

Abstract
Objectives
The relationship of immune dysregulation and autoantibody production that may contribute to systemic lupus erythematosus (SLE) pathogenesis is unknown. This study evaluates the individual and combined contributions of autoantibodies, type I interferon (IFN-α) activity, and IFN-associated soluble mediators to disease development leading to SLE.
Methods
Serial serum specimens from 55 individuals collected prior to SLE classification (average timespan = 4.3 years) and unaffected healthy controls matched by age (± 5 years), gender, race, and time of sample procurement were obtained from the Department of Defense Serum Repository. Levels of serum IFN-α activity, IFN-associated mediators, and autoantibodies were evaluated and temporal relationships assessed by growth curve modeling, path analysis, Analysis of Covariance, and random forest models.
Results
In cases, but not matched controls, autoantibody specificities and IFN-associated mediators accumulated over a period of years, plateauing near the time of disease classification (p<0.001). Autoantibody positivity coincided with or followed type II IFN dysregulation, preceding IFN-α activity in growth curve models, with elevated IFN-α activity and BLyS levels occurring shortly before SLE classification (p≤0.005). Cases were distinguished by multivariate random forest models incorporating IFN-γ, MCP-3, anti-chromatin and anti-spliceosome antibodies (accuracy 93% > 4 years pre-classification; 97% within 2 years of SLE classification).
Conclusions
Years before SLE classification, enhancement of the type II IFN pathway allows for accumulation of autoantibodies and subsequent elevations in IFN-α activity immediately preceding SLE classification. Perturbations in select immunological processes may help identify at-risk individuals for further clinical evaluation or participation in prospective intervention trials.
Keywords (MeSH): Systemic Lupus Erythematosus, Pre-clinical Autoimmunity, Humoral Immunity, Autoantibodies, Interferons
INTRODUCTION
Systemic lupus erythematosus (SLE) is marked by pathogenic autoantibody production and immune dysregulation, resulting in organ damage. Patients may accrue organ damage by the time they reach disease classification,[1] making early intervention an attractive approach to curtailing excessive morbidity. Anti-nuclear autoantibodies (ANA) might help identify future patients, since SLE-associated autoantibodies accumulate prior to clinical disease.[2, 3] However, up to 14% of healthy individuals, particularly women aged 40–49 years, are ANA positive yet never develop lupus or another autoimmune rheumatic disorder.[4] Therefore, ANA positivity is likely accompanied by other immunological changes contributing to pathogenesis and may help distinguish patients at risk of disease transition.
Elevated type I IFN (IFN-α) activity is associated with SLE pathogenesis,[5] including DNA- and RNA-protein binding autoantibody specificities[6] and B-lymphocyte stimulator (BLyS) induction.[7] BLyS, a key regulator of B cell survival and differentiation, contributes to autoantibody production and class switching.[8] Both IFN-α[9] and BLyS[8] have been recent lupus therapeutic targets. Although BLyS blockade may reduce disease activity in some lupus patients,[8] clinical trials utilizing IFN-α blocking agents have had modest clinical outcomes to date, despite evidence of decreasing IFN-α gene signatures.[9]
In addition to type I IFN, multiple genes that contribute to activation of type II IFN (IFN-γ) pathways are associated with SLE.[10] Furthermore, IFN-γ can drive both type I IFN[11] and BLyS production.[12] Bridging innate and adaptive immunity, IFN-γ is expressed by multiple innate cell types[13] and primes macrophages and dendritic cells to respond to TLR signals.[11] IFN-γ perpetuates Th1-type adaptive cellular responses, recruiting cells to sites of inflammation by stimulating the secretion of such chemokines as IP-10 (CXCL10), MIG (CXCL9), and MCP-3 (CCL7).[11] Treating SLE patients with the anti-IFN-γ monoclonal antibody AMG 811 reduces IP-10 levels,[14] which itself has become a potential rheumatic disease therapeutic target.[15]
Together, these observations suggest that dysregulation of type I/type II IFNs and autoantibody accumulation may contribute to SLE pathogenesis. However, levels of IFN-associated mediators in preclinical SLE and possible interactions between IFN-α activity, type II IFN pathways, and autoantibody accumulation in the transition to clinical disease are not understood. We sought to address this knowledge gap by analyzing longitudinal serum samples collected prior to and concurrent with SLE classification. In this first preclinical longitudinal study to assess the temporal relationship between IFN-associated soluble mediators, the accrual of lupus-associated autoantibodies, and increased type I IFN activity, our findings suggest that perturbations in combined immunological processes precede clinical classification and can help identify individuals at risk of lupus development.
METHODS
Study population and serum samples
Experiments were performed in accordance with the Helsinki Declaration and approved by the Institutional Review Boards of the Oklahoma Medical Research Foundation and the Walter Reed National Military Medical Center. A subset of individuals in the Department of Defense Serum Repository (DODSR, n=55) was selected for this study based on availability of serum samples collected prior to and at/after disease classification. Demographic and clinical information were collected at the time of sample procurement. Two-thirds of subjects were female, 55% were African-American (25% European-American), and mean (SD) age at SLE classification was 29.4 (6.0) years.
