

Abstract
Background
Syncope is a sudden transient loss of consciousness and postural tone with spontaneous recovery; the most common form is vasovagal syncope (VVS). During VVS gravitational pooling excessively reduces central blood volume and cardiac output. In VVS, as in hemorrhage, impaired adrenergic vasoconstriction and venoconstriction result in hypotension. We hypothesized that impaired adrenergic responsiveness due to excess nitric oxide (NO) can be reversed by reducing NO.
Methods and Results
We recorded cardio-pulmonary dynamics in supine syncope patients and healthy volunteers, (ages 15–27) challenged with a dose-response (DR) using the α1- agonist phenylephrine (PE), with and without the NO synthase inhibitor L-NMMA. Systolic and diastolic pressures among control and VVS were the same, although increased after L-NMMA and Saline+PE (volume and pressor control for L-NMMA). HR was significantly reduced by L-NMMA (P<0.05) for control and VVS compared to baseline but there was no significant difference in HR between L-NMMA and Saline+PE. Cardiac output and splanchnic blood flow were reduced by L-NMMA for control and VVS (P<0.05) compared to baseline while total peripheral resistance (TPR) increased (P<0.05). Phenylephrine DR for splanchnic flow and resistance were blunted for VVS compared to control after Saline+PE, but enhanced after L-NMMA (P<0.001). Post-synaptic α1-adrenergic vasoconstrictive impairment was greatest in the splanchnic vasculature, and splanchnic blood flow was unaffected by PE. Forearm and calf α1-adrenergic vasoconstriction were unimpaired in VVS and unaffected by L-NMMA.
Conclusions
Impaired post-synaptic α1-adrenergic vasoconstriction in young adults with VVS can be corrected by NO synthase inhibition, demonstrated with our use of L-NMMA.
Keywords: syncope (fainting), nitric oxide, nitric oxide synthase, adrenergic regulation, phenylephrine, dose-response, L-NMMA
Introduction
Syncope (fainting) is defined by a sudden transient loss of consciousness and postural tone due to cerebral hypoperfusion with spontaneous recovery1. The most common form of syncope in the young is simple postural faint, denoted “vasovagal syncope” (VVS)2, and is associated with vasodilatation and vagal induced bradycardia causing hypotension and loss of postural tone. VVS is extremely common with lifetime incidence approaching 50%3 and can be induced in most people at different thresholds of orthostatic stress4.
VVS due to upright positioning is initiated by subdiaphragmatic gravitational blood pooling primarily within the venous system, thereby excessively reducing central blood volume5. In the absence of skeletal muscle pump activity, venous return and cardiac output (CO) decreases. In healthy subjects, baroreceptors detect a postural decrease in arterial and cardiopulmonary stretch, and maintain blood pressure (BP) by a compensatory increase in total peripheral resistance (TPR) which provokes passive elastic recoil of venous blood, and also by active splanchnic venoconstriction6. Maintenance of BP is also aided by an increase in heart rate (HR), which increases venous return and cardiac output provided that stroke volume (SV) does not fall excessively. This is the normal reflex response to orthostasis7.
Most adults with VVS exhibit excessive pooling in the splanchnic circulation and lower extremities that contributes to increased central hypovolemia, reduced CO and a sustained increase in TPR8–10. In younger adults and children, however, TPR initially increases but then decreases while upright, producing a fall in BP with or without large changes in CO11–13. This is followed by a rapid decrease in BP and HR with circulatory collapse. Impaired adrenergic vasoconstriction has been consistently demonstrated in VVS in the young involving abnormalities of the splanchnic regional vasculature14;15.
VVS likely evolved as a defense against excessive blood loss during hemorrhage16;17. The time course of hemodynamic impairment during gradual hemorrhage mirror the stages of initial BP stability, with a slow fall in BP associated with tachycardia, and a rapid decrease in BP and HR with circulatory collapse. Studies of hemorrhage in animals showed adequate endogenous norepinephrine production but the post-synaptic α1-adrenergic response to norepinephrine and exogenous adrenergic vasoconstrictors was impaired. This impairment was reversed by nitric oxide synthase (NOS) inhibition18–20. We therefore hypothesized that young adult patients with recurrent VVS may also have an impaired post-synaptic α1-adrenergic response that can be reversed by NOS inhibition.
Methods
Subjects
To test this hypothesis we recruited 10 subjects with a history of recurrent fainting (6 female, 21.2 ± 1.2 years) and 12 healthy non-fainting control subjects (8 female, 23.0 ± 1.1 years), all between 15–27 years of age. There were no differences in the ages, weight and body mass index comparing both groups. Fainters were referred to our center for investigation after experiencing at least 3 episodes of fainting within the last 12 months. Fainters gave a medical history and underwent a physical examination, electrocardiography, echocardiography, and prolonged monitoring as needed to exclude cardiac and other medical causes of their fainting. Control subjects were recruited from among age and BMI matched volunteers. Control subjects reported no clinical illness, no orthostatic intolerance, and had never fainted.
The diagnosis of VVS was primarily based on the clinical history. Key diagnostic features encompassed predisposing situations, prodromal symptoms, physical signs, and postdrome recovery and symptoms21. In all patients, past fainting was induced after prolonged standing and in two patients it was also triggered by noxious stimuli. Prodromal features included pallor, lightheadedness, nausea with abdominal discomfort, diaphoresis, a feeling of warmth, visual scotomata or frank loss of vision. Following the faint, unconsciousness lasted less than 30 seconds in all when supine and most patients felt profoundly fatigued afterwards. Exclusion criteria for participation in this study included any infectious or systemic disease (including cardiovascular disease), other forms of orthostatic intolerance, competitive athletic training, recent long-term bed rest, use of nicotine containing products or pregnancy within the last year. Prior medication for syncope, if any, was stopped for at least 2 weeks prior to participation in this study. No subjects were taking neurally active or vasoactive drugs. VVS and healthy control subjects previously underwent an upright tilt table test to 70° in which symptoms and signs of real-world syncope were confirmed in VVS patients, and to ensure absence of orthostatic intolerance in controls under test conditions.
Because prolonged upright tilt may invoke a fainting response in young healthy subjects, we limited the head-up tilt (HUT) testing to 10 min. We10;12 have previously demonstrated that this is a sufficient tilt time for the comparison of orthostatic changes between control and syncopal subjects. All subjects in the syncope group fainted during the 10-min HUT without any provocation, whereas no subjects in the control group fainted or experienced presyncopal symptoms.
All subjects were required to refrain from caffeine and xanthine-containing products for at least 72 hours prior to testing. All subjects were instructed to fast for at least 4 hours prior to testing. This study was approved by the Institutional Review Board of New York Medical College. All subjects 18 or older signed an informed consent; those younger than 18 assented to participate and their parent or legal guardian signed an informed consent.
Protocol
All patients and control volunteers were instrumented on two separate days. On the first day they received intravenous administration of the nitric oxide synthase inhibitor, NG-Monomethyl-L-arginine, monoacetate salt (L-NMMA). On another day they received an equivolumic intravenous infusion of normal saline as a volume control. An amount of phenylephrine (PE), individualized for each subject, was added to the saline in order to produce blood pressure and heart rate changes equivalent to the reflex changes caused by the infusion of the L-NMMA. This is referred to as Saline+PE.
