

Summary
Background and Purpose
Control of nematode parasite infections relies largely on anthelmintic drugs, several of which act on nicotinic ACh receptors (nAChRs), and there are concerns about the development of resistance. There is an urgent need for development of new compounds to overcome resistance and novel anthelmintic drug targets. We describe the functional expression and pharmacological characterization of a homomeric nAChR, ACR‐16, from a nematode parasite.
Experimental Approach
Using RT‐PCR, molecular cloning and two‐electrode voltage clamp electrophysiology, we localized acr‐16 mRNA in Ascaris suum (Asu) and then cloned and expressed acr‐16 cRNA in Xenopus oocytes. Sensitivity of these receptors to cholinergic anthelmintics and a range of nicotinic agonists was tested.
Key Results
Amino acid sequence comparison with vertebrate nAChR subunits revealed ACR‐16 to be most closely related to α7 receptors, but with some striking distinctions. acr‐16 mRNA was recovered from Asu somatic muscle, pharynx, ovijector, head and intestine. In electrophysiological experiments, the existing cholinergic anthelmintic agonists (morantel, levamisole, methyridine, thenium, bephenium, tribendimidine and pyrantel) did not activate Asu‐ACR‐16 (except for a small response to oxantel). Other nAChR agonists: nicotine, ACh, cytisine, 3‐bromocytisine and epibatidine, produced robust current responses which desensitized at a rate varying with the agonists. Unlike α7, Asu‐ACR‐16 was insensitive to α‐bungarotoxin and did not respond to genistein or other α7 positive allosteric modulators. Asu‐ACR‐16 had lower calcium permeability than α7 receptors.
Conclusions and Implications
We suggest that ACR‐16 has diverse tissue‐dependent functions in nematode parasites and is a suitable drug target for development of novel anthelmintic compounds.
Abbreviations
- α‐BTX
α‐bungarotoxin
- BAPTA‐AM
1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid tetrakis (acetoxymethyl ester)
- blastP
protein–protein blast
- DHβE
dihydro‐β‐erythroidine
- DMPP
dimethyl‐4‐phenyllpiperazinium iodide
- dTC
d‐tubocurarine
- GHK
Goldman Hodgkin Katz
- MLA
methyllycaconitine
- nAChR
nicotinic ACh receptor
- PAM
positive allosteric modulator
Tables of Links
| TARGETS |
|---|
| Ligand–gated ion channels |
| Asu‐ACR‐16 |
| ACR‐16 |
| Nicotinic ACh receptor α7 subunit |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) or the PubChem or Wormbase databases. Entries in the IUPHAR database are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Soil‐transmitted helminths (gastrointestinal nematode parasites) cause significant global health problems in humans and animals. It is estimated that at least one‐quarter of the world's human population (Brooker, 2010) and most animal species are infected with parasitic worms. Nematode parasite infections have a high morbidity and cause debilitating conditions such as weight loss, anaemia, compromised immunity, impaired learning ability and, in severe cases, death (Hotez et al., 2007). No current vaccine against nematode parasite infections of humans is effective (Hewitson and Maizels, 2014), so control of these infections relies on chemotherapy. Regrettably, there are a limited number of classes of anthelmintics (Martin, 1997; Kaminsky et al., 2008), which, with the repeated large scale use of the drugs, has led to the development of resistance in animals (Wolstenholme et al., 2004), and concerns about the development of resistance in humans (Taman and Azab, 2014). The increasing resistance means that novel drug targets are required to overcome this resistance.
Nicotinic ACh receptors (nAChRs) of vertebrates and invertebrates serve a very wide range of functions. For example, in excitable cells, they are involved in neuronal and neuromuscular transmission and, in non‐excitable cells, are involved in modulation, development of growth and differentiation (Wessler and Kirkpatrick, 2008). Nematode parasite nAChRs are pharmacologically different to their host nAChRs and are validated and exploited anthelmintic drug targets (Gopalakrishnan et al., 2007). All nAChRs are five‐subunit ligand‐gated ion channels which open in the presence of ACh or choline, and are found on muscles, nerves, secretory cells and a wide range of non‐excitable tissues in both vertebrates and invertebrates. The pharmacology of individual nAChRs depends on key amino acid sequences of each nAChR subunit of the pentamer and the composition of the subunits that form the receptor. The subunits that make up vertebrate nAChRs are derived from at least 17 genes (α1–α10, β1–β4, γ, δ, ϵ) (Karlin, 1993; Millar and Gotti, 2009) and from 29 genes in the free‐living nematode, Caenorhabditis elegans (Jones et al., 2007). The subunits of the pentameric receptor channel may be composed of five identical subunits (homomeric) or a mixture of different subunits (heteromeric) arranged around a central ion conducting pore (Chen, 2010).
The older, cholinergic anthelmintics, levamisole and pyrantel, and the more recently introduced anthelmintics, tribendimidine and derquantel, activate heterogeneous pentameric nAChRs composed of a mixture of UNC‐29, UNC‐38, ACR‐8 or UNC‐63 subunits (Buxton et al., 2014). The more recently introduced cholinergic anthelmintic, monepantel, activates nAChRs composed of a mixture of DEG‐3‐like subunits, including ACR‐23, ACR‐20 and MPTL‐1 (Baur et al., 2015). Resistance to some of these anthelmintics has been observed in animal parasites (McMahon et al., 2013; Scott et al., 2013), and although the mechanism(s) of field resistance have not been well characterized, there is evidence that the resistance can involve altered subunit sequence or subunit truncation (Fauvin et al., 2010; Neveu et al., 2010).
Here, we have cloned and expressed, in Xenopus oocytes, acr‐16 cRNA from Ascaris suum, a clade III nematode parasite, which is very similar to the significant human parasite, A. lumbricoides. This receptor was expressed as a homomeric receptor in Xenopus oocytes, and transcripts are found in muscle, intestine, reproductive tract and other tissues of the Ascaris body, suggesting diverse physiological functions in addition to neuromuscular transmission. Knowing that ACR‐16 is a nicotine‐sensitive nAChR and that nicotine has in the past been used as the drug of choice for treatment of Ascaridia galli infections (Kerr and Cavett, 1952), a selective drug targeted against ACR‐16 could affect motility, digestion and reproduction of parasites and be an effective anthelmintic.
Methods
Animals
No vertebrate animals were used directly in the study. Adult female A.suum worms were collected from Marshalltown Pork Plant, Marshalltown, IA, USA. Defolliculated Xenopus laevis oocytes were obtained from Ecocyte Bioscience (Austin, TX, USA).
Sequence analysis
For convenience, the sources of the AChR subunits are indicated, in this article, by three letter prefixes. Thus, Ace, Asu, Cel, Hco, Llo, Nam, Sra, Tca and Xle, refer to Ancylostoma ceylanicum, Ascaris suum, Caenorhabditis elegans, Haemonchus contortus, Loa loa, Necator americanus, Strongyloides ratti, Toxocara canis and Xenopus laevis respectively.
Database searches around the sequence of Asu‐ACR‐16 were performed with the blast Network Service (NCBI), using the protein–protein blast (blastP) algorithm (Altschul et al., 1997). Signal peptide predictions were carried out using the SignalP 4.0 server (Bendtsen et al., 2004), and membrane‐spanning regions were predicted using TMpred (Hofmann and Stoffel, 1993). Distance trees were generated on the full‐length deduced amino acid sequences aligned with the MUSCLE programme (Edgar, 2004). The alignment was analysed using the SEAVIEW suite (Gouy et al., 2010) with the neighbour‐joining method and bootstrapped with 1000 replicates. The resulting tree was visualized and edited using FigTree 1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Asu‐ACR‐16 receptor localization
Total RNA was extracted with TRIzol (Invitrogen™, Carlsbad, CA, USA) from different body tissues of five dissected adult female Asu worms. The tissues were the gut, ovijector, head, single muscle cells (RNA extracted via micropipette), bulk somatic muscle, single muscle cells of the pharynx (RNA extracted via micropipette) and bulk muscle cells of the pharynx. Specific primers were designed for PCR for acr‐16 (forward primer GACTTGCAACCAGGCAAAGG; reverse primer ACGGGTCGTTATGCCCATTT) and gapdh (positive control) (forward primer CTGCTGGACCAATGAAGGGT; reverse primer CACTCCACTCACAGCCACTT) using the online Primer‐blast tool. With these primer sets, the expected product size for acr‐16 was 468 and 411 bp for gapdh. The presence or absence of acr‐16 in the RNA samples was detected using RT‐PCR and single‐cell RT‐PCR (QIAGEN OneStep RT‐PCR Kit; Valencia, CA, USA) at an annealing temperature of 55°C and for 45 PCR cycles. The PCR products from both RT‐PCR and single‐cell RT‐PCR were run on a 1% agarose gel, at the end of which the gel was viewed and captured on a UVP BioSpectrum Multi Spectral Imaging System (UVP LLC, Upland, CA, USA). Representative bands were sequenced to confirm the identity of acr‐16.
cRNA preparation
Total RNA samples were extracted with TRIzol (Invitrogen™) from a 1 cm muscle flap and dissected whole pharynx of Asu. RT‐PCR was used to synthesize first‐strand cDNA from muscle and pharynx total RNA with both oligo (dT) RACER primer and Random Hexamer, and superscript III reverse transcriptase (Invitrogen). To amplify the full‐length coding sequence of Asu‐ACR‐16, specific primer pairs were designed on the putative 5′‐ and 3′‐UTRs mRNA sequence deduced from Asu whole genome shotgun contig N°AEUI02000378 (Asu‐acr‐16‐F0 ATCACGCATTACGGTTGATG, Asu‐acr‐16‐F1 TTGATGTAGTGGCGTCGTGT, Asu‐acr‐16‐R0 ATTAGCGTCCCAAGTGGTTG, Asu‐acr‐16‐R1 GCATTGATGTTCCCTCACCT) for a first round of PCRs. We used a classical nested PCR approach with F0/R0, F0/R1, F1/R0 and F1/R1 respectively to perform four separate PCR reactions for the first round to increase the chance of amplification success. These four PCR products were subsequently used as templates for a second round of four separate PCR reactions using the following specific primers containing HindIII and ApaI restriction enzyme sites (Asu‐acr‐16‐F‐Hind3AAAAAGCTTATGAGCGTGCAGCGGGCATT, Asu‐acr‐16‐R‐ApaI TTTGGGCCCTAGAGTGCTGATGATGTGCTA) to facilitate directional cloning into the PTB‐207 expression vector. PCR products were then digested with HindIII and ApaI restriction enzymes, purified using the NucleoSpin® Gel and PCR Clean‐up kit (Macherey‐Nagel Inc. Bethlehem, PA, USA), cloned into PTB‐207 as previously described (Boulin et al., 2011) and sequenced. A positive clone was selected and linearized with NheI for subsequent in vitro transcription with the mMessage mMachine T7 transcription kit (Ambion). The cRNA was precipitated with lithium chloride, resuspended in RNAse‐free water, aliquoted and stored at −80°C.
Oocyte microinjection
The Xenopus oocytes were kept at 19°C for ~3 h prior to injections in incubation solution (100 mM NaCl, 2 mM KCl, 1.8 mM CaCl2.2H20, 1 mM MgCl2.6H20, 5 mM HEPES, 2.5 mM Na pyruvate, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin, pH 7.5). Depending on the cRNA mix, 10–25 ng of acr‐16 cRNA either alone or in combination with 2.5–15 ng of each ancillary (ric‐3, unc‐50 and unc‐74), in a total volume of 50 nL in RNAse‐free water, was injected into the animal pole of the oocytes using a nanoject II microinjector (Drummond Scientific, Broomall, PA, USA). The injected oocytes were transferred into 96‐well culture plates containing 200 μL incubation solution per well; each well contained one oocyte. Oocytes were incubated at 19°C for 2–7 days to allow for receptor expression; incubation solution was changed daily. Oocytes with membrane potentials less than −15 mV were excluded from recording. Oocyte recordings that failed (shown by a change in the holding current following wash) before the complete series of drug applications were excluded from the analysis.
