

Abstract
Background
In utero and early-life experienced environmental exposures are suggested to play an important role in many multifactorial diseases potentially mediated through lasting effects on the epigenome. As the epigenome in addition remains modifiable throughout life, identifying specific disease-relevant biomarkers may prove challenging. This has led to an increased interest in epigenome-wide association studies using dried blood spots (DBS) routinely collected in perinatal screening programs. Such programs are in place in numerous countries around the world producing large and unique biobanks. However, availability of this biological material is highly limited as each DBS is made only from a few droplets of blood and storage conditions may be suboptimal for epigenetic studies. Furthermore, as relevant markers may reside outside gene bodies, epigenome-wide interrogation is needed.
Results
Here we demonstrate, as a proof of principle, that genome-wide interrogation of the methylome based on methylated DNA immunoprecipitation coupled with next-generation sequencing (MeDIP-seq) is feasible using a single 3.2 mm DBS punch (60 ng DNA) from filter cards archived for up to 16 years. The enrichment profile, sequence quality and distribution of reads across genetic regions were comparable between samples archived 16 years, 4 years and a freshly prepared control sample.
Conclusions
In summary, we show that high-quality MeDIP-seq data is achievable from neonatal screening filter cards stored at room temperature, thereby providing information on annotated as well as on non-RefSeq genes and repetitive elements. Moreover, the quantity of DNA from one DBS punch proved sufficient allowing for multiple epigenome studies using one single DBS.
Electronic supplementary material
The online version of this article (doi:10.1186/s13148-016-0242-1) contains supplementary material, which is available to authorized users.
Keywords: DNA methylation, Archival dried blood spots, MeDIP-seq, Low input, Genome-wide
Background
Epigenetic regulation has proven important in numerous cellular mechanisms including cell differentiation, gene expression and genome stability [1, 2]. With mounting evidence of epigenetic variations playing an important role in the etiology of common complex diseases such as cancer, diabetes and psychiatric disorders, the interest in epigenome-wide association studies (EWAS), specifically with regard to DNA methylation, has increased extensively in recent years [3–6]. However, identification of epigenetic risk variants with small effect sizes in a heterogenic population requires sample sets of considerable sizes. Additionally, given the modifiable nature of the epigenome distinguishing causality from consequence can prove challenging in adult samples, as marks dynamically change according to cellular environment and differentiation but also as a consequence of adverse exposures and disease. In fact, exposures experienced prenatally have shown to be able to inflict, even long-lasting, epigenetic changes. For example, maternal smoking or stress during pregnancy has shown to result in aberrant epigenetic changes in the offspring [7–9]. In addition, the inherent plasticity of the epigenome leads to changes not only over time per se but also in response to the use of certain pharmaceuticals and other potentially “epitoxic” substances. For instance, DNA methylation changes have been associated with the use of anti-psychotic drugs such as clozapine and haloperidol [10, 11]. A variety of common diseases including type II diabetes and multiple sclerosis have been suggested to include a component of in utero origin [12, 13]. The epigenome is especially vulnerable at this stage, laying the foundation for the “developmental origins of health and disease hypothesis”, suggesting that in utero experienced exposures affect the epigenome [14]. Interrogation of the epigenetic profile is, therefore, preferably done before disease onset and ideally perinatally.
Screening for metabolic disorders in newborns is routinely undertaken in many countries, where blood from heel-pricks is collected on filter paper cards, also known as Guthrie cards. Subsequent storage of the cards essentially produces a population-wide biobank. Notable, since prenatal exposures are believed to have impact on multiple cell lineages, mirror effects may arise. Hence, methylome profiling in DNA extracted from dried blood spot (DBS) samples can potentially be informative both in the search for biomarkers and to delineate the etiology of diseases manifested in other tissues [15, 16]. Also, whereas the intra-individual variation in the epigenetic landscape is considerable, the inter-individual variation is relatively small, making not only longitudinal but also cross-individual studies feasible [17].
Nested case-control approaches taking advantage of large biobanks containing DBS filter cards can potentially satisfy both abovementioned needs of large and early-life sample sets provided that the epigenome remains stable under the storage conditions of the particular biobank and that sufficient biological material can be obtained. Previous studies have shown that DNA can be extracted from DBSs stored at −20°C for decades without considerable loss of quality paving the way for genome-wide association studies (GWAS) and whole genome sequencing (WGS) [18, 19]. Along the same line, both the use of methylation arrays and methylome-wide sequencing using DNA extracted from DBSs has previously shown to be feasible, however, requiring whole genome amplification of bisulfite converted DNA or large input amounts [20–24]. However, amplification of the bisulfitome (bisulfite-converted genomic DNA) has proven to introduce biases [25, 26]. Methylated DNA immunoprecipitation coupled with next-generation sequencing (MeDIP-seq) is a cost-effective methylome alternative, where methylated genomic DNA is immunoprecipitated with an antibody followed by sequencing of the enriched fragments. MeDIP-seq displays good positive correlation with array-based methods such as the HumanMethylation450 BeadChip [21, 27] and also provides information on non-RefSeq genes and repetitive elements, both of which may be of importance in disease development and not interrogated with current methylation arrays [28, 29]. The method of choice is purpose dependent. Arrays may be the appropriate choice for disease studies searching for common variants, whereas MeDIP-seq provides agnostic information on the methylation profile and can be used for a broader characterization of cell development and the impact of environmental exposure outside annotated genomic regions. The minimum DNA input requirements by the different methods is another limiting factor in the case of biobanked DBS filter cards (Additional file 1: Figure S1). With 50 ng of DNA sufficient for MeDIP-seq, this method is one of the least consuming whole genome approaches.
