

Summary
In this study we have explored the pathogenesis of the hepatic alterations which occur in diabetes and the modulation of these complications by the combination of metformin adjunct treatment and insulin monotherapy. For this purpose, diabetic rats were treated with insulin (DM + Ins) or metformin plus insulin (DM + Met + Ins). Biochemical and cardiometabolic parameters were analysed by spectrophotometry. Intravital microscopy was used to study the hepatic microcirculation. In the liver tissue, real‐time PCR was used to analyse oxidative stress enzymes, inflammatory markers and receptors for advanced glycation end products (AGE) (RAGE) gene expression. Lipid peroxidation was assessed by thiobarbituric acid reactive species (TBARs) analyses. AGE deposition and RAGE protein expression were studied by fluorescence spectrophotometry and Western blot respectively. Body weight, %HbA1c, urea, total proteins and oxidative stress parameters were found to be similarly improved by insulin or Met + Ins treatments. On the other hand, Met + Ins treatment showed a more pronounced effect on fasting blood glucose level than insulin monotherapy. Fructosamine, uric acid, creatinine, albumin and amylase levels and daily insulin dose requirements were found to be only improved by the combined Met + Ins treatment. Liver, renal and pancreatic dysfunction markers were found to be more positively affected by metformin adjunct therapy when compared to insulin treatment. Liver microcirculation damage was found to be completely protected by Met + Ins treatment, while insulin monotherapy showed no effect. Our results suggest that oxidative stress, microcirculatory damage and glycated proteins could be involved in the aetiology of liver disease due to diabetes. Additionally, metformin adjunct treatment improved systemic and liver injury in induced diabetes and showed a more pronounced effect than insulin monotherapy.
Keywords: AGE, inflammation, liver, metformin, Microcirculation, oxidative stress, RAGE, Type 1 diabetes
Type 1 diabetes (T1D) is accompanied by long‐term macro‐ and microvascular complications, including retinopathy, nephropathy, neuropathy and cardiovascular disease (CVD) (de Ferranti et al. 2014). Although the pathophysiological basis of T1D complications remains uncertain, hyperglycaemia, oxidative stress and inflammation appear to play a central role in the process (Forbes & Cooper 2013). Despite improvements in technology and glycaemic control, adolescents and adults with T1D present increasing rates of insulin resistance and obesity (Cleland et al. 2013). Adding complexity to the T1D pathophysiology, several recent studies have demonstrated a link between T1D and liver disease (Targher et al. 2010, 2012; Regnell & Lernmark 2011). Moreover, liver alterations have been suggested to be associated with other diabetic complications, such as chronic kidney disease (Targher et al. 2014).
Intensive insulin treatment was shown to reduce the rates of microvascular complications in T1D in the Diabetes Control and Complication Trial and was later associated with a lower all‐cause mortality rate (DCCT, 1993; Orchard et al. 2015); however, achieving and maintaining a tight glycaemic control using standard insulin therapy requires a high level of discipline and is associated with an increasing risk of hypoglycaemia, weight gain and, in some patients, worse CVD and metabolic profiles (Purnell et al. 1998; Sibley et al. 2003; Devries et al. 2004). Based on that, several studies proposed the use of metformin adjunct therapy to overcome the drawbacks of insulin monotherapy both in patients with T1D and in patients with type 2 diabetes (T2D) (Lund et al. 2008; (Janssen et al. 1991; Sarnblad et al. 2003). Metformin is a low‐cost and well‐established oral glucose‐lowering agent that is a first‐line oral pharmacotherapy for T2D. The main metabolic effect of metformin is the increase in the sensitivity of peripheral tissues and the liver to insulin (Giannarelli et al. 2003). Pharmacological treatment with metformin in T2D is also associated with a reduction in hepatic glucose production, fasting plasma glucose, HbA1c level, weight gain, serum triglycerides and pro‐inflammatory profiles (Wulffele et al. 2004; Isoda et al. 2006; Bailey 2008). Metformin adjunct therapy was associated with a significant reduction in the incidence of myocardial infarction in people with T2D in the UK Prospective Diabetes Study (UKPDS), a result that was sustained 10 years after the end of randomization (Holman et al. 2008). Importantly, the activation of the energy‐regulating enzyme adenosine monophosphate‐activated protein kinase (AMPK), mainly in the muscle and the liver, is considered a major mode of action of metformin (Schimmack et al. 2006). The activation of AMPK suggests that metformin may have a direct effect on the cardiovascular system that is independent of the glucose‐lowering effect. In an animal model of diabetes, metformin has been shown to improve aortic function by a mechanism dependent on the modulation of oxidative stress, inflammation and advanced glycation end products (AGE) pathway (Sena et al. 2011). Additionally, metformin has been shown to restore the endothelial function of neonatal streptozotocin‐induced diabetic rats in response to endothelium‐dependent and endothelium‐independent agents (Sartoretto et al. 2005). Metformin also reduces hepatic and circulating lipids in obese mice, ultimately leading to fatty liver disease reversal (Lin et al. 2000).
The treatment of T1D (insulin‐dependent diabetes mellitus) patients with metformin has been shown to improve insulin resistance (Gin et al. 1985; Moon et al. 2007) and lower platelet aggregation (Gin et al. 1989); however, it has shown controversial effects on circulating lipid levels (Schatz et al. 1975; Janssen et al. 1991; Kearney et al. 2008; Lund et al. 2009), percentage of glycated haemoglobin (%HbA1c) (Meyer et al. 2002; Sarnblad et al. 2003; Vella et al. 2010), fasting blood glucose level (Hamilton et al. 2003; Khan et al. 2006; Burchardt et al. 2013) and the daily insulin dose requirements (Schatz et al. 1975; Janssen et al. 1991; Meyer et al. 2002; Sarnblad et al. 2003; Khan et al. 2006; Vella et al. 2010). The use of metformin in patients with T1D in clinic is still a matter of debate, although some studies pointed out its beneficial effects on metabolic control, especially in overweight or obese subjects (Meyer et al. 2002; Hamilton et al. 2003; Burchardt et al. 2013).
Thus, the elucidation of the liver alterations due to diabetes and the usefulness of metformin in this complication would lead to further advances in the management of patients with T1D. To further investigate these issues, we studied several aspects of liver function including biochemical, oxidative stress, inflammatory, microcirculatory and AGE pathway parameters. Additionally, we tested the effect of metformin adjunct treatment on liver alterations associated with diabetes.
Methods
Animals and experimental protocol
Fifty‐five male Wistar rats from the central animal facilities of the Oswaldo Cruz Foundation, Brazil, were used in the study. The rats (12 weeks, 200–250 g) were housed in standard cages in a temperature‐controlled room (22 ± 1°C) with a 12‐h light/dark cycle. The experimental model of diabetes was induced in fifty animals by 4 weeks of feeding with a high‐fat diet (HFD) plus the administration of a single low dose of streptozotocin (STZ) (35 mg/kg, intraperitoneal injection) in 0.1 m citrate buffer (pH 4.5) as previously described in Srinivasan et al. (2005). Diabetes was confirmed by fasting blood glucose levels >300 mg/dl measured using an automatic glucose monitor (One Touch Ultra2, Johnson & Johnson Medical S.A., Buenos Aires, Argentina). The high‐fat diet consisted of a standard rat diet modified to contain 30% fat, 56% carbohydrate and 14% protein (% g) (Nascimento et al. 2013). The non‐diabetic control (CTL) animals received an intraperitoneal administration of buffer alone and standard rat diet (n = 20). The diabetic animals were further divided into three groups: untreated animals, which received vehicle for 2 weeks (DM, n = 20), and treated animals, which received only insulin (2‐6U/day) (DM + Ins, n = 15) or metformin (300 mg/Kg/day) plus insulin (2–6U/day) for 2 weeks (DM + Met + Ins, n = 15). Insulin was used in association with metformin as the metformin itself is not able to induce insulin production. All of the tissues and samples were collected and analyses were carried out at the end of the experimental protocol.
Ethical approval
All of the experimental procedures were conducted in accordance with the internationally accepted principles for the Care and Use of Laboratory Animals and were approved by the Oswaldo Cruz Foundation Animal Welfare Committee (CEUA licence # LW‐52/13).