Two‐electrode voltage clamp electrophysiology in Xenopus oocytes
Two‐electrode voltage clamp electrophysiology was used to record currents produced by activation of the expressed Asu‐ACR‐16 receptor (Buxton et al., 2014). Four hours prior to recording, 100 μM BAPTA‐AM (final concentration), a cell‐permeant calcium chelator, was added to the oocyte incubation solution to prevent activation of endogenous calcium‐activated chloride channels during recordings. Recordings from non‐injected oocytes served as control experiments. Recordings were made using an Axoclamp 2B amplifier (Warner Instruments, Hamden, CT, USA) with the oocytes voltage clamped at −60 mV and data acquired on a computer with Clampex 9.2 (Molecular Devices, Sunnyvale, CA, USA). The microelectrodes used to impale the oocytes were pulled using a Flaming/Brown horizontal electrode puller (Model P‐97; Sutter Instruments, Novato, CA, USA) set to pull micropipettes that when filled with 3 M KCl had a resistance of 20–30 MΩ. The micropipettes tips were carefully broken with a piece of tissue paper in order to achieve a resistance of 2–5 MΩ in recording solution (100 mM NaCl, 2.5 mM KCl, 1 mM CaCl2.2H2O and 5 mM HEPES, pH 7.3). The low resistance pipettes allowed large currents to be passed to maintain adequate voltage clamp.
Assessment of agonists and antagonists
Except where indicated, agonists were used at a final concentration of 100 μM, while the antagonists were used at a final concentration of 10 μM for rank order potency experiments and 1 μM for the dose–response experiments. Effects of the positive allosteric modulators (PAMs): ivermectin (10 μM), genistein (3 μM) and PNU120596 (3 μM), were tested. Control responses were obtained first with agonists applied for 10 s. Responses were again observed after the antagonists or PAMs were applied for 2 min and observed in the continued presence of antagonist or PAM. In the case of the antagonist rank order potency experiments, a control application of 100 μM ACh was first applied for 10 s, immediately followed by a test 10 s application of antagonist in the continued presence of 100 μM ACh and then a final 10 s application of 100 μM ACh. Note that, with this approach, the potency for each antagonist is likely to be slightly underestimated due to the short time of drug application. In order to minimize desensitization effects, at least 2 min was allowed for drug wash off between applications.
Dose–response studies were conducted in ascending order of concentrations and were not presented randomly. This approach minimizes desensitization by high concentrations of agonist. The application of the sequence of agonists for determining the potency series was random and not predetermined.
Blinding was not used because it was necessary to allow the appropriate technique and study methods to be used to test if the drug was a potential agonist, potential antagonist or potential PAM. In all our recordings, we applied an initial 100 μM ACh control, and all other responses were normalized to this control 100 μM ACh current. The normalized ACh current (100%) was not used for statistical analysis. Log dose–response plots are used for display and determination of the EC50, nH and maximum response.
Permeability of receptors to calcium
Calcium permeability experiments were conducted by increasing the external Ca2 + concentration from 1 to 10 mM in the recording solution without changing the concentration of other cations. Oocytes were challenged with 30 μM ACh in the recording solution, with the membrane of the oocytes held at different potentials between −60 and +30 mV. The Goldman Hodgkin Katz (GHK) constant field equation was used to calculate the permeability ratio, P Ca/P Na. Because the oocytes were BAPTA‐treated prior to recording, the assumptions made in calculating the permeability ratio of the expressed Asu‐ACR‐16 receptor were that the internal [Ca2 +] is negligible and permeability of Na and K, P Na and P K is equal. Ionic activity coefficients used for the calculations were 0.56 for Ca2 + and 0.72 for Na+ and K+. The GHK equation (with ionic activity inserted) used in calculating the P Ca/P Na was
where E rev is the reversal potential (potential at which current changes direction), R is the universal gas constant (8.314 J·K−1·mol−1), T is room temperature in Kelvin (298 K), F is Faraday's constant (96 485 C·mol−1) and P′ = P Ca/P Na{1/(1 + e FErev/RT)}.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The acquired data from electrophysiological recordings were analysed with Clampfit 9.2 (Molecular Devices, Sunnyvale, CA, USA) and GraphPad Prism 5.0 (Graphpad Software Inc., La Jolla, CA, USA). In all recordings, the peak currents in response to applied drugs were measured, and except where otherwise indicated, peak current values obtained were normalized to 100 μM ACh and expressed as mean ± SEM. Dose–response relationships were analysed by fitting log dose–response data points with the Hill equation as previously described (Boulin et al., 2008), while desensitization kinetics in response to the potent agonists were fitted using a single exponential decay fit:
where n is the number of components, A the amplitude, t the time, Ʈ the time constant and C the constant y‐offset for each i component.
The group sizes varied with the type of experiment performed, as described below. To optimize our expression and recording conditions, we injected 10 ng of Asu‐acr‐16 alone and with 1.8 ng each of the ancillary factors, ric‐3, unc‐50 and unc‐74 from A.suum, H. contortus and X.laevis (n = 21 for Asu‐acr‐16 alone; n = 15 for Asu‐acr‐16 + Hco‐ric‐3, Hco‐unc‐50 and Hco‐unc‐74; n = 15 for Asu‐acr‐16 + Asu‐ric‐3, Asu‐unc‐50 and Asu‐unc‐74; n = 17 for Asu‐acr‐16 + Hco‐ric‐3; n = 20 for Asu‐acr‐16 + Xle‐ric‐3; and n = 23 for Asu‐acr‐16 + Asu‐ric‐3). We found that Asu‐unc‐50 and Asu‐unc‐74 were not required for expression and that Asu‐acr‐16 + Asu‐ric‐3 produced the large robust currents (290.6 ± 63.7, n = 23). We also tested different amounts of Asu‐acr‐16 and Asu‐ric‐3 (n = 6 for 25 ng Asu‐acr‐16 + 5 ng Asu‐ric‐3, n = 6 for 10 ng Asu‐acr‐16 + 5 ng Asu‐ric‐3, n = 6 for 10 ng Asu‐acr‐16 + 10 ng Asu‐ric‐3 and n = 6 for 15 ng Asu‐acr‐16 + 15 ng Asu‐ric‐3) and observed that the size of the currents depended on the amount of Asu‐acr‐16 and Asu‐ric‐3 injected.
Thereafter, we injected 25 ng Asu‐acr‐16 + 5 ng Asu‐ric‐3 (which produced the biggest currents) for all subsequent experiments. For nAChR agonists and cholinergic anthelmintics, we tested larger numbers of oocytes (n = 21 for nicotine, n = 21 for cytisine, n = 15 for 3‐bromocytisine, n = 15 for epibatidine, n = 21 for DMPP, n = 36 for oxantel, n = 15 for choline, n = 15 for betaine, n = 15 for lobeline, n = 15 for A844606, n = 21 for morantel, n = 21 for levamisole, n = 15 for methyridine, n = 15 for thenium, n = 15 for bephenium, n = 15 for tribendimidine and n = 15 for pyrantel) to measure and examine variability of responses between oocytes. Once we had confirmed the reproducible nature of the responses (the mean and standard errors for each of the nAChR agonist and cholinergic anthelmintics shown in Figure 4), we reduced our n number to 6 for all other experiments, except the antagonist experiment where the n number was 5.
Figure 4.
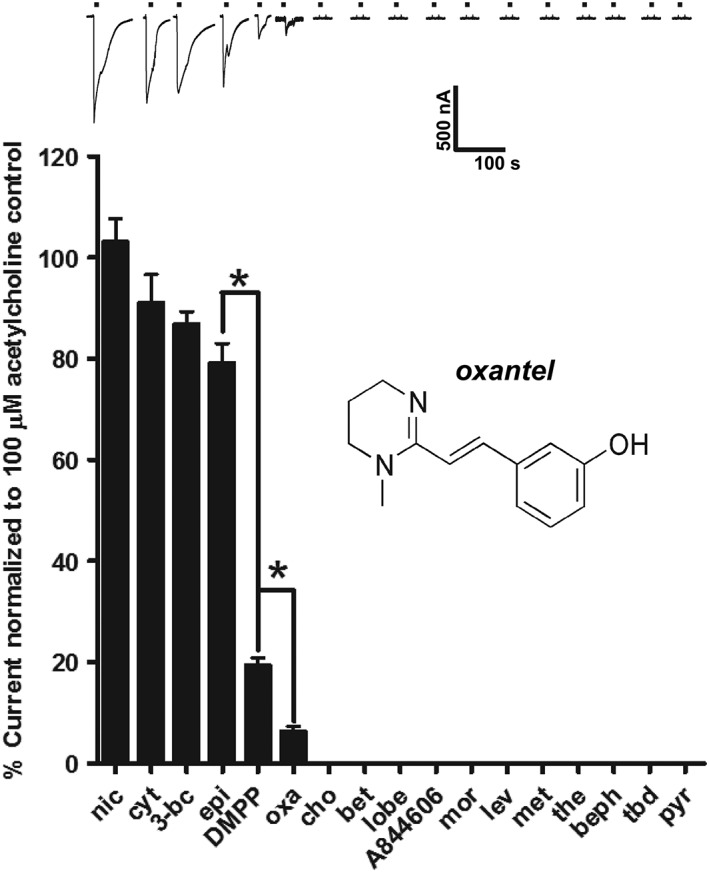
Effects of nAChR agonists and anthelmintics on Asu‐ACR‐16. Sample traces and bar chart (mean ± SEM) showing rank order potency series for nAChR agonists: nicotine (nic), cytisine (cyt), 3‐bromocytisine (3‐bc), epibatidine (epi), DMPP, choline (cho), betaine (bet), lobeline (lobe) and A844606; and cholinergic anthelmintics: oxantel (oxa), morantel (mor), levamisole (lev), methyridine (met), thenium (the), bephenium (beph), tribendimidine (tbd) and pyrantel (pyr); on Asu‐ACR‐16. Overall, the rank order potency series for agonists and anthelmintics on Asu‐ACR‐16 when normalized to the control 100 μM ACh current was as follows: 100 μM nic (n = 21) ~ 100 μM cyt (n = 21) ~ 100 μM 3‐bc (n = 15) ~ 100 μM epi (n = 15) > 100 μM DMPP (n = 21) > 100 μM oxa (n = 36 ) >>> 100 μM cho (n = 15) = 100 μM bet (n = 15) = 100 μM lobe (n = 15) = 100 μM A844606 (n = 15) = mor (n = 21) = 100 μM lev (n = 21) = 100 μM met (n = 15) = 100 μM the (n = 15) = 100 μM beph (n = 15) = 30 μM tbd (n = 15) = 100 μM pyr (n = 15). * P < 0.05; significantly different as indicated; Tukey's multiple comparison tests.
Data were analysed statistically as follows. All completed drug application sequences on the oocytes were used for analysis without exclusion. If the recording became unstable, indicated by a change in the baseline holding current, all of that recording was rejected for analysis. Statistical analyses were performed on groups of values using ANOVA to determine if the group means are similar and if there was variance inhomogeneity (Bartlett's test). Post hoc tests (Tukey's multiple comparison test for Figures 3B, 4 and 7A; Dunnett's test to compare with control, for Figure 5) were used to determine significance of differences between groups. We analysed the effects of PAMs on Asu‐ACR‐16 using two‐way ANOVA. We defined P < 0.05 as showing statistical significance.
Figure 3.
