

Abstract
Background:
Previous studies have suggested that β1-receptor blockers benefit septic shock patients. This study aimed to determine whether β1-receptor blockers benefit tissue perfusion in sepsis and to identify parameters to reduce the risk of this drug in sepsis.
Methods:
Consecutive septic shock patients were recruited from the Intensive Care Unit of Peking Union Medical College Hospital within 48 h of diagnosis. All patients were hemodynamically stable and satisfactorily sedated with a heart rate (HR) ≥100 beats/min. Esmolol therapy achieved the target HR of 10–15% lower than the baseline HR. Clinical and physiological data of patients were collected prospectively within 1 h prior to esmolol therapy and 2 h after achieving the targeted HR.
Results:
Sixty-three patients were recruited. After esmolol therapy, blood pressure was unaltered, whereas stroke volume (SV) was increased compared with before esmolol therapy (43.6 ± 22.7 vs. 49.9 ± 23.7 ml, t = −2.3, P = 0.047). Tissue perfusion, including lactate levels (1.4 ± 0.8 vs. 1.1 ± 0.6 mmol/L, t = 2.6, P = 0.015) and the central venous-to-arterial carbon dioxide difference (5.6 ± 3.3 vs. 4.3 ± 2.2 mmHg, t = 2.6 P = 0.016), was also significantly decreased after esmolol therapy. For patients with increased SV (n = 42), cardiac efficiency improved, and esmolol therapy had a lower risk for a decrease in cardiac output (CO). Therefore, pretreatment cardiac systolic and diastolic parameters with (n = 42)/without (n = 21) an increase in SV were compared. Mitral lateral annular plane systolic excursion (MAPSElat) in patients with increased SV was significantly higher than that in those without increased SV (1.3 ± 0.3 vs. 1.1 ± 0.2 cm, t = 2.4, P = 0.034).
Conclusions:
SV of septic shock patients is increased following esmolol therapy. Although CO is also decreased with HR, tissue perfusion is not worse. MAPSElat can be used to predict an increase in SV before esmolol use.
Trial Registration:
ClinicalTrials.gov, NCT01920776; https://clinicaltrials.gov/ct2/show/NCT01920776?term=NCT01920776&rank=1.
Keywords: Echocardiography, Esmolol, Myocardial Depression, Septic Shock
INTRODUCTION
Septic shock can elicit a wide range of stress-induced activation responses of the sympathetic nervous system, as well as humoral responses. The autonomic nervous system link between the nervous and immune systems plays a role in the pathogenesis of septic shock.[1,2] Tachycardia persisting after fluid resuscitation and control of agitation and pain may indicate an inappropriate degree of sympathetic activation.[3]
β-blockers may be effective in controlling heart rate (HR) by modulating the intrinsic response to β-adrenergic overstimulation.[4] An increasing number of reports have been published suggesting the advantageous effects of β-blockers in septic shock[5] and other acute critical illnesses, such as severe trauma,[6] traumatic brain injury,[7,8] or burns,[9] indicating a beneficial effect on morbidity and mortality.
In light of the growing evidence of an association between high sympathetic stress and sepsis-induced myocardial depression,[3,10] administration of β-blocking agents could be beneficial. However, excessive β-blocker dosing may have negative inotropic effects and may lead to inappropriately low cardiac output (CO) and pulmonary congestion. Hence, there is much debate over the use of β-blocker.[11,12] Therefore, β-blockers should be titrated in septic patients, and close hemodynamic monitoring is warranted for the early detection of potentially negative effects. β1-receptor blockade appears to be superior to a nonselective blockade because β2-receptor activation has cardioprotective properties.
It has been reported that β1-receptor blockers are beneficial for patients with septic shock.[5] The present study aimed to determine whether β1-receptor blockers are also beneficial for tissue perfusion in patients. In addition, negative inotropic effects induced by drugs concern patients and clinicians. Therefore, this study also aimed to identify parameters that could help reduce the risk of using drugs in septic patients. This prospective study investigated the effects of using a β1-adrenoceptor blocker to reduce HR in septic shock patients below a predefined threshold and used transthoracic echocardiography (TTE) monitoring to identify parameters for reducing the risk of using β1-adrenoceptor blockers.
METHODS
Patients
Ethical approval for this study (No. S-617) was obtained by the Ethics Committee of Peking Union Medical College Hospital, Beijing, China (Chairman Prof. Jie Chen) on November 5, 2012. Written informed consent was obtained from the patients or their families. The study was registered in http://www. ClinicalTrials.gov. Trial registration: NCT01920776, the study was one of the most important parts. All patients (aged 18 years or older) with septic shock who were admitted to the 30-bed general Intensive Care Unit (ICU) of Peking Union Medical College Hospital between August 2013 and January 2015 were consecutively included in this study.
Patients were included in the study if they met all the following criteria: (1) the diagnosis of septic shock was within 48 h after septic shock; (2) patients were in a relatively stable period of hemodynamics, as defined by no change in the mean arterial pressure of >10% with the same dosage of vasopressors for at least 3 h, and they failed to respond to fluids, which were required for a rapid volume challenge (8 ml/kg of fluid over 30 min) according to the clinical attending physician (except for hypovolemia because HR increases fast); (3) patients were assessed by thoracic echocardiography with initially preserved left ventricular ejection function (LVEF) >45%; (4) patients were sedated with a Richmond agitation–sedation scale score of 0–−3; and (5) HR was ≥100 beats/min. The criteria for exclusion from the study were age <18 years, pregnancy, and using a β-agonist, such as dobutamine.