Storage conditions vary among the biobanks, thus, where the Danish National Screening Biobank (DNSB), the California Research-Ready Biospecimen Bank store the filter cards at around −20°C, the Swedish PKU-Biobank, the Michigan Neonatal Biobank and the Danish Twin Registry store the majority of their samples at room temperature.
In this study, we profiled the methylome of DNA stored on room temperature archived filter cards by the use MeDIP-seq using a single 3.2 mm punch. A freshly prepared filter card served as benchmark control. Despite prolonged (4–16 years) storage at room temperature the methylome on a global level appeared largely unchanged. These results underline that DNA extracted from only a small punch of an invaluable archival DBS on a filter card, can be used for genome-wide methylome analyses including methylome-wide association studies (MWAS).
Results
MeDIP sequencing quality from DBS samples
Genomic DNA was extracted from one 3.2 mm punch from each of the three Whatman 903 filter cards (Table 1), being a freshly prepared “homemade” sample (hDBS), a short-term (4 years at room temperature) stored sample (rDBS) and a long-term (16 years at room temperature) stored sample (oDBS). All three are independent adult samples from two males and one female. Employing a purpose-specialized protocol, DNA was extracted in triplicates at three separate occasions, routinely yielding on average ~13 ng/mm2 (average yield in ng/mm2; hDBS 12.4 (±2.4 SD), rDBS 13.4 (±2.6 SD), oDBS 15.1 (±5.3 SD)) (Additional file 2: Figure S2). The DNA was sonicated to a mean fragment length of 180 bp (Additional file 3: Figure S3). Sequencing of MeDIP enriched libraries yielded on average ~68 million clean reads (Additional file 4: Table S1). Sequencing statistics revealed that all three samples performed well, with mean Phred scores above 30 along the entire read, and the anticipated GC content skewedness (Additional file 5: Figure S4).
Table 1.
Sample overview
| Samples | ID | Gender | Storage | Disk size | DNA extracted | MeDIP |
|---|---|---|---|---|---|---|
| 1 | hDBS (“homemade dried blood spot”) | Male | 1 month −20 °C | 2.3 mm | 111 ng | 70 ng |
| 2 | rDBS (“recent dried blood spot”) | Female | 4 years RT | 2.3 mm | 94 ng | 60 ng |
| 3 | oDBS (“old dried blood spot”) | Male | 16 years RT | 2.3 mm | 93 ng | 60 ng |
Sample description and the amount of genomic DNA extracted and used for MeDIP
RT room temperature
Clean reads were aligned to the human genome (hg38) using Burrows-Wheeler aligner (BWA) algorithm after which duplicated and unmapped reads were removed resulting in a final mean library of ~57 (±2) million mapped reads. Quality and validity check of the mapped MeDIP-seq data was performed using MEDIPS excluding sex chromosomes. Saturation plots showed that all three sets of reads had sufficient complexity and depth to saturate the coverage profile of the reference genome and that this was reproducible (Additional file 6: Figure S5A–C). Assessing the enrichment of methylated regions showed that the relative frequency of CpGs (freqCpG) and the observed/expected ratio of CpGs (ratioCpG) in the genomic regions sequenced compared to the reference genome were above 1 in all cases, indicating a successful enrichment of methylated fragments in the data sets (Fig. 1a). Along this line, the normalization of read counts to CpG density followed the expected linear trajectory, i.e. increased read counts as a function of increased CpG density at CpG poor regions and a deflating relationship at CpG rich regions, which is likely explained by the tendency of CpG islands to be unmethylated (Additional file 6: Figure S5D–E). In the end, 94 ± 2 % of the mapped reads overlapped with a least one CpG, whereas 20.8 ±0.8 % of the in total ~28 million CpGs interrogated were not covered. Conversely, 49.6 ±1.3 % of all CpGs were covered at least five times (Fig. 1b–d). As an additional sequencing quality control for poor alignment and incorrectly mapped reads, visual inspection of the repeat-rich gene SFI1 was performed (Additional file 7: Figure S6).
Fig. 1.
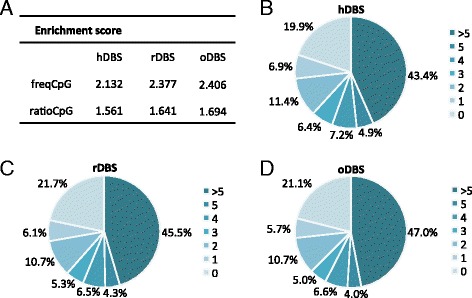
Sequencing quality. a Enrichment of CpG sites shown as the calculated frequency of CpGs (fregCpG) and ratio of CpGs (ratioCpG) in the three samples (hDBS, rDBS, oDBS) compared to the reference genome. b-d Methylome-wide coverage depicted as the percentage of the methylome (28 million CpGs) covered by the sequenced reads in the three samples (hDBS, rDBS, oDBS)
Correlation between the three DBS samples revealed no global changes in methylation patterns
With the intend to assess the large scale global differences in DNA methylation patterns in DBS samples as a consequence of prolonged storage at ambient temperatures, a pair-wise Pearson’s correlation was performed between the three DBS samples using MEDIPS excluding sex chromosomes. Since all DBS samples had good saturation profiles it was expected that the read coverage profiles were rather similar although cell composition and genetic variation was unaccounted for. The correlation coefficient ranged from 0.86 to 0.88, indicating that the samples overall had, even without correction for sex specific autosomal DNA methylation differences and age [17, 30], a very similar distribution of reads along the genome and thus an overall similar methylation profile (Fig. 2a, Additional file 8: Figure S7). The degree of correlation falls within a range observed in previous inter-individual methylome profiling studies, especially considering the whole genome MeDIP-seq approach [31–35]. Whereas this suggests that there are no gross changes in the methylation profile, it does not imply that there are no loci with genetic or environmental driven methylation differences.