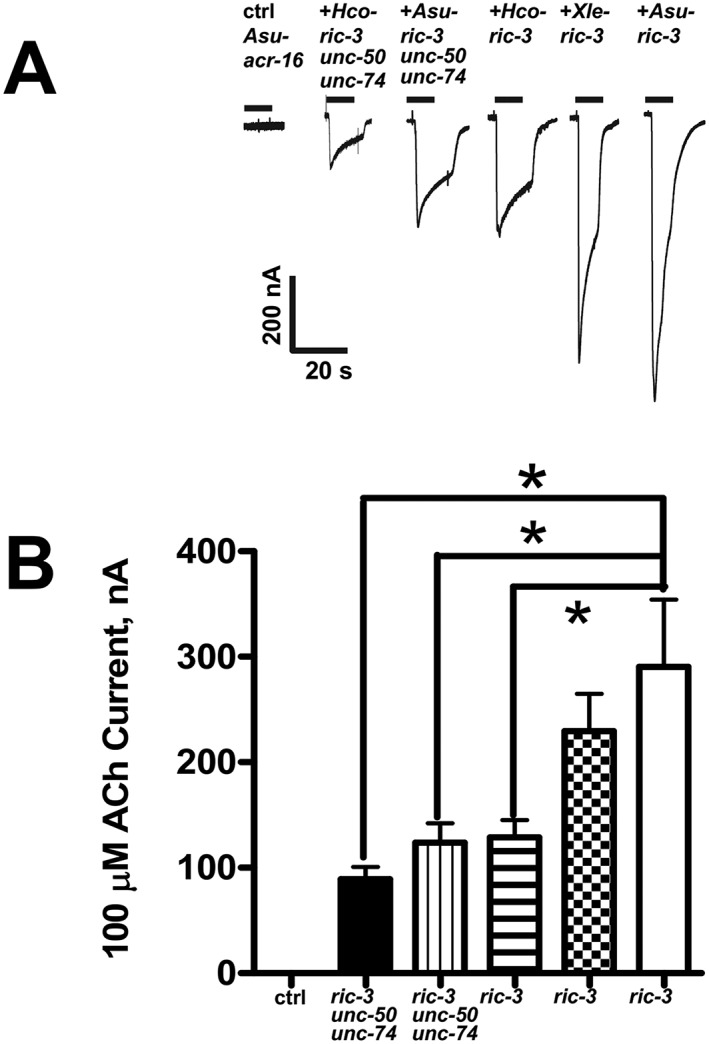
Effects of the ancillary proteins, RIC‐3, UNC‐50 and UNC‐74, from different nematode species, on Asu‐ACR‐16 expression. (A) Sample traces represented as inward currents produced in response to 100 μM ACh. (B) Bar chart (mean ± SEM) showing (left to right) current (in nA) generated in response to 100 μM ACh produced. Control (ctrl): Asu‐acr‐16 alone (n = 21). Black bar: Asu‐acr‐16 plus Hco ‐ric‐3, unc‐50 and unc‐74 (n = 15). Vertical line fill: Asu‐acr‐16 plus Asu ric‐3, unc‐50 and unc‐74 (n = 15). Horizontal line fill: Asu‐acr‐16 plus Hco ‐ric‐3 (n = 17). Checkered fill: Asu‐acr‐16 plus Xle ‐ric‐3 (n = 20). No fill: Asu‐acr‐16 plus Asu‐ric‐3 (n = 23). Asu‐acr‐16 on its own did not respond to ACh, and the largest current size was obtained when Asu‐acr‐16 was co‐injected with Asu‐ric‐3. * P < 0.05; significantly different as indicated; Tukey's multiple comparison tests.
Figure 7.
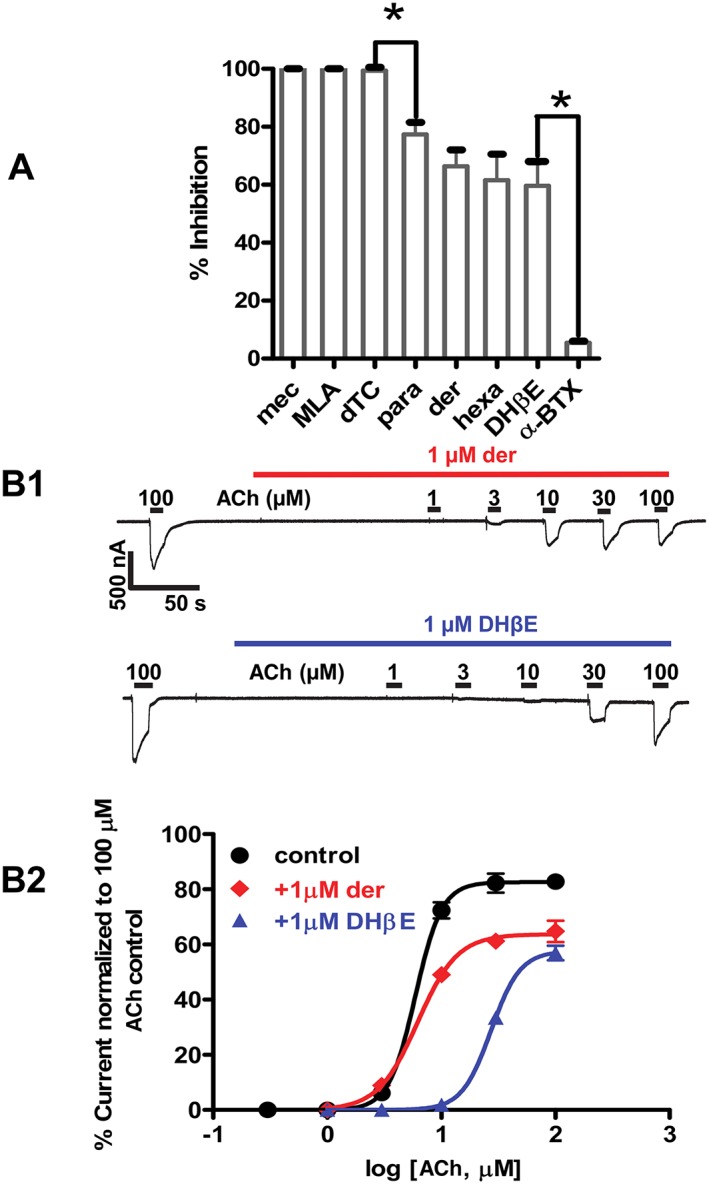
(A) Effects of selected nAChR antagonists on Asu‐ACR‐16‐mediated ACh responses. Bar chart showing effects of selected nAChR antagonists on Asu‐ACR‐16. Results were expressed as mean ( ± SEM) % inhibition of currents elicited by 100 μM ACh, n = 6, for all antagonists. dTC, mecamylamine (mec) and MLA completely blocked Asu‐ACR‐16‐mediated ACh responses, while paraherquamide (para), derquantel (der), hexamethonium (hexa) and DHβE only produced a partial block of Asu‐ACR‐16‐mediated ACh responses and α‐BTX produced an almost insignificant block of Asu‐ACR‐16‐mediated ACh responses. Rank order potency series for the nAChR antagonists each tested at a concentration of 10 μM was as follows: mecamylamine (n = 6) = MLA (n = 6) ≈ dTC (n = 6) > paraherquamide (n = 6) ~ derquantel (n = 6) ~ hexamethonium (hexa) (n = 6) ~ DHβE (n = 6) > α‐BTX (n = 6). * P < 0.05; significantly different as indicated; Tukey's multiple comparison tests. We used ANOVA and Bartlett's test for variance inhomogeneity and found no significant difference and Tukey's multiple comparison tests. (B) Dose–response relationships for Asu‐ACR‐16 in the presence of antagonists. (B1) Sample traces for ACh concentration–response relationships for Asu‐ACR‐16 in the presence of 1 μM derquantel and 1 μM DHβE. (B2) ACh concentration–response plots for Asu‐ACR‐16 in the presence of 1 μM derquantel and 1 μM DHβE. Derquantel caused a reduction in the maximum response, but no change in EC50, whereas DHβE caused both a reduction in the maximum response and a right shift in the EC50.
Figure 5.
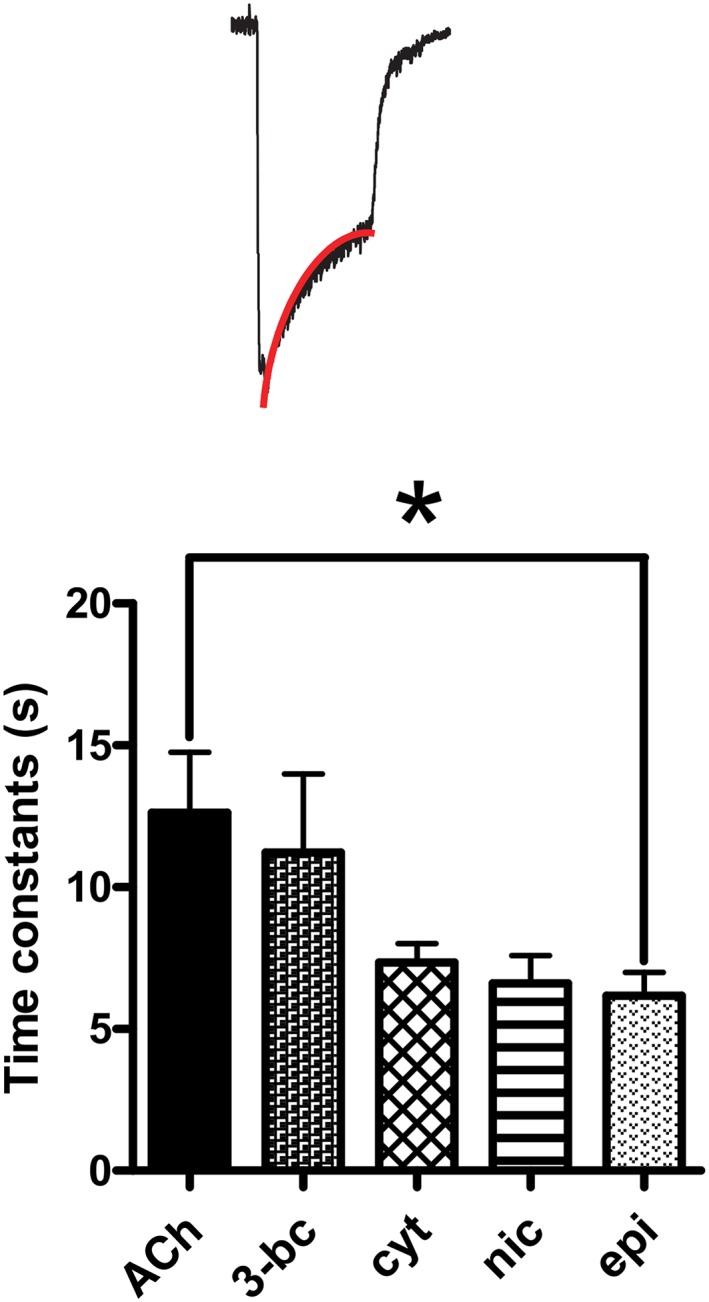
Asu‐ACR‐16 desensitization rate constant fit. Bar chart (mean ± SEM) showing Asu‐ACR‐16 desensitization in response to ACh, 3‐bromocytisine (3‐bc), cytisine (cyt), nicotine (nic) and epibatidine (epi). The rank order for Asu‐ACR‐16 time constants of desensitization was as follows: 100 μM ACh (12.6 ± 2.1 s, n = 6) ~ 100 μM 3‐bc (11.2 ± 2.8 s, n = 6) ~ 100 μM cyt (7.3 ± 0.7 s, n = 6) ~ 100 μM nic (6.6 ± 1.0 s, n = 4) ~ 100 μM epi (6.2 ± 0.8 s, n = 6). Insert: Sample trace with red line signifying desensitization fit. * P < 0.05; significantly different as indicated; Tukey's multiple comparison tests.