Cases were matched to healthy controls by age (± 5 years), gender, race, and time of sample procurement. Demographics are shown in supplementary table 1.
Serum autoantibody detection, IFN-α activity measurement, and soluble mediator detection
Autoantibodies to dsDNA, chromatin, Ro/SSA, La/SSB, Sm, SmRNP complex, and RNP were detected using the xMAP BioPlex 2200 multiplex system (Bio-Rad Technologies, Hercules, CA).[16] WISH Reporter cells (CAT# CCL125; ATCC) were used to measure the ability of sera to induce type I IFN-related gene transcription.[6] Serum BLyS levels were determined by ELISA (R&D Systems, Minneapolis, MN) and IP-10, IFN-γ, MCP-3, MIP-1α, and MIG, assessed by xMAP multiplex assays (Affymetrix-eBioscience, Santa Clara, CA).[17] See supplementary methods.
Statistical analyses
Associations between time to SLE classification vs. IFN-α activity, autoantibodies, and IFN-associated mediators were analyzed by Kruskal-Wallis testing with Dunn’s multiple comparison. Differences between cases and controls with respect to accumulation of autoantibody specificities and IFN-associated mediators were determined by Wilcoxon matched-pairs test. Within-individual correlations between autoantibody specificities or IFN-associated mediator levels and IFN-α activity were computed by comparing the residual variance from an unconditional means model to that of a Gaussian mixed model, where the variable of interest was included as a predictor. The resulting pseudo-R2 value is defined as (var(unconditional)-var(growth))/var(unconditional). Growth curve modeling, path analysis, analysis of covariance (ANCOVA), and random forest (RF) predictive modeling assessments were performed (supplementary methods). Analyses were performed using GraphPad Prism 6.02 for Windows (GraphPad Software, San Diego, CA, USA), SAS STAT 9.3 (SAS Institute Inc., Cary, NC, USA), and R 2.15.3 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
IFN-α activity increases before SLE disease classification
IFN-α is genetically and serologically associated with SLE pathogenesis.[5, 6, 18] We evaluated longitudinal changes in serum IFN-α activity over three longitudinal serum samples procured from 55 cases in the DODSR, covering symptom-free, pre-classification, and at/after SLE classification periods. IFN-α activity increased as individuals approached disease classification (p<0.001 [median difference=2.32, 95%CI 0.51 to 2.94], figure 1A). Only 20% of cases had increased serum IFN-α activity ≥4 years before SLE classification, while 44% had elevated serum IFN-α activity ≤2 years before classification, and 75% had increased serum IFN-α activity at classification.
Figure 1.

IFN-α reporter activity levels increase as individuals move toward SLE classification. Increases in serum IFN-α activity score by time before SLE classification (A) and by autoantibody status (B) are shown (mean ± SEM). IFN-α activity score ≥ 1 is considered positive (dashed line). Percent of case samples positive for IFN-α activity is listed below each time point. *p<0.05, ***p<0.001, Kruskal-Wallis with Dunn’s multiple comparison. Demographics (C) and number of positive autoantibodies (D, mean ± SEM; *p<0.05, unpaired t-test) in SLE cases that are ever IFN-α activity positive vs SLE cases that remain IFN-α activity negative are also shown. Other = Asian/Pacific Islander, American Indian/Alaskan Native, or multiracial
Since patients approaching SLE classification accumulate DNA- and RNA-associated autoantibodies,[3] we examined the relationship between serum IFN-α activity and autoantibody positivity (figure 1B). Compared to the autoantibody negative time point, serum IFN-α activity was increased with autoantibody positivity (p=0.055 [median difference=0.58, 95%CI 0.0 to 1.64]) and further increased at/after disease classification (p<0.001 [median difference=2.34, 95%CI 0.65 to 3.26]). Of the 55 cases, 41 (75%) had at least one sample with elevated IFN-α activity, with no significant differences in gender, ethnicity, or SLE classification age (figure 1C). Cases with positive serum IFN-α activity had more DNA- and RNA-associated autoantibody specificities (3.8±2.1) than cases without elevated IFN-α activity (2.4±2.6) (p=0.050; figure 1D), and the number of autoantibody specificities correlated with serum IFN-α activity level (Spearman r=0.462, p<0.001). These results imply a biological relationship between IFN-α activity and autoantibody accrual during preclinical SLE.
Elevation of IFN-α activity follows autoantibody accumulation
The number of autoantibody specificities increased preclinically and plateaued near disease classification in individuals who developed classified SLE, but not matched healthy controls, (p<0.001; figure 2A). The levels of each autoantibody specificity showed significant within-individual correlations with IFN-α activity (figure 2B). We evaluated the temporal relationship between IFN-α activity and DNA- and RNA-associated autoantibody production using growth curve linear mixed models, which account for the contribution of the longitudinal samples acquired at varying times relative to disease classification. All RNA- (figure 2C) and DNA- associated (figure 2D) autoantibodies, except anti-dsDNA, appeared before elevation of IFN-α activity and SLE classification in all cases (supplementary figure 1); no differences in the frequency of autoantibody specificities and IFN-α activity were noted between African-American (AA) and non-AA cases (supplementary table 2). Anti-dsDNA antibodies typically coincided with elevated IFN-α activity (mean appearance at −2.40 vs −2.49 years), although only 37 (67%) of cases were dually positive for serum IFN-α activity and anti-dsDNA. Therefore, autoantibody positivity and accumulation of lupus-associated autoantibody specificities can be detected prior to increases in IFN-α activity.