Biochemical parameters
Fructosamine, %HbA1c, cholesterol, triglycerides, total proteins, bilirubin, urea, uric acid, albumin and vitamin D and the enzyme activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), gamma‐glutamyl transpeptidase (GGT) and amylase were analysed using commercial kits with the Cobas c system (Roche Molecular Biochemicals, Indianapolis, IN, USA).
Blood pressure
Systolic blood pressure (SBP) was measured in each rat at the end of the experimental protocol by non‐invasive measurement of the tail pressure (BP‐2000, Visitech Blood Pressure Analysis System, Visitech Systems Inc., Apex, NC, USA).
Insulin quantification
The plasma insulin level was measured using an enzyme‐linked immunosorbent assay (ELISA) kit (Millipore, Bedford, MA, USA) following the manufacturer's guidelines.
RT‐PCR
Total RNA was isolated from the liver using the RNeasy Mini Kit (Qiagen, Hilden, Germany). cDNA was synthesized with a high‐capacity cDNA reverse transcription Kit (Applied Biosystems, Foster City, CA, USA) from 1 μg of total RNA in a final volume of 20 μl. The primers used for PCR amplification were as follows: NADPH oxidase p22 subunit (N22), forward 5′‐GGTGAGCAGTGGACTCCCATT‐3′ and reverse 5′‐TGGTAGGTGGCTGCTTGATG ‐3′; NADPH oxidase p47 subunit (N47), forward 5′‐GTGAAGCCATCGAGGTCATTC‐3′ and reverse 5′‐CCCGCGGCTTCTAATCTGT‐3′; CuZn superoxide dismutase (SOD), forward 5′‐CGGCTTCTGTCGTCTCCT‐3′ and reverse 5′ GTTCACCGCTTGCCTTCT‐ ‐3′; catalase (CAT), forward 5′‐ ACTCAGGTGCGGACATTC ‐3′ and reverse 5′‐ GGAGTTGTACTGGTCCAGAAGAGCC ‐3′; RAGE (RAGE), forward 5′‐ CAGGGTCACAGAAACCGG ‐3′ and reverse 5′‐ ATTCAGCTCTGCACGTTCCT ‐3′ and beta‐actin (β‐a), forward 5′‐ CCACCCGCGAGTACAACCTTCTT ‐3′ and reverse 5′‐ GAAGCCGGCCTTGCACATGCC ‐3′. Real‐time PCR was performed with the power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's instructions. All of the PCR amplifications were carried out using a 7500 real‐time PCR system (Applied Biosystems). The expression of target genes was normalized to the expression of β‐actin, and the ∆∆Ct method was used for the determination of gene expression.
Thiobarbituric acid reactive species (TBARs)
Lipid peroxidation in the liver was assessed by measuring malondialdehyde (MDA) concentrations using thiobarbituric acid reactive species (TBARs) analyses (Draper & Hadley 1990). The livers were homogenized in cold phosphate buffer, pH 7.4 with BHT (final concentration of 0.2%). The samples (0.5 ml) were mixed with an equal volume of 0.67% of thiobarbituric acid (Sigma Chemical Co., St. Louis, MO, USA) and then heated at 96°C for 30 min. TBARs were determined by measuring the absorbance at 535 nm. The results are expressed as malondialdehyde (MDA, ε = 1.56 × 105 M−1 cm−1) (Draper & Hadley 1990).
Western blot
The liver tissue was homogenized in lysis buffer (20 mm Trizma/137 mm NaCl/10% glycerol/1% Nonidet P‐40/2 mm EDTA) and centrifuged for 45 min at 14,000 g at 4°C. The protein content was assayed by the bicinchoninic acid method with bovine serum albumin (BSA) as the standard (BCA Protein Assay Kit, Thermo Scientific, Waltham, MA, USA). Then, 30 μg of protein per lane was separated on a 12% sodium dodecyl sulphate gel and transferred to a nitrocellulose membrane (Bio‐Rad Laboratories, Munich, Germany). After blocking with 5% BSA (Sigma‐Aldrich, St. Louis, MO, USA), the membranes were incubated overnight at 4°C with a goat polyclonal anti‐RAGE antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by a secondary RDye 680RD Donkey‐anti‐Goat antibody (1:15,000; LI‐COR Biosciences, Cambridge, UK). The bound complex was detected using the Odyssey Infrared Imaging System (Li‐Cor; Lincoln, NE, USA). The images were analysed using the Image Studio Lite Software version 4.0.21 (Li‐Cor) to obtain the integrated intensities and normalized with respect to the GAPDH signals (mouse monoclonal anti‐GAPDH antibody, 1:30,000; Fitzgerald Industries International, Acton, MA, USA) followed by a secondary IRDye 800CW Goat‐anti‐Mouse antibody (1:20,000; LI‐COR Biosciences).
Quantification of advanced glycation end products (AGEs)
Liver concentrations of fluorescent AGEs were determined by the method of Nakayama et al. (1993). Briefly, the fluorescence of AGEs in the liver samples was measured at an emission wavelength of 440 nm and at an excitation wavelength of 370 nm against a blank of 0.1 N NaOH solution on a SpectraMax M5 ELISA Microplate Reader (Molecular Devices, Acton, MA, USA). A native BSA preparation (1 mg/ml of 0.1 N NaOH) was used as a reference, and its fluorescent intensity was defined as one unit of fluorescence. The fluorescence values of the samples were measured at a protein concentration of 1 mg/ml and are expressed in AU compared with the native BSA preparation.
Intravital microscopy
For intravital microscopy of the liver, overnight fasted animals were anesthetized via intraperitoneal (i.p.) injection with ketamine (100 mg/kg) and xylazine (10 mg/kg). A midline and left subcostal incision was made to exteriorize the liver. The hepatic ligaments were dissected and the intestine was covered with a saline‐soaked gauze to minimize the tissue dehydration. The left liver lobe was then exteriorized and placed on a glass disc, and then the glass disc was covered with a glass slide for microcirculation analyses. With the use of a 10× ocular and 10× objective (Olympus BX150WI; Center Valley, PA, USA), images were displayed on a television monitor and recorded by a digital video recorder (DP73; Olympus, MA, USA) for offline analysis with the CellSens standard 1.9 software program (Olympus, Waltham, MA, USA). The leucocyte–endothelial interaction was evaluated by counting the number of labelled leucocytes (0.3 mg/kg rhodamine 6G, i.v.) rolling or adhering to sinusoids and postsinusoidal venules. Rolling leucocytes were defined as white blood cells with a slower velocity than the erythrocytes and with a detectable rolling motion. Leucocytes that remained stationary on the sinusoidal or venular endothelium for 30 s or longer were considered adherent cells. Rolling and adherent cells were counted in a 170‐μm2 area comprising sinusoids and postsinusoidal venules.
Catalase activity
The livers were collected and homogenized in KPE buffer (100 mm + EDTA 5 mm) in the proportion of 1:9 w/v liver/buffer. The samples were centrifuged at 600 g for 10 min at 4°C, and the supernatant was used in 1:10 dilution for the estimation of catalase activity. For this purpose, the enzymatic decomposition of hydrogen peroxide was followed continuously at 240 nm. Briefly, 1 μl of samples was added to a microplate, and 97 μl of 0.16% of hydrogen peroxide in PBS (pH 7.0) was used as the substrate. Enzymatic activity was expressed as mmol hydrogen peroxide decomposed/min/mg protein, according to the method described by Aebi (1984).
AMPK activity
AMPK‐α activation was assessed using the AMPK ELISA Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturers' instructions.
Histopathology
The livers were fixed in Millonig's phosphate‐buffered formalin solution at pH 7.2, embedded in paraffin and processed for light microscopy (haematoxylin and eosin (H&E) staining). Steatosis was analysed by the presence of macrovesicles in the H&E‐stained sections. The infiltration of polymorphonuclear cells was assessed in H&E‐stained sections, based upon the localization of the cell and morphology of the nucleus of the cell. For each parameter, five histological sections from six animals per group were analysed.
Statistical analysis
The results are expressed as the mean ± SEM for each group. One‐way anova including all of the groups was used to assess the overall significance of the differences between the groups. Subsequently, post hoc pairwise comparison test, including Bonferroni correction for multiple comparisons, was applied to identify which group differed from the others. Differences with P values of less than 0.05 are considered significant.