Materials
The drugs used in this study, with the exception of paraherquamide, tribendimidine and derquantel were purchased from Sigma‐Aldrich (St Louis, MO, USA), Tocris Bioscience (Ellisville, MO, USA) or Calbiochem (San Diego, CA, USA). The drugs were solubilized in recording solution, DMSO or ethanol. Paraherquamide and derquantel were gifts from Zoetis (Kalamazoo, MI, USA) and tribendimidine was a gift of Prof Shu Hua Xiao (National Institute of Parasitic Diseases, China).
Results
Identification of Asu‐ACR‐16 sequence
We used the Cel‐ACR‐16 sequence (Supporting Information Fig. S1) as a query in a blastP search in protein databases from nematodes which allowed the identification of a potential complete coding sequence of an ACR‐16 homologue in the pig parasite Asu (ERG81952.1). RT‐PCR experiments using primers designed using this predicted sequence led to the amplification of a full coding sequence (Figure 1A), which was submitted to GenBank under the accession number KP756901.
Figure 1.
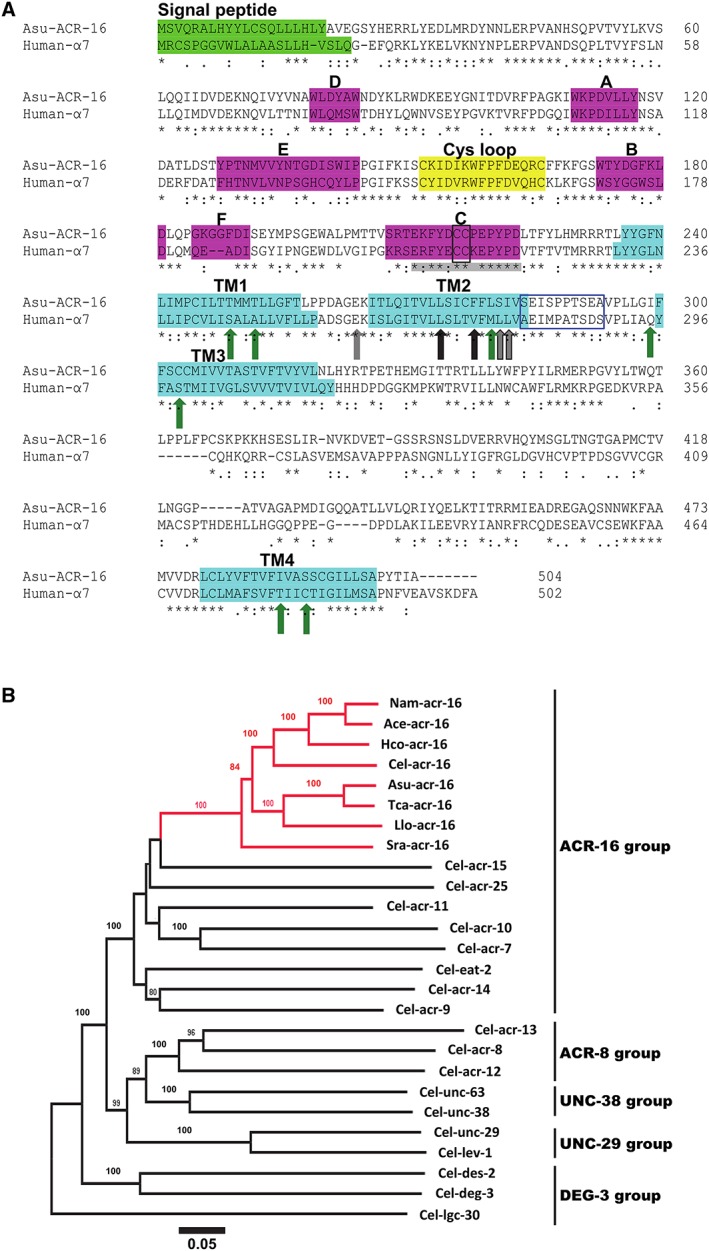
(A) Amino acid sequence alignment of Asu‐ACR‐16 and human‐α7 nAChR subunits. The signal peptide (bright green box), ACh‐binding loops A–F (pink boxes), cys‐loop (yellow box) and transmembrane regions TM1–TM4 (turquoise boxes) are indicated. The vicinal cysteines (black‐edged box) that characterize an α‐subunit are present in the C‐binding loop. The blue‐edged box between TM2 and TM3 represents the region where PNU120596 acts on α7 nAChRs. Green arrows are residues important for positive allosteric modulation of α7 receptors by ivermectin. Grey (and grey outline) arrows are residues important for permeability of α7 receptors to Ca2 +. Black (and black outline) arrows are residues affecting α7 receptor desensitization. Residues in C‐binding loop of α7 nAChRs that bind α‐BTX are highlighted in grey. (B) Distance tree showing relationships of ACR‐16 homologues in parasitic nematode species with AChR subunit sequences from C.elegans. A neighbour joining tree was generated with deduced amino acid sequence from AChR subunits representative from the ACR‐16, ACR‐8, UNC‐38, UNC‐29 and DEG‐3 group as defined by Mongan et al., (1998). Three letter prefixes in AChR subunit names: Ace, Asu, Cel, Hco, Llo, Nam, Sra and Tca, refer to A. ceylanicum, A. suum, C. elegans, H. contortus, L. loa, N. americanus, S. ratti and T. canis respectively. ACR‐16 orthologues are highlighted in red. Numbers at each branch indicate percentage bootstrap values (>80%) corresponding to 1000 replicates. The scale bar represents substitutions per site. The Cel‐lgc‐30 subunit sequence was used as an outgroup.
When we used the Asu‐acr‐16 transcript sequence as a query, a blastP search resulted in the identification of highly conserved (complete or partial) homologous sequences from other parasitic nematode species belonging to Clade III (T. canis and L. loa), Clade IV (S. ratti) and Clade V (H. contortus, A. ceylanicum and Necator americanus) (Figure 1B and Supporting Information S2). The distance tree presented in Figure 1B indicates the orthologous relationship of the ACR‐16 sequences from parasitic nematodes with the Cel‐ACR‐16 sequence and also shows the orthologous relationship to the other nAChR subunits of C elegans. The predicted complete sequences corresponding to ACR‐16 orthologues in parasitic nematodes from Clade III and V were further analysed in a structural alignment. All amino acid sequences were found to share typical features of nAChR subunits including a predicted signal peptide, four transmembrane domains, a Cys‐loop motif and a cysteine doublet in the potential agonist site defining them as α‐subunits. Of note is that, despite their phylogenetic separation, ACR‐16 sequences from Clade III and Clade V species were found to be highly conserved (i.e. 80% identities between Asu‐ACR‐16 and Hco‐ACR‐16), suggesting that their pharmacology may also be similar.
Asu‐ACR‐16 sequence suggests pharmacological differences to host mammalian α7 nAChRs
Alignment of the Asu‐ACR‐16 amino acid sequence with that of human‐α7 nAChRs using the Clustal Omega online alignment tool (Sievers et al., 2011) showed Asu‐ACR‐16 and human‐α7 receptors share 44.90% identity in their amino acid sequences. Figure 1A shows the amino acid sequence alignment of Asu‐ACR‐16, which is compared with that of human‐α7 receptors (Raymond et al., 2000; Touroutine et al., 2005). Asu‐ACR‐16 has the characteristic vicinal cysteine residues (Y‐x‐C‐C) found in the C‐loop of most nAChR α‐subunits; these C‐C residues play an essential role in ACh binding (Kao et al., 1984). Significantly, the Y‐x‐C‐C residues of the C‐loop of Asu‐ACR‐16 (Supporting Information S2) differ from the C‐loop of the levamisole receptor α‐subunits, Cel‐UNC‐38 and Cel‐ACR‐13, which have a Y‐x‐x‐C‐C motif (Mongan et al., 1998); this C‐loop difference encouraged our view that the Asu‐ACR‐16 nAChR would differ in its pharmacology from that of the levamisole receptors.
We were further encouraged to investigate the pharmacology of Asu‐ACR‐16 because of other significant differences in amino acid residues which are known to affect activation, desensitization and modulation in α7 nAChRs. For example, Figure 1A shows four amino acids (black and black outline arrows in the TM2 region known as L247, V251, L254 and L255 when mature peptide numbering of α7 subunits is used) that change the desensitization of α7 receptors (Revah et al., 1991; Bertrand et al., 1992; Corringer et al., 1999b). Note that only the first of these (L247) is conserved in Asu‐ACR‐16. Consequently, we chose to investigate the rate of desensitization of Asu‐ACR‐16. Other amino acid differences between the α7 and the levamisole α‐receptor subunits, which we describe subsequently, suggest that the Asu‐ACR‐16 receptor pharmacology will be different.
Ubiquitous tissue and single‐cell expression of ACR‐16 in Asu
We examined the distribution of Asu‐ACR‐16 in dissected adult female Asu tissues using RT‐PCR. We found reproducible and clear evidence of expression of Asu‐ACR‐16 message in strips of intestine (Figure 2A; g, gut), sections of the reproductive tract (Figure 2A; o, oviduct, uterus), whole dissected pharynx (Figure 2A; p), body muscle strips (Figure 2A; m) and head (Figure 2A; h) region. We observed similar expression patterns of Asu‐ACR‐16 message in the gut, oviduct, whole pharynx, body muscle strip and head from four other adult female Asu. The expression of Asu‐ACR‐16 in tissues other than nerves and body muscle tissue, like the non‐excitable tissues of the intestine and reproductive tract (uterus), was not anticipated. But it was reproducible and may be part of an ACh‐mediated paracrine system (Proskocil et al., 2004; Bschleipfer et al., 2007) rather than part of a neurotransmitter process. The Asu‐ACR‐16 ACh receptor may serve multiple roles in reproduction, digestion, etc., as well as neuromuscular transmission in the parasite.
Figure 2.
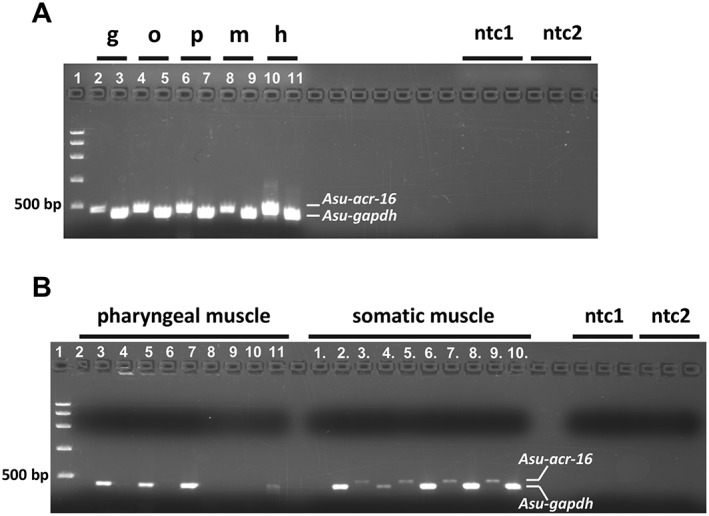
Localization of Asu‐ACR‐16 in different body tissues of the A.suum worm using RT‐PCR and single‐cell RT‐PCR (n = 5). (A) RT‐PCR analysis of Asu‐acr‐16 (lanes 2, 4, 6, 8, 10) and gapdh control (lanes 3, 5, 8, 9, 11) in gut (g), oviduct (o), pharynx (p), somatic muscle strip (m) and head region (h). The PCR products size for acr‐16 and gapdh is 468 and 411 bp respectively. (B) Single‐cell RT‐PCR of Asu‐ACR‐16 in pharyngeal muscle (2, 4, 6, 8, 10) and in somatic muscle (1., 3., 5., 7., 9.). RT‐PCR of gapdh control in pharyngeal muscle (3, 5, 7, 9, 11) and in somatic muscle (2., 4., 6., 8., 10.). 1, FastRuler High Range DNA ladder; ntc1, no‐template controls for acr‐16; ntc2, no‐template controls for gapdh.