Treatment
This study was conducted during routine treatment of other patients in the critical care department. Routine arterial and central venous catheterization was performed, and antibiotics were administered at the discretion of the treating clinicians. Critical care clinicians (intensivists, fellows, and residents providing 24-h in-house coverage) cared for all the patients. The study investigators did not influence patient care in the ICU.
A β1-receptor blocker was used (esmolol hydrochloride injection, Qilu Pharmaceutical, Jinan, China) to control HR.
The aim of the interventional therapy was to control the HR of the patients (the target HR was 10–15% lower than the baseline HR). The treatment strategy included an esmolol hydrochloride injection with a loading dose of 20 mg, which was intravenously injected within 1 min. Intravenous pumping of the maintenance dose of the esmolol hydrochloride injection was performed with an initial dose of 25 mg/h, and the HR of patients was evaluated for every 20 min. For patients whose target HR was not achieved, a loading dose of esmolol hydrochloride injection was injected for 1 min and an additional 25 mg/h was added to the maintenance dose until 1 h after the patient achieved the target HR. The clinicians then decided whether the patient should continue the use of esmolol and its dose. Treatment was discontinued if the patients had one or more of the following features: HR was decreased to lower than 70 beats/min; systolic blood pressure was decreased by 30 mmHg compared with the baseline level; mean arterial pressure was decreased by 10 mmHg; severe arrhythmia; and the inability to achieve the target HR after being treated with esmolol at a dose of 400 mg/h for 1 h. The initial treatment (including the infusion speed, doses of vasoactive agents, parameters of the ventilator, and the strategies of using sedatives and analgesics) of the patients was not changed throughout the study. The primary desired outcome was to determine whether esmolol could reduce HR to a lower value than the predefined goal. Secondary desired outcomes included observing the effects of esmolol on CO, stroke volume (SV), and tissue perfusion, and we would be according to the SV increase or not further grouping.
Measurements
Echocardiography
The investigators who performed consecutive measurements (n = 2) were intensive care specialists with an advanced level of experience (Level 2) in critical care echocardiography. Their measurement results were averaged using GE Vivid Q (GE Medical Systems, Fairfield, CN, USA). The levels of echocardiography skills and knowledge were those described in the guidelines issued by the American College of Chest Physicians, the French Society of Intensive Care,[13] and the international expert statement on training standards for critical care ultrasonography.[14]
Left ventricular ejection function and left ventricular shortening fraction
The LVEF and left ventricular shortening fraction were determined using the Teichholz method. This method has less inter-observer variation and is less dependent on an optimal echocardiographic window to obtain measurements than the Simpson method. Left ventricular volume at end diastole (EDV) was obtained from the formula calculated by the echocardiography machine.
Cardiac output, cardiac index, and stroke volume
CO was obtained by multiplication of SV by HR in the left ventricular outflow tract using the Doppler method. SV was determined as follows: SAo × velocity time integral (VTI) of the blood flow signal in the left ventricular outflow tract. The aortic area was calculated as follows: SAo (cm2) = (π × AoD2)/4, where the aortic annulus diameter (AoD) was measured on the long-axis parasternal view. The aortic velocity-time integral (VTIAo) was measured with pulsed Doppler on the five-chamber apical view. SV was calculated by the following formula: SV (ml) = VTIAo × SAo. CO was calculated using the following formula: CO (ml/min) = SV × HR. The cardiac index (CI) was calculated by the following formula: CI (ml·min−1·m−2) = CO/body surface area.
Mitral lateral annular plane systolic excursion and tricuspid annular plane systolic excursion
Left ventricular systolic function was assessed by mitral lateral annular plane systolic excursion (MAPSElat) in the M-Mode. Right ventricular systolic function was assessed by recording the tricuspid annular plane systolic excursion in the M-Mode.
The E-wave, A-wave, and E-to-A ratio
The pulse Doppler profile peak velocities of E and A waves (cm/s) and the E/A ratio were calculated.
E-wave deceleration time (ms)
The E-wave was calculated as the time interval between the peak E-wave and the zero intercept of the regression line of the deceleration profile.
Ea and Aa waves
Ea and Aa waves (cm/s) were assessed and recorded by tissue Doppler imaging at the mitral lateral annulus in the four-chamber apical view for diastolic peak E and A. The E/Ea ratio was calculated.
Tei index
The Tei index was measured as follows: E- and A-waves were obtained by pulse Doppler at the four-chamber apical view. The time from the end of the A-wave to the start of the E-wave of the next cardiac cycle was recorded as “a.” The ejection time in the frequency spectrum of the left ventricular outflow tract was measured with pulse Doppler at the five-chamber apical view and recorded as “b.” Tei was then calculated as (a − b)/b.
Isovolumetric relaxation time
The isovolumetric relaxation time was measured at the five-chamber apical view as follows: Pulse Doppler was used to measure the frequency spectrum at the systolic and diastolic phases. The time from the end of the systolic phase (the end of the frequency spectrum of the left ventricular outflow tract) to the start of the diastolic phase (start of the E-wave) was calculated and recorded as the isovolumetric relaxation time.