Figure 2.

Most autoantibodies are detected prior to increases in IFN-α activity in SLE patients. (A) Increase in the number of serum autoantibodies by time before SLE in 55 cases compared to matched healthy controls, p=0.0002 by Wilcoxon matched-pairs test. Time of SLE classification (dotted line, down arrow) is shown. Within-individual correlations of IFN-α activity vs. level of autoantibody positivity are presented, as described in the Methods (B). Temporal relationships, using growth models, of IFN-α activity with Ro/SSA, La/SSB, Sm, SmRNP, and RNP (C) and with dsDNA and chromatin autoantibodies (D) are shown. The positive cut-off values are dsDNA >10 IU/mL and >1 AI (autoantibody index) for all other specificities by xMAP multiplex assays. Time of positivity (years pre-classification): dsDNA (−2.40), 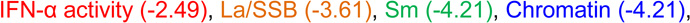 ,
, 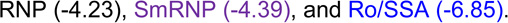
IFN-associated soluble mediators correlate with IFN-α activity and accumulation of autoantibodies
Increasing serum IFN-α activity observed in patients approaching SLE classification (figure 1A–B) may reflect inflammatory processes that stimulate IFN target gene expression.[19] We evaluated serum levels of IFN-associated mediators in pre-classification cases compared to time-of-procurement matched controls. Significant within-individual correlations existed between IFN-α activity and levels of IFN-γ (p<0.001, PseudoR2=0.151), IP-10 (p<0.001, PseudoR2=0.243), BLyS (p<0.001, PseudoR2=0.322), and MCP-3 (p=0.002, PseudoR2=0.150), with a moderate correlation between IFN-α activity and MIG levels (p=0.040, PseudoR2=0.096; supplementary figure 2A). Further, serum IFN-γ (p<0.001 [median difference=−7.65, 95%CI −8.04 to −6.45]), IP-10 (p=0.002 [median difference=−26.45, 95%CI −41.95 to −9.96]), and MCP-3 (p<0.001 [median difference=−344.3, 95%CI −351.5 to −192.9]) concentrations were significantly higher in cases ≥4 years pre-classification compared to matched controls, with additional increased levels as patients moved toward classification (p≤0.001; supplementary figure 2B–E). Control samples maintained consistent levels of the IFN-associated soluble mediators, whether assessed with respect to time of SLE classification in matched cases, relative time of sample procurement, or order of sample procurement (supplementary figure 3).
Given that type II IFN-associated mediators were elevated several years prior to SLE classification, we hypothesized that they may increase prior to or concurrent with the detection of SLE-associated autoantibodies. Elevated IFN-associated soluble mediators in each longitudinal sample were defined using cutoffs established by receiver operating characteristic (ROC) curves that optimally differentiated cases vs. controls (supplementary figure 4). The number of elevated IFN-associated inflammatory soluble mediators increased prior to and plateaued near disease classification in cases, but not matched controls (p<0.001 [median difference=−0.81, 95%CI −1.88 to −0.49]; figure 3A). Cases had a mean of 4.7 elevated mediators at the time of classification, compared to 1.8 in controls. IFN-α activity and levels of IP-10, IFN-γ, BLyS, and MIG showed significant within-individual correlations with the number of positive autoantibody specificities (p≤0.001; figure 3B), with an increased number of elevated IFN-associated mediators as cases moved from autoantibody negative status (2.4 elevated mediators) to disease classification (4.7 elevated mediators; p≤0.001 [median difference=−2.41, 95%CI −2.53 to −1.62]). Importantly, levels of IFN-γ (p<0.001 [median difference=−7.49, 95%CI −9.03 to −5.08], figure 3C) and IP-10 (p=0.007 [median difference=−29.46, 95%CI −74.49 to −5.76], figure 3D) were significantly higher in autoantibody negative cases compared to controls. However, levels of BLyS and MIG were not significantly elevated in cases until at/near the time of SLE classification (figure 3E–F). Because case samples were procured concurrently with matched control samples, longitudinal changes in serum IFN-associated soluble mediators in cases were not a consequence of time, but instead were likely related to the preclinical mechanisms that precipitated SLE disease classification (figure 3; supplementary figures 2–3). Together, these results provide further support that, like IFN-α activity, IFN-associated soluble mediators are dysregulated prior to clinical SLE. In addition, they suggest that IFN-γ and IP-10 increase prior to SLE-associated autoantibodies.
Figure 3.