Single‐cell RT‐PCR was conducted to further investigate the localization of Asu‐ACR‐16 in the pharyngeal and somatic muscle cells of A.suum. We used intracellular micropipettes to collect cytoplasm from individual somatic and pharyngeal muscle cells for PCR analysis. We recovered Asu‐ACR‐16 message from single body muscle cells but not from pharyngeal muscle cells (Figure 2B). The presence of Asu‐ACR‐16 positive bands in the whole pharynx and absence in single‐cell RT‐PCR samples suggests that Asu‐ACR‐16 is present in the neurons of the circum‐pharyngeal nerve ring but not present on pharyngeal muscle cells. In contrast to the pharyngeal muscle cells, we observed that Asu‐ACR‐16 expression is widespread in somatic muscle as well as other tissues of the Ascaris body, implying tissue‐related functions in addition to a neuromuscular function.
ric‐3 is required for functional expression of Asu‐ACR‐16
After we cloned Asu‐ACR‐16, we examined ancillary protein requirements for its expression in Xenopus oocytes. Figure 3 shows the effects of co‐injecting the cRNA for the ancillary proteins: ric‐3, unc‐50 and unc‐74 from A.suum; ric‐3, unc‐50 and unc‐74 from H. contortus ; and ric‐3 from X. laevis. We measured oocyte responses to 100 μM ACh. Figure 3A shows representative traces of inward currents induced by the different combinations of ancillary proteins and Asu‐acr‐16. Figure 3B is a bar chart of the mean ± SEM of the 100 μM ACh responses. Oocytes injected with Asu‐acr‐16 cRNA alone did not respond to 100 μM ACh (n = 21). The largest currents (I max = 290.6 ± 63.7 nA, n = 23) were seen in oocytes where just ric‐3 from A.suum was co‐injected with Asu‐acr‐16: these oocytes had larger current responses to 100 μM ACh than oocytes in which all three of the ancillaries (ric‐3, unc‐50 and unc‐74 from either H. contortus or A.suum) were injected with Asu‐acr‐16. The currents were also larger than when just Hco ‐ric‐3 was co‐injected with Asu‐acr‐16, suggesting that there is a species‐selective interaction between Asu‐ACR‐16 and Asu‐RIC‐3. Interestingly, Xle ‐ric‐3 produced mean currents that lay between those produced by Ascaris and Haemonchus ric‐3. We concluded that the robust expression of Asu‐ACR‐16 in Xenopus oocytes requires ric‐3, but not unc‐50 and unc‐74.
We also varied the amount of cRNA of Asu‐acr‐16 (10–25 ng) and cRNA of Asu‐ric‐3 (5–15 ng) injected to test the effects on expression. Supporting Information S3A, B shows representative current traces and bar charts of mean ± SEM of the current responses. The largest response (1062 ± 94.1 nA, n = 6) was obtained from oocytes injected with 25 ng Asu‐acr‐16 and 5 ng Asu‐ric‐3. We observed that the responses were dependent on the amounts of Asu‐acr‐16 and Asc‐ric‐3 injected and that increased ric‐3 did not overcome the effects of reduced acr‐16. All subsequent recordings were carried out on oocytes in which 25 ng Asu‐acr‐16 was co‐injected with 5 ng Asu‐ric‐3.
Asu‐ACR‐16 forms a nicotine‐sensitive but levamisole‐insensitive nAChR
Figure 4 shows the rank order potency series for a selection of nine nicotinic agonists and eight cholinergic anthelmintic agonists tested on the expressed Asu‐ACR‐16 nAChR. The nicotinic and anthelmintic agonists were tested at 100 μM, except for tribendimidine, which was tested at a concentration of 30 μM due to its limited solubility. Representative traces of inward currents induced by each nicotinic or anthelmintic agonist are shown along with their mean ± SEM normalized values as bar charts. Nicotine, cytisine, 3‐bromocytisine and epibatidine were the most potent (>80% of the control ACh current), whereas DMPP was least potent (<20% of the control ACh current). Choline, betaine, lobeline and A844606 were not active on the Asu‐ACR‐16 nAChR. With the exception of oxantel, which showed a very weak agonist effect (<10% of the control ACh current), the Asu‐ACR‐16 nAChR was not activated by any of the other anthelmintic drugs tested (morantel, levamisole, methyridine, thenium, bephenium, tribendimidine and pyrantel). The rank order potency series for agonists and anthelmintics on Asu‐ACR‐16 when normalized to the internal standard control 100 μM ACh current was as follows: nicotine ~ cytisine ~ 3‐bromocytisine ~ epibatidine > DMPP > oxantel >> > choline = betaine = lobeline = A844606 = morantel = levamisole = methyridine = thenium = bephenium = tribendimidine = pyrantel. Nicotine was among the most potent agonists, while none of the cholinomimetic anthelmintics produced any effect, except for the small effect of oxantel. This agonist potency series shows that this nAChR is distinct from the levamisole receptors of nematodes.
Asu‐ACR‐16 desensitization
Desensitization was a feature of the Asu‐ACR‐16 receptor responses and was characterized by the peak and waning current responses observed during maintained (for 10 s) agonist applications. We observed receptor desensitization with all potent agonists. The time constants for desensitization observed with different agonists are shown in Figure 5: the mean time constants for desensitization rates ranged between 6.2 and 12.6 s, with the time constant for desensitization depending on the agonist and significantly higher for ACh than for epibatidine. However, these time constants were markedly longer than the vertebrate α7 receptor, which has desensitization time constants in the 230–1300 ms range (McCormack et al., 2010): the slower desensitization rates may be explained in part by the absence of the amino acids: V251, L254 and L255 in Asu‐ACR‐16.
Nicotine as a potent agonist
Figure 6 shows representative recordings and concentration–response relationships of inward currents induced by the application of different concentrations of ACh (Figure 6A), top trace, and nicotine (Figure 6A), lower trace. The first application of an agonist for each new recording was always 100 μM ACh, which was used as the internal standard for normalization. The EC50 for nicotine (4.5 ± 0.2 μM, n = 6) was statistically significantly smaller than that for ACh (EC50 of 5.9 ± 0.1 μM, n = 6), which, on this basis, nicotine is more potent than ACh. Interestingly, there was no significant difference between the maximum response for nicotine (I max of 82.5 ± 3.4%, n = 6) and that for ACh (I max of 82.7 ± 2.4%, n = 6). There was also no significant difference between the Hill slopes, n H, for both nicotine and ACh. However, both the nicotine and ACh concentration–response curves were very steep, with Hill slopes, N H, of 3.4 ± 0.2, n = 6, for nicotine and 3.9 ± 0.3, n = 6, for ACh, showing strong cooperativity consistent with homomeric receptors containing multiple ligand binding sites.
Figure 6.
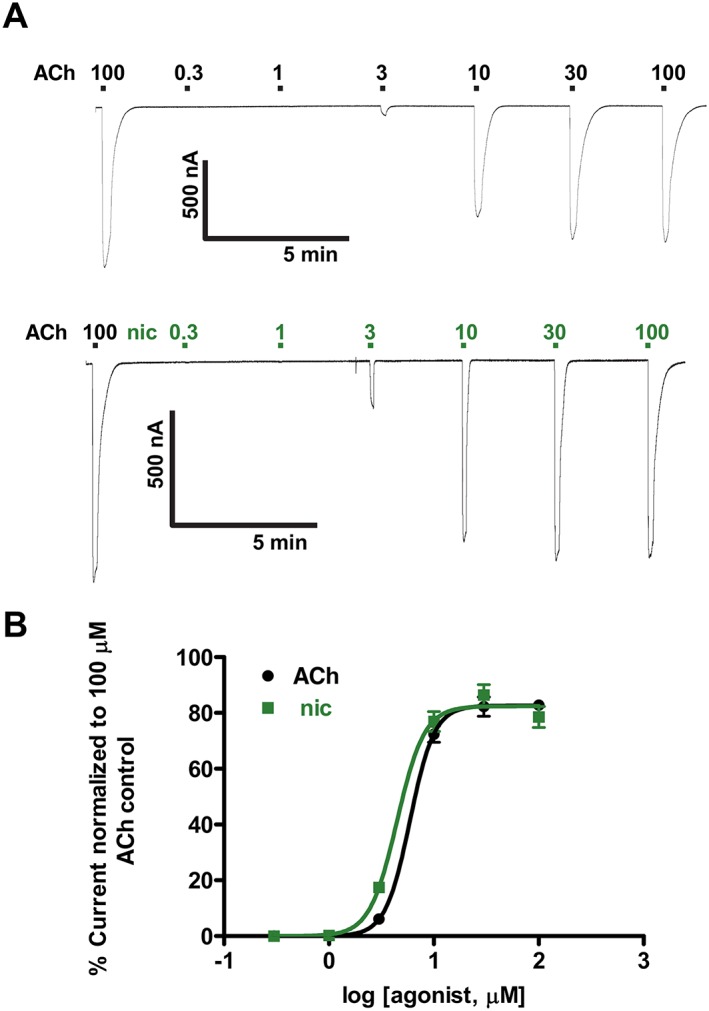
Nicotine (nic) and ACh concentration–response relationships for Asu‐ACR‐16 in the absence of antagonists. (A) Sample traces for ACh (top trace) and nicotine (lower trace) dose–response relationships for Asu‐ACR‐16. (B) ACh and nic dose–response curves for Asu‐ACR‐16. EC50 values (mean ± SEM, μM) were 5.9 ± 0.1 for ACh, Hill slope (n H) = 3.9 ± 0.3, n = 6, and 4.5 ± 0.2 for nic, n H = 3.4 ± 0.2, n = 6.
Rank order antagonist potencies: Asu‐ACR‐16 is not sensitive to α‐BTX
We tested 10 μM concentrations of eight nAChR antagonists of mammalian or nematode receptors on the expressed Asu‐ACR‐16 nAChR. The antagonists were α‐bungarotoxin (α‐BTX), dihydro‐β‐erythroidine (DHβE), d‐tubocurarine (dTC), hexamethonium, mecamylamine, methyllycaconitine (MLA), derquantel, and paraherquamide. The mean % inhibition of the control 100 μM ACh current response was used to determine the rank order potency of the antagonists. α‐BTX, an α7 receptor selective antagonist, had little effect on the Asu‐ACR‐16 receptor and was the least potent (inhibition, 5.5 ± 0.8% n = 6) antagonist in the rank order series. In this respect, the Asu‐ACR‐16 receptor was unlike the mammalian α7 nAChR. Mecamylamine, MLA and dTC were the most potent. The rank order of potency (Figure 7A) was mecamylamine = MLA = dTC > paraherquamide ~ derquantel ~ hexamethonium ~ DHβE > α‐BTX.
Non‐competitive derquantel antagonism and mixed antagonism of DHβE
We tested the effects of the nAChR antagonists, derquantel and DHβE on ACh (Figure 7B1). The concentration–response plots (Figure 7B2) show that derquantel acts as a non‐competitive antagonist, with 1 μM derquantel producing a statistically significant reduction in the maximum response, but no significant change in the EC50. I max and EC50 values were respectively 82.7 ± 2.4% and 5.9 ± 0.1 μM, n = 6, for ACh and 63.8 ± 2.8% and 6.2 ± 0.5 μM, n = 6, for derquantel. DHβE produced mixed competitive and non‐competitive antagonism (Figure 7B2) characterized by a significant right shift in the EC50 and a statistically significant reduction of the maximum response. EC50 and I max values were respectively 5.9 ± 0.1 μM and 82.7 ± 2.4%, n = 6, for ACh and 29.0 ± 1.0 μM and 58.0 ± 3.1%, n = 6, for DHβE. These observations suggest that antagonists of this receptor act at more than one site that may include the agonist‐binding site and/or a different allosteric site.