Instrumentation
Subjects arrived at our climate-controlled center at 9:30 AM and were instructed about the tests and instrumentation. Subjects were then instrumented while supine. A left antecubital vein catheter was placed for infusion of L-NMMA or for saline + PE. Beat-to-beat blood pressure was measured by Finometer photoplethysmograph (FMS, Amsterdam, The Netherlands) on the right forefinger or middle finger calibrated to the brachial artery. The Finometer uses the Modelflow algorithm to estimate beat-to-beat relative cardiac output (CO) by pulse-wave analysis. Before experiments began, ModelFlow CO was calibrated against an Innocor inert gas rebreathing CO (Innovision, Denmark). We then placed paired electrodes using anatomic landmarks to estimate thoracic, splanchnic, pelvic, and calf segmental blood volumes, blood flows and vascular resistance by impedance plethysmography9. Forearm and calf blood flow was also measured by venous occlusion plethysmography every 5 min22. Respiratory plethysmography (Respitrace, NIMS Scientific, Miami Beach, FL) and capnography (Smith Medical PM, Waukesha, WI) measured changes in respiration and end tidal carbon dioxide (ETCO2). An electrocardiograph measured HR from the beat-to-beat cardiac electrical interval. Signals were acquired at 200 samples/s, multiplexed, and A/D converted using custom software. These measurements are routine for our usual “tilt table” studies. Following instrumentation, subjects remained awake and supine for 30 minutes to acclimate. Baseline data were acquired. Thereafter either L-NMMA or saline was given by infusion on different days.
L-NMMA infusion
VVS patients and control subjects received the non-isoform specific NOS inhibitor L-NMMA delivered as a 500µg/kg/min intravenous loading dose for 15 min, followed by a 50µg/kg/min maintenance infusion. L-NMMA is the only parenteral experimental NOS inhibitor available for human use (Bachem, Switzerland; FDA IND exemption #76,314, J. Stewart). In all subjects, a steady state for HR, BP, TPR, and CO was reached during the maintenance L-NMMA infusion within 40 minutes. Maintenance L-NMMA continued throughout all subsequent measurements.
Saline + phenylephrine infusion (denoted Saline+PE)
On another day, separated by at least two days to allow for the elimination of L-NMMA, VVS patients and control subjects received saline delivered to simulate the fluid volume and timing of a loading dose of L-NMMA. During this maintenance phase a low dose of phenylephrine was slowly titrated until HR and BP were similar to HR and BP in the same subject after loading L-NMMA. This dose of phenylephrine was maintained throughout the experiment. The amount of phenylephrine required to compensate for HR and BP never exceeded 0.2µg/kg/min in any subject.
Phenylephrine Dose-Response
After either L-NMMA or Saline+PE were infused and HR and BP stabilized, new post-L-NMMA (or post-Saline+PE) baseline hemodynamic data were acquired. A phenylephrine dose response (Phenyl DR) was then obtained in each subject23 by infusing additional phenylephrine at 0.5, 1.0, 2.0, 3.0, and 4.0 µg/kg/min for 10 min. At each dose we measured ModelFlow CO, arm and leg blood flows by venous occlusion plethysmography, cuff BP and HR at minutes 8–10 at each concentration. At each phenylephrine dose, data were also obtained using all beat-to-beat measurement modalities (e.g. impedance, HR, etc). Stopping criteria for the use of phenylephrine included a HR<40 bpm, a BP>160/85, or the subject’s request to stop. Not all subjects were able to receive all doses of phenylephrine before reaching stopping criteria. However, all patients were able to receive 0.5, 1.0, and 2.0 µg/kg/min infusions of phenylephrine each for 10 minutes in succession, and these doses were therefore used for comparison. These data were depicted graphically and used for analysis. Statistics were computed by including post-loading data (L-NMMA or Saline+PE) and the responses to 0.5, 1.0 and 2.0 µg/kg/min.
Data Analysis
During experiments mean arterial pressure (MAP) was calculated as (systolic BP +2*diastolic BP)/3 and total peripheral resistance (TPR) was calculated as MAP/CO. The primary outcome variables were BP, HR, CO, and TPR; secondary outcome variables were changes in splanchnic, forearm and calf blood flows and related regional resistances calculated as MAP/flow. Thoracic (central) blood flow was measured as the CO. Paired t-tests were used to compare the response to drug loading shown in Table 1.
Table 1.
Baseline and Post-Drug-Load Characteristics
| Control-Baseline | Control Post- Load |
VVS Baseline | VVS Post-Load | |
|---|---|---|---|---|
| Systolic BP (mmHg) | ||||
| LNMMA | 117±2 | 123±2 | 121±4 | 120±6 |
| Saline+PE | 115±2 | 120±2 | 118±2 | 122±3 |
| Diastolic BP (mmHg) | ||||
| LNMMA | 62±2 | 69±2 | 63±4 | 68±3 |
| Saline+PE | 65±2 | 70±2 | 63±4 | 65±3 |
| MAP (mmHg) | ||||
| LNMMA | 79±1 | 87±2* | 80±2 | 85±1* |
| Saline+PE | 82±2 | 87±2* | 81±1 | 84±2* |
| HR (bpm) | ||||
| LNMMA | 62±2 | 57±1* | 62±4 | 58±1* |
| Saline+PE | 64±2 | 62±3 | 62±4 | 64±3 |
| CO (L/min) | ||||
| LNMMA | 4.8±0.4 | 4.2±0.4* | 4.9±0.4 | 4.2±0.4* |
| Saline+PE | 5.2±0.4 | 4.6±0.5 | 4.8±0.4 | 4.6±0.5 |
| TPR (mmHg/L/min) | ||||
| LNMMA | 16.4±1.8 | 20.3±2.2* | 16.4±1.0 | 18.4±1.1* |
| Saline+PE | 16.1±1.1 | 18.1±1.5 | 15.8±1.2 | 17.1±1.0 |
| Splanchnic BF (L/min) | ||||
| LNMMA | 1.5±0.2 | 1.2±0.1* | 1.3±0.1 | 1.0±0.1* |
| Saline+PE | 1.2±0.2 | 1.2±0.2 | 1.2±0.1 | 1.2±0.1 |
| Forearm BF (ml%/min) | ||||
| LNMMA | 1.87±0.32 | 1.65±0.33 | 1.81±0.19 | 1.61±0.14 |
| Saline+PE | 2.05±0.33 | 2.10±0.27 | 2.22±0.25 | 2.18±0.12 |
| Calf BF (ml%/min) | ||||
| LNMMA | 2.41±0.57 | 2.33±0.54 | 1.70±0.60 | 1.74±0.58 |
| Saline+PE | 1.93±0.59 | 1.82±0.55 | 1.57±0.32 | 1.54±0.58 |
(Post-Load = following L-NMMA or Saline+PE)
=p<0.05 compared to baseline
The dose-response data were analyzed as two separate experiments (L-NMMA and Saline + PE), using a repeated measures analysis of variance (RM-ANOVA). For each experiment, there was one between factor (group, i.e., patient vs. control) and one within factor (concentration). Within patient, outcomes were measured at four concentrations: pre-challenge baseline (0), 0.5, 1, and 2 ug/kg/min of PE concentration (CONC), which constituted the repeated measures. The study was conducted on two separate days, with the L-NMMA conducted on the first day and Saline + PE on the second (separated by at least two days). This was done to insure that the loading dose of Saline + PE matched the patient’s fluid volume and pressor response to L-NMMA.
The primary relationship of interest for both experiments was the GROUP by CONC interaction, which describes how patients and controls differ in their response to the concentration challenge under each experimental setting. For these analyses, we assumed a covariance structure of compound symmetry. Reported p-values reflect the interaction term using the Greenhouse-Geisser correction. Further, since we assumed that, if there were a response to the concentration challenge for either group, the response would increase or decrease monotonically. Thus, our interest was in the difference in slopes between the study groups, rather than the group difference at any particular concentration level. As such, no post-hoc pairwise comparisons of groups at concentration specific concentration levels were conducted. Values are presented as means ± SEM. Statistical significance was set at p ≤ 0.05. NCSS 2007 (NCSS, LCC, and Kaysville) statistical software was used for statistical analyses.