Vital signs
The vital signs of the patients, including blood pressure, HR, urine volume, and the peripheral perfusion index (PFi),[15] were monitored before and after the administration of esmolol. A change in finger PFi is due to blood volume pulsations, dispensability of the vascular wall, and intravascular pulse pressure. The PFi was measured in the finger by using the IntelliVue MP70 monitor (Philips Medical Systems, Boblingen, Germany). Arterial blood was collected to measure the levels of cardiac troponin I, creatine kinase MB isoenzyme, creatine kinase, and N-terminal pro-brain natriuretic peptide. Arterial blood gases and superior vena cava blood gases of the patients were also monitored. Parameters of the acid–base equilibrium, including arterial blood lactate, pH, HCO3−, and anion gap (AG) were measured. The AG corrected for albumin (AGcorrected) was calculated as follows: AGcorrected (mmol/L) = AG + 0.25 × (40 − ALB) (g/L). Superior vena cava oxygen saturation was measured. The central venous-to-arterial carbon dioxide difference was calculated as follows:[16] Pcv-a CO2 = PvCO2 − PaCO2.
The results of TTE, biochemical tests, and blood gas analysis for all patients were collected at 1 h before drug therapy and within 2 h after achieving the target HR.
Statistical analysis
SPSS 16.0 (SPSS Inc., Chicago, IL, USA) software was used for statistical analysis. All quantitative data had a normal distribution and are shown as the mean and standard deviation. The paired t-test was used to compare data before and after drug therapy. The independent t-test was used to compare data between patients with and without an increase in SV. The receiver operating characteristic curve (ROC) was used to analyze the parameters that could predict an increase in SV and estimate the cutoff values. An area under the curve (AUC) >0.7 was considered significant and an AUC > 0.9 was considered as having a high accuracy.
RESULTS
Patients
Among 116 patients with septic shock, 11 who had unstable hemodynamics within 48 h, 21 who had an HR <100 beats/min, and 17 who had an LVEF ≤45% were excluded. The remaining 67 patients met the inclusion criteria, among whom two refused to participate and two withdrew from the study because their echocardiographic parameters were immeasurable because of technical difficulties. Therefore, 63 patients were finally included in the present study [Figure 1]. The general characteristics of the patients are shown in Table 1.
Figure 1.
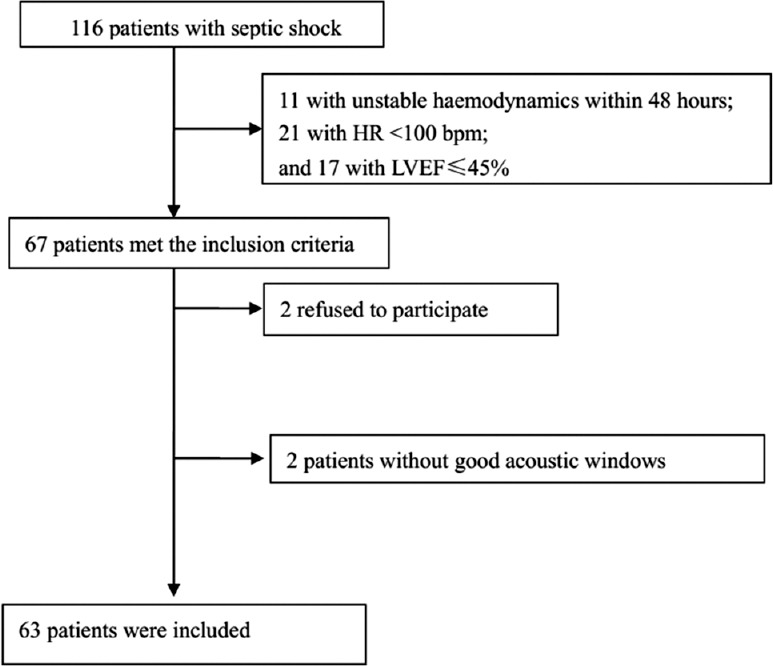
Flow chart of the study.
Table 1.
General characteristics and outcomes of the patients
| Characteristics | Results |
|---|---|
| Number of the patients | 63 |
| Age (years) | 50.4 ± 19.0 |
| Sex (male, %) | 47.6 |
| APACHE II | 15.0 ± 5.1 |
| Mechanical ventilation (%) | 68.8 |
| Infections (%) | |
| Lung | 18.8 |
| Blood | 12.5 |
| Abdominal cavity | 43.8 |
| Urinary system | 9.4 |
| Others | 15.6 |
| Baseline cardiac function (%) | |
| NYHA I | 54.0 |
| NYHA II | 36.5 |
| NYHA III | 9.5 |
| NE dose (µg·kg−1·min−1) | 0.47 ± 0.17 |
| Maximum dose of esmolol (mg/h) | 113.6 ± 89.7 |
| Total dose of esmolol (mg) | 187.8 ± 110.7 |
| Time to achieve target heart rate (h) | 1.7 ± 0.3 |
| Outcomes | |
| Hospital stay (days) | 22.7 ± 17.0 |
| ICU stay (days) | 12.0 ± 16.5 |
| 28-day mortality rate (%) | 6.3 |
| In-hospital mortality rate (%) | 7.8 |
ICU: Intensive Care Unit; APACHE: Acute Physiology and Chronic Health Evaluation; NYHA: New York Heart Association; NE: Norepinephrine.