PAMs of human α7 nAChRs inhibit Asu‐ACR‐16 responses
Ion channel receptor opening may be increased (agonists) or decreased (competitive antagonists) by drugs binding to the ligand binding site, or the opening can be increased by PAMs or decreased by negative allosteric modulators binding to a site other than the ligand binding site (allosteric sites). Ivermectin and genistein are α7 receptor type 1 PAMs (Sattelle et al., 2009), which increase the response to a fixed concentration of an agonist by increasing the amplitude of the current response; PNU120596 is an α7 receptor type 2 PAM (Kalappa and Uteshev, 2013) whose action is characterized by both an increased amplitude of response and the reduction of desensitization of the current response to a fixed concentration of agonist.
We were interested to see if ivermectin, genistein and PNU120596 were PAMs of the Asu‐ACR‐16. We tested the effects of ivermectin (10 μM), genistein (3 μM) and PNU120596 (3 μM) separately on responses to 10, 30 and 100 μM ACh (Supporting Information S4). We did not observe potentiation in any experiment, but we saw modest reductions in the amplitudes of the responses. Figure 8 shows bar charts of the mean ± SEM [n = 6 (ivermectin), n = 6 (genistein), n = 6 (PNU120596)] currents of the control and test responses in the presence of the allosteric modulator. Two‐way ANOVA showed that ivermectin, genistein and PNU120596 had a statistically significant inhibitory effect on Asu‐ACR‐16 ACh responses. This inhibitory effect of ivermectin, genistein and PNU120596 was opposite to the stimulatory effect on mammalian α7 receptors (Sattelle et al., 2009).
Figure 8.
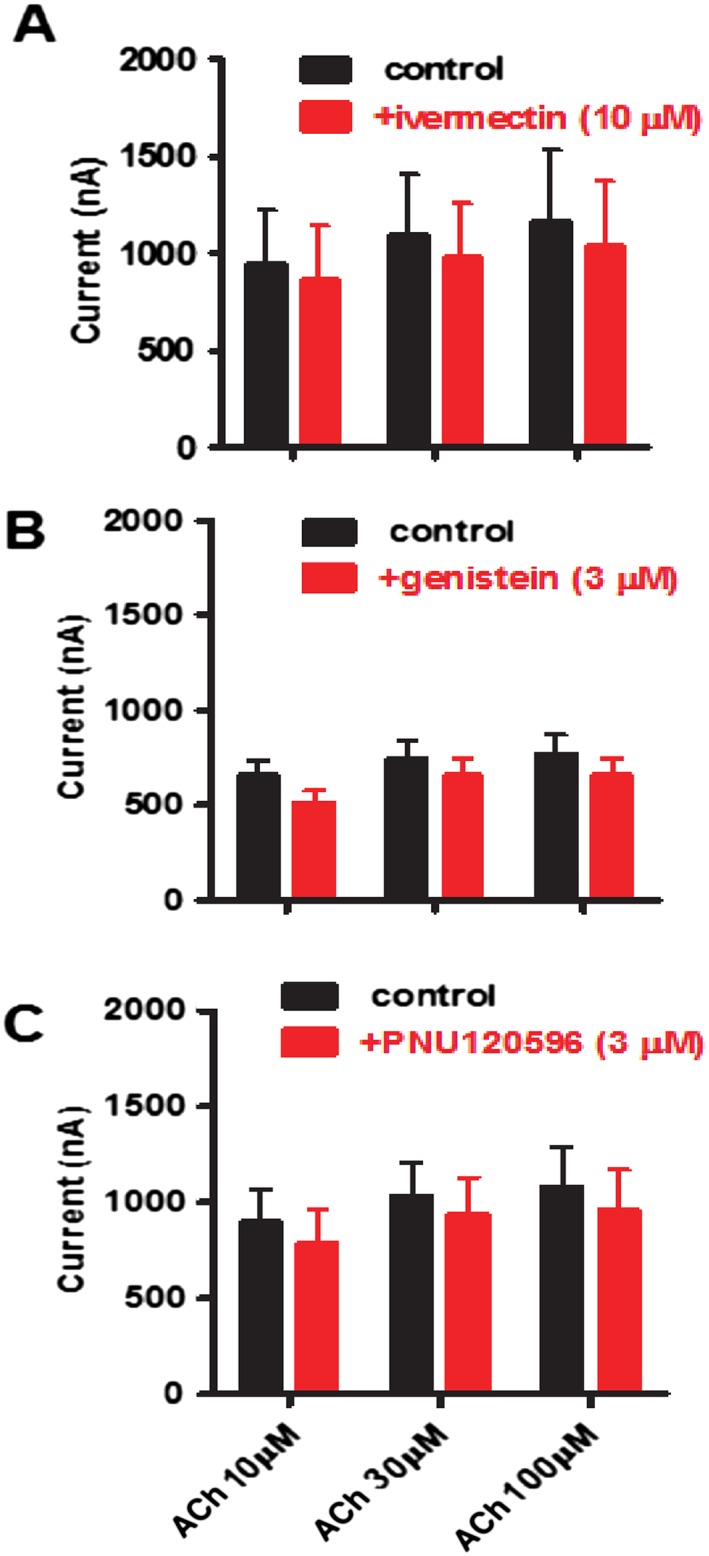
Effects of PAMs of human‐α7 on Asu‐ACR‐16‐mediated ACh responses. Bar charts showing blocking actions of human‐α7 PAMs; (A) 10 μM ivermectin (n = 6), (B) 3 μM genistein (n = 6) and (C) 3 μM PNU120596 (n = 6), on the responses of Asu‐ACR‐16 to ACh. The type 1 PAMs, ivermectin and genistein, as well as the type 2 PAM, PNU120596, caused a reduction in Asu‐ACR‐16 responses to 10, 30 and 100 μM ACh, with the reduction being more pronounced at 10 than at 30 or 100 μM ACh.
Calcium permeability of Asu‐ACR‐16
The calcium permeability of nAChRs varies and relates to the channel pore region formed by the TM2 domain of the subunit (Corringer et al., 1999a; Tapia et al., 2007). We tested the calcium permeability of the Asu‐ACR‐16 receptor by determining the shift in the reversal potential of the current–voltage (I–V) plot following a change in the concentration of Ca2 + in the extracellular solution (Supporting Information S5). Increasing the concentration of Ca2 + from 1 to 10 mM produced an inward rectification of the current through the Asu‐ACR‐16 receptor and a small positive shift of the I–V plot of 2.4 ± 2.1 mV (n = 6). A shift of 2.4 mV indicates relative calcium permeability (P Ca/P Na) of 0.4 when calculated from the GHK equation. The α7 receptor is much more permeable to calcium than the Asu‐ACR‐16 receptor. The P Ca/P Na reported for α7 nAChRs is 20, calculated from a 29 ± 3 mV shift under similar conditions (Séquéla et al., 1993), which was significantly (two‐tailed t‐test) bigger than the 2.4 mV shift observed for Asu‐ACR‐16.
Discussion
This study reports for the first time the reconstitution in the Xenopus oocyte expression system of a fully functional homomeric nAChR, ACR‐16, from a parasitic nematode. This finding is of significant importance in anthelmintic drug discovery in that it provides an efficient platform for screening anthelmintic compounds.
Comparison of pharmacology of Asu‐ACR‐16 with Cel‐ACR‐16
Previous work on the functional reconstitution in Xenopus oocytes and pharmacological characterization of ACR‐16 has focused on the model nematode, C. elegans (Ballivet et al., 1996). In agreement with Ballivet et al. (1996), Asu‐ACR‐16 was more sensitive to nicotine than ACh, but insensitive to levamisole and pyrantel. However, the Hill slopes for nicotine (n H = 3.4 ± 0.2, n = 6) and ACh (n H = 3.9 ± 0.3, n = 6) concentration–response curves for Asu‐ACR‐16 were slightly steeper than those reported for Cel‐ACR‐16 (n H = 2.2 for nicotine and n H = 2.1 for ACh). In any case, the Hill slopes for both Asu‐ACR‐16 and Cel‐ACR‐16 reveal strong cooperativity associated with homomeric receptors. The steeper Hill slopes for Asu‐ACR‐16 may account for its higher sensitivity (as shown by EC50 values) to nicotine and ACh when compared with those for Cel‐ACR‐16 (Asu‐ACR‐16's EC50 values were 4.5 ± 0.2 μM, n = 6, for nicotine and 5.9 ± 0.1 μM, n = 6, for ACh. On the other hand, for Cel‐ACR‐16, EC50 values were 12.6 μM for nicotine and 55.4 μM for ACh. Despite the high degree of sequence identity (74.75% identity) between Asu‐ACR‐16 and Cel‐ACR‐16 (Ce21), there exist significant differences in terms of the antagonist pharmacology of these two receptors. In accordance with Ballivet et al. (1996), Asu‐ACR‐16 was nearly insensitive to α‐BTX, but highly sensitive to dTC. In contrast, Asu‐ACR‐16 was highly sensitive to MLA, but moderately sensitive to hexamethonium and DHβE.
Comparison of pharmacology of Asu‐ACR‐16 with α7 nAChRs
Alignment of Asu‐ACR‐16 with human α7 nAChRs shows similarities but important differences in amino acid residues, suggesting differences in their pharmacologies. We findsimilarities in agonist sensitivity to ACh, nicotine and epibatidine of Asu‐ACR‐16 and α7 receptors (Raymond et al., 2000; Li et al., 2011). However, Asu‐ACR‐16 was insensitive to the α7‐selective agonist, A844606. Asu‐ACR‐16 was highly sensitive to MLA just like α7 (Briggs et al., 1995), but in contrast to the sensitivity of α7 receptors to α‐BTX (Zhao et al., 2003), we observed Asu‐ACR‐16 to be quite insensitive to α‐BTX. The PAMs of α7 (ivermectin, genistein and PNU120596) were inhibitory on Asu‐ACR‐16. Lastly, we found that the relative calcium permeability ratio of Asu‐ACR‐16 was about 50× smaller than that of the α7 receptor (Séquéla et al., 1993). The pharmacology of Asu‐ACR‐16 receptors is thus different to that of α7 nAChRs, indicating that the Asu‐ACR‐16 receptor has potential as a drug target site.
Comparison of pharmacology of Asu‐ACR‐16 with levamisole receptors
Asu‐ACR‐16 is different from the classical cholinergic levamisole receptor. The levamisole receptor subtypes are composed of different mixtures of heterologous subunits (UNC‐29, UNC‐38, ACR‐8, LEV‐1, ACR‐13 and UNC‐63) (Martin et al., 2012), whereas Asu‐ACR‐16 consists of only one subunit (ACR‐16). Unlike the levamisole receptors, which are sensitive to levamisole but insensitive to nicotine, Asu‐ACR‐16 is sensitive to nicotine but insensitive to levamisole. All three ancillary proteins, RIC‐3, UNC‐50 and UNC‐74, are required for robust expression of levamisole‐sensitive receptors (Boulin et al., 2008), while ACR‐16 requires only RIC‐3 for optimal expression in Xenopus oocytes (Sattelle et al., 2009; Bennett et al., 2012).
Another important difference between Asu‐ACR‐16 and the levamisole receptor can be seen in their antagonist pharmacologies. The levamisole receptor was only slightly inhibited by the nAChR antagonist, DHβE (Richmond and Jorgensen, 1999), whereas Asu‐ACR‐16 was strongly inhibited by DHβE. MLA and hexamethonium are also less potent on levamisole receptors. Because the pharmacology of ACR‐16 is different from the levamisole receptors, a selective anthelmintic directed at ACR‐16 may have the appropriate properties to bypass resistance.