Results
Baseline and Post-Drug Data
These data are tabulated in Table 1. There was no significant difference in systolic blood pressure or diastolic blood pressure among control and VVS groups, although these pressures tended to be increased after L-NMMA and Saline+PE. Mean arterial blood pressure (MAP) however was significantly increased (P<0.05). HR was not different at baseline, but was significantly reduced by L-NMMA (P<0.05) for control and VVS compared to baseline. Nevertheless, there was no significant difference in HR between L-NMMA and Saline+PE. Neither respiratory rate nor ETCO2 (not shown) were affected by either L-NMMA or Saline+PE in both control and VVS subjects. CO (thoracic blood flow) and splanchnic blood flow were reduced by L-NMMA for control and VVS (P<0.05) compared to baseline while TPR was increased (P<0.05) compared to baseline. There was no significant difference between L-NMMA and Saline+PE CO or TPR for either group. There was no effect of drugs on forearm or calf blood flow for controls and VVS.
MAP and HR responses to phenylephrine
MAP (top panels), HR (middle panels) and SV during Phenyl DR are shown in Figure 1. MAP monotonically increased for all subjects. MAP increased more in control than in VVS patients (P<0.01) during phenylephrine infusion (right panels). Loading with L-NMMA (left panels) increased the MAP Phenyl DR for both control and VVS and their respective response curves became similar. HR decreased following infusion of phenylephrine, was similar for both groups, and was shifted significantly and similarly for both groups after L-NMMA (P<0.001). SV increased significantly in VVS during phenylephrine infusions (P<0.01) but remained unchanged in controls. After L-NMMA, SV decreased in a similar manner for both control and VVS with higher concentrations of PE (P=0.56). Thus, Phenyl DR for MAP at each dose tested was decreased in VVS after Saline+PE loading but was similar to control after L-NMMA loading. HR response was similar for both groups and decreased with L-NMMA.
Figure 1.
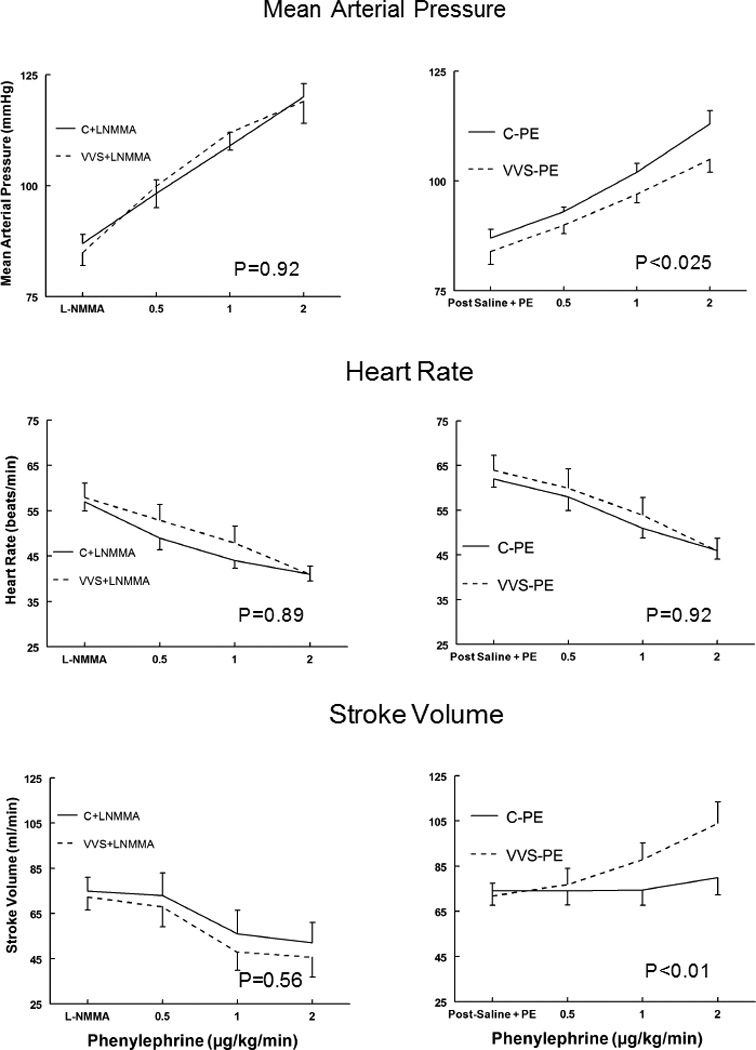
Mean Arterial Pressure (MAP), upper panels, heart rate (HR), middle panels, and stroke volume (SV) lower panels during phenylephrine dose response for control subjects (solid lines) and vasovagal syncope subjects (dashed lines). Saline and low dose phenylephrine were infused to simulate the fluid volume and maintained (right panels) to assure that HR and MAP were similar to HR and MAP in each subject after loading L-NMMA (left panels). This is depicted as Post Saline+PE and is shown for control subjects who received saline and phenylephrine (C-PE), control subjects who received L-NMMA (C+LNMMA), patients with vasovagal syncope subjects who received saline and phenylephrine (VVS-PE) and patients with VVS who received L-NMMA (VVS+LNMMA). Following that, increasing concentrations of phenylephrine were infused each for 10 minutes. P values represent the group by concentration interaction effect.
CO and TPR responses to phenylephrine
CO and TPR during Phenyl DR are shown in Figure 2. CO monotonically decreased with phenylephrine for control subjects (C-PE) (P<0.001), but was not different compared to VVS subjects (VVS-PE). The presence of L-NMMA resulted in a decrease of CO with phenylephrine that was similar for both control and VVS.
Figure 2.