Changes in vital signs, tissue oxygen metabolism, and myocardial enzymes after using esmolol
Blood pressure of the patients did not significantly change after the use of esmolol. In addition, there were no significant changes in systolic blood pressure, diastolic blood pressure, mean arterial pressure, and pulse pressure with esmolol therapy. HR of the patients was significantly decreased (107.8 ± 8.7 vs. 86.2 ± 10.2 beats/min, t = 20.8, P < 0.001), as well as CO (5.0 ± 1.8 vs. 4.3 ± 1.7 L/min, t = 5.2, P < 0.001), whereas central venous pressure was significantly increased (6.5 ± 2.1 vs. 7.5 ± 2.5 mmHg, t = −3.0, P = 0.007) after esmolol therapy compared with before esmolol therapy. In terms of tissue oxygen metabolism, mean lactate levels were normal before esmolol therapy, but were significantly decreased after esmolol therapy (1.4 ± 0.8 vs. 1.1 ± 0.6 mmol/L, t = 2.6, P = 0.015). The Pcv-a CO2 of patients was also significantly decreased after esmolol therapy compared with before esmolol therapy (5.6 ± 3.3 vs. 4.3 ± 2.2 mmHg, t = 2.6, P = 0.019). Other parameters reflecting tissue oxygen metabolism were not significantly changed after esmolol therapy. Cardiac enzymes of the patients were not significantly changed after esmolol use [Table 2].
Table 2.
Changes in vital signs, tissue oxygen metabolism, and myocardial enzymes after the usage of esmolol
| Variables | Before the usage of esmolol (n = 63) | After the usage of esmolol (n = 63) | t | P |
|---|---|---|---|---|
| Systolic blood pressure (mmHg) | 130.5 ± 16.7 | 126.4 ± 18.8 | 1.5 | 0.146 |
| Diastolic blood pressure (mmHg) | 72.8 ± 12.3 | 72.6 ± 12.4 | 0.1 | 0.926 |
| Mean arterial pressure (mmHg) | 90.0 ± 13.8 | 88.7 ± 13.9 | 0.6 | 0.548 |
| Pulse pressure (mmHg) | 57.7 ± 13.3 | 53.8 ± 16.8 | 1.7 | 0.109 |
| Central venous pressure (mmHg) | 6.5 ± 2.1 | 7.5 ± 2.5 | −3.0 | 0.007 |
| Heart rate (beats/min) | 107.8 ± 8.7 | 86.2 ± 10.2 | 20.8 | <0.001 |
| Left ventricular outflow tract cardiac output (L/min) | 5.0 ± 1.8 | 4.3 ± 1.7 | 5.2 | <0.001 |
| Lactate (mmol/L) | 1.4 ± 0.8 | 1.1 ± 0.6 | 2.6 | 0.015 |
| Pcv-a CO2 (mmHg) | 5.6 ± 3.3 | 4.3 ± 2.2 | 2.6 | 0.019 |
| ScvO2 (%) | 78.99 ± 7.95 | 78.06 ± 7.59 | 0.5 | 0.480 |
| Arterial pH | 7.42 ± 0.06 | 7.44 ± 0.06 | −1.3 | 0.192 |
| HCO3− (mmol/L) | 26.4 ± 4.7 | 26.6 ± 4.5 | −0.6 | 0.537 |
| Anion gap correction (mmol/L) | 10.8 ± 4.0 | 10.2 ± 4.4 | 1.0 | 0.362 |
| Urine output (ml/h) | 102.5 ± 88.6 | 106.8 ± 107.7 | −0.3 | 0.760 |
| Peripheral perfusion index | 2.4 ± 1.6 | 2.1 ± 1.4 | 1.8 | 0.079 |
| Creatine kinase (U/L) | 316.1 ± 522.7 | 321.9 ± 580.8 | −0.2 | 0.880 |
| Creatine kinase isoenzyme (ng/ml) | 1.8 ± 2.0 | 2.8 ± 6.1 | −0.9 | 0.403 |
| Troponin I (ng/ml) | 1.5 ± 7.0 | 1.5 ± 7.0 | −0.9 | 0.370 |
| NT-proBNP (pg/ml) | 8825.8 ± 6583.1 | 9313.3 ± 7049.8 | −0.6 | 0.553 |
Pcv-a CO2: Central venous-to-arterial carbon dioxide difference; NT-proBNP: N-terminal pro-brain natriuretic peptide.
Overall effects of esmolol therapy on cardiac function
The VTI of the left ventricular outflow tract was significantly increased after esmolol therapy compared with before esmolol therapy (15.2 ± 3.6 vs. 16.1 ± 4.0 cm, t = −2.6, P = 0.031). In addition, SV, which was calculated according to the VTI, was significantly increased after esmolol therapy compared with before esmolol therapy (43.6 ± 22.7 vs. 49.9 ± 23.7 ml, t = −2.3, P = 0.047) [Table 3]. HR of the patients was significantly decreased after esmolol therapy compared with before esmolol therapy (107.8 ± 8.7 vs. 86.2 ± 10.2 beats/min, t = 20.8, P < 0.001), and thus CO was also significantly decreased (5.0 ± 1.8 vs. 4.3 ± 1.7 L/min, t = 5.2, P < 0.001) after esmolol was used. Systolic function of the patients was decreased after esmolol therapy compared with before esmolol therapy, and this was especially represented by a decrease in ejection fraction (63.8 ± 13.9 vs. 59.6 ± 13.5%, t = 2.4, P = 0.023) and shortening fraction (34.7 ± 10.2 vs. 31.1 ± 8.1%, t = 2.6, P = 0.015), while other parameters that reflected systolic function (e.g., mitral annular displacement and tricuspid annular displacement) were not significantly changed.
Table 3.