Results
Body weight, fasting blood glucose (FBG), %HbA1c, insulin, fructosamine, triglycerides, cholesterol concentrations and arterial blood pressure of control (CTL) animals, diabetic (DM) animals and diabetic animals treated with insulin (DM + Ins) or diabetic animals treated with insulin plus metformin (DM + Met + Ins) are listed in Table 1. The body weight in the DM group was found to be significantly decreased compared to the CTL group; however, it was found to be reverted to normal values in the DM + Ins and DM + Met + Ins groups (Table 1). The FBG increase in DM group was partially protected by the insulin monotherapy and was completely prevented by Met + Ins treatment (Table 1). The %HbA1c of the DM group was higher compared to the normal rats, whereas the treated groups presented a partial prevention of the %HbA1c increase (Table 1). The decrease in insulin levels observed in the DM group was normalized by the Met + Ins therapy (Table 1). There was a significant increase in fructosamine levels in the DM group as compared to CTL animals, which were partially prevented by Met + Ins therapy and not affected by insulin monotherapy (Table 1). None of the treatments were able to control the 2.89‐fold increase observed in serum triglycerides concentration in DM group (Table 1). The cholesterol levels and arterial blood pressure were found to be not altered in the DM, DM + Ins and DM + Met + Ins groups (Table 1). We found a significant reduction in the daily dose of insulin necessary to control the glucose levels between the 3rd–7th and 8th–14th days in the metformin adjunct treatment group (6.2 vs. 3.9 units, P < 0.001, data not shown); however, there was no difference in the daily dose of insulin necessary to control the glucose levels between the first and the second week of treatment in the DM + Ins group (data not shown).
Table 1.
Effects of metformin adjunct treatment on cardiometabolic parameters of high‐fat diet‐/streptozotocin‐induced diabetic rats
| Parameters | CTL (n = 20) | DM (n = 20) | DM + Ins (n = 15) | DM + Met + Ins (n = 15) |
|---|---|---|---|---|
| Weight (g) | 406.9 ± 34.99 | 337.2 ± 64.82*** | 401.7 ± 22.92### | 383.8 ± 38.85### |
| Fasting blood glucose (mg/dl) | 92.9 ± 19.8 | 377.5 ± 76.3*** | 265.9 ± 80.62*** , ## | 152.6 ± 129.9### , $$ |
| % HbA1c | 3.9 ± 0.1 | 6.7 ± 0.6*** | 4.5 ± 0.3** , ### | 4.7 ± 0.4*** , ### |
| Insulin | 1.2 ± 0.9 | 0.5 ± 0.1** | ND | 1.6 ± 1.1### |
| Fructosamine | 226.4 ± 14.9 | 279.7 ± 26.9** | 245.8 ± 23.95 | 212.3 ± 29.1## |
| Triglycerides | 71.4 ± 28.6 | 206.6 ± 135.8*** | 217.6 ± 134.6** | 181.7 ± 114.2** |
| Cholesterol | 74.8 ± 12.3 | 66.1 ± 12.8 | 63.91 ± 7.1 | 76.4 ± 14.1 |
| Arterial blood pressure (ABP) | 140.1 ± 8.7 | 130.9 ± 13.4 | ND | 137.3 ± 0.51 |
*P < 0.05 vs. CTL, **P < 0.01 vs. CTL, ***P < 0.001 vs. CTL, # P < 0.05 vs. DM, # # P < 0.01 vs. DM, # # # P < 0.001 vs. DM, $$ P < 0.01 vs. DM+Ins.
The serum levels of biomarkers of renal injury (urea, creatinine, uric acid and albumin concentrations) and amylase and the effect of the metformin adjunct treatment and insulin monotherapy on those values are shown in Table 2. Urea was found to be significantly higher in the DM group compared to CTL animals, whereas this metabolic alteration was prevented by the insulin monotherapy and Met + Ins treatment (Table 2). Additionally, the serum concentrations of creatinine, uric acid and albumin were found to be decreased in the DM rats and were found to be not altered in the insulin‐treated group; however, their decrease was partially protected by Met + Ins dual therapy (Table 2). Total protein content diminished in the DM group when compared to CTL animals and was partially reverted to values similar to the normal rats in the DM + Met + Ins and insulin monotherapy groups (Table 2). In parallel, pancreatic damage was also evident in the DM rats, as shown by a 1.6‐fold decrease in amylase enzyme activity, which was protected by the metformin adjunct therapy and not by the insulin treatment (Table 2).
Table 2.
Effect of metformin adjunct treatment on serum biochemical markers of organ function of high‐fat diet‐/streptozotocin‐induced diabetic rats
| Parameters | CTL (n = 20) | DM (n = 20) | DM + Ins (n = 10) | DM + Met + Ins (n = 15) |
|---|---|---|---|---|
| Urea (mg/dl) | 61.2 ± 8.9 | 96.5 ± 19.1*** | 59.1 ± 6.4### | 60.7 ± 15### |
| Creatinine (mg/dl) | 0.6 ± 0.12 | 0.5 ± 0.09*** | 0.5 ± 0.07* | 0.6 ± 0.09# |
| Uric acid (mg/dl) | 2.5 ± 1.0 | 1.2 ± 0.6*** | 1.9 ± 0.7 | 2.0 ± 0.7# |
| Albumin (mg/dl) | 4.4 ± 0.3 | 3.6 ± 0.3*** | 3.8 ± 0.2*** | 4.0 ± 0.4** , ### |
| Total protein (mg/dl) | 6.3 ± 0.3 | 5.0 ± 0.4*** | 5.6 ± 0.2*** , ### | 5.6 ± 0.4*** , ### |
| Amylase (mg/dl) | 1803 ± 314.0 | 1128 ± 374.8*** | 1323 ± 260.2** | 1940 ± 411.0### , $$$ |
*P < 0.05 vs. CTL, **P < 0.01 vs. CTL, ***P < 0.001 vs. CTL, # P < 0.05 vs. DM, ## P < 0.01 vs. DM, ### P < 0.001 vs. DM, $$$ P < 0.001 vs. DM + Ins.
We evaluated the serum levels of biomarkers of liver injury (ALT, AST, ALP, GGT, bilirubin and vitamin D) in the high‐fat diet‐/streptozotocin‐induced diabetic rats and their modulation by the adjunct or insulin treatments. There was a significant increase in the serum levels of ALT, ALP, GGT and total bilirubin concentration in the DM group compared to the CTL group, which was not observed in the DM + Met + Ins group (Figure 1a–e). The insulin monotherapy was also protective of the increase in the serum biomarkers of liver injury, with the exception of the ALT enzyme activity which was only protected by Met + Ins treatment (Figure 1a). The AST enzyme activity was found to be not altered in all the animal groups analysed (Figure 1b). Vitamin D concentration was found to be diminished in the DM and DM + Ins groups as compared to control group and was found to be maintained similar to normal values in the DM + Met + Ins group (Figure 1f).
Figure 1.
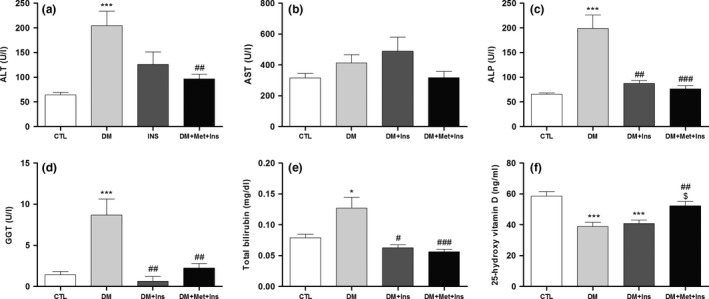
Serum levels of biomarkers of liver function in control (CTL, n = 20), diabetic (DM, n = 20), diabetic rats treated with insulin (Met + Ins, n = 15) or metformin plus insulin (DM + Met + Ins, n = 15). (a) Alanine aminotransferase (ALT); (b) aspartate aminotransferase (AST); (c) alkaline phosphatase (ALP); (d) gamma‐glutamyl transpeptidase (GGT); (e) total bilirubin; and (f) vitamin D. ***P < 0.001 vs. CTL, *P < 0.05 vs. CTL, # P < 0.05 vs. DM, ## P < 0.01 vs. DM, ### P < 0.001 vs. DM and $ P < 0.01 vs. DM + Ins.