Fig. 2.
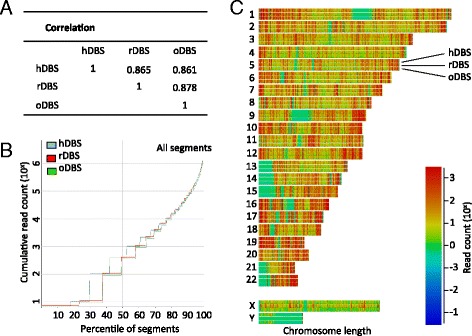
Global methylome comparison. a Three-way Pearson’s correlation between hDBS, rDBS and oDBS. b Segment trend plot depicting the accumulated number of reads (log transformed) as a function of all segments ranging from the lowest to the highest covered percentile for each of the three samples (hDBS, rDBS and oDBS). c Genome-wide domainogram showing the absolute number of all segments in the hDBS, rDBS and oDBS samples along the chromosomes using a color scheme from no segments (green) to multiple segments (dark red)
We looked in greater detail at the distribution of reads along genomic segments defined as 500 bp sliding windows with an overlap of 250 bp. The correlation was normalized only to the total number of reads and log transformed. Cumulating the number of reads from the lowest covered percentile of the genome to the highest should be largely identical between sets of the same type despite defined regional differences such as differently methylated regions (DMRs). The cumulative paths for all three sets appeared much alike, suggesting that no major technically or biologically introduced differences were present, such as large uncovered regions (Fig. 2b). The inconsistency at the lower end of the plot can likely be attributed to the low number of absolute counts of reads. Depicting the read density along the chromosomes in a domainogram again indicated that no systematic bias was evident comparing the three DBS samples. That is, the regional density of reads per segment indicated by a continuum from green (read deserts) to red is overall identical (Fig. 2c). As a check for biological validity, the X chromosome in rDBS (female) had a higher read density as expected, due to X chromosome inactivation, compared to hDBS and oDBS (both males). In addition, no coverage on the Y chromosome in the rDBS sample was observed, as expected.
Canonical DNA methylation patterns at defined genomic regions
An additional quality control step included a more detailed investigation of the specific methylation patterns around known genomic features. This was achieved by calculating the average read count over regions of interest using the 500 bp sliding windows with a 250 bp overlap. Profiling over mRNA transcribed genomic regions ±2 kb showed that the pattern was largely indistinguishable between the three sets. Moreover, a sharp and canonical drop in methylation level around transcription start sites (TSSs) was evident as well as a gradual increase in the methylation throughout the gene body (Fig. 3a). Likewise, displaying mean read count over CpG islands (CGIs) ±1 kb illustrated an overall hypomethylation at CGIs (Fig. 3b).
Fig. 3.
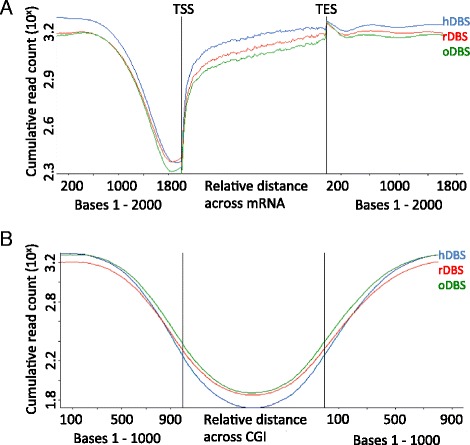
Comparison of the methylation pattern at transcription start site and CpG islands. Read count of 500 bp sliding segments (250 bp overlap) across genetic regions corresponding to a mRNA ±2 kb on either side and b CGI ±1 kb on either side. TSS transcription start site, TES transcription end site
About one-third of the genomic DNA methylation occurs in repetitive elements such as retrotransposons [36]. Thus, analysis of the methylation level in such elements can be viewed as a proxy for the global genomic methylation status. Bisulfite pyrosequencing of three promoter CpG sites within the highly repetitive long interspersed element 1 (LINE-1) was performed on all three samples (Additional file 9: Figure S8). The LINE-1 CpG sites are normally heavily methylated to prevent retrotransposition. In all our DSB samples, all three sites were highly methylated and within the range of previously reported LINE-1 methylation levels in blood, approximately 80 % [32, 37].
No apparent systematic change in methylation observed between the DBS samples when clustering based on significant different segments
With the intent to look at regions with significant variation in methylation levels between the three DBS samples, intensity differences with an multiple testing adjusted p value below 0.05 was calculated in a three-way comparison for 500 bp segments individually (Fig. 4a). Of the 1.2 × 107 segments, 5947 (0.05 %) were significantly different between at least two of the three DBS samples and did not appear to aggregate at specific genomic locations (Additional file 10: Figure S9). Segments were grouped to cover promoter regions defined as a 2 kb stretch upstream of the TSS, segments overlapping with mRNA transcribed regions and CGIs. A percent-wise equivalent number of significantly different segments was found in each group. Hence, of the 4.2 × 105 segments covering promoter regions, 287 (0.07 %) were significantly different. Of these, 74 were located on the sex chromosomes.
Fig. 4.
