed] [Google Scholar]
- 21.Azhdarinia A, et al. A peptide probe for targeted brown adipose tissue imaging. Nat Commun. 2013;4:e86351. doi: 10.1038/ncomms3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolonin MG, Saha PK, Chan L, Pasqualini R, Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nat Med. 2004;10(6):625–632. doi: 10.1038/nm1048. [DOI] [PubMed] [Google Scholar]
- 23.Kim DH, et al. Rapid and weight-independent improvement of glucose tolerance induced by a peptide designed to elicit apoptosis in adipose tissue endothelium. Diabetes. 2012;61(9):2299–2310. doi: 10.2337/db11-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnhart KF, et al. A peptidomimetic targeting white fat causes weight loss and improved insulin resistance in obese monkeys. Sci Transl Med. 2011;3(108):108–112. doi: 10.1126/scitranslmed.3002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim DH, Woods SC, Seeley RJ. Peptide designed to elicit apoptosis in adipose tissue endothelium reduces food intake and body weight. Diabetes. 2010;59(4):907–915. doi: 10.2337/db09-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bickel PE, Scherer PE, Schnitzer JE, Oh P, Lisanti MP, Lodish HF. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J Biol Chem. 1997;272(21):13793–13802. doi: 10.1074/jbc.272.21.13793. [DOI] [PubMed] [Google Scholar]
- 27.Mishra S, Murphy LC, Nyomba BL, Murphy LJ. Prohibitin: a potential target for new therapeutics. Trends Mol Med. 2005;11(4):192–197. doi: 10.1016/j.molmed.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Sharma A, Qadri A. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc Natl Acad Sci U S A. 2004;101(50):17492–17497. doi: 10.1073/pnas.0407536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garin J, et al. The phagosome proteome: insight into phagosome functions. J Cell Biol. 2001;152(1):165–180. doi: 10.1083/jcb.152.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staquicini FI, et al. Vascular ligand-receptor mapping by direct combinatorial selection in cancer patients. Proc Natl Acad Sci U S A. 2011;108(46):18637–18642. doi: 10.1073/pnas.1114503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bharadwaj A, Bydoun M, Holloway R, Waisman D. Annexin A2 heterotetramer: structure and function. Int J Mol Sci. 2013;14(3):6259–6305. doi: 10.3390/ijms14036259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6(6):449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 33.Siever DA, Erickson HP. Extracellular annexin II. Int J Biochem Cell Biol. 1997;29(11):1219–1223. doi: 10.1016/S1357-2725(97)00057-5. [DOI] [PubMed] [Google Scholar]
- 34.Luo M, Hajjar KA. Annexin A2 system in human biology: cell surface and beyond. Semin Thromb Hemost. 2013;39(4):338–346. doi: 10.1055/s-0033-1334143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacher S, Achatz G, Schmitz ML, Lamers MC. Prohibitin and prohibitone are contained in high-molecular weight complexes and interact with alpha-actinin and annexin A2. Biochimie. 2002;84(12):1207–1220. doi: 10.1016/s0300-9084(02)00027-5. [DOI] [PubMed] [Google Scholar]
- 36.Rescher U, Gerke V. Annexins — unique membrane binding proteins with diverse functions. J Cell Sci. 2004;117(pt 13):2631–2639. doi: 10.1242/jcs.01245. [DOI] [PubMed] [Google Scholar]
- 37.Liu D, et al. Mitochondrial dysfunction and adipogenic reduction by prohibitin silencing in 3T3-L1 cells. PLoS One. 2012;7(3):e86351. doi: 10.1371/journal.pone.0034315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schleicher M, et al. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J Cell Biol. 2008;180(1):101–112. doi: 10.1083/jcb.200706072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ling Q, et al. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest. 2004;113(1):38–48. doi: 10.1172/JCI19684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brakenhielm E, Cao Y. Angiogenesis in adipose tissue. Methods Mol Biol. 2008;456:65–81. doi: 10.1007/978-1-59745-245-8_5. [DOI] [PubMed] [Google Scholar]
- 41.Ande SR, Xu Z, Gu Y, Mishra S. Prohibitin has an important role in adipocyte differentiation. Int J Obes (Lond) 2012;36(9):1236–1244. doi: 10.1038/ijo.2011.227. [DOI] [PubMed] [Google Scholar]