Intravital microscopy was used to evaluate sinusoidal and postsinusoidal venular leucocyte rolling and adherence (Figure 2, VideoS1–S4). Compared to the CTL group (Video S1), there was a significant increase in the number of rolling and adherent leucocytes in the diabetic rats (Video S2), which was prevented by the Met + Ins treatment (Figure 2a–f and Video S4). On the other hand, the insulin monotherapy was not able to protect from the increase in the rolling and adhesion of leucocytes in the liver vasculature (Figure 2d–f, Video S3).
Figure 2.
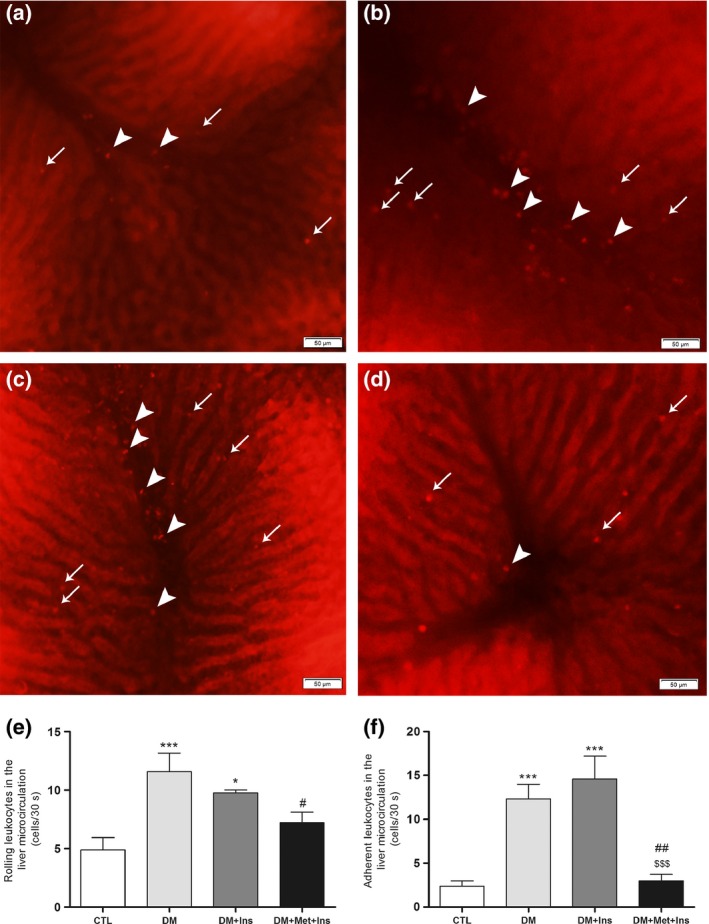
Effect of metformin adjunct treatment on hepatic microcirculation. Representative image of the liver microcirculation of CTL (a) rats, diabetic rats (b), diabetic rats treated with insulin (DM + Ins) (c) and diabetic rats treated with metformin plus insulin (DM + Met + Ins) (d), assessed by intravital microscopy; quantification of rolling (e) and adhesion (f) of leucocytes in the sinusoids and postsinusoidal venules. Values are presented as the mean (±SD). *P < 0.05 vs. CTL, ***P < 0.001 vs. CTL, # P < 0.05 vs. DM, ## P < 0.01 vs. DM and $$$ P < 0.001 vs. DM + Ins. Images are representative of at least six animals of each experimental group. Arrows indicate rolling/adherent leucocytes on sinusoids and arrowheads rolling/adherent leucocytes on postsinusoidal venules.
We further evaluated the level of lipid peroxidation in the liver of the diabetic rats and evaluated the modulation of this parameter by the metformin adjunct treatment and insulin monotherapy (Figure 3a). There was a statistically significant increase in the MDA content in the liver tissue of the diabetic rats compared to the healthy animals, which was not observed in Met+Ins and insulin treatments (Figure 3a). Histopathological analyses revealed no differences between the studied groups concerning the infiltration of leucocytes and steatoses in the liver (Video S4). The presence of fibroses was also discarded by H&E‐stained sections. The gene expression of the antioxidant enzymes NADPH oxidase p22 (N22) and p47 (N47) subunits, superoxide dismutase (SOD) and catalase (CAT) was assessed by real‐time PCR (Figure 3b). The gene expression of N22, N47 and SOD was found to be not altered in the DM and DM + Met + Ins groups compared to the healthy animals, while CAT gene expression was found to be significantly decreased in the diabetic rats (Figure 3b). Interestingly, the decrease in CAT gene expression was prevented by the metformin adjunct treatment (Figure 3b). We also analysed the gene expression of the inflammatory marker monocyte chemoattractant protein‐1 (MCP‐1), which was found to be augmented in the DM rats and was found to be not altered in the rats treated with the metformin adjunct therapy (Figure 3b). Catalase enzyme activity was also found to be diminished in DM group when compared do CTL animals, and this decrease was partially protected by Met + Ins and insulin treatments (Figure 3c). AMPK activation was shown to be reduced in DM, DM + Ins and DM + Met + Ins groups as compared to CTL animals (data not shown).
Figure 3.
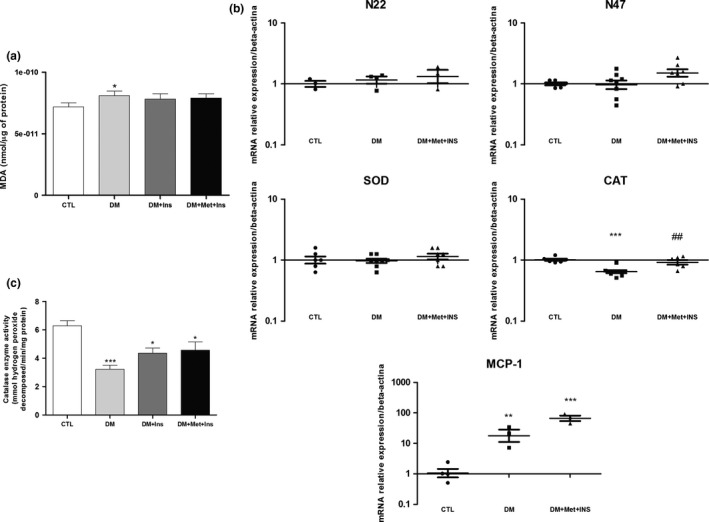
Evaluation of oxidative stress and inflammatory parameters in the liver of control (CTL) rats, diabetic (DM) rats, diabetic rats treated animals with insulin (DM + Ins) or metformin plus insulin (DM + Met + Ins). (a) Levels of malondialdehyde indicating lipid peroxidation assessed by thiobarbituric acid reactive species (TBARs); (b) Real‐time PCR analyses of mRNA transcript levels of genes coding for antioxidant enzymes (NADPH oxidase p22 subunit, NADPH oxidase p47 subunit, superoxide dismutase and catalase) and inflammatory marker monocyte chemoattractant protein‐1 (MCP‐1); (c) catalase enzyme activity in the liver of control (CTL) rats, diabetic (DM) rats, diabetic rats treated with insulin (DM + Ins) and diabetic rats treated with metformin plus insulin (DM + Met + Ins).*P < 0.05 vs. CTL, **P < 0.01 vs. CTL, ***P < 0.001 vs. CTL, # P < 0.05 vs. DM and ## P < 0.01 vs. DM. For RT‐PCR and TBARs, at least six animals of each group were analysed and three independent experiments performed. For catalase enzyme activity, ten animals of each group were analysed and three independent experiments performed.
Finally, we studied the possible participation of the AGE‐RAGE pathway in liver injury due to diabetes and its modulation by Met‐Ins or insulin treatments (Figure 4a–d). There was a significant increase in the AGE concentration in the liver of the diabetic rats, which was not observed in Met‐Ins‐ and insulin‐treated groups (Figure 4a). The RAGE gene transcripts (Figure 4b) and protein expression levels (Figure 4c and d) were not altered in any of the studied groups.