Effects of esmolol on cardiac contractility and overall cardiac function
| Variables | Before the usage of esmolol (n = 63) | After the usage of esmolol (n = 63) | t | P |
|---|---|---|---|---|
| Parameter reflecting cardiac contractility and overall cardiac functions | ||||
| Left ventricular Tei index | 0.43 ± 0.27 | 0.56 ± 0.34 | −1.9 | 0.062 |
| Left ventricular outflow tract VTI (cm) | 15.2 ± 3.6 | 16.1 ± 4.0 | −2.6 | 0.031 |
| SV (ml) | 43.6 ± 22.7 | 49.9 ± 23.7 | −2.3 | 0.047 |
| Left ventricular outflow tract CO (L/min) | 5.0 ± 1.8 | 4.3 ± 1.7 | 5.2 | <0.001 |
| EF % | 63.8 ± 13.9 | 59.6 ± 13.5 | 2.4 | 0.023 |
| FS % | 34.7 ± 10.2 | 31.1 ± 8.1 | 2.6 | 0.015 |
| TAPSE (cm) | 2.1 ± 0.7 | 1.9 ± 0.6 | 1.7 | 0.106 |
| MAPSElat (cm) | 1.3 ± 0.3 | 1.3 ± 0.3 | 0.7 | 0.209 |
| MAPSEmed (cm) | 1.3 ± 0.3 | 1.2 ± 0.3 | 1.3 | 0.213 |
| Parameters reflecting cardiac preload | ||||
| Lateral E/Ea | 7.1 ± 2.6 | 6.9 ± 2.3 | 0.5 | 0.601 |
| Septal E/Ea | 8.9 ± 4.2 | 9.1 ± 3.3 | −0.4 | 0.712 |
| LVIDd (cm) | 4.7 ± 0.6 | 4.9 ± 0.7 | −2.8 | 0.009 |
| EDV (ml) | 103.3 ± 29.7 | 119.2 ± 34.1 | −3.0 | 0.005 |
SV: Stroke volume; CO: Cardiac output; EF: Ejection fraction; FS: Shortening fraction; TAPSE: Tricuspid annular plane systolic excursion; MAPSElat: Mitral lateral annular plane systolic excursion; MAPSEmed: Mitral septal annular plane systolic excursion; Ea: Early diastolic velocity by tissue Doppler; LVIDd: Left ventricular end-diastolic diameter; EDV: End-diastolic volume; VTI: Velocity time integral.
With regard to the parameters reflecting the volume of cardiac preload as measured by TTE, the left ventricular end-diastolic diameter (4.7 ± 0.6 vs. 4.9 ± 0.7 cm, t = −2.8, P = 0.009) and left ventricular end-diastolic volume (103.3 ± 29.7 vs. 119.2 ± 34.1 ml, t = −3.0, P = 0.005) were significantly increased after esmolol therapy compared with before esmolol therapy [Table 3].
In summary, SV and cardiac preload of the patients were significantly increased after using esmolol, whereas myocardial contractility was significantly decreased.
Identifying the predictors of an increase in stroke volume
Baseline data and general characteristics of the patients were not significantly different between the two subgroups [Table 4]. Before esmolol use, CO was similar between the subgroup with an increase in SV and the subgroup without an increase in SV (5.2 ± 1.9 vs. 5.3 ± 1.2 L/min, t = −0.1, P = 0.465). After esmolol use, CO in the subgroup with an increase in SV was significantly higher than that in the subgroup without an increase in SV (4.6 ± 1.7 vs. 3.4 ± 1.5 L/min, t = 2.8, P = 0.044). The findings of the present study suggested that patients with increased SV after using esmolol (SVafter − SVbefore > 0) had a lower risk of a decrease in CO, as well as the potential for further improving cardiac efficiency. Ultrasound data before esmolol administration could be used to compare patients with and without an increase in SV to help identify patients that could benefit from this treatment.
Table 4.
General characteristics and outcomes of the patients with and without an increase in stroke volume
| Variables | With SV increase (n = 42) | Without SV increase (n = 21) | t | P |
|---|---|---|---|---|
| Age (years) | 49.4 ± 17.0 | 52.4 ± 13.0 | −0.2 | 0.474 |
| Sex (male, %) | 47.6 | 47.6 | −0.1 | 0.924 |
| APACHE II | 16.1 ± 5.5 | 14.3 ± 4.2 | 0.3 | 0.542 |
| Mechanical ventilation (%) | 73.5 | 66.9 | 0.1 | 0.679 |
| Baseline cardiac function (%) | −0.1 | 0.982 | ||
| NYHA I | 54.8 | 52.4 | ||
| NYHA II | 35.7 | 38.1 | ||
| NYHA III | 9.5 | 9.5 | ||
| NE dose (µg·kg−1·min−1) | 0.45 ± 0.18 | 0.48 ± 0.19 | −0.1 | 0.487 |
| Maximum dose of esmolol (mg/h) | 110.6 ± 76.8 | 116.4 ± 89.9 | −0.2 | 0.237 |
| Total dose of esmolol (mg) | 197.6 ± 108.7 | 184.5 ± 113.9 | 0.2 | 0.458 |
| Time to achieve target heart rate (h) | 1.7 ± 0.3 | 1.7 ± 0.5 | 0.1 | 0.786 |
SV: Stroke volume; APACHE: Acute Physiology and Chronic Health Evaluation; NYHA: New York Heart Association; NE: Norepinephrine.