- 42.Daquinag AC, Souza GR, Kolonin MG. Adipose tissue engineering in three-dimensional levitation tissue culture system based on magnetic nanoparticles. Tissue Eng Part C Methods. 2013;19(5):336–344. doi: 10.1089/ten.tec.2012.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Souza GR, et al. Three-dimensional tissue culture based on magnetic cell levitation. Nat Nanotechnol. 2010;5(4):291–296. doi: 10.1038/nnano.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2(72):e86351. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goudriaan JR, et al. CD36 deficiency in mice impairs lipoprotein lipase-mediated triglyceride clearance. J Lipid Res. 2005;46(10):2175–2181. doi: 10.1194/jlr.M500112-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Vessal M, Mishra S, Moulik S, Murphy LJ. Prohibitin attenuates insulin-stimulated glucose and fatty acid oxidation in adipose tissue by inhibition of pyruvate carboxylase. FEBS J. 2006;273(3):568–576. doi: 10.1111/j.1742-4658.2005.05090.x. [DOI] [PubMed] [Google Scholar]
- 47.Kuda O, et al. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J Biol Chem. 2013;288(22):15547–15555. doi: 10.1074/jbc.M113.473298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Razani B, et al. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277(10):8635–8647. doi: 10.1074/jbc.M110970200. [DOI] [PubMed] [Google Scholar]
- 49.Zhao H, Hardy RW. Long-chain saturated fatty acids induce annexin II translocation to detergent-resistant membranes. Biochem J. 2004;381(pt 2):463–469. doi: 10.1042/BJ20031083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pohl J, Ring A, Korkmaz U, Ehehalt R, Stremmel W. FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Mol Biol Cell. 2005;16(1):24–31. doi: 10.1091/mbc.E04-07-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ande SR, Nguyen KH, Padilla-Meier GP, Wahida W, Nyomba BL, Mishra S. Prohibitin overexpression in adipocytes induces mitochondrial biogenesis, leads to obesity development, and affects glucose homeostasis in a sex-specific manner. Diabetes. 2014;63(11):3734–3741. doi: 10.2337/db13-1807. [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, O’Brien WG, 3rd, Lee CC, Myers MG., Jr and Tong Q. Role of GABA release from leptin receptor-expressing neurons in body weight regulation. Endocrinology. 2012;153(5):2223–2233. doi: 10.1210/en.2011-2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kotas ME, et al. Role of caspase-1 in regulation of triglyceride metabolism. Proc Natl Acad Sci U S A. 2013;110(12):4810–4815. doi: 10.1073/pnas.1301996110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Daquinag AC, Amaya-Manzanares F, Sirin O, Tseng C, Kolonin MG. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012;72(20):5198–5208. doi: 10.1158/0008-5472.CAN-12-0294. [DOI] [PubMed] [Google Scholar]
- 55.Cardó-Vila M, et al. A ligand peptide motif selected from a cancer patient is a receptor-interacting site within human interleukin-11. PLoS One. 2008;3(10):e86351. doi: 10.1371/journal.pone.0003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salameh A, Galvagni F, Bardelli M, Bussolino F, Oliviero S. Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood. 2005;106(10):3423–3431. doi: 10.1182/blood-2005-04-1388. [DOI] [PubMed] [Google Scholar]
- 57.Helming L, Winter J, Gordon S. The scavenger receptor CD36 plays a role in cytokine-induced macrophage fusion. J Cell Sci. 2009;122(pt 4):453–459. doi: 10.1242/jcs.037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nie J, et al. IFATS collection: Combinatorial peptides identify alpha5beta1 integrin as a receptor for the matricellular protein SPARC on adipose stromal cells. Stem Cells. 2008;26(10):2735–2745. doi: 10.1634/stemcells.2008-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salameh A, et al. PRUNE2 is a human prostate cancer suppressor regulated by the intronic long noncoding RNA PCA3. Proc Natl Acad Sci U S A. 2015;112(27):8403–8408. doi: 10.1073/pnas.1507882112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.