Figure 4.
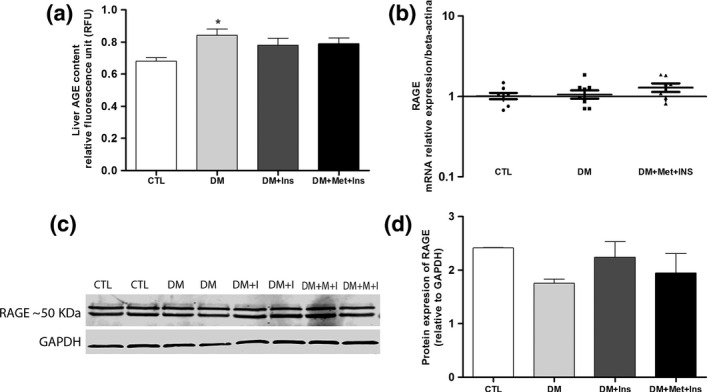
Participation of advanced glycation end product (AGE) and the receptor for advanced glycation end products (RAGE) in liver pathology. AGE deposition (a), RAGE mRNA (b) and RAGE protein levels (c and d) were compared between control (CTL) rats, diabetic (DM) rats, diabetic rats treated with insulin (DM + Ins) and diabetic animals treated with metformin plus insulin (DM + Met + Ins). *P < 0.05 vs. CTL. For AGE deposition assay, ten animals of each group were analysed in three independent experiments, while for RAGE mRNA and protein levels five animals in three independent experiments were evaluated.
Discussion
In this study we have shown that there are important systemic and hepatic alterations in HFD‐/STZ‐induced diabetes in rats, such as elevated (i) metabolic and organ dysfunction, (ii) oxidative stress, (iii) inflammation, (iv) microvascular damage and (v) AGE deposition. Additionally, we showed that metformin adjunct treatment improved systemic and liver protection in HFD‐/STZ‐induced diabetes in rats by modulating all the analysed parameters, and we showed that metformin adjunct treatment had a more pronounced effects than insulin monotherapy.
Diabetes is one of the most common causes of liver diseases, including non‐alcoholic fatty liver disease (NAFLD), such as fatty liver, steatohepatitis, liver fibrosis and cirrhosis (Baig et al. 2001), which are important causes of death in this population (Tolman et al. 2007). Although those complications are commonly associated with T2D, increasing evidences show that T1D is also linked to an increased risk of chronic liver injury (Guven et al. 2006; Torbenson et al. 2006). Patients with diabetes are at higher risk of developing hepatocellular carcinoma and chronic liver disease (El‐Serag et al. 2004; Fracanzani et al. 2008). Therefore, research aimed at developing therapeutic interventions for liver disease due to diabetes is of great interest.
Metformin is an oral antidiabetic drug that has been successfully used to treat T2D with substantial beneficial effects in T2D complications (Bailey 2008; Kooy et al. 2009). Metformin has been shown to present several properties that make it an attractive potential adjunct pharmacological agent in T1D. Previous studies have demonstrated an insulin‐sparing effect of metformin treatment as an adjunct therapy to ongoing insulin treatment in patients with type 1 diabetes. On the other hand, the results regarding glycaemic and other metabolism‐related variables are controversial. Therefore, the effect of metformin treatment on glycaemic control and other cardiovascular risk factors in patients with T1D remains a matter of controversy. Several authors hypothesized that the combined therapy with metformin and insulin would be a reasonable therapeutic approach in T1D, as it could increase the peripheral insulin sensitivity, improve the outcome of diabetic complications and diminish the daily insulin dose requirement (Meyer et al. 2002; Hamilton et al. 2003; Sarnblad et al. 2003; Lund et al. 2008; Abdelghaffar & Attia 2009; An & He 2016). However, few studies covered the different aspects of metformin adjunct treatment in T1D, which can be achieved using animal models. More importantly, the effects of metformin adjunct treatment on the liver are unclear.
In this study metformin adjunct therapy showed significant beneficial effects systemically and in the liver of HFD‐/STZ‐induced T1D in rats. The body weight and fasting blood glucose levels were found to be maintained similar to normal values after the adjunct therapy, which also improved hypertriglyceridaemia and glycated protein levels, an important finding as both are predictors of worse CVD prognoses (Misciagna et al. 2007; Nordestgaard & Varbo 2014; Scherer & Nicholls 2015). Interestingly, the insulin monotherapy was not able to control FBG and fructosamine levels, suggesting an additional effect of the metformin adjunct therapy, as previously reported by other authors (Devries et al. 2004; Lund et al. 2008). The increase in HbA1c observed in the DM group was partially protected by the metformin adjunct treatment (Lachin et al. 2008). Insulin monotherapy has shown similar effects on HbA1c although the effect of insulin monotherapy on this parameter is still controversial in the literature (Devries et al. 2004; Lachin et al. 2008; Vella et al. 2010). Importantly, the added therapy was also able to reduce the daily dose of insulin necessary to achieve a glycaemic control in the second week of treatment when compared to the first week, which was not observed in the insulin monotherapy group, which suggests additional effects of the dual therapy. The insulin‐sparing effect of metformin adjunct treatment has been shown previously by different authors (Sarnblad et al. 2003; Lund et al. 2008) and is considered an important finding as increasing doses of insulin could lead to insulin resistance and body mass gain in patients with diabetes (DeFronzo et al. 1982).
The development of T1D usually begins years before the diagnosis, which occurs only when evident clinical signs and symptoms are present (Gorsuch et al. 1981). In agreement, in the present study, we showed that the signs of organ dysfunction in T1D rats appear early, as shown by alterations in many serum biochemical markers of kidney, pancreas and liver functions. We observed that the diabetic rats had (i) renal dysfunction, (ii) liver disease and (iii) impairment of pancreatic exocrine function after 2 weeks of diabetes induction. Interestingly, metformin adjunct therapy was able to protect from those functional alterations in the target organs, showing more pronounced beneficial effects than the insulin therapy. We propose that glycaemic control may be one of the main causes of the organ function protection presently observed, as hyperglycaemia is the diabetic disturbance most commonly associated with diabetic complications (DCCT, 1993; Giacco & Brownlee 2010).
Concerning the hepatic microcirculation, we observed that the endothelium becomes adhesive to circulating leucocytes, which is one of the first steps in vascular wall damage and inflammation (Schroder et al. 1991). The presence of the intrahepatic inflammatory cell response, reflected by an increased activation of the rolling and adhesion of leucocytes, and the increased serum biomarkers of organ dysfunction could indicate the beginning of long‐term damage to the microcirculation and other tissues in the diabetic animals (Creager et al. 2003). Previous studies from our group have shown microvascular alterations in the brain and the skeletal muscle of experimental rat models of diabetes and metabolic syndrome (Estato et al. 2013; Nascimento et al. 2013); however, this is the first report of hepatic microvascular damage in a T1D experimental model. Importantly, while metformin adjunct therapy improved the microcirculation damage observed in diabetic animals, the insulin monotherapy showed no effect. We believe that the increased rolling and adhesion of leucocytes in the hepatic endothelium could be triggered by cell damage induced by hyperglycaemia‐derived oxidative stress and inflammation; however, further studies are necessary to explain the exact mechanism of action of the liver microvascular damage. In this context, the improvement in glycaemic control using the metformin adjunct therapy could explain its higher protection against liver microcirculation injury compared to insulin monotherapy.
We found an increase in ALT, ALP and bilirubin levels in parallel with a decrease in vitamin D, which were found to be involved in liver damage due to diabetes. The metformin adjunct therapy had an effect on the levels of serum ALT, ALP, GGT, bilirubin and vitamin D, which are markers of liver function (Lips 2006; Targher et al. 2007), while only ALP, GGT and bilirubin levels were found to be maintained similar to CTL values in the insulin monotherapy group. Although the protection from liver damage was previously reported by insulin therapy (Hemmings & Pekush 1994), our data suggest a significant additional effect of metformin adjunct treatment on liver damage and dysfunction in diabetic rats.