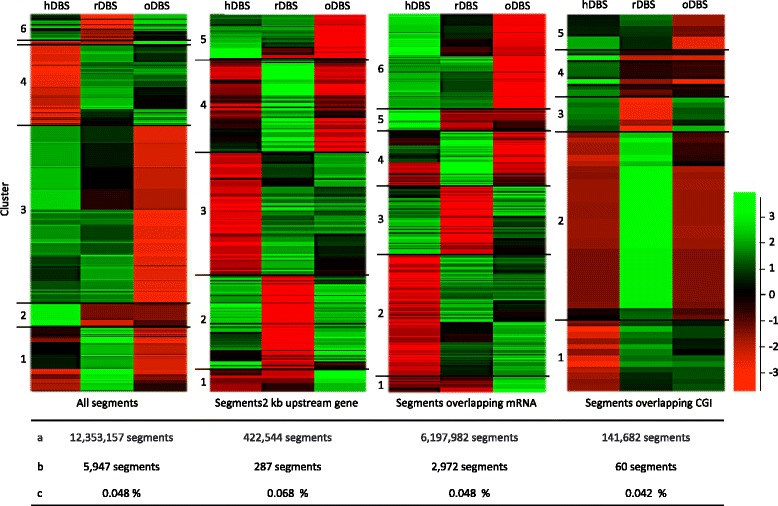
Clustering of segments with significantly different coverage. Three-way unsupervised correlation based clustering of 500 bp segments with a significantly different coverage between at least two of the three samples. Hierarchical clusters are shown with R > 0.7. The segments are per-segment normalized (actual segment values minus the median value of that segment-set across all data stores). (A) Total number of segments in category; (B) number of segments in cluster.; (C) percentage of cluster containing segments of all segments in category
An unassisted hierarchical clustering of the significantly different segments of each genomic region did not show a clear systematic grouping. There was no general tendency of reduced read density in the archived DBSs compared to the control. Subdividing the clusters, based on a threshold for correlation set to 0.7, divided the cluster into five-six groups. In none of the groups did the segments aggregate at specific genomic locations.
Only few differentially methylated windows are common for the archived DBS samples
To further analyze the occurrence of systematic changes to the methylome upon storage, we calculated the differentially methylated windows between hDBS and a merged rDBS/oDBS set using MEDIPS excluding sex chromosomes. Applying an adjusted p value ≤0.1 returned 149 significant differently methylated windows, which where reduced to 97 after merging of adjacent windows. A further collapse of the 97 regions spaced less than 10 kb apart produced 57 loci (Fig. 5a). Even at this relaxed threshold with increased risk of false-positives, a modest amount of significant differently windows were detected. For comparison, a more stringent threshold with p value ≤0.01 generated 70 significant differently methylated windows. Annotating the 97 merged windows showed that about three-quarters were located in intergenic regions, whereas ten windows were located in rRNA gene clusters and nine intragenic of which three were hypermethylated in hDBS (Fig. 5b, c).
Fig. 5.
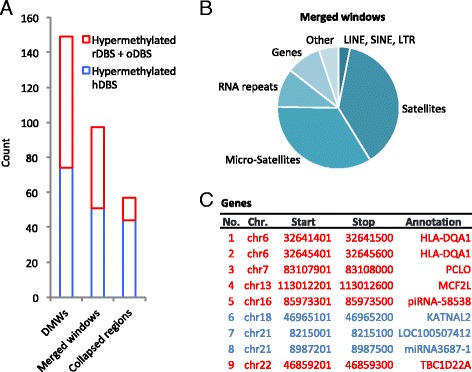
Differentially methylated windows. a Bar chart displaying windows of 100 bp with significantly different coverage between hDBS and the merged sample set rDBS + oDBS individually (149), merged if adjacent (97), collapsed if ≤10 kb apart (57). b Subdivision of merged windows into genetic categories. c Chromosomal position and description of the intragenic merged windows. Marked in red indicates hypermethylated in the combined rDBS + oDBS sample set and marked in blue hypermethylated in hDBS. HLA-DQA1: Major histocompatibility complex, class II, DQ alpha 1; PCLO: Piccolo presynaptic cytomatrix protein; MCF2L: MCF.2 cell line derived transforming sequence like; KATNAL2: katanin p60 subunit A like 2; TBC1D22A : TBC1 domain family member 22A; LOC100507412: uncharacterized long non-coding RNA LOC100507412; piRNA-58538: piwiRNA 58538; miRNA3687-1: microRNA 3687-1
Discussion
Retrospective methylome studies based on DBSs from biobanked filter cards hold great promise but are potentially hampered by poorer DNA quality and limited availability. In this study, we show that prolonged storage of DBSs, even at room temperature, has little overall influence on the methylome and that methylome-wide coverage can be obtained using DNA extracted from a single 3.2 mm punch, allowing for multiple studies on the same DBS. It has previously been shown that 50 ng of input DNA is sufficient for generating methylation libraries with adequate complexity using MeDIP-seq [38]. A further decrease to 1 ng input is possible, but at the expense of an increased rate of duplicates and immunoprecipitation bias, which leads to loss of data and potentially false-positive findings [22, 39].
From single 3.2 mm DBS punches, we consistently extracted good quality DNA and with no quantitatively loss following prolonged storage, emphasizing the robustness of the extraction methodology applied. In the agnostic search for epigenetic markers or risk variants, a genome-wide approach such as MeDIP may prove valuable. Importantly, DNA extracted in this work proved adequate for MeDIP-seq. The genomic distribution of reads and coverage showed a high positive correlation between the three DBS samples. Moreover, the expected sharp and canonical drop in methylation level around the transcription start sites, as well as the gradual increase in methylation throughout the gene body was evident in all samples [40, 41].