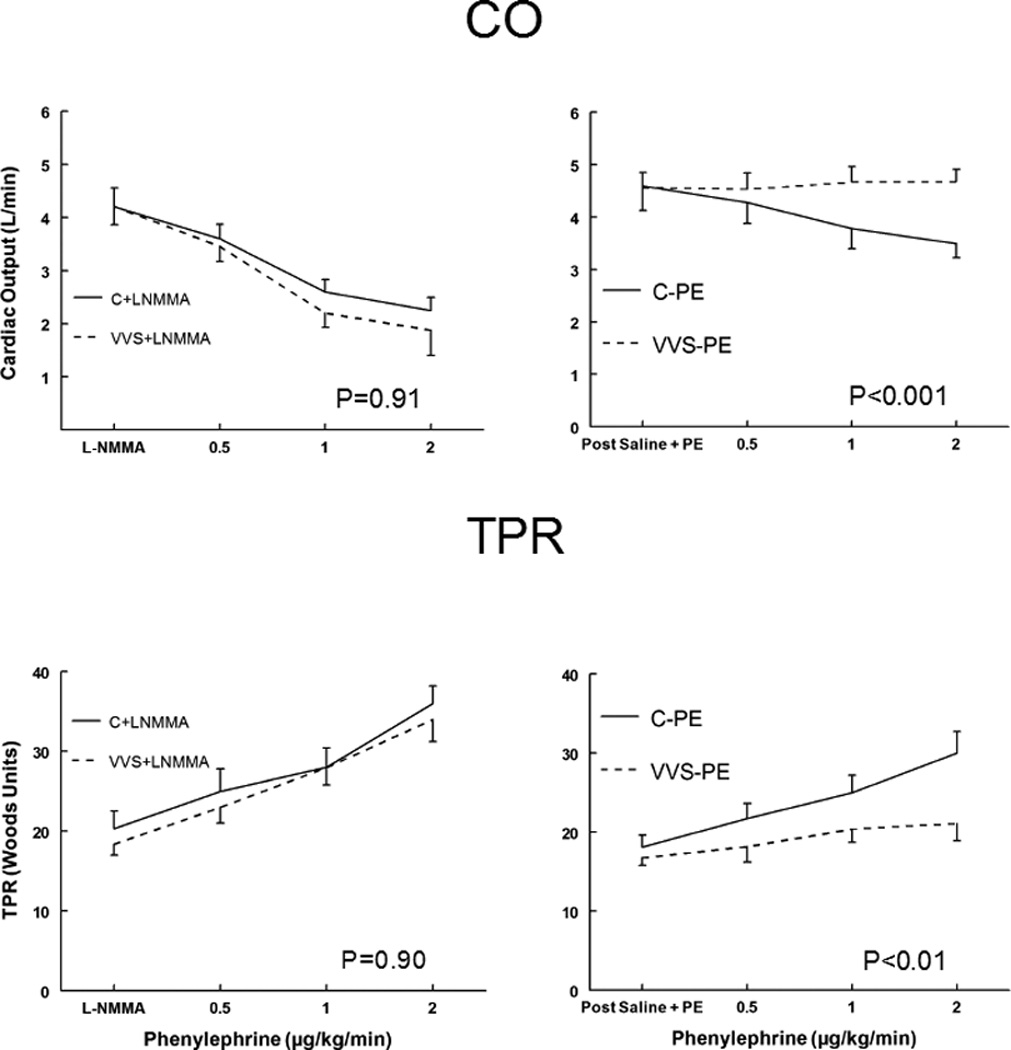
Cardiac Output (CO), upper panels, and Total Peripheral Resistance (TPR), lower panels, during phenylephrine dose response for control subjects (solid lines) and vasovagal syncope subjects (dashed lines). Saline and low dose phenylephrine were infused to simulate the fluid volume and maintained (right panels) to assure that HR and MAP were similar to HR and MAP in each subject after loading L-NMMA (left panels). This is depicted as Post Saline+PE and is shown for control subjects (C-PE), control subjects who received L-NMMA (C+LNMMA), patients with vasovagal syncope subjects who received saline and phenylephrine (VVS-PE) and patients with VVS who received L-NMMA (VVS+LNMMA). Following that, increasing concentrations of phenylephrine were infused each for 10 minutes. P values represent the group by concentration interaction effect.
The TPR with Phenyl DR was the mirror image of CO. After Saline+PE loading TPR increased significantly for control during phenyl DR (P<0.001), but was not different from Saline+PE for VVS during phenyl DR. The Phenyl DR for TPR was similar for both groups in the presence of L-NMMA and was shifted significantly upwards for VVS compared to VVS without L-NMMA (P<0.001). Thus, Phenyl DR was blunted in VVS after Saline+PE but was similar to control after L-NMMA loading.
Splanchnic blood flow (Fsplanchnic) and splanchnic vascular resistance (Rsplanchnic) responses to phenylephrine
Fsplanchnic and Rsplanchnic during Phenyl DR are shown in Figure 3. While there was almost no change in Fsplanchnic and Rsplanchnic to increasing doses of phenylephrine for VVS after Saline+PE, these values decreased significantly for Controls (P< 0.01). There was however a markedly enhanced response to phenylephrine after L-NMMA (P<0.001). Thus for VVS in the presence of L-NMMA, Fsplanchnic decreased to a great extent, while Rsplanchnic increased to a greater extent than control in response to increasing doses of phenylephrine. In controls Fsplanchnic and Rsplanchnic were unaffected by L-NMMA. However in VVS after L-NMMA, Fsplanchnic was excessively decreased and Rsplanchnic was excessively increased.
Figure 3.
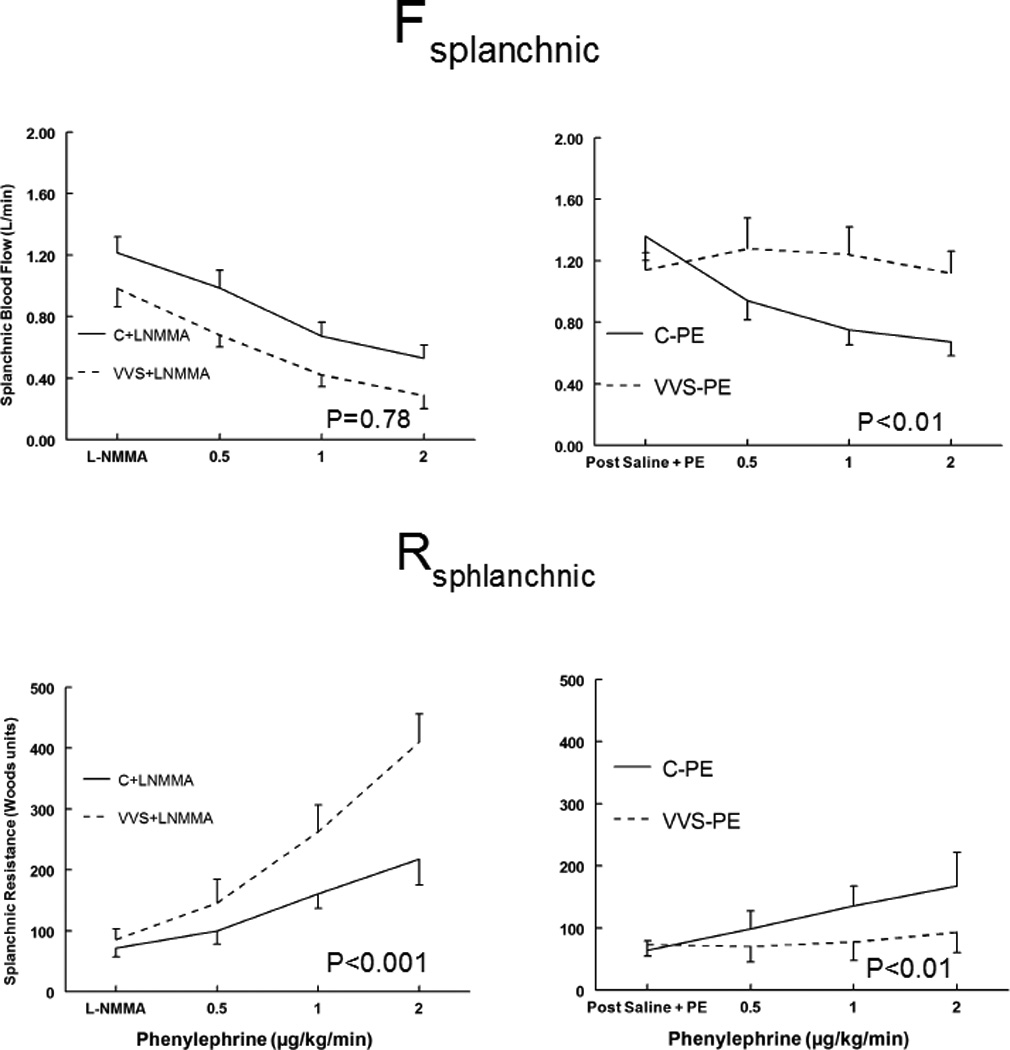
Splanchnic Blood Flow (Fsplanchnic), upper panels, and Splanchnic Resistance (Rsplanchnic) lower panels, during phenylephrine dose response for control subjects (solid lines) and vasovagal syncope subjects (dashed lines). Saline and low dose phenylephrine were infused to simulate the fluid volume and maintained (right panels) to assure that HR and MAP were similar to HR and MAP in each subject after loading L-NMMA (left panels). This is depicted as Post Saline+PE and is shown for control subjects who received saline and phenylephrine (C-PE), control subjects who received L-NMMA (C+LNMMA), patients with vasovagal syncope subjects who received saline and phenylephrine (VVS-PE) and patients with VVS who received L-NMMA (VVS+LNMMA). Following that, increasing concentrations of phenylephrine were infused each for 10 minutes. P values represent the group by concentration interaction effect.