IFN-associated soluble mediators increase as individuals move toward SLE classification and correlate with number of autoantibodies. Increase in number of serum IFN-associated mediators by time before SLE classification in 55 cases vs. matched healthy controls, p = 0.0002 by Wilcoxon matched-pairs test (A). Time of SLE classification (dotted line, down arrow) is shown. Within-individual correlations of IFN-associated serum soluble mediators vs. number of positive serum DNA- and RNA-associated autoantibodies are shown, as described in Methods (B). Serum soluble mediators IFN-γ (C), IP-10 (D), BLyS (E), and MIG (F) in cases by autoantibody status and in matched controls (Ctls) are shown (mean ± SEM). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Kruskal-Wallis test with Dunn’s multiple comparison (Cases) or by Mann-Whitney (Cases vs Ctls).
Type II IFN mediators and autoantibodies precede elevated IFN-α activity and SLE classification
The above findings, largely from univariate analyses of aggregate samples, suggest the following timing of events prior to SLE classification: dysregulation of select type II IFN mediators, followed by the detection and accrual of autoantibodies, and finally the elevation of serum IFN-α activity. To evaluate this temporal relationship directly, on a per-patient basis, we performed path analysis using data from three longitudinal samples per individual to determine the relative timing of increased levels of IFN-associated soluble mediators, autoantibody positivity, and elevated IFN-α activity.
We considered three ordinal relationships between autoantibody positivity and IFN-α activity: IFN-α activity before autoantibody positivity, autoantibody positivity before IFN-α activity, and IFN-α activity concurrent with autoantibody positivity (figure 4A). Each model was ranked based on three fit indices. In the Akaike’s information criterion (AIC) and Root Mean Square Error of Approximation (RMSEA) goodness-of-fit indices, lower values (AIC, no fixed range; RMSEA, range 0–1) indicate a better model fit.[20] In the lack-of-fit (LOF) chi-square (χ2) index, larger χ2 values (smaller p-values) indicate a less parsimonious model and/or poorer fit.[21] We found that autoantibody positivity preceding IFN-α activity was the top ranking model by all fit indices for each autoantibody specificity except anti-dsDNA (figure 4B, highlighted), consistent with the growth curve models (figure 2C–D). Anti-dsDNA positivity preceded IFN-α activity per the AIC fit index and coincided with IFN-α activity per RMSEA and chi-square model fit indices (figure 4B), consistent with the growth curve models showing anti-dsDNA positivity coinciding with increased IFN-α activity (figure 2D). Moreover, in 84% of individuals positive for both IFN-α activity and autoantibodies, at least one autoantibody specificity appeared prior to or concurrent with elevation of IFN-α activity (supplementary table 3).
Figure 4.

IFN-associated mediators are elevated prior to or concurrent with autoantibody positivity, which precedes increases in IFN-α activity in SLE patients. (A) Abbreviated path analysis diagrams showing posited temporal relationships tested in 3 longitudinal samples/patient (Sample 1, Sample 2, Sample 3): positive IFN-α activity preceding autoantibody (AutoAb) detection, AutoAb detection preceding positive IFN-α activity, or the simultaneous detection of both AutoAb and IFN-α activity positivity. Results comparing the timing of positive IFN-α activity vs. the detection of lupus-associated antibody specificities are shown (B). (C) Abbreviated path analysis diagrams showing posited temporal relationships tested in 3 longitudinal samples/patient (Sample 1, Sample 2, Sample 3): positive soluble mediator preceding preceding AutoAb detection or IFN-α activity, AutoAb detection or IFN-α activity preceding soluble mediator, or the simultaneous detection of both soluble mediator and AutoAb or IFN-α activity. Results comparing the timing of positive soluble mediator vs. the detection of anti-Ro/SSA and IFN-α activity are shown (D). The highest model rank (highlighted) was determined by the lowest values of the goodness-of-fit model indices AIC (Akaike's information criterion) and RMSEA (Root Mean Square Error of Approximation) and the highest p-value for the lack-of-fit (LOF) chi-square (χ2) index. dsDNA=double stranded DNA
Next, we considered path analysis models similarly positing that IFN-associated soluble mediators preceded, followed, or appeared concurrently with autoantibody positivity (specifically anti-Ro/SSA, the first autoantibody to be detected, figure 2C and figure 4B) and elevated IFN-α activity (figure 4C). We found that type II IFN mediators, IFN-γ and IP-10, were more likely to be elevated concurrent with or prior to anti-Ro/SSA positivity, respectively, and that both were elevated prior to the increase in IFN-α activity. In contrast, BLyS was more likely to be elevated after both anti-Ro/SSA positivity and elevated IFN-α activity (figure 4D). These findings corroborate the univariate analyses showing elevated IFN-γ and IP-10 in preclinical cases prior to autoantibody positivity, and continued increases of these mediators (with later BLyS elevation) coinciding with autoantibody accrual and SLE classification (figure 3; supplementary figure 2). Further supporting these results, most individuals were positive for IFN-γ (98%) and IP-10 (87%), along with MCP-3 (92%), prior to or concurrent with autoantibody positivity, while BLyS positivity occurred concurrent with or after autoantibody detection and serum IFN-α activity (87%; supplementary table 4).