An ultrasound examination before esmolol use in patients with and without an increase in SV showed that MAPSElat in patients with increased SV was significantly higher than that in those without increased SV (1.3 ± 0.3 vs. 1.1 ± 0.2 ml, t = 2.4, P = 0.034). The area under the ROC curve of the MAPSElat was 0.911. The cutoff value of MAPSElat for predicting an increase in SV was 1.34, which suggested that the MAPSElat could predict an increase in SV [Figure 2].
Figure 2.
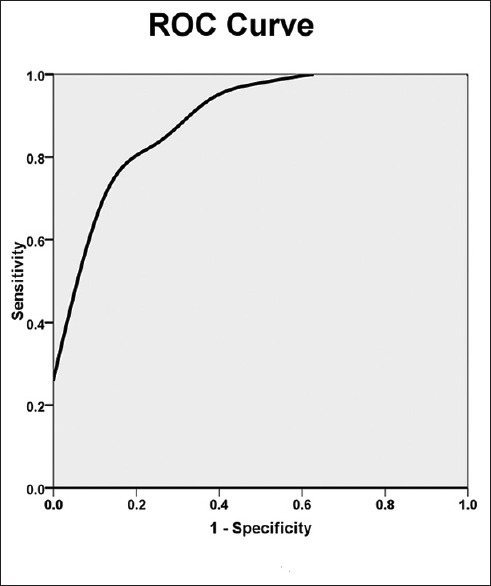
ROC curve comparing an increase in MAPSElat with an increase in SV before esmolol therapy in septic shock patients. The area under the curve was 0.911 ± 0.05 (P = 0.001). The cutoff value of MAPSElat before esmolol use for predicting an increase in SV in septic shock patients was 1.32, resulting in a sensitivity of 66.7% and a specificity of 99.9%. ROC: Receiver operating characteristic; MAPSElat: Mitral lateral annular plane systolic excursion; SV: Stroke volume.
Subgroup analysis in patients with an increase in stroke volume after esmolol use
Patients with an increased SV after using esmolol were selected for subgroup analysis. We found that parameters reflecting diastolic function were significantly changed [Table 5]. In brief, transmitral flow A-wave velocity was significantly decreased (85.7 ± 22.3 vs. 74.6 ± 20.6 cm/s, t = 3.0, P = 0.001), whereas the deceleration time (DT) of the E-wave was significantly increased after esmolol therapy compared with before esmolol therapy (105.6 ± 39.7 vs. 125.4 ± 46.2 ms, t = −2.3, P = 0.015). Tissue Doppler imaging showed that the lateral Aa was significantly decreased after esmolol therapy compared with before esmolol therapy (11.3 ± 4.2 vs. 10.1 ± 3.5 cm/s, t = 2.1, P = 0.047).
Table 5.
Subgroup analysis of diastolic function of the heart in patients with increased stroke volume
| Variables | Before the usage of esmolol (n = 42) | After the usage of esmolol (n = 42) | t | P |
|---|---|---|---|---|
| IVRT (ms) | 89.8 ± 38.4 | 79.9 ± 35.3 | 1.0 | 0.162 |
| E (cm/s) | 78.0 ± 31.7 | 74.1 ± 26.8 | 0.9 | 0.178 |
| A (cm/s) | 85.7 ± 22.3 | 74.6 ± 20.6 | 3.0 | 0.001 |
| E/A | 1.0 ± 0.7 | 1.1 ± 0.7 | −1.3 | 0.097 |
| DT (ms) | 105.6 ± 39.7 | 125.4 ± 46.2 | −2.3 | 0.015 |
| A duration (ms) | 143.3 ± 35.1 | 140.4 ± 44.3 | 0.1 | 0.769 |
| Lateral Ea (cm/s) | 11.7 ± 4.4 | 11.3 ± 3.9 | 0.1 | 0.368 |
| Septal Ea (cm/s) | 9.7 ± 3.8 | 8.8 ± 3.3 | 0.2 | 0.161 |
| Lateral Aa (cm/s) | 11.3 ± 4.2 | 10.1 ± 3.5 | 2.1 | 0.047 |
| Septal Aa (cm/s) | 11.8 ± 5.3 | 10.3 ± 4.1 | 1.8 | 0.056 |
| Lateral Ea/Aa | 1.2 ± 0.6 | 1.3 ± 0.8 | −1.1 | 0.160 |
| Septal Ea/Aa | 1.0 ± 0.9 | 1.0 ± 0.9 | 0.1 | 0.945 |
IVRT: Isovolumetric relaxation time; E: Transmitral early diastolic velocity; A: Transmitral late diastolic velocity; DT: Deceleration time; Ea: Early diastolic velocity by tissue Doppler; Aa: Late diastolic velocity by tissue Doppler.