Forearm and calf vascular resistance responses to phenylephrine
In contrast to the splanchnic vasculature, the increase in forearm and calf vascular resistance shown in Figure 4 increased similarly during the Phenyl DR for control and VVS subjects and the presence of L-NMMA did not significantly affect these relationships. These findings are consistent with data presented in Table 2, which show that there were no significant differences in calf and forearm blood flows in control and VVS patients at each concentration of phenylephrine tested.
Figure 4.
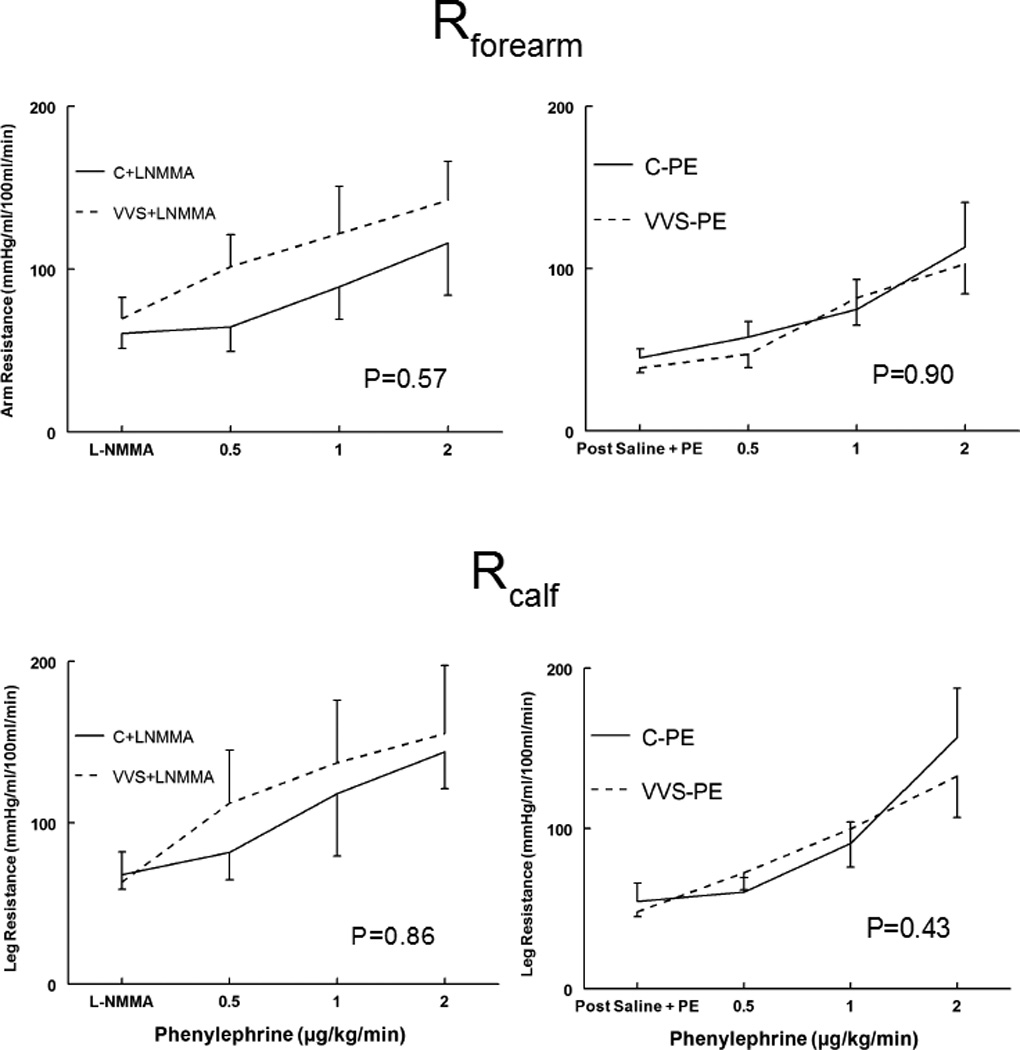
Forearm and calf vascular resistances increase similarly during the phenyl D-R for control and VVS subjects Forearm Resistance (Rforearm), upper panels, and Calf Resistance (Rcalf), lower panels, during phenylephrine dose response for control subjects (solid lines) and vasovagal syncope subjects (dashed lines). Saline and low dose phenylephrine were infused to simulate the fluid volume and maintained (right panels) to assure that HR and MAP were similar to HR and MAP in each subject after loading L-NMMA (left panels). This is depicted as Post Saline+PE and is shown for control subjects who received saline and phenylephrine (C-PE), control subjects who received L-NMMA (C+LNMMA), patients with vasovagal syncope subjects who received saline and phenylephrine (VVS-PE) and patients with VVS who received L-NMMA (VVS+LNMMA). Following that, increasing concentrations of phenylephrine were infused each for 10 minutes. P values represent the group by concentration interaction effect.
Table 2.
Cardiovascular dynamics and calf and forearm blood flows in control and VVS patients at each concentration of phenylephrine tested.
| Baseline | Post-Drug | 0.5 µg/kg/min PE | 1.0 µg/kg/min PE | 2.0 µg/kg/min PE | |
|---|---|---|---|---|---|
| Systolic BP VVS (mmHg) | |||||
| LNMMA | 121±4 | 119±6 | 140±7 | 161±6 | 174±7 |
| Saline+PE | 118±2 | 122±3 | 129±4 | 137±4 | 151±4 |
| Systolic BP Control (mmHg) | |||||
| LNMMA | 113±2 | 123±2 | 136±4 | 149±4 | 169±5 |
| Saline+PE | 115±2 | 120±2 | 128±3 | 143±4 | 160±5 |
| Diastolic BP VVS (mmHg) | |||||
| LNMMA | 63±4 | 68±3 | 80±4 | 87±4 | 92±5 |
| Saline+PE | 63±4 | 65±3 | 71±5 | 77±2 | 83±4 |
| Diastolic BP Control (mmHg) | |||||
| LNMMA | 62±2 | 69±2 | 78±3 | 90±3 | 96±3 |
| Saline+PE | 65±2 | 70±2 | 75±1 | 81±2 | 89±3 |
| HR VVS (beats/min) | |||||
| LNMMA | 62±5 | 58±3 | 53±3 | 48±2 | 41±2 |
| Saline+PE | 62±4 | 61±3 | 60±4 | 54±4 | 46±3 |
| HR Control (beats/min) | |||||
| LNMMA | 62±5 | 57±3 | 49±3 | 44±2 | 41±2 |
| Saline+PE | 64±4 | 62±3 | 58±3 | 51±2 | 46±2 |
| Forearm BF VVS (ml%/min) | |||||
| LNMMA | 1.81±0.19 | 1.61±0.14 | 1.11±0.18 | 0.87±0.18 | 0.86±0.13 |
| Saline+PE | 2.22±0.25 | 2.18±0.12 | 2.01±0.36 | 1.45±0.34 | 1.14±0.18 |
| Forearm BF Control (ml%/min) | |||||
| LNMMA | 1.87±0.32 | 1.65±0.33 | 1.58±0.58 | 1.50±0.30 | 1.38±0.34 |
| Saline+PE | 2.05±0.33 | 2.10±0.27 | 1.83±0.32 | 1.70±0.36 | 1.26±0.38 |
| Calf BF VVS (mL%/min) | |||||
| LNMMA | 1.70±0.60 | 1.74±0.58 | 1.26±0.41 | 1.16±0.38 | 1.11±0.39 |
| Saline+PE | 1.57±0.32 | 1.54±0.26 | 1.68±0.48 | 1.80±0.54 | 1.44±0.39 |
| Calf BF Control (mL%/min) | |||||
| LNMMA | 2.41±0.57 | 2.33±0.54 | 2.14±0.71 | 2.07±0.78 | 1.90±0.83 |
| Saline+PE | 1.93±0.59 | 1.82±0.55 | 1.68±0.41 | 1.32±0.38 | 0.89±0.26 |
Discussion
The results of this study in young adults indicate that there is impaired post-synaptic α1-adrenergic vasoconstriction in VVS patients compared to healthy volunteers which results in reduced TPR and CO responsiveness to phenylephrine in our subjects with a history of VVS. Interestingly, this post-synaptic α1-adrenergic vasoconstrictive impairment can be corrected following nitric oxide synthase inhibition using L-NMMA.