Type II IFN mediators and autoantibodies predict elevated IFN-α activity and SLE classification
Because elevated serum IFN-α activity occurs proximal to the time of SLE classification, we applied Analysis of Covariance (ANCOVA) analyses to determine the best predictors of IFN-α activity elevation. We evaluated whether IFN-associated soluble mediators, autoantibody specificities, gender, and/or ethnicity might predict increased serum IFN-α activity and SLE classification. We found that the number of autoantibody specificities (p<0.001), female gender (p=0.010) and number of positive IFN-associated mediators (p=0.038) were significant predictors of increased IFN-α activity (table 1).
Table 1.
Gender, Number of Positive Autoantibodies and Number of IFN-associated soluble mediators predict increase in IFN-α activitya
| SS | Df | MS | F | p-value | |
|---|---|---|---|---|---|
| No. of Pos. Autoantibodies | 225.69 | 1 | 225.69 | 40.39 | <0.001 |
| Gender | 37.57 | 1 | 37.57 | 6.72 | 0.010 |
| No. of Pos. IFN mediators | 24.38 | 1 | 24.38 | 4.36 | 0.038 |
| Gender:Ethnicity | 24.27 | 2 | 12.13 | 2.17 | 0.118 |
| Ethnicity | 19.30 | 3 | 6.43 | 1.15 | 0.331 |
| Residuals | 855 | 153 | 5.59 | ||
SS= sum of squares; Df= degrees of freedom; MS = mean squares
Modeling performed by ANCOVA
Since several IFN-associated mediators were found to be elevated prior to or concurrent with the accumulation of autoantibody specificities and serum IFN-α activity before SLE classification, we sought to determine which pre-clinical factors best predicted SLE development. We used random forest modeling [22] to evaluate the following variables as possible predictors of future SLE classification: the presence of SLE-associated autoantibodies, elevated serum IFN-α activity, and elevated levels of IFN-associated mediators. Interestingly, IFN-α activity was eliminated as a predictor variable because it did not make a significant, independent contribution to the prediction of SLE classification. Future SLE classification was best discerned by the concentrations of IFN-γ, MCP-3, anti-chromatin, and anti-spliceosomal autoantibodies, even >4 years before classification (93% accuracy, OOB error = 0.07; table 2). Thus, a combination of type II IFN mediators and autoantibodies accurately predicts SLE classification years in advance. Further, these findings suggest that early dysregulation of type II IFN mediators interact with accumulation of autoantibody specificities, leading to elevated serum IFN-α activity and ultimately resulting in clinical disease and SLE classification.
Table 2.
IFN-associated Mediators and Select Autoantibody Specificities Predict Transition to SLEa
| Sample Time | Sensitivity (95% CI) |
Specificity (95% CI) |
PPV (95% CI) |
NPV (95% CI) |
OOB error |
|---|---|---|---|---|---|
| > 4 years before Classificationb | 0.94 | 1.00 | 1.00 | 0.95 | 0.07 |
| > 2 years before Classificationc | 0.96 | 0.98 | 0.98 | 0.97 | 0.03 |
| Before Classificationd | 0.95 | 0.99 | 0.98 | 0.97 | 0.05 |
| After Classificatione | 1.00 | 1.00 | 1.00 | 1 | 0.01 |
| All Samplesf | 0.95 | 0.99 | 0.98 | 0.97 | 0.03 |
Modeling performed by Random Forest analysis
IFN-γ, MCP-3, anti-chromatin, anti-Sm, low levels of BLyS
IFN-γ, MCP-3, anti-chromatin, anti-Sm, anti-RNP, anti-Ro/SSA, anti-dsDNA
IFN-γ, MCP-3, anti-chromatin, anti-RNP, anti-Ro/SSA, anti-SmRNP
MCP-3, IFN-γ, anti-chromatin, anti-SmRNP, anti-RNP, anti-Sm, anti-Ro/SSA
IFN-γ, MCP-3, anti-chromatin, anti-RNP, anti-Ro/SSA
DISCUSSION
Understanding early lupus pathogenesis is critical for assessing and minimizing the risk of transitioning to SLE. SLE is fundamentally a disease of immune dysregulation. Utilizing a unique resource of well characterized, longitudinal serum samples collected prior to and at/after SLE classification, we found that, like autoantibody positivity, IFN-α activity and elevated IFN-associated soluble mediators arise months or years before disease classification.