DISCUSSION
The present study showed that for most septic shock patients who received esmolol at an early stage after stabilization of hemodynamics, controlling HR with esmolol could effectively increase SV. This result suggested that an increase in SV was achieved by the improvement of cardiac efficiency after esmolol therapy. However, although esmolol treatment decreased CO, no decrease in tissue perfusion was found, while lactate levels and Pcv-a CO2 were further decreased after treatment. We speculate that activity of the sympathetic nervous system could be increased in patients with septic shock. Over-increased myocardial contractility could increase CO, and thus increase oxygen delivery, which could result in excess oxygen compared with the oxygen demand of septic shock patients. In the present study, esmolol, a β-receptor blockade, was used to block overstimulation from endogenous catecholamines, which can control HR to avoid overcontraction of the myocardium. Although CO was decreased, the oxygen that was delivered still met the patients’ oxygen demands. Therefore, there was no decrease in tissue perfusion in the present study. Previous studies have shown that overdelivery of oxygen is not beneficial,[17] while tachycardia caused by overactivation of sympathetic nerves can increase oxygen consumption of the myocardium and thus aggravate myocardial ischemia. Therefore, applying a β-receptor blockade is beneficial for patients with satisfactory tissue perfusion. Our findings are in accordance with the previous clinical findings showing that β-receptor blockade can improve the mortality rate of patients with septic shock.[5]
β-receptor blockade has negative inotropic effects. However, most of the patients in the present study had increased SV after drug therapy. Therefore, changes in cardiac preload and myocardial contractility were further investigated. TTE results showed that cardiac preload (left ventricular end-diastolic diameter and EDV) and central venous pressure were significantly increased, which are in accordance with the Frank–Starling discipline. Therefore, we speculate that the increase in SV of the patients was caused by the HR-decreasing effects of esmolol, which improved left ventricular diastolic filling. This, in turn, increased cardiac preload and thus increased SV. We also found a decrease in myocardial contractility (including ejection fraction and shortening fraction) after administration of esmolol. After extensive analysis, we speculate that although the chronotropic effects of β-receptor blockade decreased the myocardial contractility, the overall effects still increased SV of the patients. Therefore, the parameters reflecting cardiac contraction and relaxation in patients with and without an increase in SV before using esmolol were further compared with identify factors that could predict this increase. We found that MAPSElat in patients with increased SV was significantly higher than that in those without increased SV. This finding suggested that patients with better cardiac contractility before treatment could be better benefited from β-receptor blockade, and patients in the early stage of septic shock who still had good cardiac contractility could also be better benefited from β-receptor blockade. We speculate that MAPSElat could be a predictor of an increase in SV after esmolol use. Further, analysis using the ROC curve was performed. The AUC was 0.911, and the cutoff value of MAPSElat in predicting the increase in SV was 1.34. These findings could help identify patients who could have a lower risk of esmolol use.
The present study also further investigated the possible reasons for the increase in cardiac preload after esmolol use by comparing the parameters reflecting cardiac diastolic function before and after treatment in patients with an increase in SV. We found that DT was increased, whereas transmitral flow A-wave velocity and lateral Aa were significantly decreased after esmolol therapy compared with before esmolol therapy. DT is a parameter that reflects myocardial compliance. Previous studies have demonstrated that when myocardial compliance is decreased, the time to achieve balance between the left ventricular and arterial pressure in the diastolic phase is the shortest, and DT is also decreased accordingly (DT <150 ms). In the present study, DT was increased after esmolol was administered, which suggested that myocardial compliance of the left ventricle was improved.[18] When analyzing changes in myocardial compliance from the aspect of the pressure–volume relationship at the left ventricular end-diastolic pressure, E/Ea is generally considered to be related to the left ventricular filling pressure. An increase in the left ventricular filling pressure decreases Ea velocity and increases E-wave velocity, thus increasing the E/Ea ratio.[19] However, no significant change in the E/Ea ratio was found in the present study after administering esmolol compared with before treatment. In light of the significant increase in EDV after esmolol therapy, we speculate that compliance of the left ventricle was improved after using esmolol. For patients with increased SV after using esmolol, transmitral flow A-wave velocity and lateral Aa were decreased, which suggested that atrial contractility was decreased. Several studies have suggested that there are linear associations between Aa and left atrial ejection fraction, left atrial systolic function, and several other parameters.[20] Contraction of the left atria is mainly associated with a compensatory increase in ventricular filling. Therefore, a decrease in the left arterial contraction could be directly associated with an improvement of the left ventricular compliance. These findings suggested that the increase in cardiac preload could have been caused by the improvement of ventricular compliance after esmolol therapy.
The present study has several limitations. This study was a single-center study with a relatively small sample size. In addition, no control group was included. Furthermore, well-designed, randomized, controlled trials with larger sample sizes are needed to validate our findings.
The most common reason for ICU admission is emergency surgery. Our department is a general ICU. Therefore, the predominant source of sepsis was the abdominal cavity. This is inconsistent with other sepsis studies.
In conclusion, the present study shows that after using esmolol in septic shock patients, the VTI of the left ventricular outflow tract and SV are increased. Although CO is also decreased with HR, tissue perfusion is not worse. This increase in SV may be caused by the improvement of ventricular compliance, which also decreases HR and increases cardiac preload. MAPSElat could be used as a parameter to predict an increase in SV before the usage of esmolol.