A limitation of the study is that the compared filter cards were not from the same individual. The samples could also not be matched for sex or age [42, 43]. A follow-up study with more and better-matched samples is, therefore, warranted. Despite this, we were able to show that the overall methylation pattern is maintained in DNA isolated from old filter cards stored at room temperature and that this good quality of data can be obtained using MeDIP-seq on low amounts of DNA input. The analysis showed that only very few regions (<0.05 %) differed significantly between at least any two of the three DBS samples. Suggesting that the level of technical or storage-introduced noise is limited. Furthermore, although the three DBS samples originate from different individuals, and thus is not a longitudinal study, the comparison did not reveal any systematic change over the time stored.
Combining the archived DBS samples into one sample set and comparing this to the freshly prepared DBS defined only 149 significant 100 bp windows indicating that even without removal of confounding effects the difference appears limited. The majority of the significant windows were located in repetitive regions. This may be attributed not only to high biological variance at these sites, especially in pericentromeric satellites and subtelomeric elements, but also to alignment issues [44–46]. A previous inter-individual comparison based on MeDIP-seq and peripheral blood monocytes found differences to be highly enriched in repetitive elements, and especially in satellites [33]. Pyrosequencing of three CpGs in the retrotransposable element LINE-1, a surrogate marker for global wide DNA methylation status, showed no apparent difference between the three DBS samples. This may again indicate that an overall loss of methylation has not taken place in the archived DBS samples.
It is important to note that defining true environment-specific or inter-individual DMRs requires that cell type composition and the genetic profile are taken into account. Means of cell composition correction in silico has been devised for array-generated data based on cell type specific methylation patterns [47, 48]. On the other hand, incorporation of genotyping data permits exploration of methylation quantitative trait loci (mQTLs), being a measure of the relationship between genetic polymorphisms and methylation [21]. Notably, a considerable overlap of mQTLs across-tissue has been documented [15, 49]. Also, this allows for examination of gene and environment interactions and the effects on the epigenome [50].
Conclusions
In conclusion, we demonstrated that the methylome profile is highly comparable between DNA from archived DBS samples and DNA from a freshly prepared DBS sample. Moreover, we show, to our knowledge, for the first time that MeDIP-seq can be performed on neonatal screening cards even with small input amounts. Together, our findings add to the notion that archival DBS samples can be used for sequencing-based epigenome studies, thereby holding great potential in epidemiological research.
Methods
Guthrie cards
hDBS: Approximately 70 μL freshly drawn peripheral blood was spotted on Whatman 903 Protein Saver cards (GE Healthcare Life Sciences, Piscataway, NJ, USA). The cards were dried and stored at −20°C.
rDBS/oDBS: Randomly selected from anonymous DBS samples stored at room temperature for 4 years (rDBS) and 16 years (oDBS), respectively. For both, peripheral blood was spotted on the Whatman 903 Protein Saver cards by means of a finger prick.
DNA extraction
Discs were punched in triplicates from the DBSs by the use of a puncher. Samples stored at RT were incubated in 180 μl PBS at 4 °C overnight in order to increase DNA extraction yield. Following the overnight incubation, PBS was removed and all samples were subjected to the DNA extraction protocol. DNA extraction was performed according to a previously published protocol [51], with introduction of additional centrifugation and ethanol washing steps. Concentration of the extracted DNA was measured using the Qubit instrument and the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Cleveland, OH, USA).
MeDIP-seq
Extracted genomic DNA (gDNA) was sonicated in a Pico Bioruptor (Diagenode, Seraing, Belgium) at 10 ng/μL for 13 cycles of 30 s on 30 s off to a mean fragment size of 180 bp. Fragment length distribution was assessed by microelectrophoresis using the Qiaxcel instrument and a high-resolution gel cartridge (Qiagen, Hilden, Germany). Sonicated gDNA (in the range of 93–111 ng) was further used for end-repair and adaptor ligation employing the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, MA, USA). The reaction mix was purified using Ampure XP beads (Beckman-Coulter, CA, USA) after which methylated DNA immunoprecipitation (MeDIP) was set up on the SX-8G-IP-Star Compact robot (Diagenode) according to manufacturer’s instructions applying the Auto MeDIP kit (Diagenode) and including unmethylated and methylated spike-in controls. Effectively between 60 and 70 ng of adaptor-ligated DNA was used in the MeDIP process. Antibody incubation was performed at 4 °C for 15 h. Immunoprecipitated samples were magnetically purified on the robot using the Auto iPure v2 kit (Diagenode) according to the manufacturer’s instructions. Recovery and enrichment was evaluated by qPCR using primer sets specific for the spike-in controls. Minimum criteria were set to 10 % recovery and 25-fold enrichment. Based on the recovery rate, samples were PCR-amplified at 13 cycles (oDBS) or 14 cycles (hDBS and rDBS) using multiplex oligos (New England Biolabs, MA, USA). Samples were size-selected with Ampure XP beads on the SX-8G-IP-Star Compact robot (Diagenode) using a total of 90 μL beads per sample and eluted in 25 μL DNase-free H2O. Purity and fragment length distribution was evaluated on the Qiaxcel instrument. Post-amplification enrichment was verified by qPCR using primer pairs targeting the endogenous hypermethylated promoter region of testis specific histone 2B (TSH2B) (Diagenode, cat.nr. C17011041) or the hypomethylated TSS of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Diagenode, cat.nr. C17011047).