Type I IFN (IFN-α)[5] and BLyS[8] have been linked to SLE pathogenesis; our data show increased IFN-α activity and BLyS levels shortly before SLE classification, which may denote a turning point in pathogenesis, where positive feed-forward mechanisms amplify immune dysregulation past the threshold for inevitable transition to classified SLE. Elevated IFN-α activity and BLyS levels may arise from early dysregulation of type II IFN mediators interacting with the accumulation of lupus-associated autoantibody specificities. This temporal relationship is supported across multiple multivariate models, including growth curve models, path analysis, and analysis of covariance, placing autoantibody positivity before or near elevated IFN-α activity. Indeed, in our investigation, the number of positive autoantibodies was the most significant (p<0.001) predictor of increased IFN-α activity in cases. A recent study demonstrated IFN-α activity associated with autoantibody positivity, a small study (n=24) showed exacerbated type I IFN signatures in incomplete lupus erythematosus patients whose autoantibody specificities had already class-switched to the pathogenic IgG isotype,[23] and a number of studies have noted the ability of autoantibody-containing immune complexes to drive type I IFN activation.[24–27] While IFN-α activity is clearly important in the transition to classified SLE, it may not be a leading factor for the initial development of lupus-associated autoantibody specificities. Indeed, roughly 25% of the patients in the current study did not have positive IFN-α activity, further highlighting the heterogeneous nature of SLE.
Preclinical detection of autoantibodies prior to increases in IFN-α activity and the modest clinical benefit from IFN-α blockade in recent clinical trials[9] infer that other forms of immune dysregulation accompany IFN-α activity to allow for clinical disease. Previous evidence in lupus-like animal models and SLE patients suggests that amplified crosstalk between innate cells and lymphocytes leads to breaks in tolerance, enabling the activation and persistence of autoreactive B cells.[28] IFN-γ, a key mediator of such crosstalk, facilitates autoantibody production by modulating TLR regulation, antigen presentation, and lymphocyte recruitment to germinal centers,[29] where IFN-γ promotes IgG class switching and pathogenic autoantibody production.[30] Our temporal analyses show elevated levels of IFN-γ and the chemokines IP-10 and MCP-3 in preclinical SLE patients prior to IFN-α activity and to positivity for most autoantibodies. Moreover, IFN-γ and MCP-3 consistently determine transition to classified SLE, even >4 years before disease classification, supporting an early role for these IFN-associated mediators in SLE pathogenesis. In longitudinal studies of patients with undifferentiated connective tissue disease, increased levels of IFN-γ signal the transition from benign to pathological disease,[31] and those who transition to SLE accumulate lupus-associated autoantibodies.[32] Finally, IFN-γ can drive the production of IFN-α[11] and BLyS,[12] thereby promoting inflammation and further enhancing B cell activation and (auto)antibody accumulation.[30, 33] Serum BLyS levels were not significantly elevated in cases compared to matched controls until shortly before SLE classification and after increases in serum IFN-γ, autoantibody positivity, and serum IFN-α activity.
This study provides critical new information to help identify ANA positive individuals at the highest risk of transition to SLE. Through the measurement of systemic soluble mediators indicative of ongoing inflammation when detected in the periphery, including IFN-γ, IP-10, MCP-3, and BLyS, in addition to the detection of DNA- and RNA-protein binding autoantibodies, we can identify individuals who may need rheumatology referral or closer monitoring, findings further supported by similar perturbations in autoantibodies and cytokines in pre-clinical rheumatoid arthritis.[34, 35] In addition, results from this study will help identify individuals at significantly increased risk of SLE development with a high level of specificity, sensitivity, and predictive accuracy, thereby providing critical information needed to design and implement prevention trials. Such prospective clinical trials would allow for parallel procurement of detailed clinical assessment and biologic specimens to address a current limitation of this study. By understanding that IFN-associated pathway dysregulation occurs years prior to clinical symptoms with further increases closer to the time of clinical disease onset, we may be able to select and test potential early therapeutic agents, such as hydroxychloroquine,[36–38] for individuals before irreversible organ damage has occurred. This may be potentially beneficial for those patients in whom organ damage occurs by the time they reach disease classification, as early damage has been shown to be predictive of further damage, co-morbidities, and early mortality.[39–41]
Supplementary Material
Acknowledgments
The authors thank Krista Bean, Jourdan Anderson, Tim Gross, and Wade DeJager for technical assistance, as well as Rebecka Bourn, PhD and Angela, Andersen, PhD for editorial assistance.
FUNDING
Research reported in this publication was supported by the National Institute of Allergy, Immunology and Infectious Diseases, Office of Research on Women’s Health, National Institute of General Medical Sciences, and the National Institute of Arthritis, Musculoskeletal and Skin Diseases under award numbers U01AI101934, U19AI082714, U54GM104938, P30GM103510, P30AR053483, and S10RR026735. This material is also the result of work supported with resources and the use of facilities through the Department of Veterans Affairs. Additional support was provided by the National Institute of Allergy, Immunology and Infectious Diseases and National Institute of Arthritis, Musculoskeletal and Skin Diseases under award numbers AI071651 and AR060861 (TBN). JBH would like to acknowledge support from the US Department of Veterans Affairs and NIH grants U01HG006828, UL1TR000077, R37AI024717, R21AI103980, P01AI083194, and P01AI049084. This publication is the sole responsibility of the authors and does not represent the views of the National Institutes of Health or the Department of Veterans Affairs. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of the Army, US Armed Forces Department of Defense, or the US Government.