Differences in α‐BTX potency, allosteric modulation and Ca2 + permeability
Site‐directed mutagenesis of nAChR subunits has been exploited as one of the tools in understanding the pharmacological differences between nAChR types. In a bid to provide reasons for the differences between the pharmacology of α7 receptors and Asu‐ACR‐16, we reviewed and compared the amino acids known to affect receptor pharmacology. In α7 for instance, the amino acids critical for α‐BTX binding are ERFYECCKEPYPD in the C‐loop (Balass et al., 1997). The corresponding residues in Asu‐ACR‐16 are EKFYDCCPEPYPD: the RK, ED and KP substitutions may explain why α‐BTX is not as potent as an antagonist of the Asu‐ACR‐16 receptor.
There are seven residues in α7 nAChRs important for positive allosteric modulation by ivermectin (Figure 1A, dark green arrows). Remarkably, none of these amino acids are conserved in Asu‐ACR‐16, which may explain why a positive allosteric effect of ivermectin for Asu‐ACR‐16 was not detected. Mutations in four of these residues (A225D, Q272, T456Y and C459) caused a significant reduction in the potency of ivermectin as a PAM, while mutations in the other three (S222M, M253L and S276V) caused ivermectin to act as an antagonist (Collins and Millar, 2010). Asu‐ACR‐16 contains the M253L substitution consistent with the observed inhibitory effect of ivermectin. Furthermore, we found that PNU120596 was not an allosteric modulator of Asu‐ACR‐16, in contrast to its action at α7 receptors. The amino acid residues between TM2 and TM3 in α7 responsible for PNU120596 positive allosteric modulation (AEIMPATSDS) (Bertrand et al., 2008) are replaced with SEISPPTSEA in Asu‐ACR‐16 (blue‐edged box, Figure 1A). These differences may account for the lack of positive allosteric modulation by PNU120596 in Asu‐ACR‐16. The amino acid differences in allosteric modulatory sites may allow for the design of selective drugs targeted at Asu‐ACR‐16.
Three residues, E237, L254 and L255, play key roles in the Ca2 + permeability of α7 nAChRs (grey and grey outline arrows, Figure 1A). In these receptors, mutations E237A, L254R/T and L255R/T/G cause a reduction in Ca2 + permeability (Bertrand et al., 1993). When compared with Asu‐ACR‐16, only one (E237) of these three residues is conserved. Amino acid differences at one or more positions may account for the reduction in the Ca2 + permeability of the Asu‐ACR‐16 compared with the α7 receptor.
Ubiquitous distribution of Asu‐ACR‐16 receptor and its function
ACR‐16 has been identified as one of the proteins required for the excitatory current at the neuromuscular junction of C. elegans (Richmond and Jorgensen, 1999; Francis et al., 2005). In fact, acr‐16 null mutants show nearly normal motor behaviour. Severe locomotion deficits were, however, seen with acr‐16:unc‐63 or acr‐16:unc‐29 double mutants (Touroutine et al., 2005; Li et al., 2014), showing that, in combination with other subunits, its function was essential.
The ubiquitous distribution of the Asu‐ACR‐16 message in tissues like the intestine and reproductive tract suggests that this receptor has functions which are not limited to regulation of fast neuromuscular transmission. The presence of Asu‐ACR‐16 in non‐excitable tissues suggests that Asu‐ACR‐16 also has slower paracrine/autocrine and homeostatic or differentiation functions associated with digestion and reproduction (Kawashima and Fujii, 2008). A paracrine and endocrine intestinal function for ACh is also supported by the presence of an organized distribution of acetylcholinesterase just beneath and adjacent to the intestinal epithelium (Lee, 1996). The wide tissue distribution of Asu‐ACR‐16 message suggests that a selective agonist may have advantages as an anthelmintic.
Consideration of Asu‐ACR‐16 as a drug target
Our findings show the Asu‐ACR‐16 receptor is pharmacologically different from previously characterized nAChRs and may serve as a potential target for therapeutic drug discovery. Given that Asu‐ACR‐16 shares some similarities with the mammalian α7 nAChR in terms of agonist pharmacology, it seems more likely that a drug directed at allosteric modulation sites may be more selective because the actions of α7 PAMs on the Asu‐ACR‐16 receptor were clearly different from those observed on the mammalian α7 receptor. This is particularly of interest because in recent years, PAMs have received considerable attention as nAChR‐targeted therapeutic agents (Williams et al., 2011). The α7 nAChR has been more exploited in this regard because of its implication in cognitive disorders such as Alzheimer's disease and schizophrenia, and drugs targeting α7 PAM sites have been suggested to have therapeutic potential for these disorders (Bertrand and Gopalakrishnan, 2007). Therefore, targeting the corresponding sites in Asu‐ACR‐16 may give rise to potential therapeutics which will not affect the mammalian host. It may therefore be possible to identify a suitable novel cholinergic anthelmintic which is ‘resistance busting’ if it were to activate a vital cholinergic receptor that was composed of different novel nAChR subunits.
Author contributions
M.A. conceived and designed the research study, performed the research, analysed the data, contributed reagents/materials/analysis tools and wrote the paper. S.K.B. performed the research. E.C. performed the research, analysed the data and contributed reagents/materials/analysis tools. C.L.C. wrote the paper. C.N. analysed the data, contributed reagents/materials/analysis tools and wrote the paper. C.J.M. performed the research. S.V. performed the research. A.P.R. conceived and designed the research study, analysed the data, contributed reagents/materials/analysis tools and wrote the paper. R.J.M. conceived and designed the research study, analysed the data, contributed reagents/materials/analysis tools and wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Amino acid sequence alignment of Asu‐ACR‐16 and Cel‐ACR‐16 nAChR subunits. The signal peptide (bright green box), ACh‐binding loops A–F (pink boxes), cys‐loop (yellow box) and transmembrane regions TM1–TM4 (turquoise boxes) are indicated. The vicinal cysteines (black‐edged box) that characterize an α‐subunit are present in the C‐binding loop. The blue‐edged box between TM2 and TM3 represents the region where PNU120596 acts on α7.
Figure S2. Alignment of ACR‐16 sequences from Asu, Toxocara canis, Loa loa, Hco, Ancylostoma ceylanicum and C.elegans. Predicted signal peptide sequences are shaded in grey. Amino acids conserved between the different ACR‐16 sequences are highlighted in blue. The Cys‐loop, the four transmembrane regions (TM1–TM4) and the primary agonist‐binding site are noted above the sequence.
Figure S3. Effects of varied amounts of Asu‐acr‐16 and Asu‐ric‐3 on Asu‐ACR‐16 expression. (A) Sample traces represented as inward currents produced in response to 100 μM ACh. (B) Bar chart (mean ± SEM) showing current sizes produced by 25 ng Asu‐acr‐16 and 5 ng Asu‐ric‐3 (1062 ± 94.1, n = 6), 10 ng Asu‐acr‐16 and 5 ng Asu‐ric‐3 (848.8 ± 155.4, n = 6), 10 ng Asu‐acr‐16 and 10 ng Asu‐ric‐3 (727.3 ± 63.1, n = 6); 15 ng Asu‐acr‐16 and 15 ng Asu‐ric‐3 (602.8 ± 106.8, n = 6) in response to 100 μM ACh. * P < 0.05, Tukey's multiple comparison tests.
Figure S4. Sample traces showing the effects of PAMs of α7; (A) 10 μM ivermectin, (B) 3 μM genistein and (C) 3 μM PNU120596, on Asu‐ACR‐16‐mediated ACh responses.
Figure S5. Calcium permeability of Asu‐ACR‐16 with 30 μM ACh currents: representative I–V plot for oocytes expressing Asu‐ACR‐16, showing current change with voltage in 1 mM (black line) and 10 mM (red line) Ca2+recording solutions. I–V relationship was plotted using a cubic polynomial equation and interpolated to measure the Erev. The mean ± SEM for the positive shift of the I–V plot for six observations was 2.4 ± 2.1 mV, and this corresponded to a relative calcium permeability ratio of 0.4. Insert: magnified view of the I–V fitted line from −10 to 10 mV, showing the Erev in 1 and 10 mM Ca2+.
Supporting info item
Supporting info item
Acknowledgements
We would like to express our gratitude to Debra J Woods, Zoetis Animal Health, Kalamazoo, MI, USA, for the generous supply of derquantel. We would also like to thank Professor Shu Hua Xiao, National Institute of Parasitic Diseases, Shanghai, Peoples' Republic of China, for the gift of tribendimidine. Research funding was by The Hatch Act, State of Iowa; NIH grant R01 AI047194 National Institute of Allergy and Infectious Diseases to R.J.M.; NIH grant R21AI092185 National Institute of Allergy and Infectious Diseases to A.P.R.; and the Schlumberger “Faculty for the Future” programme to M.A. and A.P.R. The funding agencies had no role in the design, execution or publication of this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases.
Abongwa, M. , Buxton, S. K. , Courtot, E. , Charvet, C. L. , Neveu, C. , McCoy, C. J. , Verma, S. , Robertson, A. P. , and Martin, R. J. (2016) Pharmacological profile of Ascaris suum ACR‐16, a new homomeric nicotinic acetylcholine receptor widely distributed in Ascaris tissues. British Journal of Pharmacology, 173: 2463–2477. doi: 10.1111/bph.13524.