We show that post-synaptic α1-adrenergic vasoconstrictive impairment is greatest in the splanchnic vasculature, based on our demonstration that splanchnic blood flow in the absence of nitric oxide synthase inhibition by L-NMMA is relatively unaffected by phenylephrine. However, following L-NMMA, splanchnic flow decreased in a dose-dependent manner in both control and VVS subjects following phenylephrine administration. In contrast, forearm and calf α1-adrenergic vasoconstriction are unimpaired in VVS patients and apparently unaffected by L-NMMA.
In the present investigation we were only interested in whether phenylephrine had an effect on group differences; whether the response slopes differed as concentrations of phenylephrine were increased. Thus we were not interested in differences at each concentration or in identifying the concentration at which the departure became significant as this was deemed to be relatively unimportant.
These observations are consistent with findings of nitric oxide potentiated splanchnic hyporeactivity24, blunted chemoreflex25, abnormal cerebral autoregulation26, and impaired cardiovagal baroreflex27 which occur during the later presyncopal phase of VVS.
Evidence from the literature that nitric oxide blunts adrenergic neurotransmission
Nitric oxide is a fundamental signaling molecule with pleiotropic effects and ubiquitous distribution. Three separate NOS isoforms are recognized: neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS). nNOS and eNOS are constitutively expressed and depend on calcium-calmodulin28.
nNOS is found in the brain and in peripheral tissues such as skin, kidney, and splanchnic vasculature where neuronal NO acts to modulate adrenergic neurotransmission29. nNOS has been localized to “nitrergic” nerves that proliferate within the gastrointestinal tract and distribute with the parasympathetic nervous system29. While local eNOS exerts endothelial mediated vascular effects which could directly mediate vasodilation in VVS30;31, NO released from nitrergic nerves can act at pre-junctional and post-junctional sites to reduce sympathetic transduction32. Effects are largest in the splanchnic vasculature33 and in the kidney34 where the density of nitrergic nerves is greatest, and are most potent during sympathetic activation.
Our previous work demonstrated nNOS dependence of the local skin heating response35 while co-release of NO from cholinergic nerves in part mediates active cutaneous vasodilation36. Heat-induced NO vasodilation informs on its ability to blunt adrenergic vasoconstriction37. The sympatholytic effect of NO in man may be less potent in skeletal muscle38 although combined prostaglandin and NO inhibition augments adrenergic vasoconstriction during contraction39. Excessive NO may play a role in autonomic failure40.
Splanchnic hyperemia occurs in real or simulated microgravity (bedrest in man, hindlimb suspension in rats) where it is associated with orthostatic intolerance, adrenergic hyporeactivity41;42, and enhanced microvascular production of NO through increased transcription of NOS isoforms43;44. Increased NO has been reported in both postural tachycardia syndrome and VVS45–47 but those results are controversial due to measurement methods and lack of subsetting46.
Nevertheless, nitrergic NO modulation of adrenergic vasoconstriction causes splanchnic hyperemia and our data indicate that our patients with vasovagal syncope overproduce nitric oxide within the splanchnic circulation by pathways as yet undefined, resulting in a post-synaptic defect in adrenergic vasoconstriction. If present during orthostasis, such a defect could lead to a blunted response to sympathetic stimulation and potentiate VVS in this cohort of subjects.
The effects of NO on the adrenergic system are protean. In this paper we examined only post-synaptic effects, which could include decreased number of adrenergic receptors – unlikely given the fairly rapid normalizing effects of L-NMMA. Post-synaptic effects could also include interference with adrenergic binding and in the transduction process making them likely candidates for NO-induced vasoconstrictive downregulation. These candidate mechanisms cannot be distinguished based on current data. In addition, other unmeasured NO effects such as reduction of central sympathetic activation and ganglionic transmission may be important, and are subjects for subsequent investigation.
These results, obtained in young adults, ages 15–27 years, may be different than those obtained from older adults. There is an increase in the reporting of VVS with age and in females, there is a first peak of syncopal incidents occurring at 15–24 years, with another peak occurring at age 40. The mechanisms of VVS at various ages are unclear, but this increased incidence has been attributed to the increased use of vasoactive and cardioactive medications, decreased vascular compliance, diminished cardiac function, physical deconditioning and poorer health with age. Therefore our findings of impaired post-synaptic α1-adrenergic vasoconstriction in VVS, which can be corrected by NO synthase inhibition, may be a finding that is unique to young adults.
Limitations
The study may be underpowered with respect to measurements of forearm and calf vascular resistance. Because of this we were unable to demonstrate changes in resistance after loading L-NMMA, although these noisy data could reflect a trend towards increased limb resistances with L -NMMA. However, the results do show that limb resistance following saline+PE loading is not different for VVS compared to control subjects. In addition, due to the small sample size, significant findings may be products of type I errors given that there are multiple outcomes in addition to multiple testing for each outcome.
Since the incidence of syncope is dramatically higher in older vs. younger adults, the findings of this study are likely not applicable across all age groups. Sex differences and menstrual phase were not distinguished. There were insufficient subjects for this purpose. However, no obvious difference in sympathetic nerve activity or vasoconstrictive ability by sex or menstrual phase has been found, although differences in total blood volume reduce blood pressure in women48;49.
WHAT IS KNOWN
A common syncope in the young, vasovagal syncope (VVS), is associated with hypotension and loss of postural tone. Normally, baroreceptors maintain blood pressure (BP) by a compensatory increase in total peripheral resistance (TPR), passive elastic recoil of venous blood, by active splanchnic venoconstriction. BP is also maintained by an increase in heart rate (HR), which increases venous return and cardiac output (CO).
During VVS in younger adults and children, TPR initially increases but then decreases while upright, producing a fall in BP followed by a rapid decrease in BP and HR with circulatory collapse. VVS in the young has been associated with impaired adrenergic vasoconstriction of the splanchnic regional vasculature.
Studies of hemorrhage, a model of syncope, showed the post-synaptic α1-adrenergic response to norepinephrine and exogenous adrenergic vasoconstrictors was impaired which was reversed by nitric oxide synthase (NOS) inhibition.
Thus, young adult VVS patients may have an impaired post-synaptic α1-adrenergic response that can be reversed by NOS inhibition.
WHAT THE STUDY SHOWS
There is impaired post-synaptic α1-adrenergic vasoconstriction in young adult VVS patients compared to healthy volunteers. This is associated with reduced TPR and CO responsiveness to phenylephrine in VVS patients.