Financial support and sponsorship
This study was supported by grants from the National Natural Science Foundations of China (No. 81501639) and the Central Health Care Committee (No. W2013BJ19).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Li-Shao Guo
REFERENCES
- 1.Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–608. doi: 10.1097/01.CCM.0000266683.64081.02. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 2.Surbatovic M, Veljovic M, Jevdjic J, Popovic N, Djordjevic D, Radakovic S. Immunoinflammatory response in critically ill patients: Severe sepsis and/or trauma. Mediators Inflamm 2013. 2013 doi: 10.1155/2013/362793. 362793. doi: 10.1155/2013/362793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: A prospective observational study. Intensive Care Med. 2012;38:950–8. doi: 10.1007/s00134-012-2531-2. doi: 10.1007/s00134-012-2531-2. [DOI] [PubMed] [Google Scholar]
- 4.Rudiger A. Beta-block the septic heart. Crit Care Med. 2010;38(10 Suppl):S608–12. doi: 10.1097/CCM.0b013e3181f204ca. doi: 10.1097/CCM.0b013e3181f204ca. [DOI] [PubMed] [Google Scholar]
- 5.Morelli A, Ertmer C, Westphal M, Rehberg S, Kampmeier T, Ligges S, et al. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: A randomized clinical trial. JAMA. 2013;310:1683–91. doi: 10.1001/jama.2013.278477. doi: 10.1001/jama.2013.278477. [DOI] [PubMed] [Google Scholar]
- 6.Friese RS, Barber R, McBride D, Bender J, Gentilello LM. Could beta blockade improve outcome after injury by modulating inflammatory profiles? J Trauma. 2008;64:1061–8. doi: 10.1097/TA.0b013e3181684cf0. doi: 10.1097/TA.0b013e3181684cf0. [DOI] [PubMed] [Google Scholar]
- 7.Salim A, Hadjizacharia P, Brown C, Inaba K, Teixeira PG, Chan L, et al. Significance of troponin elevation after severe traumatic brain injury. J Trauma. 2008;64:46–52. doi: 10.1097/TA.0b013e31815eb15a. doi: 10.1097/TA.0b013e31815eb15a. [DOI] [PubMed] [Google Scholar]
- 8.Schroeppel TJ, Fischer PE, Zarzaur BL, Magnotti LJ, Clement LP, Fabian TC, et al. Beta-adrenergic blockade and traumatic brain injury: Protective? J Trauma. 2010;69:776–82. doi: 10.1097/TA.0b013e3181e981b8. doi: 10.1097/TA.0b013e3181e981b8. [DOI] [PubMed] [Google Scholar]
- 9.Arbabi S, Ahrns KS, Wahl WL, Hemmila MR, Wang SC, Brandt MM, et al. Beta-blocker use is associated with improved outcomes in adult burn patients. J Trauma. 2004;56:265–9. doi: 10.1097/01.TA.0000109859.91202.C8. doi: 10.1097/01.TA.0000109859.91202.C8. [DOI] [PubMed] [Google Scholar]
- 10.Rudiger A, Singer M. The heart in sepsis: From basic mechanisms to clinical management. Curr Vasc Pharmacol. 2013;11:187–95. doi: 10.2174/1570161111311020008. [PubMed] [Google Scholar]
- 11.Sanfilippo F, Santonocito C, Morelli A, Foex P. Beta-blocker use in severe sepsis and septic shock: A systematic review. Curr Med Res Opin. 2015;31:1817–25. doi: 10.1185/03007995.2015.1062357. doi: 10.1185/03007995.2015.1062357. [DOI] [PubMed] [Google Scholar]
- 12.Ince C. To beta block or not to beta block; that is the question. Crit Care. 2015;19:339. doi: 10.1186/s13054-015-1059-6. doi: 10.1186/s13054-015-1059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayo PH, Beaulieu Y, Doelken P, Feller-Kopman D, Harrod C, Kaplan A, et al. American College of Chest Physicians/La Sociétéde Réanimation de Langue Française statement on competence in critical care ultrasonography. Chest. 2009;135:1050–60. doi: 10.1378/chest.08-2305. doi: 10.1378/chest.08-2305. [DOI] [PubMed] [Google Scholar]
- 14.Expert Round Table on Ultrasound in ICU. International expert statement on training standards for critical care ultrasonography. Intensive Care Med. 2011;37:1077–83. doi: 10.1007/s00134-011-2246-9. doi: 10.1007/s00134-011-2246-9. [DOI] [PubMed] [Google Scholar]
- 15.He H, Long Y, Liu D, Wang X, Zhou X. Clinical classification of tissue perfusion based on the central venous oxygen saturation and the peripheral perfusion index. Crit Care. 2015;19:330. doi: 10.1186/s13054-015-1057-8. doi: 10.1186/s13054-015-1057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vallée F, Vallet B, Mathe O, Parraguette J, Mari A, Silva S, et al. Central venous-to-arterial carbon dioxide difference: An additional target for goal-directed therapy in septic shock? Intensive Care Med. 2008;34:2218–25. doi: 10.1007/s00134-008-1199-0. doi: 10.1007/s00134-008-1199-0. [DOI] [PubMed] [Google Scholar]
- 17.Hayes MA, Timmins AC, Yau EH, Palazzo M, Hinds CJ, Watson D. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330:1717–22. doi: 10.1056/NEJM199406163302404. doi: 10.1056/NEJM199406163302404. [DOI] [PubMed] [Google Scholar]
- 18.Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr. 2009;22:107–33. doi: 10.1016/j.echo.2008.11.023. doi: 10.1016/j.echo.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 19.Nagueh SF, Middleton KJ, Kopelen HA, Zoghbi WA, Quiñones MA. Doppler tissue imaging: A noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. 1997;30:1527–33. doi: 10.1016/s0735-1097(97)00344-6. doi: 10.2174/1570161111311020008. [DOI] [PubMed] [Google Scholar]
- 20.Khankirawatana B, Khankirawatana S, Peterson B, Mahrous H, Porter TR. Peak atrial systolic mitral annular velocity by Doppler tissue reliably predicts left atrial systolic function. J Am Soc Echocardiogr. 2004;17:353–60. doi: 10.1016/j.echo.2003.12.023. doi: 10.1016/j.echo.2003.12.023. [DOI] [PubMed] [Google Scholar]