Giacco and Brownlee (2010) postulated that the damage caused by hyperglycaemia, although originated by several mechanisms, is activated by a common mechanism: the overproduction of reactive oxygen species (ROS) (Giacco & Brownlee 2010). Confirming the central role of oxidative stress in liver damage, we found that an increase in the MDA levels and a decrease in catalase mRNA and enzyme activity were found to be involved in liver damage due to diabetes. The dual therapy and the insulin monotherapy showed similar levels of protection against increased MDA levels in the liver of diabetic rats. On the other hand, we observed a partial protection against decreased catalase enzyme activity by Met+Ins and insulin treatments. We further analysed the genes involved in oxidative stress that could explain the effect of Met + Ins therapy and found that SOD and NADPH oxidase mRNA transcript levels were not involved, as previously reported (Kukidome et al. 2006; Araki & Nishikawa 2010).
We also studied the participation of AMPK activation in the mechanism of action of Met + Ins treatment. In contrast to the previous studies (Zhou et al. 2001; Musi et al. 2002), we did not find an increase in AMPK activation after metformin adjunct therapy, showing that this pathway is not involved neither in liver damage of HFD‐/STZ‐induced diabetes in rats nor in the protection of liver injury by metformin adjunct treatment.
There was a significant increase in the MCP‐1 transcript levels in the diabetic rats, which was not modulated by the adjunct therapy. This finding suggests a possible participation of this chemokine in the aetiology of liver disease in diabetic animals. Although an increase in MCP‐1 has been associated with T1D, its specific role is still unclear (Scarpini et al. 2002; Hanifi‐Moghaddam et al. 2006; Kiyici et al. 2006; Antonelli et al. 2008; Zineh et al. 2009). Indeed, there is some evidence of a dual biological function of this chemokine, inducing both pro‐ and anti‐inflammatory effects. This anti‐inflammatory activity has been associated with the increased stimulation of IL‐4, which is the primary Th‐2 cytokine (Gu et al. 2000).
The participation of the AGE formed by the non‐enzymatic glycation of proteins, lipids or DNA in diabetic‐induced tissue alterations has been extensively explored in patients and animal models of diabetes. The presence of AGEs has been proposed as a biomarker for the status of complications (Beisswenger et al. 1995; Nishino et al. 1995; Stitt et al. 1997; Yanagisawa et al. 1998). Previous studies reported that tissue AGE accumulation preceded the appearance of diabetic complications (Beisswenger et al. 1995). The activation of the receptor for AGE (RAGE) activates the translocation of the nuclear transcription factor NF‐κB, which in turn increases the expression of several genes involved in oxidative stress and inflammation (Rauvala & Pihlaskari 1987; Goldin et al. 2006). RAGE activation is involved in the progression of many diseases, such as neuropathy, nephropathy (Yamamoto et al., 2001), macrovascular disease, rheumatoid arthritis and sepsis (Wang et al. 1999; Liliensiek et al. 2004). Although never explored in liver injury due to diabetes, some studies have determined the participation of AGE‐RAGE pathway in liver disease due to other origins, including sepsis (Kuhla et al. 2013), hepatic fibrosis (Goodwin et al. 2013), non‐alcoholic fatty liver disease (NASH) (Hyogo et al. 2007) and ischaemia/reperfusion injury (Yue et al. 2015). In the present study, the increase in AGE deposition in the liver of the diabetic animals is the first indication of the participation of AGE‐RAGE pathway in diabetic‐induced liver damage. Importantly, the Met + Ins therapy and insulin monotherapy protected the liver from this damage. However, further studies are necessary to address the exact participation of AGE adducts in liver injury.
The data presented here showed that diabetic rats have significant liver damage, which in turn results in reduced antioxidant capacity, increased inflammation and organ and microcirculation dysfunction. This could be an important contributory factor in the development of other diabetic complications. Our findings show that metformin adjunct therapy during the phase of development of diabetes protected the liver against oxidative stress and its consequences and that the AGE pathway could be involved in this process. Additionally, the dual therapy showed additive improvements in several parameters, suggesting an additional beneficial effect of metformin on the liver diseases of diabetic animals. Further detailed studies aimed at linking AGE pathway with the development of hepatic complications due to diabetes need to be undertaken in order to fully understand the pathophysiology of liver disease in patients with T1D.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Video S1–S4. Representative videos of the hepatic microcirculation assessed by intravital microscopy showing rolling and adhesion of leukocytes (in red) in sinusoids and post‐sinusoids venules of control (video S1), high‐fat diet‐/streptozotocin‐induced diabetic (video S2), diabetic treated with insulin (video S3) and diabetic treated with insulin plus metformin (video S4) animals.
Figure S1. Representative images of HE‐stained rat liver sections from the groups: Control (CTL), diabetes (DM), diabetic treated animals with insulin (DM + Ins) and treated with metformin plus insulin (DM + Met + Ins), showing no significant histopatological changes among them. Bar = 50 μm.
Acknowledgements
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 455,384/2014‐2) and PAPES/Fiocruz (401,803/2015‐5).
References
- Abdelghaffar S., Attia A.M. (2009) Metformin added to insulin therapy for type 1 diabetes mellitus in adolescents. Cochrane Database Syst. Rev. 1, CD006691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebi H. (1984) Catalase in vitro . Methods Enzymol. 105, 121–126. [DOI] [PubMed] [Google Scholar]
- An H. & He L. (2016) Current understanding of metformin effect on the control of hyperglycemia in diabetes. J. Endocrinol. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli A., Fallahi P., Ferrari S.M. et al (2008) Serum Th1 (CXCL10) and Th2 (CCL2) chemokine levels in children with newly diagnosed Type 1 diabetes: a longitudinal study. Diabet. Med. 25, 1349–1353. [DOI] [PubMed] [Google Scholar]
- Araki E. & Nishikawa T. (2010) Oxidative stress: a cause and therapeutic target of diabetic complications. J. Diabetes Investig. 1, 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig N.A., Herrine S.K. & Rubin R. (2001) Liver disease and diabetes mellitus. Clin. Lab. Med. 21, 193–207. [PubMed] [Google Scholar]
- Bailey C.J. (2008) Metformin: effects on micro and macrovascular complications in type 2 diabetes. Cardiovasc. Drugs Ther. 22, 215–224. [DOI] [PubMed] [Google Scholar]
- Beisswenger P.J., Makita Z., Curphey T.J. et al (1995) Formation of immunochemical advanced glycosylation end products precedes and correlates with early manifestations of renal and retinal disease in diabetes. Diabetes 44, 824–829. [DOI] [PubMed] [Google Scholar]
- Burchardt P., Zawada A., Tabaczewski P. et al (2013) Metformin added to intensive insulin therapy reduces plasma levels of glycated but not oxidized low‐density lipoprotein in young patients with type 1 diabetes and obesity in comparison with insulin alone: a pilot study. Pol. Arch. Med. Wewn. 123, 526–532. [DOI] [PubMed] [Google Scholar]
- Cleland S.J., Fisher B.M., Colhoun H.M., Sattar N. & Petrie J.R. (2013) Insulin resistance in type 1 diabetes: what is ‘double diabetes’ and what are the risks? Diabetologia 56, 1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creager M.A., Luscher T.F., Cosentino F. & Beckman J.A. (2003) Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 108, 1527–1532. [DOI] [PubMed] [Google Scholar]