The post-synaptic α1-adrenergic vasoconstrictive impairment was greatest in the splanchnic vasculature; forearm and calf α1-adrenergic vasoconstriction were unimpaired in VVS patients.
This post-synaptic α1-adrenergic vasoconstrictive impairment in young adults with VVS can be corrected following nitric oxide synthase inhibition using L-NMMA.
Acknowledgments
Sources of Funding: Funding for this project was provided by grants RO1 HL 112736 and RO1 HL 074873 from the National Heart Lung and Blood Institute.
Footnotes
Author contributions: JMS was responsible for the conception and design of the experiments, interpretation of data and drafting the article. MS was responsible for collection, assembly and interpretation of the data. SM was responsible for collection, assembly and interpretation of the data. RS was responsible for interpretation of the data and revising the manuscript. CT was responsible for collection, assembly and interpretation of the data. PV was responsible for biostatistical analysis and interpretation of the data and critically revising intellectual content of the article. MSM was responsible for design of the experiments, analysis and interpretation of the data and critically revising intellectual content of the article.
Disclosures: None.
References
- 1.Folino AF, Russo G, Porta A, Buja G, Cerutti S, Iliceto S. Modulations of autonomic activity leading to tilt-mediated syncope. Int J Cardiol. 2007;120:102–107. doi: 10.1016/j.ijcard.2006.03.093. [DOI] [PubMed] [Google Scholar]
- 2.Gowers WR. A lecture on vagal and vasovagal attacks. Lancet. 1907;173:716–724. [Google Scholar]
- 3.Moya A, Sutton R, Ammirati F, Blanc JJ, Brignole M, Dahm JB, Deharo JC, Gajek J, Gjesdal K, Krahn A, Massin M, Pepi M, Pezawas T, Ruiz GR, Sarasin F, Ungar A, van Dijk JG, Walma EP, Wieling W. Guidelines for the diagnosis and management of syncope (version 2009) Eur Heart J. 2009;30:2631–2671. doi: 10.1093/eurheartj/ehp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.el Bedawi KM, Hainsworth R. Combined head-up tilt and lower body suction: a test of orthostatic tolerance. Clin Auton Res. 1994;4:41–47. doi: 10.1007/BF01828837. [DOI] [PubMed] [Google Scholar]
- 5.Rowell LB. Human cardiovascular control. New York, NY: Oxford University Press; 1993. [Google Scholar]
- 6.Donald DE, Rowlands DJ, Ferguson DA. Similarity of blood flow in the normal and the sympathectomized dog hind limb during graded exercise. Circ Res. 1970;26:185–199. doi: 10.1161/01.res.26.2.185. [DOI] [PubMed] [Google Scholar]
- 7.O'Leary DD, Kimmerly DS, Cechetto AD, Shoemaker JK. Differential effect of head-up tilt on cardiovagal and sympathetic baroreflex sensitivity in humans. Exp Physiol. 2003;88:769–774. doi: 10.1113/eph8802632. [DOI] [PubMed] [Google Scholar]
- 8.Mosqueda-Garcia R, Furlan R, Md JT, Fernandez-Violante R. The elusive pathophysiology of neurally mediated syncope. Circulation. 2000;102:2898–2906. doi: 10.1161/01.cir.102.23.2898. [In Process Citation] [DOI] [PubMed] [Google Scholar]
- 9.Stewart JM, Medow MS, Glover JL, Montgomery LD. Persistent splanchnic hyperemia during upright tilt in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2006;290:H665–H673. doi: 10.1152/ajpheart.00784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taneja I, Medow MS, Glover JL, Raghunath NK, Stewart JM. Increased vasoconstriction predisposes to hyperpnea and postural faint. Am J Physiol Heart Circ Physiol. 2008;295:H372–H381. doi: 10.1152/ajpheart.00101.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Jong-de Vos van Steenwijk CC, Wieling W, Johannes JM, Harms MP, Kuis W, Wesseling KH. Incidence and hemodynamic characteristics of near-fainting in healthy 6- to 16-year old subjects. J Am Coll Cardiol. 1995;25:1615–1621. doi: 10.1016/0735-1097(95)00056-a. [DOI] [PubMed] [Google Scholar]
- 12.Nowak JA, Ocon A, Taneja I, Medow MS, Stewart JM. Multiresolution wavelet analysis of time-dependent physiological responses in syncopal youths. Am J Physiol Heart Circ Physiol. 2009;296:H171–H179. doi: 10.1152/ajpheart.00963.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verheyden B, Liu J, van Dijk N, Westerhof BE, Reybrouck T, Aubert AE, Wieling W. Steep fall in cardiac output is main determinant of hypotension during drug-free and nitroglycerine-induced orthostatic vasovagal syncope. Heart Rhythm. 2008;5:1695–1701. doi: 10.1016/j.hrthm.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Dietz NM, Halliwill JR, Spielmann JM, Lawler LA, Papouchado BG, Eickhoff TJ, Joyner MJ. Sympathetic withdrawal and forearm vasodilation during vasovagal syncope in humans. J Appl Physiol (1985) 1997;82:1785–1793. doi: 10.1152/jappl.1997.82.6.1785. [DOI] [PubMed] [Google Scholar]
- 15.Stewart JM, McLeod KJ, Sanyal S, Herzberg G, Montgomery LD. Relation of postural vasovagal syncope to splanchnic hypervolemia in adolescents. Circulation. 2004;110:2575–2581. doi: 10.1161/01.CIR.0000145543.88293.21. [DOI] [PubMed] [Google Scholar]
- 16.Barcroft H, McMichael J. Posthaemorrhagic fainting. Study by cardiac output and forearm flow. Lancet. 1944;1:489–491. [Google Scholar]
- 17.Diehl RR. Vasovagal syncope and Darwinian fitness. Clin Auton Res. 2005;15:126–129. doi: 10.1007/s10286-005-0244-0. [DOI] [PubMed] [Google Scholar]
- 18.Liu LM, Ward JA, Dubick MA. Hemorrhage-induced vascular hyporeactivity to norepinephrine in select vasculatures of rats and the roles of nitric oxide and endothelin. Shock. 2003;19:208–214. doi: 10.1097/00024382-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Pieber D, Horina G, Sandner-Kiesling A, Pieber TR, Heinemann A. Pressor and mesenteric arterial hyporesponsiveness to angiotensin II is an early event in haemorrhagic hypotension in anaesthetised rats. Cardiovasc Res. 1999;44:166–175. doi: 10.1016/s0008-6363(99)00194-7. [DOI] [PubMed] [Google Scholar]
- 20.Thiemermann C, Szabo C, Mitchell JA, Vane JR. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc Natl Acad Sci U S A. 1993;90:267–271. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheldon RS, Grubb BP, Olshansky B, Shen WK, Calkins H, Brignole M, Raj SR, Krahn AD, Morillo CA, Stewart JM, Sutton R, Sandroni P, Friday KJ, Hachul DT, Cohen MI, Lau DH, Mayuga KA, Moak JP, Sandhu RK, Kanjwal K. 2015 heart rhythm society expert consensus statement on the diagnosis and treatment of postural tachycardia syndrome, inappropriate sinus tachycardia, and vasovagal syncope. Heart Rhythm. 2015;12:e41–e63. doi: 10.1016/j.hrthm.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart JM. Transient orthostatic hypotension is common in adolescents. J Pediatr. 2002;140:418–424. doi: 10.1067/mpd.2002.122643. [DOI] [PubMed] [Google Scholar]