Sequencing and bioinformatics
Samples were PE50 sequenced on a single lane on a HiSeq2000 instrument (Illumina, CA, USA), generating around 64–71 million clean reads per sample. The reads were cleaned using Soapnuke 1.5.0 tool (BGI, Shenzhen, China) applying the following filters: remove reads with adaptors, remove reads with >10 % unknown reads and remove reads with >50 % low quality bases (Q score <5). Clean reads only qualify if Q20 ≥ 85 %.
Using the Galaxy platform, clean reads were groomed and aligned to the human genome (build hg38) using BWA. SAM files were converted to BAM files and filtered to include only de-duplicated uniquely mapped reads resulting in ~54–58 million reads per sample. BAM files were imported to R and SeqMonk v0.32.1 (Barbraham Institute). The R package MEDIPS was used for quality control, genomic coverage estimation and differential coverage analysis [52]. In SeqMonk segments were generated by quantitation of read counts in genetic windows corrected for total counts and with duplicates ignored. Filters include subdivision based on defined features (e.g. genes and CGIs) on any strand and calculating significantly different segments in pair-wise comparisons with a minimum intensity difference p value threshold of 0.05 with multiple testing correction (Bonferroni) applied. UCSC Genome Browser was used for visualization and interpretation of genomic regions.
Pyrosequencing
Genomic DNA from two 3.2 mm DBS punches were pooled and bisulfite-treated using the EpiTect Bisulfite Kit (Qiagen) following the manufacturer’s protocol. A 146 bp fragment of the LINE-1 promoter was amplified by PCR from 100 ng bisulfite-treated DNA and preprocessed for sequencing using the PyroMark Q24 LINE-1 kit (Qiagen) following the manufacturer’s instructions. Prepared PCR products were sequenced on a PyroMark Q24 Advanced and analyzed on the appertaining software (Qiagen) according to the manufacturer’s instructions.
Abbreviations
BWA, Burrows-Wheeler aligner; CGI, CpG Island; DBS, dried blood spots; DMR, differentially methylated region; DNSB, Danish Neonatal Screening Biobank; EWAS, epigenome-wide association study; GWAS, genome-wide association study; LINE-1, long interspersed nuclear element 1 ; MeDIP, methylated DNA immunoprecipitation; mQTLs, methylation quantitative trait loci; MWAS, methylome-wide association study; TSS, transcription start site; WGS, whole genome sequencing
Acknowledgements
We would like to thank Marit Nielsen and Tina Fuglsang Daugaard for the expert technical assistance at Translational Neuropsychiatric Unit, Aarhus University Hospital and Department of Biomedicine, University of Aarhus, respectively.
Funding
This work was funded by the Toyota Foundation and The Lundbeck Foundation, Denmark.
Availability of data and materials
The MeDIP-seq dataset supporting the conclusions of this article is available upon request.
Authors’ contributions
NHS, AB and OM conceived and designed the experiments. LC collected and supplied samples. NHS and AS performed the experiments. NHS, ALN, MN and LC analyzed the data. NHS drafted the manuscript. All authors read and approved the final manuscript.
Competing interest
The authors declare that they have no competing interests.
Consent for publication
Not applicable
Ethics approval and consent to participate
All procedures were performed in accordance with relevant regulation and approved by the Danish health research ethical committee (license no.: S-VF-19980072).
Additional files
Number of experiments achievable per DBS by different methods. Part of a DBS necessary to meet the minimum input requirements of commonly used DNA methylation assessing methods based on an average yield of 13 ng DNA/mm2 DBS. (PDF 38 kb)
Robust extraction of genomic DNA from hDBS, rDBS and oDBS. Box-whisker plot depicting median and 1.5 interquartile range (IQR) of gDNA extractions in triplicates at three independent runs for all three filter cards hDBS, rDBS and oDBS. (PDF 42 kb)
Capillary gel electrophoresis electropherograms. DBS extracted DNA was sonicated to a mean length of ~180 bp (upper left). Final hDBS, rDBS and oDBS MeDIP libraries assessed for fragment length distribution and purity. Region of interest mark the targeted length interval of 200 to 500 bp. (PDF 239 kb)
Filtering applied to raw reads. Raw reads were filtered by removal of reads i) containing adaptor sequence (“filter adaptor”), ii) with ambiguous bases (≥10 % N per read; “filter N”) and iii) with poor quality (≥50 % bases with Q score <5; “filter low quality”). For a sample to qualify the Q score must be ≥20 for ≥85 % of the reads. (PDF 31 kb)
Quality of sequence data: (A–C) Data output overview. (D–F) Sequencing quality (Q) score across the PE50 reads. (G–I) GC content in the MeDIP enriched samples (red line) compared to the theoretical distribution (blue line). (A, D, G) oDBS, (B, F, H) rDBS, and (C, F, I) hDBS. (PDF 619 kb)
Sequencing quality. (A–C) Saturation analysis indicating adequate complexity and reproducibility of the mapped reads in the hDBS, rDBS and oDBS sample sets compared to the reference genome. (D–F) Calibration plot showing correct normalization of reads per window in the hDBS, rDBS and oDBS sample sets as a function of CpG density (chromosome 1 only). (PDF 380 kb)
Segment distribution along the repeat-rich gene SFI1. Visualization of 500 bp segments (250 bp sliding window) at the SFI1 locus for hDBS, rDBS and oDBS. (PDF 113 kb)
Pearson’s correlation analysis of hDBS, rDBS and oDBS. Scatter plot depicting the pair-wise Pearson’s correlation of the genome-wide coverage (log transformed number of reads) of hDBS, rDBS and oDBS. (PDF 124 kb)
Pyrosequencing of three promoter CpGs of LINE-1. (A) Table listing methylation percentage at each CpG. (B) Representative diagram of a single hDBS pyrosequencing reaction. All reactions were performed in triplicate and data is shown as mean ± SD. (PDF 89 kb)
Domainogram of all significantly different segments genome. Domainogram showing the genomic distribution of segments constituting the “all segments” hierarchical cluster. (PDF 859 kb)
Contributor Information
Nicklas H. Staunstrup, Email: nhs@clin.au.dk
Anna Starnawska, Email: as@biomed.au.dk.