- DCCT (1993) The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N. Engl. J. Med. 329, 977–986. [DOI] [PubMed] [Google Scholar]
- DeFronzo R.A., Simonson D. & Ferrannini E. (1982) Hepatic and peripheral insulin resistance: a common feature of type 2 (non‐insulin‐dependent) and type 1 (insulin‐dependent) diabetes mellitus. Diabetologia 23, 313–319. [DOI] [PubMed] [Google Scholar]
- Devries J.H., Snoek F.J. & Heine R.J. (2004) Persistent poor glycaemic control in adult Type 1 diabetes. A closer look at the problem. Diabet. Med. 21, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Draper H.H. & Hadley M. (1990) Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol. 186, 421–431. [DOI] [PubMed] [Google Scholar]
- El‐Serag H.B., Tran T. & Everhart J.E. (2004) Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 126, 460–468. [DOI] [PubMed] [Google Scholar]
- Estato V., Obadia N., Carvalho‐Tavares J. et al (2013) Blockade of the renin‐angiotensin system improves cerebral microcirculatory perfusion in diabetic hypertensive rats. Microvasc. Res. 87, 41–49. [DOI] [PubMed] [Google Scholar]
- de Ferranti S.D., de Boer I.H., Fonseca V. et al (2014) Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Diabetes Care 37, 2843–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes J.M. & Cooper M.E. (2013) Mechanisms of diabetic complications. Physiol. Rev. 93, 137–188. [DOI] [PubMed] [Google Scholar]
- Fracanzani A.L., Valenti L., Bugianesi E. et al (2008) Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: a role for insulin resistance and diabetes. Hepatology 48, 792–798. [DOI] [PubMed] [Google Scholar]
- Giacco F. & Brownlee M. (2010) Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannarelli R., Aragona M., Coppelli A., Del P.S. (2003) Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 29(4 Pt 2), 6S28–6S35. [DOI] [PubMed] [Google Scholar]
- Gin H., Messerchmitt C., Brottier E. & Aubertin J. (1985) Metformin improved insulin resistance in type I, insulin‐dependent, diabetic patients. Metabolism 34, 923–925. [DOI] [PubMed] [Google Scholar]
- Gin H., Freyburger G., Boisseau M. & Aubertin J. (1989) Study of the effect of metformin on platelet aggregation in insulin‐dependent diabetics. Diabetes Res. Clin. Pract. 6, 61–67. [DOI] [PubMed] [Google Scholar]
- Goldin A., Beckman J.A., Schmidt A.M. & Creager M.A. (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114, 597–605. [DOI] [PubMed] [Google Scholar]
- Goodwin M., Herath C., Jia Z. et al (2013) Advanced glycation end products augment experimental hepatic fibrosis. J. Gastroenterol. Hepatol. 28, 369–376. [DOI] [PubMed] [Google Scholar]
- Gorsuch A.N., Spencer K.M., Lister J. et al (1981) Evidence for a long prediabetic period in type I (insulin‐dependent) diabetes mellitus. Lancet 2(8260–61), 1363–1365. [DOI] [PubMed] [Google Scholar]
- Gu L., Tseng S., Horner R.M., Tam C., Loda M. & Rollins B.J. (2000) Control of TH2 polarization by the chemokine monocyte chemoattractant protein‐1. Nature 404, 407–411. [DOI] [PubMed] [Google Scholar]
- Guven A., Yavuz O., Cam M. et al (2006) Effects of melatonin on streptozotocin‐induced diabetic liver injury in rats. Acta Histochem. 108, 85–93. [DOI] [PubMed] [Google Scholar]
- Hamilton J., Cummings E., Zdravkovic V., Finegood D. & Daneman D. (2003) Metformin as an adjunct therapy in adolescents with type 1 diabetes and insulin resistance: a randomized controlled trial. Diabetes Care 26, 138–143. [DOI] [PubMed] [Google Scholar]
- Hanifi‐Moghaddam P., Kappler S., Seissler J. et al (2006) Altered chemokine levels in individuals at risk of Type 1 diabetes mellitus. Diabet. Med. 23, 156–163. [DOI] [PubMed] [Google Scholar]
- Hemmings S.J. & Pekush R.D. (1994) The impact of type I diabetes on rat liver gamma‐glutamyltranspeptidase. Mol. Cell. Biochem. 139, 131–140. [DOI] [PubMed] [Google Scholar]
- Holman R.R., Paul S.K., Bethel M.A., Matthews D.R. & Neil H.A. (2008) 10‐year follow‐up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 359, 1577–1589. [DOI] [PubMed] [Google Scholar]
- Hyogo H., Yamagishi S., Iwamoto K. et al (2007) Elevated levels of serum advanced glycation end products in patients with non‐alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 22, 1112–1119. [DOI] [PubMed] [Google Scholar]
- Isoda K., Young J.L., Zirlik A. et al (2006) Metformin inhibits proinflammatory responses and nuclear factor‐kappaB in human vascular wall cells. Arterioscler. Thromb. Vasc. Biol. 26, 611–617. [DOI] [PubMed] [Google Scholar]
- Janssen M., Rillaerts E. & De Leeuw I. (1991) Effects of metformin on haemorheology, lipid parameters and insulin resistance in insulin‐dependent diabetic patients (IDDM). Biomed. Pharmacother. 45, 363–367. [DOI] [PubMed] [Google Scholar]
- Kearney P.M., Blackwell L., Collins R. et al (2008) Efficacy of cholesterol‐lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta‐analysis. Lancet 371, 117–125. [DOI] [PubMed] [Google Scholar]
- Khan A.S., McLoughney C.R. & Ahmed A.B. (2006) The effect of metformin on blood glucose control in overweight patients with Type 1 diabetes. Diabet. Med. 23, 1079–1084. [DOI] [PubMed] [Google Scholar]
- Kiyici S., Erturk E., Budak F. et al (2006) Serum monocyte chemoattractant protein‐1 and monocyte adhesion molecules in type 1 diabetic patients with nephropathy. Arch. Med. Res. 37, 998–1003. [DOI] [PubMed] [Google Scholar]
- Kooy A., de Jager J., Lehert P. et al (2009) Long‐term effects of metformin on metabolism and microvascular and macrovascular disease in patients with type 2 diabetes mellitus. Arch. Intern. Med. 169, 616–625. [DOI] [PubMed] [Google Scholar]
- Kuhla A., Norden J., Abshagen K., Menger M.D. & Vollmar B. (2013) RAGE blockade and hepatic microcirculation in experimental endotoxaemic liver failure. Br. J. Surg. 100, 1229–1239. [DOI] [PubMed] [Google Scholar]
- Kukidome D., Nishikawa T., Sonoda K. et al (2006) Activation of AMP‐activated protein kinase reduces hyperglycemia‐induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 55, 120–127. [PubMed] [Google Scholar]
- Lachin J.M., Genuth S., Nathan D.M., Zinman B. & Rutledge B.N. (2008) Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial–revisited. Diabetes 57, 995–1001. [DOI] [PubMed] [Google Scholar]
- Liliensiek B., Weigand M.A., Bierhaus A. et al (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Invest. 113, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.Z., Yang S.Q., Chuckaree C., Kuhajda F., Ronnet G. & Diehl A.M. (2000) Metformin reverses fatty liver disease in obese, leptin‐deficient mice. Nat. Med. 6, 998–1003. [DOI] [PubMed] [Google Scholar]
- Lips P. (2006) Vitamin D physiology. Prog. Biophys. Mol. Biol. 92, 4–8. [DOI] [PubMed] [Google Scholar]
- Lund S.S., Tarnow L., Astrup A.S. et al (2008) Effect of adjunct metformin treatment in patients with type‐1 diabetes and persistent inadequate glycaemic control. A randomized study. PLoS One 3, e3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund S.S., Tarnow L., Astrup A.S. et al (2009) Effect of adjunct metformin treatment on levels of plasma lipids in patients with type 1 diabetes. Diabetes Obes. Metab. 11, 966–977. [DOI] [PubMed] [Google Scholar]
- Meyer L., Bohme P., Delbachian I. et al (2002) The benefits of metformin therapy during continuous subcutaneous insulin infusion treatment of type 1 diabetic patients. Diabetes Care 25, 2153–2158. [DOI] [PubMed] [Google Scholar]
- Misciagna G., De Michele G. & Trevisan M. (2007) Non enzymatic glycated proteins in the blood and cardiovascular disease. Curr. Pharm. Des. 13, 3688–3695. [DOI] [PubMed] [Google Scholar]
- Moon R.J., Bascombe L.A. & Holt R.I. (2007) The addition of metformin in type 1 diabetes improves insulin sensitivity, diabetic control, body composition and patient well‐being. Diabetes Obes. Metab. 9, 143–145. [DOI] [PubMed] [Google Scholar]
- Musi N., Hirshman M.F., Nygren J. et al (2002) Metformin increases AMP‐activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51, 2074–2081. [DOI] [PubMed] [Google Scholar]