- 23.Van Tassell BW, Rondina MT, Huggins F, Huggins F, Gilbert EM, Munger MA. Carvedilol increases blood pressure response to phenylephrine infusion in heart failure subjects with systolic dysfunction: evidence of improved vascular alpha1-adrenoreceptor signal transduction. Am Heart J. 2008;156:315–321. doi: 10.1016/j.ahj.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamza SM, Kaufman S. Role of spleen in integrated control of splanchnic vascular tone: physiology and pathophysiology. Can J Physiol Pharmacol. 2009;87:1–7. doi: 10.1139/Y08-103. [DOI] [PubMed] [Google Scholar]
- 25.Valdes V, Mosqueira M, Rey S, Del Rio R, Iturriaga R. Inhibitory effects of NO on carotid body: contribution of neural and endothelial nitric oxide synthase isoforms. Am J Physiol Lung Cell Mol Physiol. 2003;284:L57–L68. doi: 10.1152/ajplung.00494.2001. [DOI] [PubMed] [Google Scholar]
- 26.Lavi S, Egbarya R, Lavi R, Jacob G. Role of nitric oxide in the regulation of cerebral blood flow in humans: chemoregulation versus mechanoregulation. Circulation. 2003;107:1901–1905. doi: 10.1161/01.CIR.0000057973.99140.5A. [DOI] [PubMed] [Google Scholar]
- 27.Liu JL, Murakami H, Zucker IH. Effects of NO on baroreflex control of heart rate and renal nerve activity in conscious rabbits. Am J Physiol. 1996;270:R1361–R1370. doi: 10.1152/ajpregu.1996.270.6.R1361. [DOI] [PubMed] [Google Scholar]
- 28.Busse R, Mulsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265:133–136. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- 29.Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev. 2003;55:271–324. doi: 10.1124/pr.55.2.3. [DOI] [PubMed] [Google Scholar]
- 30.Pietrucha AZ. Endothelial function in vasovagal syncope. Expert Rev Cardiovasc Ther. 2014;12:1387–1389. doi: 10.1586/14779072.2014.982095. [DOI] [PubMed] [Google Scholar]
- 31.Santini L, Capria A, Brusca V, Violo A, Smurra F, Scarfo I, Forleo GB, Papavasileiou LP, Borzi M, Romeo F. An increased endothelial-independent vasodilation is the hallmark of the neurally mediated syncope. Clin Cardiol. 2012;35:107–110. doi: 10.1002/clc.20990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Storgaard T, Nedergaard OA. Prejunctional modulation by angiotensins of noradrenaline release from sympathetic neurons in isolated rabbit aorta. Naunyn Schmiedebergs Arch Pharmacol. 1997;356:706–711. doi: 10.1007/pl00005109. [DOI] [PubMed] [Google Scholar]
- 33.Kolo LL, Westfall TC, Macarthur H. Nitric oxide decreases the biological activity of norepinephrine resulting in altered vascular tone in the rat mesenteric arterial bed. Am J Physiol Heart Circ Physiol. 2004;286:H296–H303. doi: 10.1152/ajpheart.00668.2003. [DOI] [PubMed] [Google Scholar]
- 34.Toda N, Okamura T. Modulation of renal blood flow and vascular tone by neuronal nitric oxide synthase-derived nitric oxide. J Vasc Res. 2011;48:1–10. doi: 10.1159/000317395. [DOI] [PubMed] [Google Scholar]
- 35.Stewart JM, Medow MS, Minson CT, Taneja I. Cutaneous neuronal nitric oxide is specifically decreased in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2007;293:H2161–H2167. doi: 10.1152/ajpheart.00600.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charkoudian N. Mechanisms and modifiers of reflex induced cutaneous vasodilation and vasoconstriction in humans. J Appl Physiol (1985) 2010;109:1221–1228. doi: 10.1152/japplphysiol.00298.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wingo JE, Low DA, Keller DM, Brothers RM, Shibasaki M, Crandall CG. Effect of elevated local temperature on cutaneous vasoconstrictor responsiveness in humans. J Appl Physiol. 2009;106:571–575. doi: 10.1152/japplphysiol.91249.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dinenno FA, Joyner MJ. Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory? J Physiol. 2003;553:281–292. doi: 10.1113/jphysiol.2003.049940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dinenno FA, Joyner MJ. Combined NO and PG inhibition augments alpha-adrenergic vasoconstriction in contracting human skeletal muscle. Am J Physiol Heart Circ Physiol. 2004;287:H2576–H2584. doi: 10.1152/ajpheart.00621.2004. [DOI] [PubMed] [Google Scholar]
- 40.Gamboa A, Shibao C, Diedrich A, Paranjape SY, Farley G, Christman B, Raj SR, Robertson D, Biaggioni I. Excessive nitric oxide function and blood pressure regulation in patients with autonomic failure. Hypertension. 2008;51:1531–1536. doi: 10.1161/HYPERTENSIONAHA.107.105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arbeille PP, Besnard SS, Kerbeci PP, Mohty DM. Portal vein cross-sectional area and flow and orthostatic tolerance: a 90-day bed rest study. J Appl Physiol. 2005;99:1853–1857. doi: 10.1152/japplphysiol.00331.2005. [DOI] [PubMed] [Google Scholar]
- 42.Overton JM, Tipton CM. Effect of hindlimb suspension on cardiovascular responses to sympathomimetics and lower body negative pressure. J Appl Physiol (1985) 1990;68:355–362. doi: 10.1152/jappl.1990.68.1.355. [DOI] [PubMed] [Google Scholar]
- 43.Mueller PJ, Foley CM, Hasser EM. Hindlimb unloading alters nitric oxide and autonomic control of resting arterial pressure in conscious rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R140–R147. doi: 10.1152/ajpregu.00820.2004. [DOI] [PubMed] [Google Scholar]
- 44.Vaziri ND, Ding Y, Sangha DS, Purdy RE. Upregulation of NOS by simulated microgravity, potential cause of orthostatic intolerance. J Appl Physiol (1985) 2000;89:338–344. doi: 10.1152/jappl.2000.89.1.338. [DOI] [PubMed] [Google Scholar]
- 45.Galetta F, Franzoni F, Plantinga Y, Ghiadoni L, Merico G, Tocchini L, Braccini L, Rossi M, Carpi A, Taddei S, Santoro G. Endothelial function in young subjects with vaso-vagal syncope. Biomed Pharmacother. 2006;60:448–452. doi: 10.1016/j.biopha.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 46.Ruiz GA, Sinigaglia S, Hermes R, Chirife R, Capula M, Perfetto JC, Tentori MC, Grancelli H, Nogues M. Role of nitric oxide in young patients with vasovagal syncope. Europace. 2010;12:987–990. doi: 10.1093/europace/euq148. [DOI] [PubMed] [Google Scholar]
- 47.Shi Y, Tian H, Gui YH, He L. Association of nitric oxide and eNOS with the pathogenesis of vasovagal syncope. Zhongguo Dang Dai Er Ke Za Zhi. 2008;10:478–480. [PubMed] [Google Scholar]
- 48.Fu Q, Witkowski S, Okazaki K, Levine BD. Effects of gender and hypovolemia on sympathetic neural responses to orthostatic stress. Am J Physiol Regul Integr Comp Physiol. 2005;289:R109–R116. doi: 10.1152/ajpregu.00013.2005. [DOI] [PubMed] [Google Scholar]
- 49.Stickford AS, Vangundy TB, Levine BD, Fu Q. Menstrual cycle phase does not affect sympathetic neural activity in women with postural orthostatic tachycardia syndrome. J Physiol. 2015;593:2131–2143. doi: 10.1113/JP270088. [DOI] [PMC free article] [PubMed] [Google Scholar]