Mette Nyegaard, Email: nyegaard@biomed.au.dk.
Lene Christiansen, Email: lchristiansen@health.sdu.dk.
Anders L. Nielsen, Email: aln@biomed.au.dk
Anders Børglum, Email: anders@biomed.au.dk.
Ole Mors, Email: nielmors@rm.dk.
References
- 1.Putiri EL, Robertson KD. Epigenetic mechanisms and genome stability. Clin Epigenetics. 2011;2:299–314. doi: 10.1007/s13148-010-0017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol. 2010;28:1079–88. doi: 10.1038/nbt.1684. [DOI] [PubMed] [Google Scholar]
- 3.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 5.Nishioka M, Bundo M, Kasai K, Iwamoto K. DNA methylation in schizophrenia: progress and challenges of epigenetic studies. Genome Med. 2012;4:96. doi: 10.1186/gm397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 7.Guerrero-Preston R, Goldman LR, Brebi-Mieville P, Ili-Gangas C, Lebron C, Witter FR, Apelberg BJ, Hernandez-Roystacher M, Jaffe A, Halden RU, et al. Global DNA hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics. 2010;5:539–46. doi: 10.4161/epi.5.6.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suter M, Ma J, Harris A, Patterson L, Brown KA, Shope C, Showalter L, Abramovici A, Aagaard-Tillery KM. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6:1284–94. doi: 10.4161/epi.6.11.17819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao-Lei L, Massart R, Suderman MJ, Machnes Z, Elgbeili G, Laplante DP, Szyf M, King S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS One. 2014;9:e107653. doi: 10.1371/journal.pone.0107653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melas PA, Rogdaki M, Osby U, Schalling M, Lavebratt C, Ekstrom TJ. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012;26:2712–8. doi: 10.1096/fj.11-202069. [DOI] [PubMed] [Google Scholar]
- 11.Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci U S A. 2008;105:13614–9. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warner MJ, Ozanne SE. Mechanisms involved in the developmental programming of adulthood disease. Biochem J. 2010;427:333–47. doi: 10.1042/BJ20091861. [DOI] [PubMed] [Google Scholar]
- 13.Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meaney MJ, Ferguson-Smith AC. Epigenetic regulation of the neural transcriptome: the meaning of the marks. Nat Neurosci. 2010;13:1313–8. doi: 10.1038/nn1110-1313. [DOI] [PubMed] [Google Scholar]
- 15.Smith AK, Kilaru V, Kocak M, Almli LM, Mercer KB, Ressler KJ, Tylavsky FA, Conneely KN. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics. 2014;15:145. doi: 10.1186/1471-2164-15-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aberg KA, Xie LY, McClay JL, Nerella S, Vunck S, Snider S, Beardsley PM, van den Oord EJ. Testing two models describing how methylome-wide studies in blood are informative for psychiatric conditions. Epigenomics. 2013;5:367–77. doi: 10.2217/epi.13.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–83. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollegaard MV, Grauholm J, Nielsen R, Grove J, Mandrup S, Hougaard DM. Archived neonatal dried blood spot samples can be used for accurate whole genome and exome-targeted next-generation sequencing. Mol Genet Metab. 2013;110:65–72. doi: 10.1016/j.ymgme.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, den Dekker HT, Husby A, Sevelsted A, Faura-Tellez G, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46:51–5. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
- 20.Hollegaard MV, Grauholm J, Norgaard-Pedersen B, Hougaard DM. DNA methylome profiling using neonatal dried blood spot samples: a proof-of-principle study. Mol Genet Metab. 2013;108:225–31. doi: 10.1016/j.ymgme.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Beyan H, Down TA, Ramagopalan SV, Uvebrant K, Nilsson A, Holland ML, Gemma C, Giovannoni G, Boehm BO, Ebers GC, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 2012;22:2138–45. doi: 10.1101/gr.134304.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aberg KA, Xie LY, Nerella S, Copeland WE, Costello EJ, van den Oord EJ. High quality methylome-wide investigations through next-generation sequencing of DNA from a single archived dry blood spot. Epigenetics. 2013;8:542–7. doi: 10.4161/epi.24508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joo JE, Wong EM, Baglietto L, Jung CH, Tsimiklis H, Park DJ, Wong NC, English DR, Hopper JL, Severi G, et al. The use of DNA from archival dried blood spots with the Infinium HumanMethylation450 array. BMC Biotechnol. 2013;13:23. doi: 10.1186/1472-6750-13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruickshank MN, Oshlack A, Theda C, Davis PG, Martino D, Sheehan P, Dai Y, Saffery R, Doyle LW, Craig JM. Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Med. 2013;5:96. doi: 10.1186/gm500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bundo M, Sunaga F, Ueda J, Kasai K, Kato T, Iwamoto K. A systematic evaluation of whole genome amplification of bisulfite-modified DNA. Clin Epigenetics. 2012;4:22. doi: 10.1186/1868-7083-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghantous A, Saffery R, Cros MP, Ponsonby AL, Hirschfeld S, Kasten C, Dwyer T, Herceg Z, Hernandez-Vargas H. Optimized DNA extraction from neonatal dried blood spots: application in methylome profiling. BMC Biotechnol. 