- Nakayama H., Mitsuhashi T., Kuwajima S. et al (1993) Immunochemical detection of advanced glycation end products in lens crystallins from streptozocin‐induced diabetic rat. Diabetes 42, 345–350. [DOI] [PubMed] [Google Scholar]
- Nascimento A.R., Machado M., de Jesus N. et al (2013) Structural and functional microvascular alterations in a rat model of metabolic syndrome induced by a high‐fat diet. Obesity (Silver Spring) 21, 2046–2054. [DOI] [PubMed] [Google Scholar]
- Nishino T., Horii Y., Shiiki H. et al (1995) Immunohistochemical detection of advanced glycosylation end products within the vascular lesions and glomeruli in diabetic nephropathy. Hum. Pathol. 26, 308–313. [DOI] [PubMed] [Google Scholar]
- Nordestgaard B.G. & Varbo A. (2014) Triglycerides and cardiovascular disease. Lancet 384, 626–635. [DOI] [PubMed] [Google Scholar]
- Orchard T.J., Nathan D.M., Zinman B. et al (2015) Association between 7 years of intensive treatment of type 1 diabetes and long‐term mortality. JAMA 313, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purnell J.Q., Hokanson J.E., Marcovina S.M., Steffes M.W., Cleary P.A. & Brunzell J.D. (1998) Effect of excessive weight gain with intensive therapy of type 1 diabetes on lipid levels and blood pressure: results from the DCCT. Diabetes Control and Complications Trial. JAMA 280, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauvala H. & Pihlaskari R. (1987) Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J. Biol. Chem. 262, 16625–16635. [PubMed] [Google Scholar]
- Regnell S.E. & Lernmark A. (2011) Hepatic steatosis in type 1 diabetes. Rev. Diabet. Stud. 8, 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnblad S., Kroon M. & Aman J. (2003) Metformin as additional therapy in adolescents with poorly controlled type 1 diabetes: randomised placebo‐controlled trial with aspects on insulin sensitivity. Eur. J. Endocrinol. 149, 323–329. [DOI] [PubMed] [Google Scholar]
- Sartoretto J.L., Melo G.A., Carvalho M.H. et al (2005) Metformin treatment restores the altered microvascular reactivity in neonatal streptozotocin‐induced diabetic rats increasing NOS activity, but not NOS expression. Life Sci. 77, 2676–2689. [DOI] [PubMed] [Google Scholar]
- Scarpini E., Galimberti D., Baron P. et al (2002) IP‐10 and MCP‐1 levels in CSF and serum from multiple sclerosis patients with different clinical subtypes of the disease. J. Neurol. Sci. 195, 41–46. [DOI] [PubMed] [Google Scholar]
- Schatz H., Winkler G., Jonatha E.M. & Pfeiffer E.F. (1975) Studies on juvenile–type diabetes in children. Assessment of control under treatment with constant and variable doses of insulin with or without addition of biguanides. Diabete Metab. 1, 211–220. [PubMed] [Google Scholar]
- Scherer D.J. & Nicholls S.J. (2015) Lowering triglycerides to modify cardiovascular risk: will icosapent deliver? Vasc. Health Risk Manag. 11, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmack G., Defronzo R.A. & Musi N. (2006) AMP‐activated protein kinase: Role in metabolism and therapeutic implications. Diabetes Obes. Metab. 8, 591–602. [DOI] [PubMed] [Google Scholar]
- Schroder S., Palinski W. & Schmid‐Schonbein G.W. (1991) Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am. J. Pathol. 139, 81–100. [PMC free article] [PubMed] [Google Scholar]
- Sena C.M., Matafome P., Louro T., Nunes E., Fernandes R. & Seica R.M. (2011) Metformin restores endothelial function in aorta of diabetic rats. Br. J. Pharmacol. 163, 424–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley S.D., Palmer J.P., Hirsch I.B. & Brunzell J.D. (2003) Visceral obesity, hepatic lipase activity, and dyslipidemia in type 1 diabetes. J. Clin. Endocrinol. Metab. 88, 3379–3384. [DOI] [PubMed] [Google Scholar]
- Srinivasan K., Viswanad B., Asrat L., Kaul C.L. & Ramarao P. (2005) Combination of high‐fat diet‐fed and low‐dose streptozotocin‐treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol. Res. 52, 313–320. [DOI] [PubMed] [Google Scholar]
- Stitt A.W., Bucala R., Vlassara H. (1997). Atherogenesis and advanced glycation: promotion, progression, and prevention. Ann. N. Y. Acad. Sci. 811: 115–127; discussion 127‐119. [DOI] [PubMed] [Google Scholar]
- Targher G., Bertolini L., Scala L. et al (2007) Associations between serum 25‐hydroxyvitamin D3 concentrations and liver histology in patients with non‐alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 17, 517–524. [DOI] [PubMed] [Google Scholar]
- Targher G., Bertolini L., Padovani R. et al (2010) Prevalence of non‐alcoholic fatty liver disease and its association with cardiovascular disease in patients with type 1 diabetes. J. Hepatol. 53, 713–718. [DOI] [PubMed] [Google Scholar]
- Targher G., Pichiri I., Zoppini G., Trombetta M. & Bonora E. (2012) Increased prevalence of chronic kidney disease in patients with Type 1 diabetes and non‐alcoholic fatty liver. Diabet. Med. 29, 220–226. [DOI] [PubMed] [Google Scholar]
- Targher G., Mantovani A., Pichiri I. et al (2014) Nonalcoholic fatty liver disease is independently associated with an increased incidence of chronic kidney disease in patients with type 1 diabetes. Diabetes Care 37, 1729–1736. [DOI] [PubMed] [Google Scholar]
- Tolman K.G., Fonseca V., Dalpiaz A. & Tan M.H. (2007) Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care 30, 734–743. [DOI] [PubMed] [Google Scholar]
- Torbenson M., Chen Y.Y., Brunt E. et al (2006) Glycogenic hepatopathy: an underrecognized hepatic complication of diabetes mellitus. Am. J. Surg. Pathol. 30, 508–513. [DOI] [PubMed] [Google Scholar]
- Vella S., Buetow L., Royle P., Livingstone S., Colhoun H.M. & Petrie J.R. (2010) The use of metformin in type 1 diabetes: a systematic review of efficacy. Diabetologia 53, 809–820. [DOI] [PubMed] [Google Scholar]
- Wang H., Bloom O., Zhang M. et al (1999) HMG‐1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251. [DOI] [PubMed] [Google Scholar]
- Wulffele M.G., Kooy A., de Zeeuw D., Stehouwer C.D. & Gansevoort R.T. (2004) The effect of metformin on blood pressure, plasma cholesterol and triglycerides in type 2 diabetes mellitus: a systematic review. J. Intern. Med. 256, 1–14. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Kato I., Doi T. et al (2001) Development and prevention of advanced diabetic nephropathy in RAGE‐overexpressing mice. J. Clin. Invest. 108, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa K., Makita Z., Shiroshita K. et al (1998) Specific fluorescence assay for advanced glycation end products in blood and urine of diabetic patients. Metabolism 47, 1348–1353. [DOI] [PubMed] [Google Scholar]
- Yue S., Zhou H.M., Zhu J.J. et al (2015) Hyperglycemia and Liver Ischemia Reperfusion Injury: a Role for the Advanced Glycation Endproduct and Its Receptor Pathway. Am. J. Transplant. 15, 2877–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G., Myers R., Li Y. et al (2001) Role of AMP‐activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zineh I., Beitelshees A.L., Silverstein J.H. & Haller M.J. (2009) Serum monocyte chemoattractant protein‐1 concentrations associate with diabetes status but not arterial stiffness in children with type 1 diabetes. Diabetes Care 32, 465–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1–S4. Representative videos of the hepatic microcirculation assessed by intravital microscopy showing rolling and adhesion of leukocytes (in red) in sinusoids and post‐sinusoids venules of control (video S1), high‐fat diet‐/streptozotocin‐induced diabetic (video S2), diabetic treated with insulin (video S3) and diabetic treated with insulin plus metformin (video S4) animals.
Figure S1. Representative images of HE‐stained rat liver sections from the groups: Control (CTL), diabetes (DM), diabetic treated animals with insulin (DM + Ins) and treated with metformin plus insulin (DM + Met + Ins), showing no significant histopatological changes among them. Bar = 50 μm.