Footnotes
CONTRIBUTIONS
MEM, TBN, GCT, MPK, JBH, and JAJ designed the study. MEM, JRA, TG, JMR, BFB, TBN, and JAJ participated in data acquisition. MEM, RL, JRA, DAF, JMR, YDZ, TBN, JBH and JAJ participated in data analysis. All authors assisted with the development of the manuscript and gave final approval for publication. MEM, RL, JMR, and JAJ had full access to data for the study. JAJ had the final responsibility for the decision to submit for publication.
COMPETING INTERESTS
None.
PATIENT CONSENT
This research falls under exempt category 4 of the U.S. Department of Health and Human Services (DHHS) regulations 45 CFR 46.101(b). The research involves the collection or study of existing data, documents, records, and specimens. The information is recorded by the DODSR and the investigator in such a manner that subjects cannot be identified, directly or through identifiers linked to the subjects.
ETHICS APPROVAL
Experiments were performed in accordance with the Helsinki Declaration and approved by the Institutional Review Boards of the Oklahoma Medical Research Foundation and the Walter Reed National Military Medical Center.
REFERENCES
- 1.Stahl Hallengren C, Nived O, Sturfelt G. Outcome of incomplete systemic lupus erythematosus after 10 years. Lupus. 2004;13(2):85–88. doi: 10.1191/0961203304lu477oa. [DOI] [PubMed] [Google Scholar]
- 2.Heinlen LD, McClain MT, Merrill J, et al. Clinical criteria for systemic lupus erythematosus precede diagnosis, and associated autoantibodies are present before clinical symptoms. Arthritis Rheum. 2007 Jul;56(7):2344–2351. doi: 10.1002/art.22665. [DOI] [PubMed] [Google Scholar]
- 3.Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003 Oct 16;349(16):1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 4.Satoh M, Chan EK, Ho LA, et al. Prevalence and sociodemographic correlates of antinuclear antibodies in the United States. Arthritis Rheum. 2012 Jul;64(7):2319–2327. doi: 10.1002/art.34380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. J Interferon Cytokine Res. 2011 Dec;31(12):887–892. doi: 10.1089/jir.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weckerle CE, Franek BS, Kelly JA, et al. Network analysis of associations between serum interferon-alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011 Apr;63(4):1044–1053. doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez P, Scheel-Toellner D, Rodriguez-Carrio J, et al. Interferon-alpha-induced B-lymphocyte stimulator expression and mobilization in healthy and systemic lupus erthymatosus monocytes. Rheumatology (Oxford) 2014 Dec;53(12):2249–2258. doi: 10.1093/rheumatology/keu249. [DOI] [PubMed] [Google Scholar]
- 8.Vincent FB, Morand EF, Schneider P, et al. The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol. 2014 Mar 11; doi: 10.1038/nrrheum.2014.33. [DOI] [PubMed] [Google Scholar]
- 9.Lauwerys BR, Ducreux J, Houssiau FA. Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford) 2014 Aug;53(8):1369–1376. doi: 10.1093/rheumatology/ket403. [DOI] [PubMed] [Google Scholar]
- 10.Moser KL, Kelly JA, Lessard CJ, et al. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009 Jul;10(5):373–379. doi: 10.1038/gene.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder K, Hertzog PJ, Ravasi T, et al. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004 Feb;75(2):163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 12.Harigai M, Kawamoto M, Hara M, et al. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol. 2008 Aug 1;181(3):2211–2219. doi: 10.4049/jimmunol.181.3.2211. [DOI] [PubMed] [Google Scholar]
- 13.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol. 2013 Feb;13(2):75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 14.Chen P, Vu T, Narayanan A, et al. Pharmacokinetic and Pharmacodynamic Relationship of AMG 811, An Anti-IFN-gamma IgG Monoclonal Antibody, in Patients with Systemic Lupus Erythematosus. Pharmaceutical research. 2014 Sep 12; doi: 10.1007/s11095-014-1492-2. [DOI] [PubMed] [Google Scholar]
- 15.Yellin M, Paliienko I, Balanescu A, et al. A phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2012 Jun;64(6):1730–1739. doi: 10.1002/art.34330. [DOI] [PubMed] [Google Scholar]
- 16.Bruner BF, Guthridge JM, Lu R, et al. Comparison of autoantibody specificities between traditional and bead-based assays in a large, diverse collection of patients with systemic lupus erythematosus and family members. Arthritis Rheum. 2012 Nov;64(11):3677–3686. doi: 10.1002/art.34651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munroe ME, Vista ES, Guthridge JM, et al. Pro-inflammatory adaptive cytokines and shed tumor necrosis factor receptors are elevated preceding systemic lupus erythematosus disease flare. Arthritis & rheumatology. 2014 Jul;66(7):1888–1899. doi: 10.1002/art.38573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niewold TB, Hua J, Lehman TJ, et al. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007 Sep;8(6):492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hua J, Kirou K, Lee C, et al. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006 Jun;54(6):1906–1916. doi: 10.1002/art.21890. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto T, Mizoguchi Y, Kaneno H, et al. Serum immunoglobulin G subclass levels and estimated clinical severity caused by possible influenza A (H1N1) pdm 2009 infection. Journal of infection and chemotherapy : official journal of the Japan Society of Chemotherapy. 2013 Oct;19(5):833–842. doi: 10.1007/s10156-013-0570-4. [DOI] [PubMed] [Google Scholar]