References
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balass M, Katchalski‐Katzir E, Fuchs S (1997). The alpha‐bungarotoxin binding site on the nicotinic acetylcholine receptor: analysis using a phage‐epitope library. Proc Natl Acad Sci U S A 94: 6054–6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballivet M, Alliod C, Bertrand S, Bertrand D (1996). Nicotinic acetylcholine receptors in the nematode Caenorhabditis elegans . J Mol Biol 258: 261–269. [DOI] [PubMed] [Google Scholar]
- Baur R, Beech R, Sigel E, Rufener L (2015). Monepantel irreversibly binds to and opens Haemonchus contortus MPTL‐1 and Caenorhabditis elegans ACR‐20 receptors. Mol Pharmacol 87: 96–102. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004). Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340: 783–795. [DOI] [PubMed] [Google Scholar]
- Bennett HM, Lees K, Harper KM, Jones AK, Sattelle DB, Wonnacott S et al. (2012). Xenopus laevis RIC‐3 enhances the functional expression of the C. elegans homomeric nicotinic receptor, ACR‐16, in Xenopus oocytes. J Neurochem 123: 911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Bertrand S, Cassar S, Gubbins E, Li J, Gopalakrishnan M (2008). Positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2–M3 segment. Mol Pharmacol 74: 1407–1416. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Devillers‐Thiery A, Revah F, Galzi JL, Hussy N, Mulle C et al. (1992). Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc Natl Acad Sci U S A 89: 1261–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers‐Thiery A, Bertrand S, Changeux JP (1993). Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci U S A 90: 6971–6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M (2007). Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol 74: 1155–1163. [DOI] [PubMed] [Google Scholar]
- Boulin T, Fauvin A, Charvet CL, Cortet J, Cabaret J, Bessereau J‐L et al. (2011). Functional reconstitution of Haemonchus contortus acetylcholine receptors in Xenopus oocytes provides mechanistic insights into levamisole resistance. Br J Pharmacol 164: 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulin T, Gielen M, Richmond JE, Williams DC, Paoletti P, Bessereau JL (2008). Eight genes are required for functional reconstitution of the Caenorhabditis elegans levamisole‐sensitive acetylcholine receptor. Proc Natl Acad Sci U S A 105: 18590–18595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs CA, McKenna DG, Piattoni‐Kaplan M (1995). Human alpha 7 nicotinic acetylcholine receptor responses to novel ligands. Neuropharmacology 34: 583–590. [DOI] [PubMed] [Google Scholar]
- Brooker S (2010). Estimating the global distribution and disease burden of intestinal nematode infections: adding up the numbers – a review. Int J Parasitol 40: 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bschleipfer T, Schukowski K, Weidner W, Grando SA, Schwantes U, Kummer W et al. (2007). Expression and distribution of cholinergic receptors in the human urothelium. Life Sci 80: 2303–2307. [DOI] [PubMed] [Google Scholar]
- Buxton SK, Charvet CL, Neveu C, Cabaret J, Cortet J, Peineau N et al. (2014). Investigation of acetylcholine receptor diversity in a nematode parasite leads to characterization of tribendimidine‐ and derquantel‐sensitive nAChRs. PLoS Pathog 10 .e1003870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L (2010). In pursuit of the high‐resolution structure of nicotinic acetylcholine receptors. J Physiol 588: 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T, Millar NS (2010). Nicotinic acetylcholine receptor transmembrane mutations convert ivermectin from a positive to a negative allosteric modulator. Mol Pharmacol 78: 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corringer PJ, Bertrand S, Galzi JL, Devillers‐Thiery A, Changeux JP, Bertrand D (1999a). Molecular basis of the charge selectivity of nicotinic acetylcholine receptor and related ligand‐gated ion channels. Novartis Found Symp 225: 215–224. discussion 224‐230 [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Bertrand S, Galzi JL, Devillers‐Thiery A, Changeux JP, Bertrand D (1999b). Mutational analysis of the charge selectivity filter of the alpha7 nicotinic acetylcholine receptor. Neuron 22: 831–843. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvin A, Charvet C, Issouf M, Cortet J, Cabaret J, Neveu C (2010). cDNA–AFLP analysis in levamisole‐resistant Haemonchus contortus reveals alternative splicing in a nicotinic acetylcholine receptor subunit. Mol Biochem Parasitol 170: 105–107. [DOI] [PubMed] [Google Scholar]
- Francis MM, Evans SP, Jensen M, Madsen DM, Mancuso J, Norman KR et al. (2005). The Ror receptor tyrosine kinase CAM‐1 is required for ACR‐16‐mediated synaptic transmission at the C. elegans neuromuscular junction. Neuron 46: 581–594. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan M, Bertrand D, Williams M (2007). Nicotinic acetylcholine receptors as therapeutic targets: emerging frontiers in basic research and clinical science. Biochem Pharmacol 74: 1091. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27: 221–224. [DOI] [PubMed] [Google Scholar]
- Hewitson JP, Maizels RM (2014). Vaccination against helminth parasite infections. Expert Rev Vaccines 13: 473–487. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W (1993). TMbase – a database of membrane spanning proteins segments. Biol Chem Hoppe Seyler 374: 166. [Google Scholar]
- Hotez PJ, Molyneaux DH, Fenwick A, Kumaresan J, Sachs SE, Sachs JD et al. (2007). Control of neglected tropical diseases. N Engl J Med 357: 1018–1027. [DOI] [PubMed] [Google Scholar]
- Jones AK, Davis P, Hodgkin J, Sattelle DB (2007). The nicotinic acetylcholine receptor gene family of the nematode Caenorhabditis elegans: an update on nomenclature. Invert Neurosci 7: 129–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa BI, Uteshev VV (2013). The dual effect of PNU‐120596 on alpha7 nicotinic acetylcholine receptor channels. Eur J Pharmacol 718: 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky R, Gauvry N, Weber SS, Skripsky T, Bouvier J, Wenger A et al. (2008). Identification of the amino‐acetonitrile derivative monepantel (AAD 1566) as a new anthelmintic drug development candidate. Parasitol Res 103: 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao PN, Dwork AJ, Kaldany RR, Silver ML, Wideman J, Stein S et al. (1984). Identification of the alpha subunit half‐cystine specifically labeled by an affinity reagent for the acetylcholine receptor binding site. J Biol Chem 259: 11662–11665. [PubMed] [Google Scholar]
- Karlin A (1993). Structure of nicotinic acetylcholine receptors. Curr Opin Neurobiol 3: 299–309. [DOI] [PubMed] [Google Scholar]
- Kawashima K, Fujii T (2008). Basic and clinical aspects of non‐neuronal acetylcholine: overview of non‐neuronal cholinergic systems and their biological significance. J Pharmacol Sci 106: 167–173. [DOI] [PubMed] [Google Scholar]
- Kerr KB, Cavett JW (1952). A technic for initial evaluation of potential anthelmintics. Exp Parasitol 1: 161–167. [Google Scholar]
- Lee DL (1996). Why do some nematode parasites of the alimentary tract secrete acetylcholinesterase? Int J Parasitol 26: 499–508. [DOI] [PubMed] [Google Scholar]
- Li SX, Huang S, Bren N, Noridomi K, Dellisanti CD, Sine SM et al. (2011). Ligand‐binding domain of an alpha7‐nicotinic receptor chimera and its complex with agonist. Nat Neurosci 14: 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu J, Zheng M, Xu XZ (2014). Encoding of both analog‐ and digital‐like behavioral outputs by one C. elegans interneuron. Cell 159: 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RJ (1997). Modes of action of anthelmintic drugs. Vet J 154: 11–34. [DOI] [PubMed] [Google Scholar]
- Martin RJ, Robertson AP, Buxton SK, Beech RN, Charvet CL, Neveu C (2012). Levamisole receptors: a second awakening. Trends Parasitol 28: 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack TJ, Melis C, Colon J, Gay EA, Mike A, Karoly R et al. (2010). Rapid desensitization of the rat alpha7 nAChR is facilitated by the presence of a proline residue in the outer beta‐sheet. J Physiol 588 (Pt 22): 4415–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon C, Bartley DJ, Edgar HW, Ellison SE, Barley JP, Malone FE et al. (2013). Anthelmintic resistance in Northern Ireland (I): prevalence of resistance in ovine gastrointestinal nematodes, as determined through faecal egg count reduction testing. Vet Parasitol 195: 122–130. [DOI] [PubMed] [Google Scholar]
- Millar NS, Gotti C (2009). Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 56: 237–246. [DOI] [PubMed] [Google Scholar]
- Mongan NP, Baylis HA, Adcock C, Smith GR, Sansom MS, Sattelle DB (1998). An extensive and diverse gene family of nicotinic acetylcholine receptor alpha subunits in Caenorhabditis elegans . Receptors Channels 6: 213–228. [PubMed] [Google Scholar]
- Neveu C, Charvet CL, Fauvin A, Cortet J, Beech RN, Cabaret J (2010). Genetic diversity of levamisole receptor subunits in parasitic nematode species and abbreviated transcripts associated with resistance. Pharmacogenet Genomics 20: 414–425. [DOI] [PubMed] [Google Scholar]
- Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J et al. (2004). Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology 145: 2498–2506. [DOI] [PubMed] [Google Scholar]
- Raymond V, Mongan NP, Sattelle DB (2000). Anthelmintic actions on homomeric‐forming nicotinic acetylcholine receptor subunits: chicken α7 and ACR‐16 from the nematode Caenorhabditis elegans . Neuroscience 101: 785–791. [DOI] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Devillers‐Thiery A, Mulle C, Hussy N et al. (1991). Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 353: 846–849. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM (1999). One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2: 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattelle DB, Buckingham SD, Akamatsu M, Matsuda K, Pienaar I, Jones AK et al. (2009). Comparative pharmacology and computational modelling yield insights into allosteric modulation of human α7 nicotinic acetylcholine receptors. Biochem Pharmacol 78: 836–843. [DOI] [PubMed] [Google Scholar]
- Scott I, Pomroy WE, Kenyon PR, Smith G, Adlington B, Moss A (2013). Lack of efficacy of monepantel against Teladorsagia circumcincta and Trichostrongylus colubriformis . Vet Parasitol 198: 166–171. [DOI] [PubMed] [Google Scholar]
- Séquéla P, Wadiche J, Dineley‐Miller K, Dani JA, Patrick JW (1993). Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W et al. (2011). Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl. Acids Res. 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taman A, Azab M (2014). Present‐day anthelmintics and perspectives on future new targets. Parasitol Res 113: 2425–2433. [DOI] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J (2007). Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Mol Pharmacol 71: 769–776. [DOI] [PubMed] [Google Scholar]
- Touroutine D, Fox RM, Von Stetina SE, Burdina A, Miller DM, Richmond JE (2005). acr‐16 encodes an essential subunit of the levamisole‐resistant nicotinic receptor at the Caenorhabditis elegans neuromuscular junction. J Biol Chem 280: 27013–27021. [DOI] [PubMed] [Google Scholar]
- Wessler I, Kirkpatrick CJ (2008). Acetylcholine beyond neurons: the non‐neuronal cholinergic system in humans. Br J Pharmacol 154: 1558–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Wang J, Papke RL (2011). Positive allosteric modulators as an approach to nicotinic acetylcholine receptor‐targeted therapeutics: advantages and limitations. Biochem Pharmacol 82: 915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme AJ, Fairweather I, Prichard R, von Samson‐Himmelstjerna G, Sangster NC (2004). Drug resistance in veterinary helminths. Trends Parasitol 20: 469–476. [DOI] [PubMed] [Google Scholar]
- Zhao L, Kuo YP, George AA, Peng JH, Purandare MS, Schroederv L et al. (2003). Functional properties of homomeric, human alpha 7‐nicotinic acetylcholine receptors heterologously expressed in the SH‐EP1 human epithelial cell line. J Pharmacol Exp Ther 305: 1132–1141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Amino acid sequence alignment of Asu‐ACR‐16 and Cel‐ACR‐16 nAChR subunits. The signal peptide (bright green box), ACh‐binding loops A–F (pink boxes), cys‐loop (yellow box) and transmembrane regions TM1–TM4 (turquoise boxes) are indicated. The vicinal cysteines (black‐edged box) that characterize an α‐subunit are present in the C‐binding loop. The blue‐edged box between TM2 and TM3 represents the region where PNU120596 acts on α7.
Figure S2. Alignment of ACR‐16 sequences from Asu, Toxocara canis, Loa loa, Hco, Ancylostoma ceylanicum and C.elegans. Predicted signal peptide sequences are shaded in grey. Amino acids conserved between the different ACR‐16 sequences are highlighted in blue. The Cys‐loop, the four transmembrane regions (TM1–TM4) and the primary agonist‐binding site are noted above the sequence.
Figure S3. Effects of varied amounts of Asu‐acr‐16 and Asu‐ric‐3 on Asu‐ACR‐16 expression. (A) Sample traces represented as inward currents produced in response to 100 μM ACh. (B) Bar chart (mean ± SEM) showing current sizes produced by 25 ng Asu‐acr‐16 and 5 ng Asu‐ric‐3 (1062 ± 94.1, n = 6), 10 ng Asu‐acr‐16 and 5 ng Asu‐ric‐3 (848.8 ± 155.4, n = 6), 10 ng Asu‐acr‐16 and 10 ng Asu‐ric‐3 (727.3 ± 63.1, n = 6); 15 ng Asu‐acr‐16 and 15 ng Asu‐ric‐3 (602.8 ± 106.8, n = 6) in response to 100 μM ACh. * P < 0.05, Tukey's multiple comparison tests.
Figure S4. Sample traces showing the effects of PAMs of α7; (A) 10 μM ivermectin, (B) 3 μM genistein and (C) 3 μM PNU120596, on Asu‐ACR‐16‐mediated ACh responses.
Figure S5. Calcium permeability of Asu‐ACR‐16 with 30 μM ACh currents: representative I–V plot for oocytes expressing Asu‐ACR‐16, showing current change with voltage in 1 mM (black line) and 10 mM (red line) Ca2+recording solutions. I–V relationship was plotted using a cubic polynomial equation and interpolated to measure the Erev. The mean ± SEM for the positive shift of the I–V plot for six observations was 2.4 ± 2.1 mV, and this corresponded to a relative calcium permeability ratio of 0.4. Insert: magnified view of the I–V fitted line from −10 to 10 mV, showing the Erev in 1 and 10 mM Ca2+.
Supporting info item
Supporting info item