2014;14:60. doi: 10.1186/1472-6750-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark C, Palta P, Joyce CJ, Scott C, Grundberg E, Deloukas P, Palotie A, Coffey AJ. A comparison of the whole genome approach of MeDIP-seq to the targeted approach of the Infinium HumanMethylation450 BeadChip((R)) for methylome profiling. PLoS One. 2012;7:e50233. doi: 10.1371/journal.pone.0050233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen G, Yu D, Chen J, Cao R, Yang J, Wang H, Ji X, Ning B, Shi T. Re-annotation of presumed noncoding disease/trait-associated genetic variants by integrative analyses. Sci Rep. 2015;5:9453. doi: 10.1038/srep09453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feber A, Wilson GA, Zhang L, Presneau N, Idowu B, Down TA, Rakyan VK, Noon LA, Lloyd AC, Stupka E, et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome Res. 2011;21:515–24. doi: 10.1101/gr.109678.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Morgan M, Hutchison K, Calhoun VD. A study of the influence of sex on genome wide methylation. PLoS One. 2010;5:e10028. doi: 10.1371/journal.pone.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, Meissner A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28:1106–14. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu HC, Wang Q, Chung WK, Andrulis IL, Daly MB, John EM, Keegan TH, Knight J, Bradbury AR, Kappil MA, et al. Correlation of DNA methylation levels in blood and saliva DNA in young girls of the LEGACY Girls study. Epigenetics. 2014;9:929–33. doi: 10.4161/epi.28902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen H, Qiu C, Li J, Tian Q, Deng HW. Characterization of the DNA methylome and its interindividual variation in human peripheral blood monocytes. Epigenomics. 2013;5:255–69. doi: 10.2217/epi.13.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, Kobor MS. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci U S A. 2012;109(Suppl 2):17253–60. doi: 10.1073/pnas.1121249109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, Yang AS. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet. 2009;18:4808–17. doi: 10.1093/hmg/ddp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irahara N, Nosho K, Baba Y, Shima K, Lindeman NI, Hazra A, Schernhammer ES, Hunter DJ, Fuchs CS, Ogino S. Precision of pyrosequencing assay to measure LINE-1 methylation in colon cancer, normal colonic mucosa, and peripheral blood cells. J Mol Diagn. 2010;12:177–83. doi: 10.2353/jmoldx.2010.090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, Beck S, Butcher LM. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat Protoc. 2012;7:617–36. doi: 10.1038/nprot.2012.012. [DOI] [PubMed] [Google Scholar]
- 39.Zhao MT, Whyte JJ, Hopkins GM, Kirk MD, Prather RS. Methylated DNA immunoprecipitation and high-throughput sequencing (MeDIP-seq) using low amounts of genomic DNA. Cell Reprogram. 2014;16:175–84. doi: 10.1089/cell.2014.0002. [DOI] [PubMed] [Google Scholar]
- 40.Guo H, Zhu P, Guo F, Li X, Wu X, Fan X, Wen L, Tang F. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat Protoc. 2015;10:645–59. doi: 10.1038/nprot.2015.039. [DOI] [PubMed] [Google Scholar]
- 41.Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, Yan J, Ren X, Lin S, Li J, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–10. doi: 10.1038/nature13544. [DOI] [PubMed] [Google Scholar]
- 42.McCarthy NS, Melton PE, Cadby G, Yazar S, Franchina M, Moses EK, Mackey DA, Hewitt AW. Meta-analysis of human methylation data for evidence of sex-specific autosomal patterns. BMC Genomics. 2014;15:981. doi: 10.1186/1471-2164-15-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell. 2015;14:924–32. doi: 10.1111/acel.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flanagan JM, Popendikyte V, Pozdniakovaite N, Sobolev M, Assadzadeh A, Schumacher A, Zangeneh M, Lau L, Virtanen C, Wang SC, et al. Intra- and interindividual epigenetic variation in human germ cells. Am J Hum Genet. 2006;79:67–84. doi: 10.1086/504729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandovici I, Kassovska-Bratinova S, Loredo-Osti JC, Leppert M, Suarez A, Stewart R, Bautista FD, Schiraldi M, Sapienza C. Interindividual variability and parent of origin DNA methylation differences at specific human Alu elements. Hum Mol Genet. 2005;14:2135–43. doi: 10.1093/hmg/ddi218. [DOI] [PubMed] [Google Scholar]
- 46.Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, et al. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012;22:557–67. doi: 10.1101/gr.131383.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maksimovic J, Gagnon-Bartsch JA, Speed TP, Oshlack A. Removing unwanted variation in a differential methylation analysis of Illumina HumanMethylation450 array data. Nucleic Acids Res. 2015;43:e106. doi: 10.1093/nar/gkv526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10:1024–32. doi: 10.1080/15592294.2015.1100786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS, Bradley B, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.St Julien KR, Jelliffe-Pawlowski LL, Shaw GM, Stevenson DK, O’Brodovich HM, Krasnow MA, Stanford BPDSG High quality genome-wide genotyping from archived dried blood spots without DNA amplification. PLoS One. 2013;8:e64710. doi: 10.1371/journal.pone.0064710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 2014;30:284–6. doi: 10.1093/bioinformatics/btt650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The MeDIP-seq dataset supporting the conclusions of this article is available upon request.