- 21.Hooper D, Coughlan J, Mullen MR. Structural Equation Modelling: Guidelines for Determining Model Fit. The Electronic Journal of Business Research Methods. 2008;6(1):53–60. [Google Scholar]
- 22.Genuer R, Poggi JM, Tuleau-Malot C. Variable selection using random forests. Pattern Recogn Lett. 2010 Oct 15;31(14):2225–2236. [Google Scholar]
- 23.Li QZ, Zhou J, Lian Y, et al. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clin Exp Immunol. 2010 Mar;159(3):281–291. doi: 10.1111/j.1365-2249.2009.04057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eloranta ML, Barbasso Helmers S, Ulfgren AK, et al. A possible mechanism for endogenous activation of the type I interferon system in myositis patients with anti-Jo-1 or anti-Ro 52/anti-Ro 60 autoantibodies. Arthritis Rheum. 2007 Sep;56(9):3112–3124. doi: 10.1002/art.22860. [DOI] [PubMed] [Google Scholar]
- 25.Mathsson L, Ahlin E, Sjowall C, et al. Cytokine induction by circulating immune complexes and signs of in-vivo complement activation in systemic lupus erythematosus are associated with the occurrence of anti-Sjogren's syndrome A antibodies. Clin Exp Immunol. 2007 Mar;147(3):513–520. doi: 10.1111/j.1365-2249.2006.03313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savarese E, Chae OW, Trowitzsch S, et al. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood. 2006 Apr 15;107(8):3229–3234. doi: 10.1182/blood-2005-07-2650. [DOI] [PubMed] [Google Scholar]
- 27.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007 May;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 28.Kil LP, Hendriks RW. Aberrant B cell selection and activation in systemic lupus erythematosus. International reviews of immunology. 2013 Aug;32(4):445–470. doi: 10.3109/08830185.2013.786712. [DOI] [PubMed] [Google Scholar]
- 29.Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus-an update. Curr Opin Immunol. 2012 Dec;24(6):651–657. doi: 10.1016/j.coi.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A. 2002 Apr 16;99(8):5545–5550. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szodoray P, Nakken B, Barath S, et al. Progressive divergent shifts in natural and induced T-regulatory cells signify the transition from undifferentiated to definitive connective tissue disease. Int Immunol. 2008;20(8):971–979. doi: 10.1093/intimm/dxn056. [DOI] [PubMed] [Google Scholar]
- 32.Mosca M, Neri R, Bencivelli W, et al. Undifferentiated connective tissue disease: analysis of 83 patients with a minimum followup of 5 years. J Rheumatol. 2002 Nov;29(11):2345–2349. [PubMed] [Google Scholar]
- 33.Kiefer K, Oropallo MA, Cancro MP, et al. Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol. 2012 May;90(5):498–504. doi: 10.1038/icb.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deane KD, O'Donnell CI, Hueber W, et al. The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. 2010 Nov;62(11):3161–3172. doi: 10.1002/art.27638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes-Austin JM, Deane KD, Derber LA, et al. Multiple cytokines and chemokines are associated with rheumatoid arthritis-related autoimmunity in first-degree relatives without rheumatoid arthritis: Studies of the Aetiology of Rheumatoid Arthritis (SERA) Ann Rheum Dis. 2013 Jun;72(6):901–907. doi: 10.1136/annrheumdis-2012-201505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costedoat-Chalumeau N, Dunogue B, Morel N, et al. Hydroxychloroquine: a multifaceted treatment in lupus. Presse medicale. 2014 Jun;43(6 Pt 2):e167–e180. doi: 10.1016/j.lpm.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Akhavan PS, Su J, Lou W, et al. The early protective effect of hydroxychloroquine on the risk of cumulative damage in patients with systemic lupus erythematosus. J Rheumatol. 2013 Jun;40(6):831–841. doi: 10.3899/jrheum.120572. [DOI] [PubMed] [Google Scholar]
- 38.James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus. 2007;16(6):401–409. doi: 10.1177/0961203307078579. [DOI] [PubMed] [Google Scholar]
- 39.Rahman P, Gladman DD, Urowitz MB, et al. Early damage as measured by the SLICC/ACR damage index is a predictor of mortality in systemic lupus erythematosus. Lupus. 2001;10(2):93–96. doi: 10.1191/096120301670679959. [DOI] [PubMed] [Google Scholar]
- 40.Urowitz MB, Gladman DD, Ibanez D, et al. Evolution of disease burden over five years in a multicenter inception systemic lupus erythematosus cohort. Arthritis Care Res. 2012 Jan;64(1):132–137. doi: 10.1002/acr.20648. [DOI] [PubMed] [Google Scholar]
- 41.Urowitz MB, Gladman DD, Ibanez D, et al. American College of Rheumatology criteria at inception, and accrual over 5 years in the SLICC inception cohort. J Rheumatol. 2014 May;41(5):875–880. doi: 10.3899/jrheum.130704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.